

Abstract
AIM
To examine the predictive value of gene polymorphisms potentially linked to toxicity, clinical response, time to progression and overall survival, following cetuximab–tegafur-uracil (UFT)–irinotecan therapy.
METHODS
Fifty-two patients with advanced colorectal cancer were enrolled in an ancillary pharmacogenetic study of the phase II CETUFTIRI trial. Treatment consisted of 21 day cycles of cetuximab (day 1–day 8–day 15, 250 mg m−2 week−1 following a 400 mg m−2 initial dose) together with irinotecan (day 1, 250 mg m−2) and UFT–folinic acid (days 1–14, 250 mg m−2 day−1 UFT, 90 mg day−1 folinic acid). Analysed gene polymorphisms (blood DNA) were as follows: EGFR (CA repeats in intron 1, −216G>T, −191C>A), EGF (61A>G), FCGR2A (131Arg>His), FCGR3A (158Phe>Val), UDP-glycosyltransferase1-polypeptide A1 (TA repeats), TYMS (28 bp repeats, including the G>C mutation on the 3R allele, 6 bp deletion in 3′ UTR) and MTHFR (677C>T, 1298A>C).
RESULTS
Maximum toxicity grade was linked to EGFR−191C>A polymorphism, with 71.1% grade 3–4 toxicity in CC patients vs. 28.6% in other patients (P = 0.010). A tendency to a better response was observed in patients bearing the TYMS 3RG allele (P = 0.029) and those bearing the FCGR3A 158Val genotype (P = 0.020). The greater the score of favourable TYMS and FCGR3A genotypes, the better the response rate (P = 0.009) and the longer the overall survival (P = 0.007). In multivariate analysis, the score of favourable genotypes was a stronger survival predictor than the performance status.
CONCLUSIONS
Present data suggest the importance of FCGR3A 158Phe>Val and TYMS 5′ UTR polymorphisms in responsiveness and survival of patients receiving cetuximab–fluoropyrimidine-based therapy.
Keywords: antibody-dependent cell cytotoxicity, cetuximab, epidermal growth factor receptor, pharmacogenetics, tegafur-uracil, TYMS
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Previous pharmacogenetic studies have reported the potential predictive value of thymidylate synthase (TYMS) polymorphisms or methylenetetrahydrofolate reductase (MTHFR) polymorphisms for the efficacy of 5-fluorouracil-based therapy, even though they have not yet been fully validated. Also, functional polymorphisms of genes linked to the epidermal growth factor receptor pathway [epidermal growth factor (EGF) and epidermal growth factor receptor (EGFR)], as well as polymorphisms of genes encoding for Fcγ receptors [Fc fragment of IgG receptor 2A (FCGR2A) and 3A (FCGR3A)], which influence their affinity for the Fc fragment, have been reported to be linked to the pharmacodynamics of cetuximab in the clinical setting.
WHAT THIS STUDY ADDS
This prospective study conducted on advanced colorectal cancer patients receiving first-line tegafur-uracil–irinotecan–cetuximab therapy suggests that a favourable genotype score, considering both the TYMS 3RG allele and any Val-containing FCGR3 allele, may be an indicator of better clinical response and longer overall survival.
Introduction
Colorectal cancer (CRC) is the second highest cause of cancer death in Western countries. Tegafur-uracil (UFT) is an oral fluoropyrimidine approved in the treatment of advanced colorectal cancer in several Western countries. The combination of irinotecan with 5-fluorouracil (5FU) and folinic acid (FA) [1] or with UFT–FA [2] results in significant antitumoural activity in metastatic CRC patients. The anti-epidermal growth factor receptor (EGFR) monoclonal antibody (mAb) cetuximab has demonstrated clinical activity in metastatic CRC in combination with irinotecan or oxaliplatin [3], [4]. Cetuximab acts by means of the following two independent mechanisms: the inhibition of EGFR signal transduction; and the possible activation of antibody-dependent cell cytotoxicity (ADCC). The ADCC is mediated by the Fc fragment of IgG1 mAbs, such as cetuximab. This fragment links target cancer cells to the Fc receptors (FcγR2a, FcγR3A) carried by immune cells, causing the lysis of target cells.
In the present study, cetuximab was given with oral UFT–FA and irinotecan, as first-line treatment in patients with metastatic colorectal carcinoma. This ancillary pharmacogenetic study was conducted on 52 of the 60 patients included in the French multicentre phase II study, CETUFTIRI. Our purpose was to analyse the possible relationships between treatment efficacy, or toxicity, and germinal gene polymorphisms linked to the administered drugs. We analysed the main functional polymorphism of the UDP-glycosyltransferase1-polypeptide A1 (UGT1A1) gene (UGT1A1*28 variant), which affects the glucuronidation capacity of SN38, the active metabolite of irinotecan [5], along with the DPYD*2A variant because the DPYD gene encodes for dihydropyrimidine dehydrogenase, the key enzyme of the 5FU catabolic pathway, as well as the following other gene polymorphisms relevant for fluoropyrimidine pharmacodynamics: the TYMS gene, coding for thymidylate synthase (TS), the main 5FU pharmacological target; and the MTHFR gene, coding for the methylenetetrahydrofolate reductase enzyme, controlling the intracellular reduced folate concentration, which is an essential cofactor for enhancing TS inhibition mediated by 5FU. Numerous studies have reported the potential predictive value of TYMS polymorphisms [6] or MTHFR polymorphisms [7] for the efficacy of 5FU-based therapy, even though they are not yet fully validated [8]. In our study, we also analysed functional polymorphisms of genes linked to the EGFR pathway, namely EGF and EGFR genes, as well as polymorphisms of genes encoding for Fcγ receptors (FCGR2A and FCGR3A genes), which influence their affinity for the Fc fragment. In fact, previous studies have suggested that EGFR and/or EGF polymorphisms [9]–[11] as well as FCGR2A and FCGR3A gene polymorphisms [11], [12] may explain interpatient variability in the pharmacodynamics of cetuximab.
Materials and methods
Patients and treatment
Patient recruitment was performed between December 2005 and December 2006, before KRAS-mutation testing was introduced as a requirement for cetuximab treatment. Inclusion criteria included patient age ≥18 years, histologically or cytologically confirmed, bidimensionally measured metastatic, unresectable CRC, Eastern Cooperative Oncology Group performance status 0 or 1, no prior chemotherapy, and adequate bone marrow and renal and hepatic function. The study was carried out with ethics committee approval. All patients received first-line therapy consisting of 21 day cycles of cetuximab (400 mg m−2 as initial dose, 250 mg m−2 for subsequent doses, i.v. over 2 h on days 1, 8 and 15), together with irinotecan (250 mg m−2 i.v. over 90 min on day 1) and UFT (250 mg m−2 day−1) plus leucovorin (90 mg day−1) daily from days 1 to 14. Treatment was administered until disease progression or unacceptable toxicity, for a maximum of eight cycles. A description of the 52 analysed patients is given in Table 1.
Table 1.
