

Abstract
AIMS
To compare the O-demethylation (CYP2D6-mediated), N-demethylation (CYP3A4-mediated) and 6-glucuronidation (UGT2B4/7-mediated) metabolism of codeine between methadone- and buprenorphine-maintained CYP2D6 extensive metabolizer subjects.
METHODS
Ten methadone- and eight buprenorphine-maintained subjects received a single 60 mg dose of codeine phosphate. Blood was collected at 3 h and urine over 6 h and assayed for codeine, norcodeine, morphine, morphine-3- and -6-glucuronides and codeine-6-glucuronide.
RESULTS
The urinary metabolic ratio for O-demethylation was significantly higher (P = 0.0044) in the subjects taking methadone (mean ± SD, 2.8 ± 3.1) compared with those taking buprenorphine (0.60 ± 0.43), likewise for 6-glucuronide formation (0.31 ± 0.24 vs. 0.053 ± 0.027; P < 0.0002), but there was no significant difference (P = 0.36) in N-demethylation. Similar changes in plasma metabolic ratios were also found. In plasma, compared with those maintained on buprenorphine, the methadone-maintained subjects had increased codeine and norcodeine concentrations (P < 0.004), similar morphine (P = 0.72) and lower morphine-3- and -6- and codeine-6-glucuronide concentrations (P < 0.008).
CONCLUSION
Methadone is associated with inhibition of CYP2D6 and UGTs 2B4 and 2B7 reactions in vivo, even though it is not a substrate for these enzymes. Plasma morphine was not altered, owing to the opposing effects of inhibition of both formation and elimination; however, morphine-6-glucuronide (analgesically active) concentrations were substantially reduced. Drug interactions with methadone are likely to include drugs metabolized by various UGTs and CYP2D6.
Keywords: buprenorphine, codeine metabolism, CYP2D6 inhibition, glucuronidation inhibition, methadone
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Although not well recognized, methadone inhibits CYP2D6 in vivo and in vitro and UGT2B7 and 2B4 in vitro.
We aimed to investigate the effect of methadone on the pathways of codeine metabolism, namely O-demethylation (CYP2D6), 6-glucuronidation (UGT2B4/7) and N-demethylation (CYP3A4/2C8), in subjects maintained on methadone or buprenorphine as a control.
WHAT THIS STUDY ADDS
Compared with subjects on buprenorphine, methadone reduced the clearance of codeine to morphine and to codeine-6-glucuronide but had no effect on norcodeine formation.
Plasma morphine concentrations remained unchanged, as although its formation was reduced, its metabolism to M3G and M6G was also reduced.
Metabolic drug interactions with methadone cannot assume substrate-dependent inhibition.
Introduction
Methadone and buprenorphine maintenance therapies (MMT and BMT, respectively) are used in Australia and many other countries as treatment options for people who are addicted to opioids [1]. These therapies aim to reduce a person's use of drugs such as heroin, morphine and oxycodone, resulting in a reduced risk of death due to drug overdose, improved mental health and a lowered risk of developing diseases such as HIV and hepatitis [1].
Experience of pain is common in MMT patients. Rosenblum et al. Found that 80% of patients on methadone had experienced pain of any type or duration in the week preceding their study and 37% had experienced chronic severe pain [2]. Eleven per cent of MMT patients who then presented to their doctor in this situation were prescribed codeine alone and 24% were prescribed codeine in combination with paracetamol [2].
Codeine is a weak opioid drug used widely as an analgesic for the relief of mild to moderate pain [3], [4]. Up to 10% of the administered dose is oxidatively metabolized in the liver via O-demethylation to form morphine by CYP2D6 [5]–[9]. Morphine crosses the blood–brain barrier and binds to the µ-opioid receptor, at which it has a 200 times higher affinity than codeine [9]–[11]. The binding of morphine to the µ-opioid receptor is believed to be responsible, to a large degree, for the analgesia that occurs when taking codeine [3], [12]. Thus, the conversion of codeine to morphine is considered crucial for the analgesic effectiveness of codeine. Once morphine is formed, it is rapidly glucuronidated at the 3- or 6-positions to form the analgesically inactive morphine-3-glucuronide (M3G) and analgesically active morphine-6-glucuronide (M6G), respectively [9], [13]–[15]. The analgesic effect of codeine, therefore, depends on two metabolites, morphine and M6G. In the presence of methadone, however, the predominant enzyme responsible for the formation of both of these conjugated metabolites, UGT2B7 [16], [17], has been found to be noncompetitively inhibited in vitro, with the formation of M6G being more strongly inhibited than that of M3G [18]. Thus, giving morphine on its own, in the presence of methadone, may result in increased plasma morphine concentrations and reduced plasma M3G and M6G concentrations, the clinical consequences of which remain speculative.
