

Body size determination is tightly linked with developmental maturation, and ecdysone, an insect maturation hormone, contributes to this process by antagonizing insulin signaling and suppressing juvenile growth. However, how ecdysone regulates insulin signaling to attain appropriate body size remains unclear. In this study, Hyun and colleagues found that miR-8 is transcriptionally regulated by ecdysone's early response genes and that this regulation of miR-8 is essential for ecdysone-induced control of insulin signaling and fly growth. In addition, the authors demonstrate that miR-8 targets USH during body size determination. Therefore, this study delineates a novel function for miR-8 as a molecular rheostat that adjusts organismal growth rate in response to a developmental signal.
Keywords: body size, ecdysone, insulin signaling, maturation, mir-8, ush
Abstract
Body size determination is a process that is tightly linked with developmental maturation. Ecdysone, an insect maturation hormone, contributes to this process by antagonizing insulin signaling and thereby suppressing juvenile growth. Here, we report that the microRNA miR-8 and its target, u-shaped (USH), a conserved microRNA/target axis that regulates insulin signaling, are critical for ecdysone-induced body size determination in Drosophila. We found that the miR-8 level is reduced in response to ecdysone, while the USH level is up-regulated reciprocally, and that miR-8 is transcriptionally repressed by ecdysone's early response genes. Furthermore, modulating the miR-8 level correlatively changes the fly body size; either overexpression or deletion of miR-8 abrogates ecdysone-induced growth control. Consistently, perturbation of USH impedes ecdysone's effect on body growth. Thus, miR-8 acts as a molecular rheostat that tunes organismal growth in response to a developmental maturation signal.
One of the most interesting yet still largely unresolved biological questions is how an animal achieves its final body size. Animal growth is not a continuous process. Exponential growth occurs in the juvenile period, and when sexual maturation is completed, growth stops, and the body size is virtually fixed (Edgar 2006; Mirth and Riddiford 2007). Insulin signaling is the key effector pathway in organismal growth, sensing and transducing organismal energy conditions to promote growth (Ikeya et al. 2002; Rulifson et al. 2002; Demontis and Perrimon 2009). Development from larva to puparium in Drosophila is thought to have intriguing parallels to mammalian development from adolescence to adulthood (McBrayer et al. 2007; Rewitz et al. 2010; Tennessen and Thummel 2011). Several studies in Drosophila have revealed that spatiotemporal regulation of insulin signaling during the larval stage is important for proper development and timing of pupariation, suggesting that dynamic and elaborate regulation of larval insulin signaling is important for the coordination of juvenile growth and maturation (Brogiolo et al. 2001; Shingleton et al. 2005; Walkiewicz and Stern 2009). Interestingly, ecdysone, the key steroid hormone directing metamorphosis and maturation in insects, impedes insulin signaling and organismal growth in Drosophila larvae and thus may contribute to the coordination between growth and maturation (Caldwell et al. 2005; Colombani et al. 2005; Mirth et al. 2005; Delanoue et al. 2010). However, the mechanism by which ecdysone controls insulin signaling remains unknown.
MicroRNAs (miRNAs) are ∼22-nucleotide (nt) noncoding RNAs that act as post-transcriptional repressors by base-pairing to the 3′ untranslated region (UTR) of their target mRNAs (Brennecke et al. 2005; Bartel 2009). In contrast to transcription factors, many miRNAs have been proposed to function as fine-tuners, rather than on–off switches for the target gene expression. Nonetheless, as miRNAs modulate a large number of target genes, they can play integral roles in organismal physiology, such as maintaining homeostasis or ensuring robust development, by counteracting environmental fluctuations/stresses (Karres et al. 2007; Flynt and Lai 2008; Li et al. 2009; Hilgers et al. 2010; Staton et al. 2011).
We previously showed that a conserved miRNA, miR-8, promotes insulin signaling and body growth by targeting a conserved gene, u-shaped (ush), in flies (Hyun et al. 2009). We further found that its human orthologs (the miR-200 family of miRNAs) play equivalent roles by targeting Friend of Gata 2 (FOG2), a human homolog of ush (Hyun et al. 2009). In the present study, we report that this conserved miRNA/target axis is controlled by ecdysone in Drosophila, thereby providing a link between steroid signaling and insulin signaling.