Patient characteristics (n = 52)
| Age (years) | Mean | 63.3 |
| Range | 36–84 | |
| Gender | Men | 33 |
| Women | 19 | |
| Performance status (ECOG) | 0 | 33 |
| 1 | 19 | |
| Surgery on primary | No | 13 |
| Yes | 36 | |
| Adjuvant chemo- and/or radiotherapy | No | 43 |
| Yes | 9 | |
| Primary localization | Colon | 34 |
| Rectosigmoid | 7 | |
| Rectum | 11 | |
| Metastasis site | Liver | 20 |
| Lung | 1 | |
| Peritoneum | 1 | |
| Lymph node | 1 | |
| Multiple sites | 29 | |
| Number of cycles | Mean | 5.8 |
| Median | 7 | |
| Range | 1–8 | |
| Cumulative cetuximab dose (g m−2) | Mean | 4.0 |
| Median | 4.7 | |
| Range | 0.4–6.2 | |
| Cumulative UFT dose (g m−2) | Mean | 19.3 |
| Median | 22.4 | |
| Range | 0–28.0 | |
| Cumulative irinotecan dose (g m−2) | Mean | 1.4 |
| Median | 1.6 | |
| Range | 0–2.0 |
ECOG, Eastern Cooperative Oncology Group.
Toxicity evaluation
For each toxicity (leukopenia, neutropenia, thrombocytopenia, diarrhoea, nausea, vomiting, mucositis, asthenia, alopecia, acneiform rash, paronychia, hand–foot syndrome, anaphylactic shock, septic shock), the maximum observed toxicity grade was recorded (NCI-Common Terminology Criteria for Adverse Events v3.0). Then, for each patient, we considered the maximum observed toxicity grade (whatever the toxic pattern). In addition, we focused on the following factors: (i) the maximum observed neutropenia grade as a relevant indicator of irinotecan toxicity; and (ii) the score corresponding to the sum of rash and paronychia grades (score 0, 1 or 2 vs. score 3, 4, 5 or 6) as a relevant indicator of cetuximab-related cutaneous toxicity.
Efficacy evaluation
Best clinical response was assessed according to modified Response Evaluation Criteria in Solid Tumors [complete response (CR), partial response (PR), stable disease (SD), progressive disease (PD)]. Time to progression (TTP) and survival were computed from day 1 of treatment. At the time of analysis, 51 patients of the 52 had progressed and 41 had died. As all 41 recorded deaths were cancer related, overall survival corresponded to specific survival. Median follow-up was 32.4 months (reverse Kaplan–Meier method).
KRAS mutation analysis
KRAS mutation analysis was performed retrospectively. Formalin-fixed, paraffin-embedded tumour material was collected from different pathology laboratories. In total, tumour material from 38 patients was collected. The percentage of tumour cells in analysed samples was ≥30%. DNA extraction (RecoverAll™ Kit from AMBION, Applied Biosystems, Courtaboeuf, France) and mutation analysis were centralized at the Centre Antoine Lacassagne, Nice. KRAS mutations at codon 12 and codon 13 were analysed according to a single-base extension multiplex assay adapted from Di Fiore et al. [13], on a Beckman CEQ 8000 sequencer. The following KRAS-characterized cell lines were used as controls: CCRF-CEM (mutated G12D), HCT116 (mutated G13D) and WiDr (wild-type). Nineteen patients of the 38 exhibited a KRAS mutation.
Pharmacogenetic analyses
On completion of patient recruitment, frozen blood samples (9 ml) were sent to the Centre Antoine Lacassagne (Nice), where DNA extractions were performed (Paxgene Blood DNA kit; QIAGEN, Courtaboeuf, France). Germinal polymorphisms of TYMS, MTHFR, DPYD, EGFR, EGF, FCGR2A and FCGR3A genes were analysed at the Centre Antoine Lacassagne, and UGT1A1 polymorphism was analysed at the Centre Claudius Regaud (Toulouse).
The 28 bp repeat polymorphisms (2R or 3R, rs34743033) in the promoter region of the TYMS gene, along with the G>C mutation in the second repeat of the 3R allele (rs11540151), were analysed by means of polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP), as previously described [14]. TYMS genotype was classified as a function of the number of theoretical E-box binding sites likely to bind Upstream Stimulatory Factor proteins, as follows: class 2 (2R2R or 2R3RC or 3RC3RC), class 3 (2R3RG or 3RC3RG) or class 4 (3RG3RG). The 6 bp deletion at position 1494 of the TYMS gene (rs11280056) was analysed by PCR and electrophoresis [15]. Polymorphisms at positions 677C>T (rs1801133) and 1298A>C (rs1801131) of the MTHFR gene were analysed according to melting curve analysis on LightCycler (Roche, Meylan, France) as previously described [15]. The DPYD IVS14+1G>A mutation (DPYD*2A variant, rs3918290) was analysed with PCR-RFLP using the NdeI restriction enzyme [14]. The EGFR−216G>T (rs712829) and −191C>A (rs712830) polymorphisms were analysed by PCR-RFLP [16]. The CA repeats polymorphism in intron 1 of the EGFR gene (rs11568315) was investigated by means of fragment length analysis [17]. Owing to the large number of genotypes (between 15 and 22 CA repeats), patients were split into the following three groups: patients with both alleles <17 vs. patients with both alleles ≥17 vs. others. The EGF 61A>G (rs4444903), FCGR2A 131Arg>His (rs1801274) and FCGR3A 158Phe>Val (rs396991) gene polymorphisms were analysed by validated PCR-RFLP methods [18], [19]. The TA tandem repeat in the UGT1A1 gene promoter was analysed by PCR using 5′-GCCAGTTCAACTGTTGTTGCC-3′ as forward primer and 5′-CCACTGGGATCAACAGTATCT-3′ as reverse primer. The UGT1A1*28 variant (rs8175347) corresponds to the [A(TA)7TAA] sequence, while UGT1A1*1 (wild-type allele) corresponds to the [A(TA)6TAA] sequence. The expected fragments (320 bp) were subjected to direct sequencing analysis with the Big dye terminator v3.1 cycle kit (Applied Biosystems, Warrington, UK). For UGT1A1 genotype, DNA samples from three patients with known TA6/TA6, TA6/TA7 and TA7/TA7 were used as controls. For other genotypes, wild-type and mutated cell lines were used as controls.
Statistics
The exact P values for Hardy–Weinberg equilibrium were tested on http://innateimmunity.net/IIPGA2. The nonparametric Kruskal–Wallis test was used to examine the influence of UGT1A1 genotype on irinotecan dose. Fisher's exact test was applied to test the links between analysed genotypes and clinical end-points (CR + PR vs. SD + PD; toxicity grade or score ≤2 vs. toxicity grade or score >2), or between responsiveness and acneiform rash or KRAS status, or between toxicity and patient's characteristics. A logistic model was applied to estimate the odds ratio associated with toxicity markers (1 = grade or score >2, 0 = grade or score ≤2), response markers (1 = CR + PR, 0 = SD + PD) and for multivariate analysis. The TTP and survival curves were plotted according to the Kaplan–Meier method. The influence of the various tested parameters on TTP and survival was assessed by means of log rank test or Cox analysis (for continuous variables or multivariate analysis). Owing to the large number of tests performed, a P value of less than or equal to 0.010 was considered statistically significant (two-sided tests). Statistics were performed using SPSS software (v15.0; SPSS Inc., Chicago, IL, USA).