Codeine also undergoes N-demethylation by CYP3A4/2C8 [19] to form norcodeine and glucuronidation via UDP-glucuronosyltransferases (UGTs) 2B4 and 2B7 [16], [17] to form codeine-6-glucuronide (C6G). N-Demethylation is a minor elimination pathway that accounts for ∼10% of the administered codeine dose, whereas glucuronidation is the major metabolic pathway and accounts for 60–80% [6], [8], [20], [21].
Mutations in the gene encoding for CYP2D6 are common, and their phenotypical outcome varies from no enzyme function to ultrarapid metabolism [22]. Approximately 7–10% of the Caucasian population are poor metabolizers (PM) and thus are unable to metabolize CYP2D6 substrates effectively, with clinical consequences [12], [22]–[24]. Although not a substrate, in both in vivo and in vitro human studies, methadone inhibits the CYP2D6-mediated O-demethylation of dextromethorphan to dextrorphan [25], [26] and inhibits the conversion of codeine to morphine in human liver microsomes (G. Mikus, F. Bochner, A.A. Somogyi unpublished observations).
Methadone is predominantly metabolized by CYP3A4 [27], CYP2B6 and, to a minor extent, by CYP2C19 [28], [29]. Buprenorphine is metabolized by UGT2B7 to the 3-glucuronide and by CYP3A4 to norbuprenorphine [29]–[31]. Buprenorphine is not a clinically significant inhibitor of CYP2D6 and therefore subjects in maintenance treatment would be a suitable population to evaluate whether methadone inhibits codeine O-demethylation and/or the glucuronidations of codeine or morphine in humans. The aim of this study was to compare the metabolism of codeine in methadone- and buprenorphine-maintained patients.
Materials and methods
The study was conducted as an open-label, parallel group investigation. The subjects were patients maintained on methadone or buprenorphine. The study was approved by the Royal Adelaide Hospital Research Ethics Committee (RAH protocol no. 060523a), and signed informed consent was obtained from each participant.
Subjects were recruited from Warinilla Clinic (Drug and Alcohol Services SA, Adelaide, Australia).
Both men and women, between the ages of 18 and 50 years, who had been on their current maintenance medication for at least 4 weeks and were on a stable dose for at least the past week, were eligible for inclusion in the study. Exclusion criteria included taking any known CYP2D6 inhibitor medication in the week prior to the study or having a positive urine drug screen for opioids (excluding their maintenance medication; Microcheck Multidrug Screening Test, Thermo Fisher Scientific, Scoresby, Victoria, Australia; limit of detection 300 ng ml−1). Genotypic CYP2D6 poor metabolizers were also excluded from analysis after the completion of the study. Likewise, those who had liver function test results (alanine aminotransferase, alkaline phosphatase and γ-glutamyl transferase) greater than three times the upper normal limit were excluded from analysis.
Subjects received their prescribed daily dose of methadone or buprenorphine according to the normal clinic protocol. Upon submitting a urine sample for testing and it being confirmed to be negative for opioids other than methadone or buprenorphine, they received a single, orally administered 60 mg dose of codeine phosphate as two 30 mg codeine phosphate tablets (Fawns & McAllan Pty Ltd, Belmont, Western Australia, Australia) with 200 ml water.
Biological sample collection
A 15 ml venous blood sample was taken by venepuncture at 3 h (approximate time to maximum plasma methadone concentration) for CYP2D6 genotype, liver function tests and plasma concentrations of codeine and metabolites. All urine passed for 6 h after codeine dosing was collected, volume and pH recorded and an aliquot retained for analysis. Samples were stored at −20°C until required for analysis.
Genotyping
Extraction of genomic DNA, PCRs, DNA sequencing and the subsequent CYP2D6 genotype (identifiable alleles *1-*10,*16,*33,*41,*45A/*45B/*46) was determined as previously described by us [32].
Analysis of plasma and urine concentrations
Urine
The urine samples were thawed and centrifuged, and a 10 µl aliquot was taken from each subject's sample and then 990 µl of mobile phase and 100 µl each of the internal standards d3-morphine (0.25 µg ml−1) and 100 µl of d3-morphine-6-glucuronide (0.25 µg ml−1) added; the tubes were vortex mixed and 200 µl placed in individual injection vials. A calibration curve of five standards prepared in drug-free urine containing codeine, C6G, norcodeine, morphine, M3G and M6G with concentrations ranging from 0.01 to 100 µg ml−1 for all six substances plus the internal standards were also prepared. The six analytes were prepared from individual weighings of pure substances. All analytes were quantified by the use of calibration curves (r2 > 0.99 for all analytes) of peak area ratios of analyte to d3-morphine for codeine, morphine and norcodeine and analyte to d3-M6G for M3G, M6G and C6G.
