

Abstract
AIMS
To investigate the effect of inhibitors of cytochrome P450 (CYP) 3A4 and glucuronidation (UGT2B7) on the pharmacokinetics of lersivirine (UK-453,061), a next generation non-nucleoside reverse transcriptase inhibitor with a unique resistance profile, and to investigate the safety and tolerability of co-administration of lersivirine with these inhibitors.
METHODS
Two open-label, randomized, placebo-controlled, crossover studies were conducted in healthy subjects. Study 1 investigated the effect of ketoconazole (400 mg once daily) on the pharmacokinetics of lersivirine (250 mg once daily). Subjects received ketoconazole 400 mg once daily or placebo on days 1–2 and received lersivirine 250 mg once daily and ketoconazole 400 mg once daily or placebo on days 3–9. Study 2 investigated the effect of valproic acid (VPA, sodium valproate, 1000 mg once daily) on the PK of lersivirine (500 mg once daily). On days 1–7, subjects received lersivirine 500 mg once daily plus either VPA 1000 mg or placebo.
RESULTS
Compared with lersivirine alone, co-administration with ketoconazole increased the lersivirine mean area under the curve (AUC(0,24 h)) and maximum plasma concentration (Cmax) by 82% (90% CI 74%, 91%) and 61% (90% CI 41%, 83%), respectively. VPA increased the mean lersivirine AUC(0,24 h) by 25% (90% CI 16%, 35%), with little effect on Cmax (2.5%, 90% CI −9%, 16%). There were no serious adverse events and no treatment-related discontinuations from either study.
CONCLUSIONS
Inhibition of CYP3A4 and UGT2B7 by ketoconazole increased lersivirine exposure. Inhibition of UGT2B7-mediated glucuronidation by VPA had a modest effect on lersivirine exposure. Co-administration of lersivirine with either ketoconazole or VPA appeared to be well tolerated.
Keywords: ketoconazole; lersivirine; pharmacokinetics; UK-453,061; valproic acid
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Lersivirine (UK-453,061) is predominantly cleared by glucuronidation (UGT2B7) and oxidation via cytochrome P450 (CYP) 3A4.
Lersivirine metabolism, and thus pharmacokinetics, may be affected by concomitant administration of drugs affecting CYP3A4 and UGT2B7.
Ketoconazole is a potent inhibitor of CYP3A4 and an inhibitor of UGT2B7. Valproic acid is a potent inhibitor of UGT2B7.
WHAT THIS STUDY ADDS
Combined inhibition of CYP3A4 and UGT2B7 with ketoconazole increases the exposure of lersivirine, and may warrant lersivirine dose adjustment to compensate for this interaction.
Inhibition of UGT2B7 with valproic acid does not have a substantial effect on lersivirine pharmacokinetics.
Introduction
Patients with HIV are treated with a combination of antiretroviral medications and frequently receive additional medications for the treatment of co-morbidities [1]. Furthermore, patients will receive treatment for their entire lifetime. Hence, new compounds for the treatment of HIV must be submitted to thorough investigation for potential interactions with other drugs and food, which may necessitate dose alterations or make co-administration impossible.
Lersivirine (UK-453,061) is a second generation non-nucleoside reverse transcriptase inhibitor (NNRTI), currently in phase IIb clinical development for the treatment of HIV-1 infection. Lersivirine has a unique binding interaction within the NNRTI binding pocket [2], and has demonstrated potent antiviral activity against wild-type and clinically relevant drug-resistant viruses in vitro[3]. The protein-adjusted in vitro IC90 for lersivirine against wild-type virus is 5.28 ng ml−1. In a phase IIa, dose-ranging, randomized, double-blind, placebo-controlled monotherapy study in 48 asymptomatic HIV-1 infected NNRTI-naïve subjects, treatment with lersivirine for 7 days achieved mean viral RNA reductions of 1.7 log10 copies ml−1 and 1.8 log10 copies ml−1 after receiving 500 and 750 mg once daily, respectively, and 1.6 log10 copies ml−1 after receiving 500 mg twice daily [4]. Common AEs observed for lersivirine at clinical doses include nausea, vomiting, headache, dizziness and abdominal pain [5], [6] with nausea and vomiting occurring at higher frequencies with higher clinical doses. This AE profile for lersivirine is based on emergent AEs observed at the current stage of development (phase IIb).