Results and Discussion
While investigating upstream factors that regulate the expression of miR-8, we came across several reports showing that the miR-8 level is reduced in the prepupa/pupa stage of Drosophila development (Aravin et al. 2003; Sempere et al. 2003). Because ecdysone signaling is activated on pupal development, these data raise the possibility that miR-8 expression may be regulated by ecdysone. We also noticed from our own experiments that the miR-8 level correlates inversely with those of ecdysone response genes during larval development (Fig. 1A). Close examination of the temporal expression pattern revealed that miR-8 is elevated in the late embryo/first larval instar (10∼24 h after egg laying [AEL]), followed by a gradual decrease (Fig. 1A,B), which is similar to a previous observation (Aravin et al. 2003). Notably, the levels of E74 and Broad Complex (BR-C), the early response genes of ecdysone signaling (Thummel 1996), start to increase after the late embryo/first larval instar (Fig. 1A), and as the expressions of these genes gradually increase during the third larval instar, the level of pri-miR-8 decreases correlatively (Supplemental Fig. 1). Consistent with the changes in miR-8, the miR-8 target mRNA ush decreased in the late embryo/first larval instar and gradually increased until the third larval instar (Fig. 1C). These data raised a possibility that miR-8 and USH may be temporally and reciprocally modulated by ecdysone signaling during larval development, where most of the organismal growth occurs (Edgar 2006).
Figure 1.

Regulation of miR-8 and USH by ecdysone signaling in vivo and in vitro. (A, top) Temporal expression pattern of miR-8 during larval development. (Bottom) Temporal expression patterns of E74 and BR-C during larval development. Harvesting times (AEL) are denoted above the blot. (B) Temporal expressions of miR-8 from two independent experiments are individually plotted as blue and red lines. (C) Temporal expression of ush transcripts from two independent experiments are individually plotted as blue and red lines. (D) Feeding with 20-hydroxyecdysone (20E) decreases the level of miR-8 in the whole larvae. Newly hatched larvae were fed 20E-containing (0.25 mg/mL) glucose–yeast paste until 3 d AEL, after which RNA was prepared. (E) Inhibition of EcR activity by expressing EcRDN in larval fat bodies increases miR-8 levels sustainably during the third instar larval period. (F) EcRDN expression in larval fat bodies sustainably decreases USH transcripts during the third instar larval period. Two biological replicates (six to nine animals from each genotype per replicate) were measured. (G) Fat cell clones overexpressing EcRDN by FLP-out GAL4 (marked by the absence of CD2; white arrows) show decreased levels of USH, as analyzed by immunostaining with USH antibody. (H) Treatment with 10 μM 20E in S2 cells progressively decreases both mature and pri-miR-8 levels in a similar pattern. pri-miR-8 levels were normalized against mitochondrial large ribosomal RNA1 (mtl rRNA1). This experiment was repeated with similar results. (I) Knockdown of EcR, E74, or BR-C impedes the repressive effect of 20E on pri-miR-8 expression in S2 cells (n = 3). (J) Promoter activity of pri-miR-8 is significantly down-regulated by 20E treatment. The inset shows a magnified graph of the pGL3-null reporter. Fivefold more pGL3-null plasmids were transfected than the other reporter plasmids (n = 3). (***) P < 0.001; (**) P < 0.02; (*) P < 0.05 compared with respective controls. (n.s.) Not significant (P > 0.3). Error bars denote SEM.
To test the possibility that ecdysone controls miR-8 in larvae, we fed larvae with 20-hydroxyecdysone (20E, an active form of ecdysone). Indeed, 20E treatment significantly reduced the level of miR-8 in the whole larva, supporting the hypothesis that 20E represses miR-8 expression in vivo (Fig. 1D). Previous reports indicate that the larval fat body, the Drosophila counterpart of mammalian liver and adipose tissue, is the main target organ of ecdysone in its effect on organismal growth (Colombani et al. 2005; Delanoue et al. 2010). Moreover, miR-8 in this organ is important for regulating organismal growth (Hyun et al. 2009). Thus, we examined the effect of ecdysone on miR-8 in the larval fat body. To inhibit ecdysone signaling in the larval fat body, we expressed the dominant-negative form of the ecdysone receptor (EcRDN; EcRF645A) (Colombani et al. 2005) by using a fat body-specific driver, Cg GAL4 (CgG4). Inhibition of ecdysone signaling by EcRDN increased the miR-8 level (Fig. 1E). Notably, although the observed increase in miR-8 at each time point is rather modest (∼30% increase), miR-8 remained constantly up-regulated in the fat body throughout the third instar larval period (84∼125 h AEL) (Fig. 1E; discussed below). Furthermore, expression of EcRDN in the fat body significantly reduced the level of ush mRNA throughout the third instar larval period (Fig. 1F). Mosaic clonal analysis of fat cells further confirmed the decrease of the USH protein in the presence of EcRDN (Fig. 1G). Together, these results indicate that EcR signaling regulates miR-8 and its target, USH, in vivo.