A modified assay [33] involved liquid chromatography-mass spectrometry (LC-MS) (Shimadzu, Kyoto, Japan) using a Luna 5 µm C18(2) 150 mm × 2.0 mm column; the mobile phase consisted of 25:475:2.75 methanol: 50 mm ammonium formate : formic acid. The electrospray ionization probe detection conditions were as follows: curved desolvation line (CDL) voltage 175 V, CDL temperature 250°C, block temperature 200°C, Q-array voltage DC 60 V and RF 150, probe high voltage 3.0 kC, detector gain voltage 1.8 kV, nebulizer gas (N2) 1.5 l min−1, drying gas (N2) 2 l min−1, and the run time for the assay was 20 min. The limits of quantification were as follows: codeine, morphine and norcodeine, 0.01 µg ml−1; M3G and M6G, 0.2 µg ml−1; and C6G, 0.1 µg ml−1. The assay was precise (coefficient of variation values of multiple replicates was <20%) and accurate (>85%).
Plasma
For the analysis of plasma codeine and metabolites morphine, M3G, M6G, norcodeine and C6G concentrations in the 3 h blood sample, an LC-MS-MS method was used [34] because the modified urine LC-MS method lacked sufficient sensitivity to measure the very low morphine and M6G concentrations. The lower limit of quantification was 0.05 ng ml−1, and intra- and interday precision and inaccuracy were <15%.
Plasma (R)- and (S)-methadone concentrations were quantified by LC-MS [35]. The lower limit of quantification was 0.05 ng ml−1, and intra- and interday precision and inaccuracy were <10%.
Data analysis
Urine
The percentage of the dose excreted as codeine and its five metabolites was calculated after taking into account molecular weight and drug base differences. A urinary metabolic ratio was calculated as the concentration of codeine divided by its metabolite(s) formed via each pathway as: N-demethylation, codeine/norcodeine; glucuronidation, codeine/C6G; and O-demethylation, codeine/morphine + M3G + M6G.
Plasma
A metabolic ratio for plasma was calculated as the plasma concentration of codeine divided by its metabolites formed via each pathway, as for urine.
Statistical analysis
Data for urinary recovery, plasma concentrations and metabolic ratios (urine and plasma) were statistically compared through the use of method 5 from [36] to provide an indication of the size of the effect of methadone on the metabolism of codeine. A probability score, U/mn, was calculated, where U is the Mann–Whitney U-statistic and mn the product of the two population sample sizes, with the possible score ranging between 0 and 1. A probability score of 0 or 1 indicated complete separation of the two maintenance populations' distributions and thus an effect of the methadone maintenance medication on the metabolism of codeine when compared with buprenorphine, whilst a score of 0.5, the null hypothesis value, indicated overlapping distributions and thus no effect of methadone.
The 95% confidence intervals of the probability scores were calculated through the use of an Excel spreadsheet available at http://medicine.cf.ac.uk/en/research/research-groups/clinical-epidemiology/resources/, as previously described [37].
The association between metabolic ratios and plasma methadone concentrations was assessed by the Spearman rank correlation coefficient (rs). These were all performed using GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego, CA, USA). Data are presented as means ± SD.
Results
Twenty-five patients were initially screened for eligibility for the study. Twenty-one met all initial inclusion criteria and participated in the trial, but the data from three subjects were excluded from the statistical analysis of results because two were genotypic poor metabolizers (CYP2D6*4/*4 and CYP2D6*4/*6) and one had liver function test results greater than three times the upper limit of normal. Thus, 18 subjects completed the study and were included in all statistical analyses; 10 subjects in MMT [three men and seven women; age range 23–37 years (mean 32.3 ± 4.64 years); daily dose range 35–200 mg (75 ± 54 mg)] and eight subjects in BMT [six men and two women; age range 24–32 years (mean 28.7 ± 2.31 years); daily dose range 4–34 mg (17 ± 9.6 mg)]. There was a statistically significant difference in age (P = 0.04, Mann–Whitney U-test) but not for gender (P = 0.15, Fisher's exact test) between the two groups.
Genotyping
Twenty of the twenty-one subjects who participated in the study were genotyped for CYP2D6 to determine their phenotype status. Fifteen of the subjects (eight MMT and seven BMT) were determined to be extensive metabolizers having the following genotypes: CYP2D6*1/*1 (n = 1 MMT), CYP2D6*1/*2 (n = 1 BMT), CYP2D6*1/*4 (n = 4; 3 MMT, 1 BMT), CYP2D6*1/*41 (n = 3; 2 MMT, 1BMT), CYP2D6*2/*2 (n = 2 BMT), CYP2D6*2/*4 (n = 1 MMT), CYP2D6*2/*5 (n = 1 BMT), CYP2D6*2/*33 (n = 1 BMT) and CYP2D6*2/*41 (n = 1 MMT). Two subjects were intermediate metabolizers with the CYP2D6*4/*41(MMT) genotype and another was an intermediate metabolizer with the CYP2D6*41/(*45A or *45B or *46 BMT) genotype.