In vitro studies indicate that lersivirine is predominantly cleared by metabolism, with glucuronidation (UGT2B7) and oxidation via cytochrome P450 (CYP3A4) being the major metabolic pathways [7]. Ketoconazole, an antifungal agent, is a potent inhibitor of CYP3A4 and an inhibitor of UGT2B7 [8]. Valproic acid (VPA) is an anticonvulsant and a potent inhibitor of UGT2B7 [9]–[11]. Two studies were conducted in order to investigate the effect of multiple doses of ketoconazole (study 1) or VPA (study 2) on the pharmacokinetics (PK) of lersivirine, and to investigate the safety and tolerability of co-administration of lersivirine with ketoconazole or VPA, in healthy male subjects.
Methods
The study protocols were reviewed and approved by Comité d'éthique de l'hôpital Erasme in Belgium (study 1) or Parkway Independent Ethics Committee in Singapore (study 2), and all subjects gave written, informed consent. The studies were conducted in accordance with the Declaration of Helsinki [12] and in compliance with all International Conference on Harmonization Good Clinical Practice Guidelines.
Subjects
Eligible subjects were healthy adult males aged 18–55 years (study 1) or 21–55 years (study 2), with a body mass index of 18–30 kg m−2, and a total body weight >50 kg (study 1) or >60 kg (study 2). Subjects with evidence or history of clinically significant disease or any condition possibly affecting drug absorption were excluded from both studies. Other exclusion criteria in both studies included a history of convulsions, excessive alcohol or tobacco consumption and a history of sensitivity to heparin. Additional exclusion criteria for study 2 included: febrile illness, a positive result for HIV or hepatitis, a history of Gilbert's syndrome and a family history of porphyria. The use of any prescription or non-prescription drug, vitamin or dietary supplement within 14 (study 1) or 7 (study 2) days prior to day 1, or any herbal supplement 28 days prior to day 1, was not permitted. As an exception, paracetamol (acetaminophen) could be administered at doses of ≤1 g day−1 on 2 consecutive days.
Study designs
These were phase 1, open, randomized, placebo-controlled two-way crossover studies. In both studies, pre dose screening was conducted for up to 28 days before the first dose and the follow-up visit occurred at least 7 days (study 1) or 10–14 days (study 2) after the last dose. A light breakfast was provided in the study centre approximately 30 min prior to dosing, followed by lunch approximately 4 h and dinner approximately 9 to 10 h post dose. While confined to the study centre, the total daily nutritional composition for each subject was approximately 50% carbohydrate, 35% fat and 15% protein.
Effect of ketoconazole on the PK of lersivirine (study 1)
Subjects received two 9 day treatments, with a minimum 7 day washout period. Subjects were randomized to receive either ketoconazole (400 mg once daily) or placebo on days 1–9, with lersivirine oral suspension (250 mg once daily) on days 3–9. Blood samples for PK analyses were collected pre dose on days 3–8. On day 9, blood samples were collected pre dose and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16 and 24 h post dose. Subjects were admitted to the study centre on the evening pre dose (day 0) and discharged post dose on day 1, returning each day until day 8. They were readmitted on the evening of day 8 or the morning of day 9, and discharged on day 10 after PK blood sampling.
Effect of VPA on the PK of lersivirine (study 2)
Subjects received two 7 day treatments, with a minimum 14 day washout period. Subjects were randomized to receive VPA (sodium valproate, 1000 mg once daily) or placebo on days 1–7, with lersivirine oral tablets (500 mg once daily). Blood samples for PK analyses were collected pre dose on days 1–6. On day 7, blood samples were collected pre dose and at 0.5, 1, 2, 3, 4, 6, 8, 12, 18 and 24 h post dose. Subjects remained in the study centre from day 0 until 24 h after the final administration of lersivirine.
Safety
In both studies, the investigator obtained and recorded all AEs, assessed the severity of reported AEs, and provided an opinion on the relationship of AEs to study drug. Safety laboratory tests were performed at screening, on days 0, 3 (study 1 only), 8 (study 2 only), 10 (study 1 only) and at follow-up.