Drosophila S2 cells endogenously express miR-8 (Sempere et al. 2003). Like in larvae fed with 20E, we found that 20E treatment decreased the level of miR-8 in S2 cells (Fig. 1H; Supplemental Fig. 2). The level of primary transcript (pri-miR-8) also decreased in response to 20E (Fig. 1H), suggesting that the hormone may control miR-8 at the transcriptional level. Consistently, the analysis of the promoter region of pri-miR-8 by using the P-Match and Match programs (Chekmenev et al. 2005) predicted numerous binding sites for transcription factors such as E74 and BR-C (Supplemental Fig. 3). E74 and BR-C are transcription factors that are rapidly induced by ecdysone and mediate the third instar larval and pupal development (Thummel 1996). To test whether these components of the ecdysone signaling pathway are involved in miR-8 regulation, we knocked down each of these genes in S2 cells. Knockdown of EcR by RNAi largely abrogated the suppressive effect of 20E on pri-miR-8 production (Fig. 1I). Knockdown of E74 or BR-C similarly interfered with the suppressive effect of 20E, although to a lesser extent than EcR RNAi (Fig. 1I). To directly monitor the promoter activity of pri-miR-8, we generated two reporters harboring different genomic DNA fragments upstream of the mir-8 locus. One reporter contains the region between the end of an upstream gene (CG34460) and the point where transcribed fragments of pri-miR-8 begin to appear (according to Affy Signal), as described in the University of California at Santa Cruz Genome Browser (http://genome.ucsc.edu) (pGL3 PR1-miR-8); another reporter contains a more extended region between the end of the upstream gene and the beginning of the mature miR-8 sequences (pGL3 PR2-miR-8) (Supplemental Fig. 4). The luciferase expressions from these reporters were significantly and specifically down-regulated by 20E treatment, indicating that 20E indeed controls the transcription of pri-miR-8 (Fig. 1J). Note that the expression of the pGL3 PR2-miR-8 reporter was not only higher, but also more sensitive to 20E treatment than that of pGL3 PR1-miR-8, indicating that the elements required for 20E-mediated miR-8 suppression are broadly distributed over the intergenic region between miR-8 and its upstream gene. Because >20 sites were predicted to bind to E74 and BR-C, it was difficult to validate each site by site-directed mutagenesis. However, mutations of the first two predicted binding sites for E74 in the pGL3 PR1-miR-8 reporter partially, but significantly, alleviated the repressive effects of 20E, indicating that the predicted sites may at least partially mediate EcR signaling (Supplemental Fig. 5).
To test the possibility that ecdysone controls body growth through modulation of miR-8, we first confirmed whether modulation of the miR-8 level could modify final body size. We combined the miR-8 enhancer trap GAL4 (mir-8G4) with the miR-8-null allele (mir-8Δ2), wild-type allele (+), or UAS-miR-8 (UAS-mir-8) to generate flies expressing low, intermediate, or high levels of miR-8 (Fig. 2A). The pupal size increased as a function of miR-8; the higher the miR-8 level, the bigger the pupa, demonstrating that the miR-8 concentration indeed affects the final body size.
Figure 2.

Overexpression of miR-8 makes flies bigger and insensitive to ecdysone effects of growth suppression. (A) Gradually increasing miR-8 progressively increases pupal size. (Top left) Picture of pupae whose genotypes are indicated. (Bottom left) Amounts of miR-8 in larval fat bodies in each of the indicated flies. (Right) Quantification of the pupal volumes in each of the genotypes. Approximately 20 pupae per genotype were measured. (B) Feeding larva with 20E (0.3 mg/mL) decreases final pupal size, while overexpression of miR-8 largely abolishes the suppressive effects of 20E feeding. The flies indicated as either CyOgfp/UAS-mir-8 or mir-8G4/UAS-mir-8 are siblings generated from the same parents and reared in the same vial. Relative repression folds by 20E feedings are compared between the genotypes indicated. Approximately 30 pupae per genotype were measured. (#) P < 1.6 × 10−4; (**) P < 0.01 compared with respective controls. Error bars denote SEM.