Plasma concentrations and urinary recovery
For plasma concentrations, values for M3G, M6G and C6G were significantly lower (P < 0.008) in subjects receiving methadone compared with those receiving buprenorphine and significantly higher (P < 0.0004) for codeine and norcodeine (Table 1). The greatest differences were seen in the plasma norcodeine concentrations, which were sixfold higher in the subjects maintained on methadone, and M3G and M6G, which were about 0.3 and 0.4 that of buprenorphine-maintained subjects. For urinary recovery, values for codeine and norcodeine were significantly higher (P < 0.05), whereas C6G and M6G were significantly lower (P < 0.006) in subjects receiving methadone compared with those receiving buprenorphine (Table 1).
Table 1.
Comparison of plasma concentrations at 3 h (a) and 6 h urinary recovery (b) of codeine and five metabolites between subjects on methadone (MMT) and buprenorphine maintenance treatment (BMT) after taking a single dose of 60 mg codeine phosphate
| Analyte | BMT (n = 8) | MMT (n = 10) | P value* | Probability score† (95% confidence interval) |
|---|---|---|---|---|
| (a) Plasma concentration (ng ml−1) | ||||
| Codeine | 58.5 ± 15.3 | 174 ± 45.1 | <0.0001 | 1.0 (0.78–1.00) |
| Morphine | 1.15 ± 0.58 | 1.15 ± 0.430 | 0.722 | 0.56 (0.31–0.78) |
| M6G | 8.97 ± 3.99 | 2.59 ± 1.05 | 0.0021 | 0.088 (0.02–0.35) |
| M3G | 39.1 ± 17.1 | 16.4 ± 6.90 | 0.0077 | 0.12 (0.03–0.39) |
| C6G | 1110 ± 195 | 625 ± 202 | 0.0005 | 0.05 (0.008–0.30) |
| Norcodeine | 3.02 ± 1.66 | 18.8 ± 6.99 | 0.0004 | 1.0 (0.78–1.00) |
| (b) Urinary recovery (mg) | ||||
| Codeine | 0.631 ± 0.379 | 1.27 ± 0.75 | 0.043 | 0.79 (0.51–0.92) |
| Morphine | 0.208 ± 0.523 | 0.015 ± 0.019 | 0.146 | 0.29 (0.12–0.56) |
| M6G | 0.254 ± 0.403 | 0.252 ± 0.720 | 0.006 | 0.13 (0.034–0.39) |
| M3G | 5.55 ± 11.5 | 0.63 ± 0.44 | 0.068 | 0.24 (0.09–0.51) |
| C6G | 14.1 ± 4.81 | 6.18 ± 4.27 | 0.0044 | 0.11 (0.03–0.38) |
| Norcodeine | 0.177 ± 0.11 | 0.45 ± 0.21 | 0.0117 | 0.85 (0.58–0.96) |
Data are displayed as means ± SD. Abbreviations: C6G, codeine-6-glucuronide; M3G, morphine-3-glucuronide; and M6G, morphine-6-glucuronide.
Mann–Whitney U-test.
Probability score =U/mn (methadone > buprenorphine).
The total urinary recovery of codeine and metabolites was 19.9 ± 12.9% of dose in MMT compared with 47.8 ± 28.7% of dose in BMT (P < 0.006).
Metabolic ratios
The urinary metabolic ratio for O-demethylation was significantly higher (P = 0.0044) in the subjects taking methadone compared with those taking buprenorphine [probability score 0.89; 95% confidence interval (CI) 0.62–0.97; Figure 1a]. Likewise, the urinary metabolic ratio for glucuronidation to C6G was significantly (P = 0.0002) higher in MMT compared with BMT subjects (probability score 0.98; 95% CI 0.74–1.00; Figure 1b). There was no difference in the N-demethylation ratio (P = 0.36) to norcodeine (probability score 0.36 95% CI 0.17–0.63; Figure 1c). In addition, the morphine glucuronidation ratio was not significantly different (P = 0.97; probability score 0.51; 95% CI 0.27–0.75).
Figure 1.