Sampling and analytical methodology
Within 1 h of collection blood samples were centrifuged at 4°C for 10 min and the plasma stored at −20°C. Quantification of lersivirine in serum samples was performed using solid phase extraction and a validated high performance liquid chromatography/dual mass spectrometry assay (Covance Bioanalytical Services, LLC, Indianapolis, IN, USA). Ketoconazole and VPA concentrations were not measured. The lower limit of quantification (LLOQ) was 1.0 ng ml−1 and the upper limit of quantification was 2000 ng ml−1. The calibration range included eight concentrations from 1.0 to 2000 ng ml−1. The selectivity of the method towards endogenous compounds, potential interferences and possible impurities of the internal standard was acceptable, as illustrated by the chromatograms of a blank plasma sample with internal standard (zero blank) and a blank plasma sample without internal standard (double blank), respectively. The interference, contamination or carryover in each assay batch did not exceed 20% (>5% for internal standards) of the analyte response for the lower of two duplicate LLOQ calibration standards. Assay precision and accuracy was from 4.7% to 8.0% (coefficient of variance) and −2.0 to 2.6% (relative error), respectively.
Statistical analyses
Fourteen subjects were recruited into each study and dosed with the intention of at least 12 subjects completing the study. With 12 subjects and using 5% one-sided tests, study 1 had 90% power to exclude an increase in the maximum plasma concentration (Cmax) and the area under the curve over the dosing interval (AUC(0,24 h)) greater than 200%. This was concluded for each parameter if the upper limit of the 90% confidence interval (CI) for the test : reference ratio (%) was lower than 300%. These power calculations were performed based on a hypothesized true ratio of 225%, the estimated effect when a quarter of lersivirine metabolism was through the CYP3A4 pathway. Within-subject standard deviations of 0.222 and 0.091, estimated from previous studies, were used in the power calculations for logeCmax and loge AUC(0,24 h), respectively.
In study 2, a sample size of 12 subjects was required to provide 90% CIs for the difference between treatments of ± 0.0539 and ± 0.1525 on the natural log scale for AUC(0,24 h) and Cmax, respectively, with 80% coverage probability. This estimate was based on within-subject standard deviations of 0.089 and 0.252 for loge AUC(0,24 h) and logeCmax, respectively, as obtained from study 1.
Descriptive statistics were calculated for AUC(0,24 h), Cmax, minimum plasma concentration (Cmin), and time to Cmax (tmax). Loge transformed data were analyzed using a mixed effect model with sequence, period and treatment as fixed effects and subject within sequence as a random effect.
Estimates of the adjusted mean differences (test – reference) and corresponding 90% CIs were obtained from the model. The adjusted mean differences and 90% CIs for the differences were exponentiated to provide estimates of the ratio of adjusted geometric means (test : reference) and 90% CIs for the ratios. Lersivirine plus placebo was the reference treatment and lersivirine plus ketoconazole or VPA was the test treatment.
Results
Subjects
In study 1, 14 subjects were randomly assigned to the study treatment sequences. One subject in the lersivirine plus ketoconazole group was permanently discontinued from the study due to a non-treatment-related AE of tonsillitis during the second period of study treatment. Another subject (lersivirine plus ketoconazole group) discontinued period 1 due to a limb injury, but completed period 2. Both discontinuations were prior to lersivirine dosing on day 3. No other subjects withdrew from the study. Subjects were aged between 21 and 48 years and most were white (Table 1).
Table 1.
Subject demographics and baseline characteristics (studies 1 and 2)
| Study 1 (n = 14) | Study 2 (n = 14) | |
|---|---|---|
| Age (years) | ||
| Range | 21–48 | 21–44 |
| Mean (SD) | 32.3 (8.2) | 25.6 (6.3) |
| Race (n) | ||
| Asian | 0 | 14 |
| Black | 1 | 0 |
| White | 13 | 0 |
| Weight (kg) | ||
| Range | 53–92 | 63–82 |
| Mean (SD) | 75.9 (11.0) | 71.9 (5.7) |
| Body mass index (kg m−2) | ||
| Range | 18.0–30.0 | 20.7–29.8 |
| Mean (SD) | 24.1 (3.1) | 24.3 (2.6) |
| Height (cm) | ||
| Range | 160–186 | 162–185 |
| Mean (SD) | 177.4 (7.3) | 172.6 (5.8) |
SD, standard deviation.