Feeding 20E to wild-type larvae decreased the endogenous level of miR-8 and also reduced fly body size as previously reported (Figs. 1D, 2B, 4A [see below]; Delanoue et al. 2010). We examined whether overexpression of miR-8 could retard the growth-suppressive effects of 20E feeding. Overexpression of miR-8 in larval tissues expressing endogenous miR-8 led to an increase in final pupal size (Fig. 2B). Importantly, feeding 20E to larvae overexpressing miR-8 did not substantially reduce final pupal size (Fig. 2B), indicating that excessive amounts of miR-8 make flies both bigger and resistant to ecdysone-mediated control of larval growth. We then investigated the effects of miR-8 deficiency on ecdysone-mediated growth control. We attenuated ecdysone signaling by RNAi against EcR (Fig. 3A). Reduction of the EcR activity in larvae is known to increase the final pupal size by accelerating larval growth (Colombani et al. 2005; Delanoue et al. 2010). Consistent with previous reports, ubiquitous knockdown of EcR increased pupal volume by ∼17% in the wild-type background (Fig. 3A, black bars). Importantly, the same genetic manipulation did not increase pupal volume in miR-8-null flies (Fig. 3A, gray bars), indicating that miR-8 is required for regulation. Then, we blocked the EcR activity specifically in the larval fat body (Fig. 3B–F), the major target organ of ecdysone in the control of body growth. We introduced EcRDN in the fat body by using Cg GAL4. As expected, the pupal volume of wild-type animals increased when EcRDN was expressed (Fig. 3B). Importantly, however, EcRDN failed to increase the size of miR-8-null animals (Fig. 3B). We found that, like pupal volume, the weight of wild-type adult flies was also elevated by fat body-specific expression of EcRDN, while the miR-8 mutant flies did not respond to the EcR inhibition (Fig. 3C). It is noted that although the expression of EcRDN by Cg GAL4 caused some pharate lethality in the pupal case (Delanoue et al. 2010), a significant number of adult flies could still be obtained in our experimental condition, allowing the measurement of adult size. Moreover, we found that re-expression of miR-8 in the fat body rescued the small body phenotype of the miR-8 mutant in which the fat body EcR was inhibited, implying that the increase of body size by EcR inhibition may be mediated by miR-8 in the fat body (Fig. 3D). It was previously shown that the timing of puparium formation is not significantly affected by knockdown of EcR in the whole body or in the fat body (Colombani et al. 2005; Delanoue et al. 2010). Consistently, we observed that both EcR RNAi in the whole body or EcRDN expression in the fat body affected pupariation time by only ∼4 h (Supplemental Fig. 6). Considering that the total developmental time from embryo to puparium is ∼120 h, it is likely that the increase in final animal size is mainly due to an increase of growth rate, rather than an extension of the growth period.
Figure 4.

Perturbation of USH expression impedes ecdysone-mediated growth regulation. (A) Heterozygous mutants of USH (ushvx22/+) show significant resistance to the growth-suppressive effect of 20E feeding (0.3 mg/mL). The flies indicated as CyOgfp/+ or ushvx22/+ are siblings generated from the same parents and reared in the same vial. Relative repression folds by 20E feedings are compared between the genotypes indicated. Approximately 44 pupae per genotype were measured. (B) Knockdown of EcR in USH-expressing tissues noticeably increases the adult weight, and this effect is impeded by overexpression of USH. More than 21 flies per genotype were measured. (**) P < 0.01; (*) P < 0.04 compared with respective controls. Error bars denote SEM.
Figure 3.

Deletion of miR-8 abrogates the EcR-mediated regulation of fly growth and insulin signaling. (A) Ubiquitous knockdown of EcR by expressing dsRNA targeting EcR using armadillo GAL4 (armG4) increases final pupal size. This effect is abrogated when miR-8 is removed. Knockdown experiment in the wild type or miR-8 mutant was independently performed, so the size was separately normalized in each genetic background. Approximately 25 pupae per genotype were measured. (B) Expression of EcRDN in fat bodies with Cg GAL4 (CgG4) increases final pupal size, the effect of which is abrogated when miR-8 is removed. Approximately 20 pupae per genotype were measured. (C) Expression of EcRDN in fat bodies increases final adult mass, and this genetic manipulation does not increase the final adult mass in the absence of miR-8. More than 33 flies per genotype were measured. (D) Re-expression of miR-8 in the fat body rescues the small body phenotype of the miR-8 mutant in which EcR is inhibited. Approximately 24 pupae per genotype were measured. (E) Expression of EcRDN in the fat bodies of the mid-third instar larva (∼98 h AEL) increases the level of phospho-Akt, while this effect on Akt is abolished when miR-8 is removed. (F) Expression of EcRDN in the fat bodies of the mid-third instar larva (∼98 h AEL) decreases the transcript levels of Inr and step, the target genes of dFOXO, while these effects are abolished when miR-8 is removed. Four biological replicates (six to nine animals from each genotype per replicate) were measured. (#) P < 2 × 10−3 compared with respective controls. Error bars denote SEM.