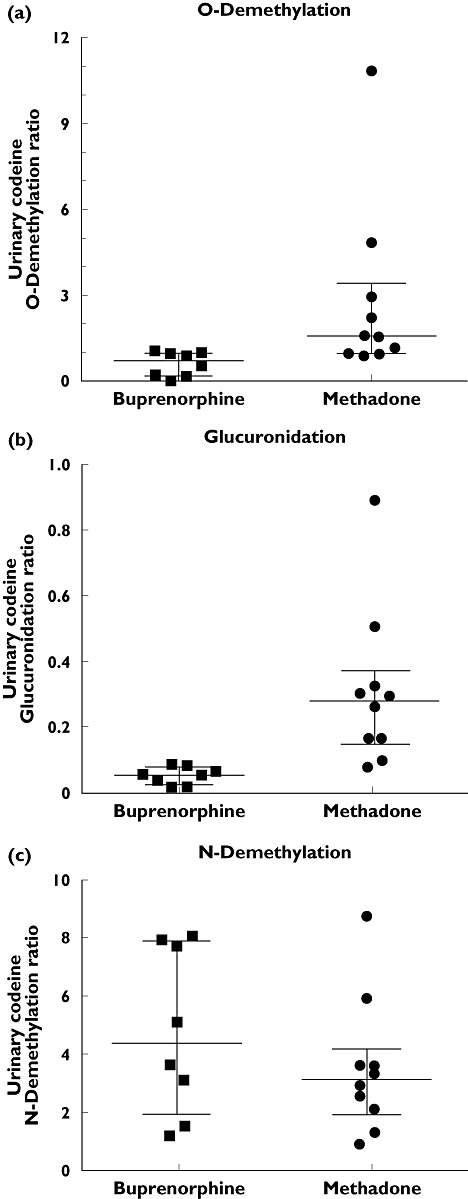
Comparison of urinary metabolic ratios for codeine O-demethylation to morphine + morphine-3-glucuronide + morphine-6-glucuronide (a), codeine glucuronidation to codeine-6-glucuronide (b) and codeine N-demethylation to norcodeine (c) between methadone- and buprenorphine-maintained subjects. Data are individual values, with the line indicating median and bars the interquartile range
The plasma metabolic ratio for O-demethylation and codeine glucuronidation were both significantly higher (O-demethylation, P < 0.0001, probability score 1.0, 95% CI 0.78–1.0; and glucuronidation, P < 0.0001, probability score 1.0, 95% CI 0.78–1.0) in MMT subjects, whilst the N-demethylation ratio was significantly (P = 0.02) lower in the MMT subjects (probability score 0.16; 95% CI 0.05–0.44). The morphine glucuronidation ratio was significantly different (P = 0.006; probability score 0.88; 95% CI 0.61–0.97).
Figure 2 summarizes the effect of methadone on codeine metabolism and plasma concentrations of codeine and metabolites.
Figure 2.
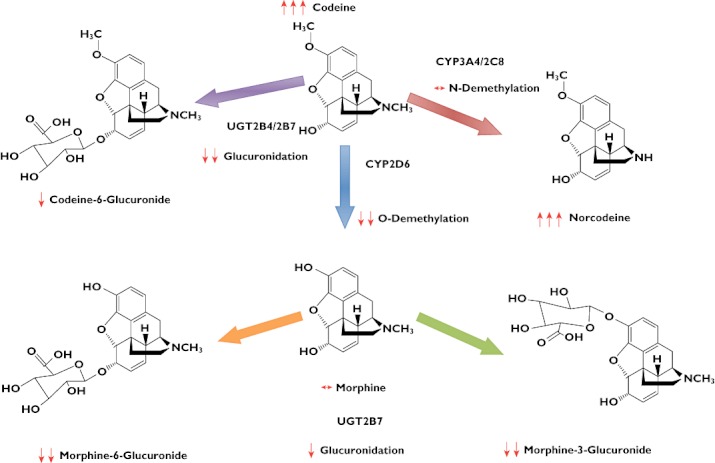
In vivo effect of methadone on the plasma concentrations of codeine and metabolites expressed as mean percentage change and overall influence on the metabolic pathways of codeine. Key: ↔, no change; ↓, 0–50% decrease; ↓↓, 51–100% decrease; and ↑↑↑, >100% increase
There was no relationship between the urinary and plasma metabolic ratios for codeine O-demethylation (MMT, rs= 0.39, P = 0.26; and BMT, rs= 0.33, P = 0.43), glucuronidation (MMT, rs= 0.53, P = 0.12; and BMT, rs= 0.071, P = 0.88) and N-demethylation (MMT, rs= 0.60, P = 0.073; and BMT, rs= 0.33, P = 0.43).
In the MMT subjects, the plasma concentrations of methadone were as follows: (R)-methadone 256 ± 163 ng ml−1 (range to 595 ng ml−1) and (S)-methadone 304 ± 166 ng ml−1 (range 104–542 ng ml−1). There was no relationship between the urinary or plasma O-demethylation metabolic ratio in the MMT subjects and plasma racemic methadone (urine, rs=−0.067, P = 0.87; and plasma, rs=−0.055, P = 0.89). Likewise, there was no relationship between the urinary or plasma glucuronidation metabolic ratio and plasma racemic methadone (urine, rs=−0.188, P = 0.45; and plasma, rs=−0.188, P = 0.32).