In study 2, 14 subjects were randomly assigned to the study treatment sequences. No subjects withdrew from the study. Subjects were aged between 21 and 44 years and all were Asian (Table 1).
Pharmacokinetics
Study 1
Daily doses of lersivirine 250 mg with steady-state ketoconazole increased the plasma exposure of lersivirine. When administered with ketoconazole the adjusted geometric means (90% CI) for lersivirine AUC(0,24 h), Cmax and Cmin increased by 82% (74%, 91%), 61% (41%, 83%) and 65% (8%, 152%), respectively (Table 2). Absorption of lersivirine was rapid when administered in the presence or absence of ketoconazole, with tmax medians of 1.5 and 2.0 h for placebo and ketoconazole, respectively (Table 2). The effect of ketoconazole on the steady-state median plasma concentration–time profiles of lersivirine is shown in Figure 1.
Table 2.
Effect of ketoconazole on the steady-state pharmacokinetics of lersivirine (study 1)
| Lersivirine (250 mg once daily) + ketoconazole (400 mg once daily) | Lersivirine (250 mg once daily) + placebo | |||||
|---|---|---|---|---|---|---|
| Parameter (units) | Geometric mean (%CV) (n = 12) | Adjusted geometric mean (A) (n = 12) | Geometric mean (%CV) (n = 14) | Adjusted geometric mean (B) (n = 12) | Ratio of adjusted geometric means (A : B) | 90% CI |
| AUC(0,24 h) (ng ml−1 h) | 4216 (24) | 4216 | 2346 (19) | 2316 | 1.82 | 1.74, 1.91 |
| Cmax (ng ml−1) | 679 (23) | 679 | 431 (18) | 423 | 1.61 | 1.41, 1.83 |
| Cmin (ng ml−1) | 25.9 (76.5) | 25.9 | 15.0 (93.8) | 15.7 | 1.65 | 1.08, 2.52 |
| Median (range) | Median (range) | |||||
| tmax (h) | 2.0 (0.5–3.0) | 1.5a (0.5–3.0) | – | – | ||
n = 14.
AUC(0,24 h), area under the curve over the dosing interval; CI, confidence interval; Cmax, maximum plasma concentration; Cmin, minimum plasma concentration; CV, coefficient of variation; tmax, time for Cmax.
Figure 1.
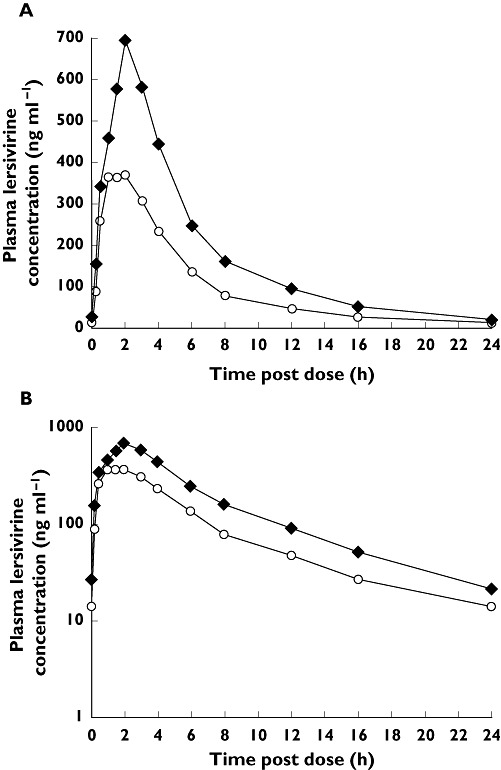
Effect of ketoconazole on the steady-state median plasma concentration–time profile of lersivirine: A) linear and B) semi-log scales (study 1). Lersivirine 250 mg once daily + placebo once daily ( ); Lersivirine 250 mg once daily + ketoconazole 400 mg once daily (
); Lersivirine 250 mg once daily + ketoconazole 400 mg once daily ( )
)
Study 2
The steady-state median plasma concentration–time profile of lersivirine in the presence of VPA is shown in Figure 2. There was a nominal difference in the median lersivirine plasma concentrations between the VPA and placebo groups; steady-state VPA resulted in a small increase in lersivirine exposure. Inhibition of UGT2B7 with VPA increased the mean AUC(0,24 h) and Cmin by 25% (90% CI 16%, 35%) and 63% (90% CI 49%, 79%), respectively, with minimal effect on Cmax (3% increase). Median tmax for lersivirine was unaffected by the inhibition of UGT2B7 by VPA (3.0 h for both groups) (Table 3).