Because we and others have previously shown that the insulin/PI3 kinase pathway in the fat body plays an important part in body size determination (Britton et al. 2002; DiAngelo et al. 2009; Hyun et al. 2009), we next questioned how insulin signaling is affected by ecdysone signaling in the absence of miR-8. In wild-type larval fat bodies, overexpression of EcRDN derepressed insulin signaling, as indicated by the up-regulation of phosphorylated Akt, a key factor that acts downstream from PI3 kinase (Fig. 3E). In contrast, EcRDN expression failed to increase phospho-Akt in the fat bodies of miR-8-null animals, indicating that miR-8 is critical for the communication between ecdysone signaling and insulin signaling (Fig. 3E). We also examined the activity of dFOXO, the downstream transcription factor repressed by insulin signaling, by monitoring its target genes, Insulin receptor (Inr) and steppke (step) (Puig and Tjian 2005; Fuss et al. 2006). As expected, in wild-type fat bodies, the Inr and step mRNA levels were down-regulated following EcRDN expression, while remaining unaltered in the fat bodies of miR-8-null larva (Fig. 3F). This result shows that the inhibition of EcR activity can activate insulin signaling only in the presence of miR-8. The Inr and step levels were higher in miR-8-null larvae than those in wild-type animals (Fig. 3F) due to positive regulation of insulin signaling by miR-8 (Hyun et al. 2009). Leopold and colleagues (Delanoue et al. 2010) recently showed that knockdown of EcR increased the dMyc transcript level in larval fat bodies, thereby promoting organismal growth. Unexpectedly, under our experimental condition, EcRDN affected dMyc expression only marginally in larval fat bodies (Supplemental Fig. 7). This discrepancy may be relevant to the previous finding that two inactivation methods (EcRDN expression and EcR RNAi) may exert partially distinct downstream effects in certain conditions (Mirth et al. 2009). Thus, EcRDN expression and EcR RNAi may promote larval growth by related but slightly distinct mechanisms, both of which require miR-8 (Fig. 3A,B).
Since USH functions downstream from miR-8 (Hyun et al. 2009), and the level of USH is increased by EcR (Fig. 1F,G), we examined whether modulation of USH expression could divert the ecdysone-mediated growth control. Because the loss of ush is lethal, we instead attenuated USH by using heterozygotes for the USH-null allele (ushvx22) (Cubadda et al. 1997). Indeed, heterozygosity for the USH-null allele conferred a significant resistance to the growth-suppressive effects of 20E feeding (Fig. 4A). These data suggest that the regulation of USH by ecdysone signaling plays a role in ecdysone-mediated growth control. Finally, we examined whether overexpression of USH could suppress the growth-promoting effect of EcR inhibition. While attenuation of EcR in the tissues endogenously expressing USH increased the final adult size, overexpression of USH in the same tissue mitigated the increase of adult weight (Fig. 4B). These body size responses measured in adult flies were also observed in pupae (Supplemental Fig. 8). Note that the effects of USH modulation are milder than those of miR-8 modulation (cf. Figs. 2 and 4). This suggests that additional targets of miR-8 may partially contribute to ecdysone-mediated growth control.
In conclusion, our study reveals the mechanism by which ecdysone suppresses insulin signaling and thereby decelerates larval growth. During larval development, ecdysone regulates the levels of miR-8 and its target, USH, a PI3 kinase inhibitor, through the EcR downstream pathway, and quantitative regulation of miR-8 by ecdysone leads to determining the final fly size. Since the ecdysone level is low during the larval stage not engaged in the molting process, a mild response of miR-8 increase by EcR inhibition in this stage would be expected (Fig. 1E). Notably, however, we found that this increase in miR-8 level by EcRDN is constantly sustained throughout the third instar larval stage, which is the period of exponential growth. Interestingly, among early response genes of EcR signaling, E74 and BR-C are also persistently expressed during this period (Fig. 1A; Thummel 1996); these gene products repress miR-8 expression at the transcriptional level (Fig. 1I,J; Supplemental Fig. 5). Thus, throughout the third instar larval period, EcR downstream signaling keeps miR-8 (and, concomitantly, insulin signaling) under control (Fig. 5). Because the duration of this regulation lasts several days, the effect of miR-8 modulation accumulates and manifests a significant impact on final body size (Fig. 5). This cumulative effect accounts for the effect of ecdysone signaling on body size despite only modest changes in the miR-8 level. When EcR signaling was hampered throughout the larval stage, leading to a sustained increase of miR-8, the final body size became noticeably bigger (Figs. 1E, 3B,C).