Discussion
The urinary metabolic ratios for codeine O-demethylation to morphine and glucuronidation to C6G were significantly elevated in the methadone-maintained subjects by factors of 4.7 and 6.3 based on medians, respectively, but no difference was found in N-demethylation. As presented here, the urinary metabolic ratio of codeine to metabolite(s) is a function of the renal clearance of codeine and inversely related to the unbound fraction of codeine in blood and the intrinsic clearance via the metabolic pathways [38]. It is highly unlikely that the renal codeine clearance should be significantly different between methadone- and buprenorphine-maintained subjects, as its value is about 180 ml min−1[6] and even if the tubular secretory pathway were to be blocked, it would result in a less than doubling of the ratio. Furthermore, urine pH was not different between the two groups, hence discounting any effect on tubular reabsorption and, finally, the unbound fraction of codeine is high, leading to the conclusion that methadone reduced the intrinsic clearance by the two pathways of O-demethylation via CYP2D6 and glucuronidation via UGTs 2B4 and 2B7. Likewise, for plasma, the metabolic ratio of AUC of codeine to metabolite(s) is a function of the unbound fraction of metabolite(s) in plasma (which are all very high) and renal clearance of codeine and is inversely related to the intrinsic clearance of codeine via the pathway [39]. Plasma metabolic ratios for codeine O-demethylation and glucuronidation were increased in methadone-maintained subjects, supporting the urine data, even though only a single plasma concentration and not AUC was used. However, the ratio for N-demethylation was significantly reduced, in contrast to the urinary metabolic ratio (which remained unchanged), and might indicate the lack of robustness of a single blood sample to estimate a metabolic ratio.
Of interest was the effect of methadone on morphine and metabolites. In plasma, morphine concentrations were the same between the two groups. This would not have been predicted, given that codeine O-demethylation to morphine was inhibited by methadone, which would predict lower plasma morphine concentrations. However, plasma M3G and M6G concentrations were reduced by 2.4- and 3.5-fold, respectively, implying that methadone also inhibited morphine glucuronidation to these active metabolites with antinociceptive (M6G) and neuroexcitatory (M3G) effects. The urinary metabolic ratio for morphine glucuronidation was not significantly altered; however, the plasma metabolic ratio was significantly higher in methadone subjects, indicating inhibition of glucuronidation. The reason for this discrepancy between plasma and urine is uncertain. Nevertheless, the plasma metabolic ratio data are consistent with a previous study that found both the (R)- and the (S)-methadone enantiomers noncompetitively inhibited UGT2B7 in vitro using liver microsomes, through reducing the formation of M3G and M6G from morphine [18].
The codeine glucuronidation ratio and individual codeine-6-glucuronide concentrations indicate that there appears to be a significant effect of methadone on the UGTs 2B7 and 2B4, which are responsible for codeine glucuronidation. Indeed, Raungrut et al. [40] showed that methadone had an inhibition constant of 0.32 µm for codeine glucuronidation. They also predicted that at a methadone dosing rate of 74 mg day−1, plasma codeine concentrations would be 1.72-fold higher. Our subjects were taking on average 75 mg day−1, and the increase in plasma codeine concentration was on average 2.97, similar to that predicted, but not taking into account the small contribution of O-demethylation inhibition or the fact that a single time point was assessed and not AUC values.
Plasma norcodeine concentrations were sixfold higher in methadone-maintained subjects, resulting in a minor metabolite now becoming predominant. Methadone did not affect the N-demethylation of codeine using urine data, even though both share a common isoenzyme for metabolism, CYP3A4. It is likely that plasma methadone concentrations were insufficiently high enough to inhibit norcodeine formation. Thus, the substantial increase in plasma norcodeine concentrations was mainly due to methadone inhibition of C6G formation, with a small overall contribution from O-demethylation inhibition, driving more of the codeine down the N-demethylation pathway.
There are several limitations to the study. Firstly, there was a lack of a control group of subjects not receiving maintenance treatment. There is no evidence that buprenorphine affects (induces or inhibits) the metabolism of other drugs even though it is metabolized by UGT2B7 and CYP3A4 [29]–[31]; lack of competitive inhibition of metabolism clinically may be due to the very low plasma concentrations (<10 ng ml−1) that are attained in maintenance subjects. In addition, the mean plasma codeine and codeine-6-glucuronide concentrations attained in the buprenorphine group at 3 h (58.5 and 1110 ng ml−1, respectively) are very similar (60–70 and 1100–1300 ng ml−1, respectively) to those estimated from reported data in healthy subjects [6], [21], [34]. Likewise, the mean plasma glucuronidation and O-demethylation ratios of 0.05 and 1.2 are comparable (0.050–0.055 and 0.42–0.74, respectively) and the O-demethylation urinary metabolic ratio of 1.19 is similar to those (0.75–1.5) estimated from those previous studies [6], [21], [34]. Hence, it is likely that the buprenorphine group could be considered to be fairly similar in their O-demethylation and glucuronidation of codeine to healthy subjects on no treatment. Blood samples were collected at only one time point, thus not allowing for full AUC values to be estimated. The poor state of venous access in these subjects prevented multiple blood sampling. The implications of one plasma concentration to represent an AUC value would be greater variability in differences between the two groups; however, the close concordance between the urinary and plasma concentration data, especially for the O-demethylation and 6-glucuronidation pathways, suggests that the latter did not lead to misinterpretation. In addition, urine was only collected for 6 h, which is reasonable in the buprenorphine group where the half-lives of all analytes would be similar to codeine (about 2 h) and as a result about 50% of the dose could be recovered. However, in the methadone-maintained subjects, the half-lives of codeine and its metabolites would be prolonged at least twofold for morphine and metabolites, resulting in only about 20% dose recovery. This could make estimates of metabolic ratios less reliable. However, given the large differences found, it is unlikely to have greatly influenced the interpretation. Also, normorphine and its glucuronides (a metabolite of both morphine and norcodeine) were not assayed. As these metabolites combined represent <5% of the codeine dose [21], it is unlikely that they significantly influenced the data analysis and interpretation.