Results
Description of toxicity and efficacy
The most frequent major toxicities were diarrhoea (2 grade 1, 10 grade 2, 8 grade 3) and acneiform rash (3 grade 1, 11 grade 2, 6 grade 3), followed by neutropenia (1 grade 1, 3 grade 2, 7 grade 3, 5 grade 4) and leukopenia (1 grade 1, 3 grade 2, 3 grade 3, 2 grade 4). Considering all toxicities, the highest toxicity recorded was grade 1 in 3 patients, grade 2 in 18 patients, grade 3 in 25 patients and grade 4 in 6 patients. Grade 3–4 toxicity was thus recorded in 59.6% of patients (31 of 52). Two patients developed anaphylactic shock at the first treatment cycle. Toxicity was not influenced by gender, age or Performance Status (PS).
Best clinical response, assessable in 49 patients, showed 3 CR, 21 PR, 11 SD and 14 PD, accounting for an overall response rate of 49%. Best response was significantly linked to the occurrence of an acneiform rash, with 65.5% response in patients developing grade 2–3 rash vs. 25% in those who did not [P = 0.007, odds ratio 5.7, 95% confidence interval (CI) 1.6–20.3]. Clinical response was higher in wild-type KRAS tumours compared with mutated KRAS tumours, even though the difference was not significant (64.7 vs. 47.4%, respectively, P = 0.29, odds ratio 2.0, 95% CI 0.53–7.8).
Median TTP was 5.7 months (95% CI 4.5–7.0). The TTP was not influenced by any demographic characteristics or treatment exposure (number of cycles, cumulative doses). Median overall survival was 18.6 months (95% CI 12.9–24.3). Overall survival was not related to demographic or therapeutic data other than PS (medians 20.9 vs. 9.9 months in patients with PS 0 and 1, respectively, P = 0.027), the number of administered cycles (median 18 months in patients with fewer than six cycles vs. 34 months in others, P < 0.001) and, as a corollary, the cumulative dose of cetuximab (P < 0.001), UFT (P < 0.001) and irinotecan (P < 0.001). The TTP and overall survival were not linked to KRAS mutation status.
Pharmacogenetic–pharmacodynamic relationships
Table 2 depicts the frequency of analysed genotypes, which were all in Hardy–Weinberg equilibrium. Of note, the irinotecan cumulative dose was not related to the UGT1A1 gene polymorphism (P = 0.54). One patient of the 52 exhibited the IVS14+1G>A mutation (heterozygous) on the DPYD gene; this patient (36-year-old man) received two chemotherapy cycles at full UFT dose (treatment stopped for progression) and developed a maximum toxicity grade 3 mucositis, associated with a grade 2 acneiform rash and grade 1 diarrhoea, nausea, vomiting, asthenia, leukopenia and neutropenia.
Table 2.
Distribution of gene polymorphisms
| Gene | Genotype | n | % | |
|---|---|---|---|---|
| TYMS | 28 bp repeats | 2R2R | 10 | 19.6 |
| 2R3R | 29 | 56.9 | ||
| 3R3R | 12 | 23.5 | ||
| Class including G>C | 2: 2R2R or 2R3RC or 3RC3RC | 23 | 52.3 | |
| 3: 2R3RG or 3RC3RG | 21 | 47.7 | ||
| 4: 3RG3RG | 0 | 0 | ||
| 6 bp deletion | wt/wt | 19 | 38.0 | |
| wt/del | 23 | 46.0 | ||
| del/del | 8 | 16.0 | ||
| MTHFR | 677C>T | CC | 20 | 40.0 |
| CT | 27 | 54.0 | ||
| TT | 3 | 6.0 | ||
| 1298A>C | AA | 24 | 48.0 | |
| AC | 24 | 48.0 | ||
| CC | 2 | 4.0 | ||
| UGT1A1 | TA repeats | 6/6 | 19 | 36.5 |
| 6/7 | 26 | 50.0 | ||
| 7/7 | 7 | 13.5 | ||
| EGFR | CA repeats (intron 1) | Both alleles <17 | 13 | 25.0 |
| One allele <17, one allele ≥17 | 22 | 42.3 | ||
| Both alleles ≥17 | 17 | 32.7 | ||
| −216G>T | GG | 17 | 33.4 | |
| GT | 27 | 52.9 | ||
| TT | 7 | 13.7 | ||
| −191C>A | CC | 38 | 73.1 | |
| CA | 14 | 26.9 | ||
| AA | 0 | 0 | ||
| EGF | 61A>G | AA | 19 | 36.5 |
| AG | 27 | 52.0 | ||
| GG | 6 | 11.5 | ||
| FCGR2A | 131Arg>His | Arg/Arg | 13 | 25.0 |
| Arg/His | 24 | 46.2 | ||
| His/His | 15 | 28.8 | ||
| FCGR3A | 158Phe>Val | Phe/Phe | 20 | 39.2 |
| Phe/Val | 25 | 49.0 | ||
| Val/Val | 6 | 11.8 |
The analysis of maximum toxicity grade, whatever the toxicity, revealed a marked tendency (P = 0.010) for patients bearing the EGFR−191C allele to develop greater toxicity, with 71.1% grade 3–4 toxicity in CC patients vs. 28.6% in CA patients (there was no AA patient), with an odds ratio of 6.13 (95% CI 1.58–23.79). In addition, an analysis focused on neutropenia demonstrated a strong tendency for deficient UGT1A1*28 patients to develop grade 3–4 neutropenia (P = 0.011); in comparison with *1/*1 patients, odds ratio was 3.13 (95% CI 0.57–17.2) for *1/*28 patients (n = 26) and 21.3 (95% CI 2.36–191.6) for *28/*28 patients (n = 7). Finally, analysis of cetuximab-related cutaneous toxicity, considering both acneiform rash and paronychia, revealed no influence of any EGFR or EGF polymorphism.
As regards efficacy, a trend was observed towards a better response in patients bearing the TYMS 3RG allele, i.e. belonging to the TYMS class 3 rather than class 2 (no class 4 in the present cohort, 65.0% response in class 3 vs. 28.6% in class 2, P = 0.029, odds ratio 4.64, 95% CI 1.24–17.37) and in patients bearing the FCGR3A 158Val genotype (62.1% response in Phe/Val or Val/Val vs. 26.3% in Phe/Phe, P = 0.020, odds ratio 4.58, 95% CI 1.29–16.27). We thus defined a favourable genotype score, considering both the class 3 TYMS genotype and any Val-containing FCGR3 genotype (Table 3). The greater the favourable genotype score, the better the response rate, with 9.1, 50.0 and 69.2% response in patients with a score of 0, 1 and 2, respectively (P = 0.009; Table 3). In a bivariate analysis including the KRAS mutation status (n = 29 patients), the favourable genotype score was no longer significant.
Table 3.