Figure 5.
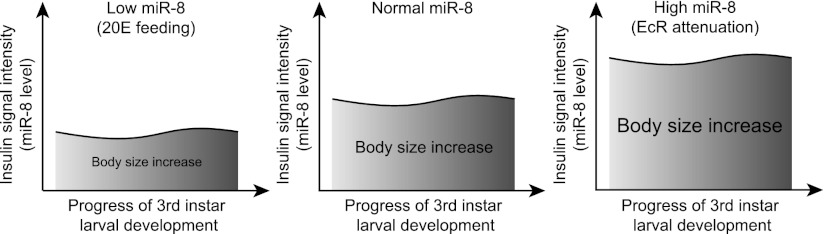
Function of miR-8 in the process of ecdysone-induced fly size determination. Ecdysone signaling continuously influences the miR-8 and, concomitantly, insulin signaling throughout the third instar larval period. When altered levels of miR-8 induced by changes in ecdysone signaling are sustained throughout this period, the effects of miR-8 alteration accumulate, leading to a significant difference in the final body size. In model figures, the increase of body size during the third instar larval period is expressed as the area under the curve (AUC).
This function of miR-8 in shaping body size provides a novel example in which an animal uses the inherent ability of miRNAs in fine-tuning target gene expression. Previously, several studies have shown that such a strategy has been used with miRNAs in diverse biological contexts. Specific examples include maintaining the optimal level of miRNA target proteins, which is critical for organismal survival, and setting the thresholds of target gene activity to prevent inappropriate development (Li et al. 2006; Karres et al. 2007; Flynt and Lai 2008). Our data show the application of this type of strategy in a continuous process of organismal growth. By tuning the activity of insulin signaling, miRNAs could regulate organismal growth and ensure the attainment of appropriate body size (Fig. 5). It is currently unclear whether similar regulatory mechanisms exist in other organisms. However, in humans and rodents, the miR-200 family of miRNAs are predominantly expressed in organs such as the pituitary, thyroid, testes, ovary, and breast, most of which are major target organs of steroid hormones (Landgraf et al. 2007). Moreover, the miR-200 family of miRNAs are significantly down-regulated by the estrogen hormone in breast cancer cells and uterus tissues (Maillot et al. 2009; Yamagata et al. 2009; Nothnick and Healy 2010), suggesting that the miR-200 family may also be controlled by steroids in mammals. It would be interesting to investigate whether a comparable regulatory axis of steroid hormone/miR-200/insulin signaling is conserved through metazoan evolution.
Materials and methods
Flies, measurement of fly size, and ecdysone treatment
UAS-EcRDN (EcRDNF645A) and armadillo (armGal4) were from the Bloomington Drosophila Stock Center. UAS-EcR RNAi was from Vienna RNAi Library Center. The Cg Gal4, mir-8Δ2, UAS-mir-8, mir-8 Gal4, ushvx2, ush Gal4, UAS-ush, and Act>CD2>Gal4 flies are described elsewhere (Karres et al. 2007; Hyun et al. 2009). To measure body size, groups of three female adult flies newly enclosed for not more than 5 h were weighed. All flies were ice-anesthetized before weight measurement. Pupae at 6 d AEL were photographed, and cross-sectional areas were measured by Adobe Photoshop. Measured values were multiplied by the power of 3/2 to transform area to volume, calculating relative pupal volume. 20E was purchased from Sigma and dissolved in ethanol at 10 mg/mL as a stock. A paste was made by mixing autoclaved yeast with 20% glucose and 20E solutions to obtain the final concentration of 0.3 mg/mL. This was fed to the second instar larvae (2 d AEL) except as otherwise indicated. S2 cells were maintained in HyQ SFX-INSECT medium (Hyclone) containing 10% fetal bovine serum (FBS) at 25°C. S2 cells grown in 6-cm dishes were treated with 10 μM 20E or an equal volume of EtOH and harvested for miRNA Northern blotting and quantitative RT–PCR at six different time points.
Other information on Materials and Methods is described in the Supplemental Material.