Figure 2.
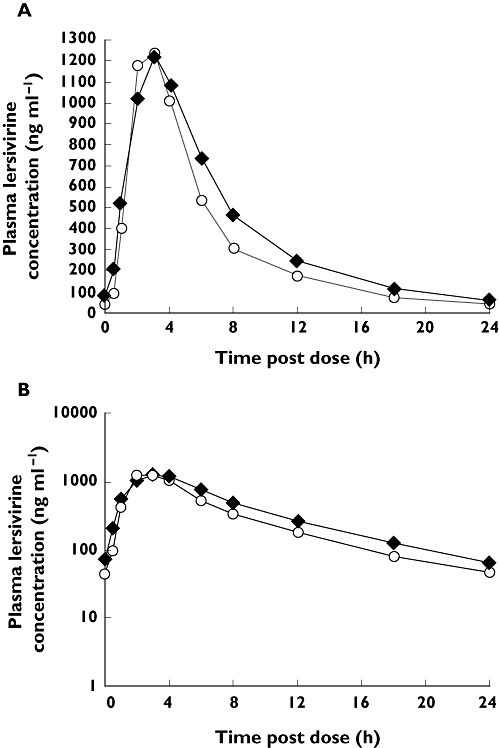
Effect of valproic acid on the steady-state median plasma concentration–time profile of lersivirine: A) linear and B) semi-log scales (study 2). Lersivirine 500 mg once daily + VPA 1000 mg once daily ( ); Lersivirine 500 mg once daily + placebo once daily (
); Lersivirine 500 mg once daily + placebo once daily ( )
)
Table 3.
Effect of valproic acid on the steady-state pharmacokinetics of lersivirine (study 2)
| Lersivirine (500 mg once daily) + valproic acid (1000 mg once daily) (n = 14) | Lersivirine (500 mg once daily) + placebo (n = 14) | |||||
|---|---|---|---|---|---|---|
| Parameter (units) | Geometric mean (%CV) | Adjusted geometric mean (A) | Geometric mean (%CV) | Adjusted geometric mean (B) | Ratio of adjusted geometric means (A : B) | 90% CI |
| AUC(0,24 h) (ng ml−1 h) | 9383 (16) | 9383 | 7498 (18) | 7498 | 1.25 | (1.16, 1.35) |
| Cmax (ng ml−1) | 1327 (18) | 1327 | 1295 (18) | 1295 | 1.03 | (0.91, 1.16) |
| Cmin, (ng ml−1) | 62 (37) | 62 | 38 (38) | 38 | 1.63 | (1.49, 1.79) |
| Median (range) | Median (range) | |||||
| tmax (h) | 3.0 (1–6) | 3.0 (2–4) | – | – | ||
AUC(0,24 h), area under the curve over the dosing interval; CI, confidence interval; Cmax, maximum plasma concentration; Cmin, minimum plasma concentration; CV, coefficient of variation; tmax, time for Cmax.
Safety and tolerability
Co-administration of lersivirine with ketoconazole, or VPA, appeared to be well tolerated. There were no clinically significant changes in vital signs or clinical laboratory parameters. The nature of the AEs reported were similar between all the treatment groups; being predominantly nervous system and gastrointestinal disorders (Table 4).
Table 4.
Treatment-emergent all-causality adverse events occurring in ≥2 subjects in any treatment group (studies 1 and 2)
| Study 1 | ||
|---|---|---|
| Lersivirine (250 mg once daily) + ketoconazole (400 mg once daily) (n = 14) | Lersivirine (250 mg once daily) + placebo (n = 14) | |
| MedDRA preferred term, n (%) | ||
| Abdominal pain | 2 (14) | 0 |
| Diarrhoea | 5 (36) | 2 (14) |
| Dyspepsia | 3 (21) | 0 |
| Nausea | 5 (36) | 0 |
| Headache | 5 (36) | 1 (7) |
| Pharyngolaryngeal pain | 2 (14) | 3 (21) |
| Study 2 | ||
|---|---|---|
| Lersivirine (500 mg once daily) + valproic acid (1000 mg once daily) (n = 14) | Lersivirine (500 mg once daily) + placebo (n = 14) | |
| MedDRA preferred term, n (%) | ||
| Nausea | 1 (7) | 2 (14) |
| Vessel puncture site haematoma | 2 (14) | 0 |
| Upper respiratory tract infection | 0 | 2 (14) |
| Headache | 2 (14) | 0 |
MedDRA, medical dictionary for regulatory activities.