Impact of the favourable genotype score* on patient outcome
| Clinical response (complete response + partial response) | Overall survival | |||||
|---|---|---|---|---|---|---|
| Favourable genotype score | Number of responsive patients/total number of patients | % Response | Odds ratio (95% confidence interval) | Number of deaths/total number of patients | Median survival (months) | Relative risk of death (95% confidence interval) |
| 0 | 1/11 | 9.1% | 1 | 11/11 | 12.4 | 1 |
| 1 | 8/16 | 50.0% | 10.0 (1.03–97.5) | 15/19 | 14.7 | 0.49 (0.22–1.09) |
| 2 | 9/13 | 69.2% | 22.5 (2.11–240) | 8/13 | 28.7 | 0.23 (0.09–0.60) |
| Overall statistics | Fischer's exact test | Log rank | ||||
| P = 0.009 | P = 0.007 | |||||
The score corresponds to the number of favourable genotypes. Favourable genotypes are the class 3 TYMS genotype and any 158Val-containing FCGR3A genotype.
The TTP was not influenced by any of the analysed gene polymorphisms, including the previously defined favourable genotype score.
In line with pharmacogenetic relationships reported on responsiveness, a longer, although nonsignificant, overall survival was observed in patients belonging to the TYMS class 3 genotype (median 26.4 months in class 3 vs. 14.4 months in class 2, P = 0.041; Figure 1) and in patients bearing the FCGR3A 158Val genotype (20.9 months in Phe/Val or Val/Val vs. 12.4 months in Phe/Phe, P = 0.032; Figure 2). As illustrated in Figure 3, the score of favourable TYMS and FCGR3A genotypes significantly influenced overall survival, with a median of 12.4 months in patients with no favourable genotype, 14.7 months in patients with one favourable genotype and 28.7 months in patients with two favourable genotypes (log rank, P = 0.007; Table 3). In addition, a bivariate Cox analysis including both the favourable genotype score and the PS showed that the genotype score (P = 0.009) was a stronger survival predictor than the PS (P = 0.086). Finally, adding KRAS mutation status in the multivariate model did not improve the above statistical significance (P values of 0.026, 0.083 and 0.61 for genotype score, PS and KRAS, respectively; n = 30 patients).
Figure 1.
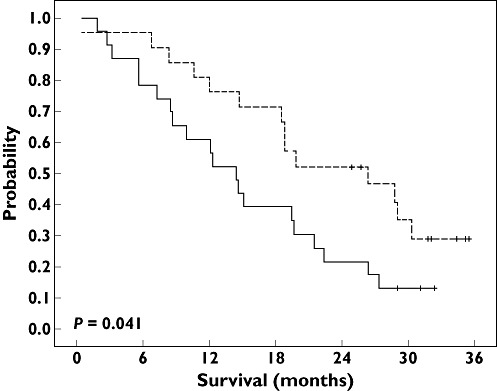
Overall survival probability according to TYMS genotype. Median overall survival was 14.4 months in class 2 patients (continuous line; 23 patients and 20 deaths) and 26.4 months in class 3 patients (dashed line; 21 patients and 14 deaths). Log rank test: P = 0.041. Class 2 ( ); class 3 (
); class 3 ( )
)
Figure 2.
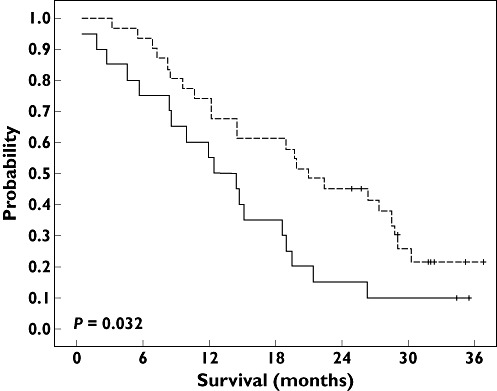
Overall survival probability according to FCGR3A 158Phe>Val genotype. Median overall survival was 12.4 months in Phe/Phe patients (continuous line; 20 patients and 18 deaths) and 20.9 months in Phe/Val or Val/Val patients (dashed line; 31 patients and 23 deaths). Log rank test: P = 0.032. Phe/Phe ( ); Phe/Val or Val/Val (
); Phe/Val or Val/Val ( )
)
Figure 3.
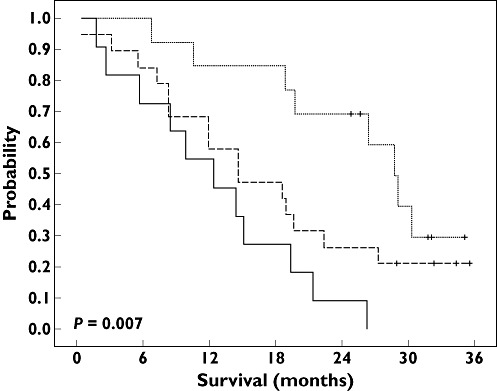
Overall survival probability according to the favourable genotype score, defined as class 3 TYMS genotype and Val-containing FCGR3A genotypes. Median overall survival was 12.4 months in patients with score 0 (continuous line; 11 patients and 11 deaths), 14.7 months in patients with score 1 (dashed line; 19 patients and 15 deaths) and 28.7 months in patients with score 2 (dotted line; 13 patients and 8 deaths). Log rank test: P = 0.007. Score 0 ( ); score 1 (
); score 1 ( ); score 2 (
); score 2 ( )
)
Discussion
The aim of this ancillary prospective study, conducted in 52 patients with metastatic CRC, was to perform a multifactorial pharmacogenetic analysis in patients receiving cetuximab in combination with irinotecan and tegafur-uracil plus folinic acid. To this end, we selected 11 relevant candidate gene polymorphisms that have previously been shown to influence the pharmacodynamics of cetuximab [9]–[12], irinotecan [5] or fluoropyrimidines [6], [7]. In contrast with the abundant literature on 5FU pharmacogenetics, little attention has been paid to the impact of DPYD, TYMS and MTHFR gene polymorphisms in the context of tegafur administration. However, such pharmacogenetic–pharmacodynamic relationships are supposed to be similar for 5FU and UFT, because p.o. UFT administration leads to 5FU systemic concentrations comparable to those observed after i.v. 5FU administration [20]. Pharmacogenetic studies on UFT have focused on functional CYP2A6 polymorphisms, because CYP2A6 is responsible for the activation of tegafur into 5FU [21]; however, CYP2A6 polymorphisms have not been included in the present study because these variants are very rare in the Caucasian population and are more common among Asians [22].