The clinical implications of the findings are such that they provide a likely additional reason for not using codeine for pain relief in opioid maintenance patients and, hence, have the potential to improve opioid prescribing practices. As these patients are opioid tolerant, codeine per se would be a less effective analgesic even at high doses. However, in methadone-maintained patients, codeine is likely to be even less effective than in buprenorphine-maintained patients, because the analgesic effect would be only from morphine, while little if any effect would result from the greatly reduced M6G concentrations. The clinical implications of the elevated concentrations of the parent drug, codeine, and substantially elevated plasma norcodeine are unknown, but both are unlikely to contribute to analgesia. Therefore, the use of codeine as a simple analgesic in methadone maintenance patients is likely to be ineffective not only due to opioid tolerance but also due to unfavourable metabolic codeine–methadone drug interaction. Furthermore, the mechanistic implications are also important. As methadone is associated with reduced functional activity of CYP2D6 and UGTs 2B4 and 7, even though it is not a substrate for these enzymes, our findings suggest that drug interactions with methadone are likely to include drugs metabolized by various UGTs and CYP2D6. Thus, the implications when methadone is used in chronic pain patients who are receiving multiple analgesic and symptom control medications cannot be readily dismissed.
In summary, this study shows a significant effect of methadone on the function of CYP2D6 and also UGTs 2B7 and 2B4. Methadone inhibits morphine formation via CYP2D6, and C6G formation via UGTs 2B4 and 2B7. This results in significantly higher plasma codeine and norcodeine concentrations, lower C6G, M3G and M6G concentrations and unchanged morphine concentrations.
Acknowledgments
We thank Aaron Farquharson for his help in organizing the trial, recruiting subjects and taking the blood samples, Martin Hurley (Drug and Alcohol Services SA) for assisting in the recruitment of subjects and for enabling the use of Warinilla Clinic facilities during the trial and Dr Benjamin Davies for his advice and for the LC-MS analysis. Thomas Sullivan provided valuable statistical advice. The study was supported by the National Health and Medical Research Council of Australia.
Competing Interests
There are no competing interests to declare.
REFERENCES
- 1.O'Brien S. Treatment Options for Heroin and Other Opioid Dependence – A Guide for Users. Sydney: National Drug and Alcohol Research Centre, University of New South Wales; 2004. [Google Scholar]
- 2.Rosenblum A, Joseph H, Fong C, Kipnis S, Cleland C, Portenoy RK. Prevalence and characteristics of chronic pain among chemically dependent patients in methadone maintenance and residential treatment facilities. JAMA. 2003;289:2370–8. doi: 10.1001/jama.289.18.2370. [DOI] [PubMed] [Google Scholar]
- 3.Mikus G, Somogyi AA, Bochner F, Eichelbaum M. Codeine O-demethylation: rat strain differences and the effects of inhibitors. Biochem Pharmacol. 1991;41:757–62. doi: 10.1016/0006-2952(91)90077-i. [DOI] [PubMed] [Google Scholar]
- 4.Rossi S. Australian Medicines Handbook. Adelaide: Australian Medicines Handbook Pty Ltd; 2011. [Google Scholar]
- 5.Chen ZR, Somogyi AA, Bochner F. Polymorphic O-demethylation of codeine. Lancet. 1988;2:914–5. doi: 10.1016/s0140-6736(88)92529-9. [DOI] [PubMed] [Google Scholar]
- 6.Chen ZR, Somogyi AA, Reynolds G, Bochner F. Disposition and metabolism of codeine after single and chronic doses in one poor and seven extensive metabolisers. Br J Clin Pharmacol. 1991;31:381–90. doi: 10.1111/j.1365-2125.1991.tb05550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dayer P, Desmeules J, Leemann T, Striberni R. Bioactivation of the narcotic drug codeine in human liver is mediated by the polymorphic monooxygenase catalyzing debrisoquine 4-hydroxylation (cytochrome P-450 db1/bufI) Biochem Biophys Res Commun. 1988;152:411–6. doi: 10.1016/s0006-291x(88)80729-0. [DOI] [PubMed] [Google Scholar]