Study 1
There were no serious AEs and no discontinuations from the study due to treatment-related AEs. All treatment-emergent AEs were mild, except for three in the ketoconazole group (nausea, headache and tonsillitis) and four in the placebo group (headache, pharyngolaryngeal pain, nasopharyngitis and skin lesion excision), which were moderate, and a limb injury in the ketoconazole group which was severe. Treatment-related AEs were reported for 11 subjects (79%) in the ketoconazole group and five subjects (36%) in the placebo group and all were mild to moderate. The AEs reported in the ketoconazole group were primarily of types typically associated with ketoconazole administration (nausea, vomiting, diarrhoea, abdominal pain and headache). Four subjects had a laboratory test result that met the sponsor-defined criteria for potential concern (ketones in urine and decrease in lymphocytes, decrease in lymphocytes and increase in total neutrophils, glucose in urine and decrease in total neutrophils).
Study 2
There were no serious or severe AEs and no discontinuations from the study. All treatment-emergent AEs were either mild or moderate. Treatment-related AEs were reported for four subjects (29%) in the VPA group and three subjects (21%) in the placebo group. No AE occurred in more than two subjects. Six and five subjects in the ketoconazole and placebo groups, respectively, had a laboratory test result out of the reference range that met the sponsor-defined criteria for potential concern (decrease in serum sodium, increase in monocytes, increase in urine blood and ketones, increase in urine ketones (two subjects), decrease in urine specific gravity, increase in basophils, increase in total bilirubin, increase in lymphocytes (two subjects) and increase in urine blood).
Discussion
Lersivirine is metabolized via glucuronidation (UGT2B7) and CYP3A4 and hence co-administration of ketoconazole may be expected to alter lersivirine PK. Study 1 demonstrated that inhibition of CYP3A4 and UGT2B7 by ketoconazole resulted in an increase in lersivirine AUC(0,24 h), Cmax and Cmin by 82%, 61% and 65%, respectively. In addition, inhibition of P-glycoprotein (P-gp) by ketoconazole may play a role as in vitro data suggests that lersivirine is also a substrate for P-gp (data on file). As the lersivirine peak : trough ratio does not significantly change whether lersivirine is dosed with or without ketoconazole, the inhibitory effect of ketoconazole on lersivirine exposures can presumably be attributed to an increase in lersivirine bioavailability.
Lersivirine was administered as an oral suspension in study 1, because a tablet formulation for lersivirine was not available at the start of the study. In a subsequent bioavailability study that assessed single 500 mg doses of lersivirine, tablets had a lower exposure than oral suspension (mean AUC(0,∞) and Cmax were 8% and 20% lower, respectively, data on file). However, the magnitude of the interaction is expected to be comparable with the tablet formulation.
The PK of other next generation NNRTIs are also affected by co-administration with ketoconazole. Etravirine is a substrate of CYP3A, CYP2C9 and CYP2C19. Although not formally studied, co-administration of ketoconazole with etravirine may increase plasma concentrations of etravirine [13]. Co-administration of ketoconazole with rilpivirine, a next generation NNRTI, increased rilpivirine AUC(0,24 h), Cmax and Cmin by 49%, 30% and 76%, respectively, likely due to inhibition of CYP3A4-mediated metabolism of rilpivirine by ketoconazole [14].
Inhibition of UGT2B7-mediated glucuronidation by VPA has the potential to affect significantly the PK of a co-administered drug which is glucuronidated via UGT2B7. An example of this is the decreased clearance of zidovudine (UGT2B7 substrate) when co-administered with VPA [10]. However, co-administration of lersivirine with VPA had only a modest effect on total lersivirine exposures, with an increase of lersivirine AUC(0,24 h) of 25%. The lack of a larger effect of VPA supports our understanding that UGT2B7 may be one of multiple clearance pathways for lersivirine, though a greater effect was seen on Cmin (63% increase) than on Cmax (3% increase), suggesting that the effect of inhibition was predominantly on elimination. The lersivirine exposures observed in this study following co-administration of lersivirine 500 mg once daily and VPA are within the range of lersivirine exposures achieved with clinical doses (500–750 mg once daily) in phase IIb (study A5271015; NCT00824421) where lersivirine was dosed in combination with tenofovir/emtricitabine [5], [15].