An intrinsic difficulty of such analyses is the multifactorial nature of both toxicity and efficacy. Analysis of the global toxicity, whatever the toxicity pattern, shows no influence of either TYMS or MTHFR polymorphisms, in agreement with the study of Tsunoda et al. [23], conducted in 99 patients receiving UFT–FA, and that of Schwab et al. [24], conducted in 683 patients receiving 5FU, suggesting that these two genes play a limited role in fluoropyrimidine-related toxicity. However, these results constrast with those of Lecomte et al. [25] and Kristensen et al. [26], both reporting a significant relationship between 2R2R TYMS genotype and an increased risk of grade 3–4 toxicity, in 90 and 68 colorectal cancer patients receiving 5FU-based treatment, respectively. Also, a recent study from Afzal et al. [27] reported that 677C>T and 1298A>C MTHFR genotypes associated with the greatest enzyme expressions were predictive of gastrointestinal toxicity after 5FU-based treatment. In the present study, the analysis of global toxicity revealed a marked influence of EGFR−191C>A polymorphism, with CC patients being more exposed to grade 3–4 toxicity compared with CA patients (odds ration 6.13, P = 0.01). Regarding cetuximab-related cutaneous toxicity, however, none of the analysed EGFR polymorphisms (−216G>T, −191C>A, CA repeats in intron 1) showed a significant predictive value. The lack of prediction of intron 1 CA repeats on skin toxicity has recently been reported in cetuximab-treated patients [28] and contrasts with previous data from Amador et al. [29] and Graziano et al. [9], who demonstrated greater skin toxicity in anti-EGFR-treated patients exhibiting fewer CA repeats in intron 1 of the EGFR gene. We also examined the impact of UGT1A1*28 polymorphism on neutropenia, because neutropenia is a limiting toxicity of irinotecan, although it may also be induced by UFT. The UGT1A1 enzyme governs the glucuronidation of SN38, the active metabolite of irinotecan, and numerous studies have shown that patients deficient for UGT1A1 enzyme, i.e. bearing the UGT1A1*28 variant, were prone to a lower SN38 glucuronidation rate and developed more severe neutropenia [5], [8], [30], [31]. Accordingly, the present data show a strong tendency for homozygous- and heterozygous-deficient patients to be at risk for developing grade 3–4 neutropenia, compared with homozygous nondeficient patients (odds ratios 21.3 and 3.13, respectively).
Patient recruitment was done before KRAS analysis was required for initiating cetuximab therapy. In line with data in the literature [32], response rate was higher in patients with a wild-type KRAS tumour compared with patients having a mutated KRAS tumour, although this difference did not reach significance (odds ratio 2.0, 95% CI 0.5–7.8). Also, response rate was significantly higher in patients developing skin toxicity (odds ratio 5.7, 95% CI 1.6–20.3), in agreement with previous studies [33], [34]. A pharmacological explanation for such a relationship may lie in pharmacokinetic variability, as suggested by Fracasso et al. [35], who reported higher cetuximab serum concentrations in patients with partial response/stable disease compared with patients having progressive disease. As for skin toxicity, we did not observe a significant link between EGFR gene polymorphisms and clinical response. This contrasts with data of Graziano et al. [9] reporting that patients with fewer CA repeats in intron 1 had both a higher response rate and skin toxicity, suggesting an additional pharmacogenetic explanation for the relationship between skin toxicity and cetuximab responsiveness.
In the present study, a trend was observed towards both a higher response rate and a longer overall survival in patients bearing the TYMS 3RG allele, as well as in patients bearing the FCGR3A 158Val allele. Interestingly, as illustrated in Table 3 and Figure 3, when combining TYMS 3RG and FCGR3A 158Val genotypes, the greater the number of favourable genotypes, the higher the response rate (P = 0.009) and the longer the overall survival (P = 0.007). Importantly, overall survival was significantly linked to PS, and a multivariate analysis including PS showed that the number of favourable genotypes was a significantly stronger survival predictor than PS.
Numerous studies have shown that elevated TS protein or mRNA expression is generally associated with poor outcome in patients receiving exclusive 5FU-based chemotherapy [36]. However, the impacts of TYMS gene polymorphisms on fluoropyrimidine pharmacodynamics are more conflicting [6], with some studies demonstrating a deleterious impact of TYMS 3RG genotypes [14], [37]–[39], whereas others do not [40], [41], and other investigators reporting a favourable impact of the 3R allele [42], [43]. In the majority of the above-mentioned studies [14], [37]–[42], as well as in the present one, TYMS genotyping was performed on blood DNA. Of note, TYMS gene is localized on the short arm of chromosome 18, which is prone to frequent deletions in colorectal cancers [44], thus resulting in loss of heterozygosity at the TYMS locus in the tumour [45]. As suggested in two clinical studies which analysed TYMS 2R3R genotype in both tumoural and blood DNA, TYMS gene polymorphism measured in blood DNA is not as relevant as TYMS gene polymorphism measured in tumour for predicting outcome of 5FU-treated patients [43], [46]. This observation may explain the inconsistency of TYMS pharmacogenetic–pharmacodynamic relationships in the literature.
Numerous in vitro studies have demonstrated that blood mononuclear cells, or NK cells, mediate cetuximab-induced ADCC against different cancer cell line types [47]–[50]. Furthermore, it has been shown that in vitro cell cytotoxicity was significantly higher with effector cells expressing FCGR3A 158Val/Val genotype compared with Phe/Val or Phe/Phe genotypes [48], [49]. This latter observation is in agreement with IgG binding experiments which demonstrated that anti-CD20 or anti-CD16 IgG1 mAbs display greater affinity for FcγR3A receptors carried by NK cells isolated from FCGR3A 158Val/Val individuals compared with FCGR3A 158Phe/Phe subjects [51], [52]. Taken together, these data suggest that FCGR3A 158Phe>Val genotype may influence the efficacy of cetuximab-based therapy, via ADCC. Accordingly, we report that patients carrying the FCGR3A 158Val allele exhibited a higher response rate and a longer survival than homozygous 158Phe/Phe patients, in line with data in the literature showing that the FCGR3A Val allele is associated with an improved outcome in patients treated with cetuximab [12] or with other IgG1 mAbs, such as rituximab [19], [53], [54] or trastuzumab [55]. This consistent finding that FCGR3A 158Val allele enhanced IgG1 mAb efficacy contrasts with a single study published by Pander et al. [11] showing the opposite pattern on 122 metastatic CRC patients receiving cetuximab together with bevacizumab, capecitabine and oxaliplatin, with longer progression-free survival in FCGR3A 158Phe/Phe patients (P = 0.025). Also, three studies did not reveal any significant relationship between FCGR3A 158Phe>Val genotype and outcome of metastatic CRC patients receiving cetuximab alone (35 and 127 patients, respectively) [10], [56] or in combination with irinotecan (110 patients) [9]. The therapeutic impact of ADCC in vivo is likely to be of secondary importance, as suggested by the lack of objective response observed in KRAS-mutated patients receiving cetuximab as monotherapy.
In conclusion, our present data suggest the importance of FCGR3A and TYMS gene polymorphisms in responsiveness and overall survival of patients receiving cetuximab–UFT based therapy. Engineering approaches of mAbs are currently being developed to enhance ADCC by increasing the affinity of mAb to Fcγ receptors. To this end, protein- and glyco-engineering of the mAb Fc region have recently been applied to cetuximab and have proved to be effective against KRAS-mutated tumours in vitro[57]. Such approaches open up promising prospects for improving anti-EGFR therapy in metastatic cancer patients and are presently being tested in clinical trials.
Acknowledgments
Acknowledgements to MERCK Serono for financial support.
Competing Interests
EF has received fees for speaking and consulting from Merck Laboratories. There are no other competing interests to declare.