Acknowledgments
We are very grateful to Drs. Steve Cohen, Bruce Edgar, and Marc Haenlin for kindly sharing fly lines. We thank Je-Keun Rhee for promoter analysis. We also thank Dr. Yoosik Kim, Dr. Mihye Lee, Dr. Young-Kook Kim, Dr. Inha Heo, and Jong-Eun Park for critical reading of the manuscript. This research was supported by the National Honor Scientist Program (20100020415 to V.N.K.), the BK21 Research Fellowships (to H.J.), and the Basic Science Research Program (2012009732 to S.H.), all through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology. S.H. was supported by a T.J. Park Junior Faculty Fellowship. This research was supported by the Chung-Ang University Research Grants in 2010.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.192872.112.
References
- Aravin AA, Lagos-Quintana M, Yalcin A, Zavolan M, Marks D, Snyder B, Gaasterland T, Meyer J, Tuschl T 2003. The small RNA profile during Drosophila melanogaster development. Dev Cell 5: 337–350 [DOI] [PubMed] [Google Scholar]
- Bartel DP 2009. MicroRNAs: Target recognition and regulatory functions. Cell 136: 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Stark A, Russell RB, Cohen SM 2005. Principles of microRNA–target recognition. PLoS Biol 3: e85 doi: 10.1371/journal.pbio.0030085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton JS, Lockwood WK, Li L, Cohen SM, Edgar BA 2002. Drosophila's insulin/PI3-kinase pathway coordinates cellular metabolism with nutritional conditions. Dev Cell 2: 239–249 [DOI] [PubMed] [Google Scholar]
- Brogiolo W, Stocker H, Ikeya T, Rintelen F, Fernandez R, Hafen E 2001. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr Biol 11: 213–221 [DOI] [PubMed] [Google Scholar]
- Caldwell PE, Walkiewicz M, Stern M 2005. Ras activity in the Drosophila prothoracic gland regulates body size and developmental rate via ecdysone release. Curr Biol 15: 1785–1795 [DOI] [PubMed] [Google Scholar]
- Chekmenev DS, Haid C, Kel AE 2005. P-Match: Transcription factor binding site search by combining patterns and weight matrices. Nucleic Acids Res 33: W432–W437 doi: 10.1093/nar/gki441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombani J, Bianchini L, Layalle S, Pondeville E, Dauphin-Villemant C, Antoniewski C, Carre C, Noselli S, Leopold P 2005. Antagonistic actions of ecdysone and insulins determine final size in Drosophila. Science 310: 667–670 [DOI] [PubMed] [Google Scholar]
- Cubadda Y, Heitzler P, Ray RP, Bourouis M, Ramain P, Gelbart W, Simpson P, Haenlin M 1997. u-shaped encodes a zinc finger protein that regulates the proneural genes achaete and scute during the formation of bristles in Drosophila. Genes Dev 11: 3083–3095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delanoue R, Slaidina M, Leopold P 2010. The steroid hormone ecdysone controls systemic growth by repressing dMyc function in Drosophila fat cells. Dev Cell 18: 1012–1021 [DOI] [PubMed] [Google Scholar]
- Demontis F, Perrimon N 2009. Integration of Insulin receptor/Foxo signaling and dMyc activity during muscle growth regulates body size in Drosophila. Development 136: 983–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAngelo JR, Bland ML, Bambina S, Cherry S, Birnbaum MJ 2009. The immune response attenuates growth and nutrient storage in Drosophila by reducing insulin signaling. Proc Natl Acad Sci 106: 20853–20858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar BA 2006. How flies get their size: Genetics meets physiology. Nat Rev Genet 7: 907–916 [DOI] [PubMed] [Google Scholar]
- Flynt AS, Lai EC 2008. Biological principles of microRNA-mediated regulation: Shared themes amid diversity. Nat Rev Genet 9: 831–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuss B, Becker T, Zinke I, Hoch M 2006. The cytohesin Steppke is essential for insulin signalling in Drosophila. Nature 444: 945–948 [DOI] [PubMed] [Google Scholar]
- Hilgers V, Bushati N, Cohen SM 2010. Drosophila microRNAs 263a/b confer robustness during development by protecting nascent sense organs from apoptosis. PLoS Biol 8: e1000396 doi: 10.1371/journal.pbio.1000396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun S, Lee JH, Jin H, Nam J, Namkoong B, Lee G, Chung J, Kim VN 2009. Conserved MicroRNA miR-8/miR-200 and its target USH/FOG2 control growth by regulating PI3K. Cell 139: 1096–1108 [DOI] [PubMed] [Google Scholar]