Etravirine is only subject to minor glucuronidation and similarly rilpivirine has not been shown to be a substrate for glucuronidation. Consequently, neither agent would be expected to be affected by co-administration with a UGT2B7 inhibitor such as VPA. Lersivirine, which is a UGT2B7 substrate, was minimally affected by VPA in this study, suggesting that the effects of UGT inhibition are likely to be negligible at clinically relevant doses of lersivirine.
In both of these short-term studies, lersivirine was generally well tolerated and AEs were consistent with the known AE profile for lersivirine.
The PK of ketoconazole and VPA were not formally assessed in these studies. However, given that lersivirine is a weak inducer of CYP3A4 and weak inhibitor of glucuronidation [7], [16] exposures of both ketoconazole and VPA are not expected to be affected significantly in the presence of lersivirine at clinical doses.
Lersivirine exposure can be substantially modified by co-administration with an inhibitor of both CYP3A4 and UGT2B7. Lersivirine dose adjustments may be used to compensate for these alterations in lersivirine exposure. However, further investigation is required to support this. In addition, as the optimal lersivirine clinical dose will not be determined until completion of phase III studies, a conclusive statement regarding dose adjustment cannot be made at this time. Study 2 suggests that no dose modifications are necessary when lersivirine is co-administered with a potent inhibitor of UGT2B7 alone. In these two trials, co-administration of lersivirine with ketoconazole or VPA over 7 or 9 days was generally well tolerated in healthy subjects.
Competing Interests
Manoli Vourvahis, John Davis, Grant Langdon, Gary Layton and Chew-Lan Chong are employees of and shareholders in Pfizer Inc. Georges Weissgerber was employed by Pfizer Inc. at the time the studies were performed. Both studies were conducted and funded by Pfizer Inc. Lersivirine is under development by Pfizer Inc. and ViiV Healthcare.
Acknowledgments
Editorial support was provided by Dr Gary Dever at Complete Medical Communications and was funded by ViiV Healthcare.
REFERENCES
- 1.Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. Available at http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Updated 2011 (last accessed 28 June 2011)
- 2.Phillips C, Irving S, Ringrose H, Corbau R, Mowbray C. HIV-1 reverse transcriptase structure-based drug design: crystals to clinic. Acta Cryst. 2007;A63:s18. [Google Scholar]
- 3.Mori J, Corbau R, Lewis D, Ellery S, Mayer H, Perros M, Westby M. In vitro characterization of UK-453,061, a non-nucleoside reverse transcriptase inhibitor. Paper presented at the 15th Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 3–6 February, 2008.
- 4.Fätkenheuer G, Staszewski S, Plettenburg A, Hackman F, Layton G, McFadyen L, Davis J, Jenkins TM. Activity, pharmacokinetics and safety of lersivirine (UK-453,061), a next-generation nonnucleoside reverse transcriptase inhibitor, during 7-day monotherapy in HIV-1 infected patients. AIDS. 2009;23:2115–22. doi: 10.1097/QAD.0b013e32832fef5b. [DOI] [PubMed] [Google Scholar]
- 5.Vernazza P, Wang C, Pozniak A, Weil E, Pulik P, Cooper DA, Kaplan R, Lazzarin A, Valdez H, Goodrich J, Craig C, Mori J, Tawadrous M. Efficacy and safety of lersivirine vs efavirenz in antiretroviral treatment-naive HIV-1-infected patients: Week 48 primary analysis results from an ongoing multicentre, randomised, double-blind, Phase IIIb Trial (Study A5271015) Paper presented at the 16th International AIDS Conference on HIV Pathogenesis, Treatment and Prevention, Rome, Italy, 17–20 July, 2011.