REFERENCES
- 1.Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, Jandik P, Iveson T, Carmichael J, Alakl M, Gruia G, Awad L, Rougier P. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet. 2000;355:1041–7. doi: 10.1016/s0140-6736(00)02034-1. [DOI] [PubMed] [Google Scholar]
- 2.Lembersky BC, Wieand HS, Petrelli NJ, O'Connell MJ, Colangelo LH, Smith RE, Seay TE, Giguere JK, Marshall ME, Jacobs AD, Colman LK, Soran A, Yothers G, Wolmark N. Oral Uracil and tegafur plus leucovorin compared with intravenous fluorouracil and leucovorin in stage II and III carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project Protocol C-06. J Clin Oncol. 2006;24:2059–64. doi: 10.1200/JCO.2005.04.7498. [DOI] [PubMed] [Google Scholar]
- 3.Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D'Haens G, Pintér T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–17. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 4.Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, Loos AH, Zubel A, Koralewski P. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–71. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- 5.Toffoli G, Cecchin E, Corona G, Russo A, Buonadonna A, D'Andrea M, Pasetto LM, Pessa S, Errante D, De Pangher V, Giusto M, Medici M, Gaion F, Sandri P, Galligioni E, Bonura S, Boccalon M, Biason P, Frustaci S. The role of UGT1A1*28 polymorphism in the pharmacodynamics and pharmacokinetics of irinotecan in patients with metastatic colorectal cancer. J Clin Oncol. 2006;24:3061–8. doi: 10.1200/JCO.2005.05.5400. [DOI] [PubMed] [Google Scholar]
- 6.Lurje G, Manegold PC, Ning Y, Pohl A, Zhang W, Lenz HJ. Thymidylate synthase gene variations: predictive and prognostic markers. Mol Cancer Ther. 2009;8:1000–7. doi: 10.1158/1535-7163.MCT-08-0219. [DOI] [PubMed] [Google Scholar]
- 7.De Mattia E, Toffoli G. C677T and A1298C MTHFR polymorphisms, a challenge for antifolate and fluoropyrimidine-based therapy personalisation. Eur J Cancer. 2009;45:1333–51. doi: 10.1016/j.ejca.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 8.McLeod HL, Sargent DJ, Marsh S, Green EM, King CR, Fuchs CS, Ramanathan RK, Williamson SK, Findlay BP, Thibodeau SN, Grothey A, Morton RF, Goldberg RM. Pharmacogenetic Predictors of adverse events and response to chemotherapy in metastatic colorectal cancer: results from North American Gastrointestinal Intergroup Trial N9741. J Clin Oncol. 2010;28:3227–33. doi: 10.1200/JCO.2009.21.7943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graziano F, Ruzzo A, Loupakis F, Canestrari E, Santini D, Catalano V, Bisonni R, Torresi U, Floriani I, Schiavon G, Andreoni F, Maltese P, Rulli E, Humar B, Falcone A, Giustini L, Tonini G, Fontana A, Masi G, Magnani M. Pharmacogenetic profiling for cetuximab plus irinotecan therapy in patients with refractory advanced colorectal cancer. J Clin Oncol. 2008;26:1427–34. doi: 10.1200/JCO.2007.12.4602. [DOI] [PubMed] [Google Scholar]
- 10.Lurje G, Nagashima F, Zhang W, Yang D, Chang HM, Gordon MA, El-Khoueiry A, Husain H, Wilson PM, Ladner RD, Mauro DJ, Langer C, Rowinsky EK, Lenz HJ. Polymorphisms in cyclooxygenase-2 and epidermal growth factor receptor are associated with progression-free survival independent of K-ras in metastatic colorectal cancer patients treated with single-agent cetuximab. Clin Cancer Res. 2008;14:7884–95. doi: 10.1158/1078-0432.CCR-07-5165. [DOI] [PubMed] [Google Scholar]
- 11.Pander J, Gelderblom H, Antonini NF, Tol J, van Krieken JHJM, van der Straaten T, Punt CJA, Guchelaar HJ. Correlation of FCGR3A and EGFR germline polymorphisms with the efficacy of cetuximab in KRAS wild-type metastatic colorectal cancer. Eur J Cancer. 2010;46:1829–34. doi: 10.1016/j.ejca.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 12.Bibeau F, Lopez-Crapez E, Di Fiore F, Thezenas S, Ychou M, Blanchard F, Lamy A, Penault-Llorca F, Frébourg T, Michel P, Sabourin JC, Boissière-Michot F. Impact of FcγRIIa-FcγRIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. 2009;27:1122–9. doi: 10.1200/JCO.2008.18.0463. [DOI] [PubMed] [Google Scholar]
- 13.Di Fiore F, Sesboüé R, Michel P, Sabourin JC, Frebourg T. Molecular determinants of anti-EGFR sensitivity and resistance in metastatic colorectal cancer. Br J Cancer. 2010;103:1765–72. doi: 10.1038/sj.bjc.6606008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Largillier R, Etienne-Grimaldi MC, Formento JL, Ciccolini J, Nebbia JF, Ginot A, Francoual M, Renée N, Ferrero JM, Foa C, Namer M, Lacarelle B, Milano G. Pharmacogenetics of capecitabine in advanced breast cancer patients. Clin Cancer Res. 2006;12:5496–502. doi: 10.1158/1078-0432.CCR-06-0320. [DOI] [PubMed] [Google Scholar]
- 15.Etienne MC, Ilc K, Formento JL, Laurent-Puig P, Formento P, Cheradame S, Fischel JL, Milano G. Thymidylate synthase and methylenetetrahydrofolate reductase gene polymorphisms: relationships with 5-fluorouracil sensitivity. Br J Cancer. 2004;90:526–34. doi: 10.1038/sj.bjc.6601523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu W, Innocenti F, Wu MH, Desai AA, Dolan ME, Cook EH, Jr, Ratain MJ. A functional common polymorphism in a Sp1 recognition site of the epidermal growth factor receptor gene promoter. Cancer Res. 2005;65:46–53. [PubMed] [Google Scholar]
- 17.Etienne-Grimaldi MC, Pereira S, Magné N, Formento JL, Francoual M, Fontana X, Demard F, Dassonville O, Poissonnet G, Santini J, Bensadoun RJ, Szepetowski P, Milano G. Analysis of the dinucleotide repeat polymorphism in the epidermal growth factor receptor (EGFR) gene in head and neck cancer patients. Ann Oncol. 2005;16:934–41. doi: 10.1093/annonc/mdi189. [DOI] [PubMed] [Google Scholar]
- 18.Amend KL, Elder JT, Tomsho LP, Bonner JD, Johnson TM, Schwartz J, Berwick M, Gruber SB. EGF gene polymorphism and the risk of incident primary melanoma. Cancer Res. 2004;64:2668–72. doi: 10.1158/0008-5472.can-03-3855. [DOI] [PubMed] [Google Scholar]