- Ikeya T, Galic M, Belawat P, Nairz K, Hafen E 2002. Nutrient-dependent expression of insulin-like peptides from neuroendocrine cells in the CNS contributes to growth regulation in Drosophila. Curr Biol 12: 1293–1300 [DOI] [PubMed] [Google Scholar]
- Karres JS, Hilgers V, Carrera I, Treisman J, Cohen SM 2007. The conserved microRNA miR-8 tunes atrophin levels to prevent neurodegeneration in Drosophila. Cell 131: 136–145 [DOI] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. 2007. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129: 1401–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang F, Lee JA, Gao FB 2006. MicroRNA-9a ensures the precise specification of sensory organ precursors in Drosophila. Genes Dev 20: 2793–2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Cassidy JJ, Reinke CA, Fischboeck S, Carthew RW 2009. A microRNA imparts robustness against environmental fluctuation during development. Cell 137: 273–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillot G, Lacroix-Triki M, Pierredon S, Gratadou L, Schmidt S, Benes V, Roche H, Dalenc F, Auboeuf D, Millevoi S, et al. 2009. Widespread estrogen-dependent repression of microRNAs involved in breast tumor cell growth. Cancer Res 69: 8332–8340 [DOI] [PubMed] [Google Scholar]
- McBrayer Z, Ono H, Shimell M, Parvy JP, Beckstead RB, Warren JT, Thummel CS, Dauphin-Villemant C, Gilbert LI, O'Connor MB 2007. Prothoracicotropic hormone regulates developmental timing and body size in Drosophila. Dev Cell 13: 857–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirth CK, Riddiford LM 2007. Size assessment and growth control: How adult size is determined in insects. Bioessays 29: 344–355 [DOI] [PubMed] [Google Scholar]
- Mirth C, Truman JW, Riddiford LM 2005. The role of the prothoracic gland in determining critical weight for metamorphosis in Drosophila melanogaster. Curr Biol 15: 1796–1807 [DOI] [PubMed] [Google Scholar]
- Mirth CK, Truman JW, Riddiford LM 2009. The ecdysone receptor controls the post-critical weight switch to nutrition-independent differentiation in Drosophila wing imaginal discs. Development 136: 2345–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothnick WB, Healy C 2010. Estrogen induces distinct patterns of microRNA expression within the mouse uterus. Reprod Sci 17: 987–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig O, Tjian R 2005. Transcriptional feedback control of insulin receptor by dFOXO/FOXO1. Genes Dev 19: 2435–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rewitz KF, Yamanaka N, O'Connor MB 2010. Steroid hormone inactivation is required during the juvenile-adult transition in Drosophila. Dev Cell 19: 895–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulifson EJ, Kim SK, Nusse R 2002. Ablation of insulin-producing neurons in flies: Growth and diabetic phenotypes. Science 296: 1118–1120 [DOI] [PubMed] [Google Scholar]
- Sempere LF, Sokol NS, Dubrovsky EB, Berger EM, Ambros V 2003. Temporal regulation of microRNA expression in Drosophila melanogaster mediated by hormonal signals and broad-Complex gene activity. Dev Biol 259: 9–18 [DOI] [PubMed] [Google Scholar]
- Shingleton AW, Das J, Vinicius L, Stern DL 2005. The temporal requirements for insulin signaling during development in Drosophila. PLoS Biol 3: e289 doi: 10.1371/journal.pbio.0030289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staton AA, Knaut H, Giraldez AJ 2011. miRNA regulation of Sdf1 chemokine signaling provides genetic robustness to germ cell migration. Nat Genet 43: 204–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JM, Thummel CS 2011. Coordinating growth and maturation—insights from Drosophila. Curr Biol 21: R750–R757 doi: 10.1016/j.cub.2011.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel CS 1996. Flies on steroids—Drosophila metamorphosis and the mechanisms of steroid hormone action. Trends Genet 12: 306–310 [DOI] [PubMed] [Google Scholar]
- Walkiewicz MA, Stern M 2009. Increased insulin/insulin growth factor signaling advances the onset of metamorphosis in Drosophila. PLoS ONE 4: e5072 doi: 10.1371/journal.pone.0005072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Fujiyama S, Ito S, Ueda T, Murata T, Naitou M, Takeyama K, Minami Y, O'Malley BW, Kato S 2009. Maturation of microRNA is hormonally regulated by a nuclear receptor. Mol Cell 36: 340–347 [DOI] [PubMed] [Google Scholar]