- 6.Davis J, Hackman F, Ndongo M-N, Choo HW, Lewis D, Tawadrous M, Goodrich J, Langdon G. Safety and tolerability of lersivirine, a nonnucleoside reverse transcriptase inhibitor, during a 28-day, randomized, placebo-controlled, phase I clinical study in healthy male volunteers. Clin Ther. 2010;32:1889–95. doi: 10.1016/j.clinthera.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 7.Vourvahis M, Gleave M, Nedderman ANR, Hyland R, Gardner I, Howard M, Kempshall S, Collins C, Labadie R. Excretion and metabolism of lersivirine (5-([3,5-diethyl-1-(2-hydroxyethyl)(3,5-14C2)-1H-pyrazol-4-yl]oxy)benzene-1,3-dicarbonitrile), a next-generation non-nucleoside reverse transcriptase inhibitor, after administration of [14C]Lersivirine to healthy volunteers. Drug Metab Dispos. 2010;38:789–800. doi: 10.1124/dmd.109.031252. [DOI] [PubMed] [Google Scholar]
- 8.Takeda S, Kitajima Y, Ishii Y, Nishimura Y, Mackenzie PI, Oguri K, Yamada H. Inhibition of UDP-glucuronosyltransferase 2b7-catalyzed morphine glucuronidation by ketoconazole: dual mechanisms involving a novel noncompetitive mode. Drug Metab Dispos. 2006;34:1277–82. doi: 10.1124/dmd.106.009738. [DOI] [PubMed] [Google Scholar]
- 9.Ethell BT, Anderson GD, Burchell B. The effect of valproic acid on drug and steroid glucuronidation by expressed human UDP-glucuronosyltransferases. Biochem Pharmacol. 2003;65:1441–9. doi: 10.1016/s0006-2952(03)00076-5. [DOI] [PubMed] [Google Scholar]
- 10.Lertora JJ, Rege AB, Greenspan DL, Akula S, George WJ, Hyslop NE, Jr, Agrawal KC. Pharmacokinetic interaction between zidovudine and valproic acid in patients infected with human immunodeficiency virus. Clin Pharmacol Ther. 1994;56:272–8. doi: 10.1038/clpt.1994.137. [DOI] [PubMed] [Google Scholar]
- 11.Trapnell CB, Klecker RW, Jamis-Dow C, Collins JM. Glucuronidation of 3′-azido-3′-deoxythymidine (zidovudine) by human liver microsomes: relevance to clinical pharmacokinetic interactions with atovaquone, fluconazole, methadone, and valproic acid. Antimicrob Agents Chemother. 1998;42:1592–6. doi: 10.1128/aac.42.7.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.World Medical Association General Assembly. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects (revised October 7, 2000) HIV Clin Trials. 2001;2:92–5. doi: 10.1310/GTFR-2DRX-M6YE-ELXR. [DOI] [PubMed] [Google Scholar]
- 13.Tibotec Inc. Intelence (etravirine) prescribing information. Available at http://www.intelence-info.com/sites/default/files/pdf/INTELENCE_Booklet_Package_Insert_hcp.pdf Updated 2011 (last accessed 28 June 2011)
- 14.Van Heeswijk R, Hoetelmans R, Kestens D, Stevens M, Peeters M, Williams P, Woodfall B, Boven K. The pharmacokinetic interaction between ketoconazole and TMC278, an investigational non-nucleoside reverse transcriptase inhibitior (NNRTI), in healthy HIV-negative subjects. Paper presented at the XVI International AIDS Conference, Toronto, Canada, 13–18 August, 2006.
- 15.McFadyen L, Weatherley B, Vourvahis M. Preliminary lersivirine population pharmacokinetics in treatment-naive HIV-1-infected subjects receiving lersivirine (UK-453,061) and tenofovir/emtricitabine in a Phase 2b study. Paper presented at the 6th International AIDS Conference on HIV Pathogenesis, Treatment and Prevention, Rome, Italy, 17–20 July, 2011.
- 16.Davis J, Langdon G, Layton G, Chong CL, Ndongo M-N, Weissgerber G, Vourvahis M. The effect of UK-453,061, a next-generation NNRTI, on the pharmacokinetics of zidovudine, midazolam, and contraceptive steroids. Paper presented at the 9th International Workshop on Clinical Pharmacology of HIV Therapy, New Orleans, LA, USA, 7–9 April, 2008.