- 19.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood. 2002;99:754–8. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- 20.Milano G, Ferrero JM, François E. Comparative pharmacology of oral fluoropyrimidines: a focus on pharmacokinetics, pharmacodynamics and pharmacomodulation. Br J Cancer. 2004;91:613–7. doi: 10.1038/sj.bjc.6601973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ozawa S, Hamada M, Murayama N, Nakajima Y, Kaniwa N, Matsumoto Y, Fukuoka M, Sawada J, Ohno Y. Cytosolic and microsomal activation of doxifluridine and tegafur to produce 5-fluorouracil in human liver. Cancer Chemother Pharmacol. 2002;50:454–8. doi: 10.1007/s00280-002-0528-1. [DOI] [PubMed] [Google Scholar]
- 22.Nakajima M, Fukami T, Yamanaka H, Higashi E, Sakai H, Yoshida R, Kwon JT, McLeod HL, Yokoi T. Comprehensive evaluation of variability in nicotine metabolism and CYP2A6 polymorphic alleles in four ethnic populations. Clin Pharmacol Ther. 2006;80:282–97. doi: 10.1016/j.clpt.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 23.Tsunoda A, Nakao K, Watanabe M, Matsui N, Ooyama A, Kusano M. Associations of various gene polymorphisms with toxicity in colorectal cancer patients receiving oral uracil and tegafur plus leucovorin: a prospective study. Ann Oncol. 2011;22:355–61. doi: 10.1093/annonc/mdq358. [DOI] [PubMed] [Google Scholar]
- 24.Schwab M, Zanger UM, Marx C, Schaeffeler E, Klein K, Dippon J, Kerb R, Blievernicht J, Fischer J, Hofmann U, Bokemeyer C, Eichelbaum M. Role of genetic and nongenetic factors for fluorouracil treatment-related severe toxicity: a prospective clinical trial by the German 5-FU Toxicity Study Group. J Clin Oncol. 2008;26:2131–8. doi: 10.1200/JCO.2006.10.4182. [DOI] [PubMed] [Google Scholar]
- 25.Lecomte T, Ferraz JM, Zinzindohoué F, Loriot MA, Tregouet DA, Landi B, Berger A, Cugnenc PH, Jian R, Beaune P, Laurent-Puig P. Thymidylate synthase gene polymorphism predicts toxicity in colorectal cancer patients receiving 5-fluorouracil-based chemotherapy. Clin Cancer Res. 2004;10:5880–8. doi: 10.1158/1078-0432.CCR-04-0169. [DOI] [PubMed] [Google Scholar]
- 26.Kristensen MH, Pedersen PL, Melsen GV, Ellehauge J, Mejer J. Variants in the dihydropyrimidine dehydrogenase, methylenetetrahydrofolate reductase and thymidylate synthase genes predict early toxicity of 5-fluorouracil in colorectal cancer patients. J Int Med Res. 2010;38:870–83. doi: 10.1177/147323001003800313. [DOI] [PubMed] [Google Scholar]
- 27.Afzal S, Gusella M, Vainer B, Vogel UB, Andersen JT, Broedbaek K, Petersen M, Jimenez-Solem E, Bertolaso L, Barile C, Padrini R, Pasini F, Jensen SA, Poulsen HE. Combinations of polymorphisms in genes involved in the 5-Fluorouracil metabolism pathway are associated with gastrointestinal toxicity in chemotherapy-treated colorectal cancer patients. Clin Cancer Res. 2011;17:3822–9. doi: 10.1158/1078-0432.CCR-11-0304. [DOI] [PubMed] [Google Scholar]
- 28.Klinghammer K, Knödler M, Schmittel A, Budach V, Keilholz U, Tinhofer I. Association of epidermal growth factor receptor polymorphism, skin toxicity, and outcome in patients with squamous cell carcinoma of the head and neck receiving cetuximab-docetaxel treatment. Clin Cancer Res. 2010;16:304–10. doi: 10.1158/1078-0432.CCR-09-1928. [DOI] [PubMed] [Google Scholar]
- 29.Amador ML, Oppenheimer D, Perea S, Maitra A, Cusati G, Iacobuzio-Donahue C, Baker SD, Ashfaq R, Takimoto C, Forastiere A, Hidalgo M. An epidermal growth factor receptor intron 1 polymorphism mediates response to epidermal growth factor receptor inhibitors. Cancer Res. 2004;64:9139–43. doi: 10.1158/0008-5472.CAN-04-1036. [DOI] [PubMed] [Google Scholar]
- 30.Iyer L, Das S, Janisch L, Wen M, Ramírez J, Karrison T, Fleming GF, Vokes EE, Schilsky RL, Ratain MJ. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2002;2:43–7. doi: 10.1038/sj.tpj.6500072. [DOI] [PubMed] [Google Scholar]
- 31.Kim TW, Innocenti F. Insights, challenges, and future directions in irinogenetics. Ther Drug Monit. 2007;29:265–70. doi: 10.1097/FTD.0b013e318068623b. [DOI] [PubMed] [Google Scholar]
- 32.Dahabreh IJ, Terasawa T, Castaldi PJ, Trikalinos TA. Systematic review: anti–epidermal growth factor receptor treatment effect modification by KRAS mutations in advanced colorectal cancer. Ann Intern Med. 2011;154:37–49. doi: 10.7326/0003-4819-154-1-201101040-00006. [DOI] [PubMed] [Google Scholar]
- 33.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 34.Peréz-Soler R, Saltz L. Cutaneous adverse effects with HER1/EGFR-targeted agents: is there a silver lining? J Clin Oncol. 2005;23:5235–46. doi: 10.1200/JCO.2005.00.6916. [DOI] [PubMed] [Google Scholar]
- 35.Fracasso PM, Burris H, III, Arquette MA, Govindan R, Gao F, Wright LP, Goodner SA, Greco FA, Jones SF, Willcut N, Chodkiewicz C, Pathak A, Springett GM, Simon GR, Sullivan DM, Marcelpoil R, Mayfield SD, Mauro D, Garrett CR. A phase 1 escalating single-dose and weekly fixed-dose study of cetuximab: pharmacokinetic and pharmacodynamic rationale for dosing. Clin Cancer Res. 2007;13:986–93. doi: 10.1158/1078-0432.CCR-06-1542. [DOI] [PubMed] [Google Scholar]
- 36.Popat S, Matakidou A, Houlston RS. Thymidylate synthase expression and prognosis in colorectal cancer: a systematic review and meta-analysis. J Clin Oncol. 2004;22:529–36. doi: 10.1200/JCO.2004.05.064. [DOI] [PubMed] [Google Scholar]
- 37.Marcuello E, Altés A, del Rio E, César A, Menoyo A, Baiget M. Single nucleotide polymorphism in the 5′ tandem repeat sequences of thymidylate synthase gene predicts for response to fluorouracil-based chemotherapy in advanced colorectal cancer patients. Int J Cancer. 2004;112:733–7. doi: 10.1002/ijc.20487. [DOI] [PubMed] [Google Scholar]
- 38.Ruzzo A, Graziano F, Kawakami K, Watanabe G, Santini D, Catalano V, Bisonni R, Canestrari E, Ficarelli R, Menichetti ET, Mari D, Testa E, Silva R, Vincenzi B, Giordani P, Cascinu S, Giustini L, Tonini G, Magnani M. Pharmacogenetic profiling and clinical outcome of patients with advanced gastric cancer treated with palliative chemotherapy. J Clin Oncol. 2006;24:1883–91. doi: 10.1200/JCO.2005.04.8322. [DOI] [PubMed] [Google Scholar]
- 39.Graziano F, Ruzzo A, Loupakis F, Santini D, Catalano V, Canestrari E, Maltese P, Bisonni R, Fornaro L, Baldi G, Masi G, Falcone A, Tonini G, Giordani P, Alessandroni P, Giustini L, Vincenzi B, Magnani M. Liver-only metastatic colorectal cancer patients and thymidylate synthase polymorphisms for predicting response to 5-fluorouracil-based chemotherapy. Br J Cancer. 2008;99:716–21. doi: 10.1038/sj.bjc.6604555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinez-Balibrea E, Abad A, Aranda E, Sastre J, Manzano JL, Díaz-Rubio E, Gómez-España A, Aparicio J, García T, Maestu I, Martínez-Cardús A, Ginés A, Guino E. Pharmacogenetic approach for capecitabine or 5-fluorouracil selection to be combined with oxaliplatin as first-line chemotherapy in advanced colorectal cancer. Eur J Cancer. 2008;44:1229–37. doi: 10.1016/j.ejca.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 41.Goekkurt E, Al-Batran SE, Hartmann JT, Mogck U, Schuch G, Kramer M, Jaeger E, Bokemeyer C, Ehninger G, Stoehlmacher J. Pharmacogenetic analyses of a phase III trial in metastatic gastroesophageal adenocarcinoma with fluorouracil and leucovorin plus either oxaliplatin or cisplatin: a study of the arbeitsgemeinschaft internistische onkologie. J Clin Oncol. 2009;27:2863–73. doi: 10.1200/JCO.2008.19.1718. [DOI] [PubMed] [Google Scholar]
- 42.Jakobsen A, Nielsen JN, Gyldenkerne N, Lindeberg J. Thymidylate synthase and methylenetetrahydrofolate reductase gene polymorphism in normal tissue as predictors of fluorouracil sensitivity. J Clin Oncol. 2005;23:1365–9. doi: 10.1200/JCO.2005.06.219. [DOI] [PubMed] [Google Scholar]
- 43.Dotor E, Cuatrecases M, Martínez-Iniesta M, Navarro M, Vilardell F, Guinó E, Pareja L, Figueras A, Molleví DG, Serrano T, de Oca J, Peinado MA, Moreno V, Germà JR, Capellá G, Villanueva A. Tumor thymidylate synthase 1494del6 genotype as a prognostic factor in colorectal cancer patients receiving fluorouracil-based adjuvant treatment. J Clin Oncol. 2006;24:1603–11. doi: 10.1200/JCO.2005.03.5253. [DOI] [PubMed] [Google Scholar]
- 44.Vogelstein B, Fearon ER, Kern SE, Hamilton SR, Preisinger AC, Nakamura Y, White R. Allelotype of colorectal carcinomas. Science. 1989;244:207–11. doi: 10.1126/science.2565047. [DOI] [PubMed] [Google Scholar]
- 45.Kawakami K, Ishida Y, Danenberg KD, Omura K, Watanabe G, Danenberg PV. Functional polymorphism of the thymidylate synthase gene in colorectal cancer accompanied by frequent loss of heterozygosity. Jpn J Cancer Res. 2002;93:1221–9. doi: 10.1111/j.1349-7006.2002.tb01227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uchida K, Hayashi K, Kawakami K, Schneider S, Yochim JM, Kuramochi H, Takasaki K, Danenberg KD, Danenberg PV. Loss of heterozygosity at the thymidylate synthase (TS) locus on chromosome 18 affects tumor response and survival in individuals heterozygous for a 28-bp polymorphism in the TS gene. Clin Cancer Res. 2004;10:433–9. doi: 10.1158/1078-0432.ccr-0200-03. [DOI] [PubMed] [Google Scholar]
- 47.Kurai J, Chikumi H, Hashimoto K, Yamaguchi K, Yamasaki A, Sako T, Touge H, Makino H, Takata M, Miyata M, Nakamoto M, Burioka N, Shimizu E. Antibody-dependent cellular cytotoxicity mediated by cetuximab against lung cancer cell lines. Clin Cancer Res. 2007;13:1552–61. doi: 10.1158/1078-0432.CCR-06-1726. [DOI] [PubMed] [Google Scholar]
- 48.Taylor RJ, Chan SL, Wood A, Voskens CJ, Wolf JS, Lin W, Chapoval A, Schulze DH, Tian G, Strome SE. FcγRIIIa polymorphisms and cetuximab induced cytotoxicity in squamous cell carcinoma of the head and neck. Cancer Immunol Immunother. 2009;58:997–1006. doi: 10.1007/s00262-008-0613-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.López-Albaitero A, Lee SC, Morgan S, Grandis JR, Gooding WE, Ferrone S, Ferris RL. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol Immunother. 2009;58:1853–64. doi: 10.1007/s00262-009-0697-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patel D, Guo X, Ng S, Melchior M, Balderes P, Burtrum D, Persaud K, Luna X, Ludwig DL, Kang X. IgG isotype, glycosylation, and EGFR expression determine the induction of antibody-dependent cellular cytotoxicity in vitro by cetuximab. Hum Antibodies. 2010;19:89–99. doi: 10.3233/HAB-2010-0232. [DOI] [PubMed] [Google Scholar]
- 51.Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M. FcγRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell FcγRIIIa, independently of the FcγRIIIa-48L/R/H phenotype. Blood. 1997;90:1109–14. [PubMed] [Google Scholar]
- 52.Dall'Ozzo S, Tartas S, Paintaud G, Cartron G, Colombat P, Bardos P, Watier H, Thibault G. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res. 2004;64:4664–9. doi: 10.1158/0008-5472.CAN-03-2862. [DOI] [PubMed] [Google Scholar]
- 53.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21:3940–7. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- 54.Treon SP, Hansen M, Branagan AR, Verselis S, Emmanouilides C, Kimby E, Frankel SR, Touroutoglou N, Turnbull B, Anderson KC, Maloney DG, Fox EA. Polymorphisms in FcγRIIIA (CD16) receptor expression are associated with clinical response to rituximab in Waldenström's macroglobulinemia. J Clin Oncol. 2005;23:474–81. doi: 10.1200/JCO.2005.06.059. [DOI] [PubMed] [Google Scholar]
- 55.Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, Laccabue D, Zerbini A, Camisa R, Bisagni G, Neri TM, Ardizzoni A. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. 2008;26:1789–96. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- 56.Zhang W, Gordon M, Schultheis AM, Yang DY, Nagashima F, Azuma M, Chang HM, Borucka E, Lurje G, Sherrod AE, Iqbal S, Groshen S, Lenz HJ. FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J Clin Oncol. 2007;25:3712–8. doi: 10.1200/JCO.2006.08.8021. [DOI] [PubMed] [Google Scholar]
- 57.Schlaeth M, Berger S, Derer S, Klausz K, Lohse S, Dechant M, Lazar GA, Schneider-Merck T, Peipp M, Valerius T. Fc-engineered EGF-R antibodies mediate improved antibody dependent cellular cytotoxicity (ADCC) against KRAS-mutated tumor cells. Cancer Sci. 2010;101:1080–8. doi: 10.1111/j.1349-7006.2010.01505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]