- 8.Poulsen L, Brøsen K, Arendt-Nielsen L, Gram LF, Elbæk K, Sindrup SH. Codeine and morphine in extensive and poor metabolisers of sparteine: pharmacokinetics, analgesic effect and side effects. Eur J Clin Pharmacol. 1996;51:289–95. doi: 10.1007/s002280050200. [DOI] [PubMed] [Google Scholar]
- 9.Coller JK, Christrup LL, Somogyi AA. Role of active metabolites in the use of opioids. Eur J Clin Pharmacol. 2009;65:121–39. doi: 10.1007/s00228-008-0570-y. [DOI] [PubMed] [Google Scholar]
- 10.Chen ZR, Irvine RJ, Somogyi AA, Bochner F. Mu receptor binding of some commonly used opioids and their metabolites. Life Sci. 1991;48:2165–71. doi: 10.1016/0024-3205(91)90150-a. [DOI] [PubMed] [Google Scholar]
- 11.Lötsch J, Skarke C, Schmidt H, Rohrbacher M, Hofmann U, Schwab M, Geisslinger G. Evidence for morphine-independent central nervous opioid effects after administration of codeine: contribution of other codeine metabolites. Clin Pharmacol Ther. 2006;79:35–48. doi: 10.1016/j.clpt.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 12.Somogyi AA, Barratt DT, Coller JK. Pharmacogenetics of opioids. Clin Pharmacol Ther. 2007;81:429–44. doi: 10.1038/sj.clpt.6100095. [DOI] [PubMed] [Google Scholar]
- 13.Shimomura K, Kamata O, Ueki S, Ida S, Oguri K. Analgesic effect of morphine glucuronides. Tohoku J Exp Med. 1971;105:45–52. doi: 10.1620/tjem.105.45. [DOI] [PubMed] [Google Scholar]
- 14.Milne RW, Nation RL, Somogyi AA. The disposition of morphine and its 3- and 6-glucuronide metabolites in humans and animals, and the importance of the metabolites to the pharmacological effects of morphine. Drug Metab Rev. 1996;28:345–472. doi: 10.3109/03602539608994011. [DOI] [PubMed] [Google Scholar]
- 15.Trescot AM, Datta S, Lee M, Hansen H. Opioid pharmacology. Pain Physician. 2008;11(2 Suppl.):S133–53. [PubMed] [Google Scholar]
- 16.Court MH, Krishnaswamy S, Hao Q, Duan SX, Patten CJ, Von Moltke LL, Greenblatt DJ. Evaluation of 3′-azido-3′-deoxythymidine, morphine, and codeine as probe substrates for UDP-glucuronosyltransferase 2B7 (UGT2B7) in human liver microsomes: specificity and influence of the UGT2B7*2 polymorphism. Drug Metab Dispos. 2003;31:1125–33. doi: 10.1124/dmd.31.9.1125. [DOI] [PubMed] [Google Scholar]
- 17.Miners JO, Mackenzie PI, Knights KM. The prediction of drug-glucuronidation parameters in humans: UDP-glucuronosyltransferase enzyme-selective substrate and inhibitor probes for reaction phenotyping and in vitro-in vivo extrapolation of drug clearance and drug-drug interaction potential. Drug Metab Rev. 2010;42:196–208. doi: 10.3109/03602530903210716. [DOI] [PubMed] [Google Scholar]
- 18.Morrish GA, Foster DJ, Somogyi AA. Differential in vitro inhibition of M3G and M6G formation from morphine by (R)- and (S)-methadone and structurally related opioids. Br J Clin Pharmacol. 2006;61:326–35. doi: 10.1111/j.1365-2125.2005.02573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caraco Y, Tateishi T, Guengerich FP, Wood AJ. Microsomal codeine N-demethylation: cosegregation with cytochrome P4503A4 activity. Drug Metab dispos. 1996;24:761–4. [PubMed] [Google Scholar]
- 20.Vree TB, Verwey-van Wissen CP. Pharmacokinetics and metabolism of codeine in humans. Biopharm Drug Dispos. 1992;13:445–60. doi: 10.1002/bdd.2510130607. [DOI] [PubMed] [Google Scholar]
- 21.Yue QY, Hasselstrom J, Svensson JO, Sawe J. Pharmacokinetics of codeine and its metabolites in Caucasian healthy volunteers: comparisons between extensive and poor hydroxylators of debrisoquine. Br J Clin Pharmacol. 1991;31:635–42. doi: 10.1111/j.1365-2125.1991.tb05585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zanger UM, Raimundo S, Eichelbaum M. Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:23–37. doi: 10.1007/s00210-003-0832-2. [DOI] [PubMed] [Google Scholar]
- 23.Sachse C, Brockmöller J, Bauer S, Roots I. Cytochrome P450 2D6 variants in a Caucasian population: allele frequencies and phenotypic consequences. Am J Hum Genet. 1997;60:284–95. [PMC free article] [PubMed] [Google Scholar]
- 24.Cascorbi I. Pharmacogenetics of cytochrome p4502D6: genetic background and clinical implication. Eur J Clin Invest. 2003;33(Suppl. 2):17–22. doi: 10.1046/j.1365-2362.33.s2.3.x. [DOI] [PubMed] [Google Scholar]
- 25.Wu D, Otton SV, Sproule BA, Busto U, Inaba T, Kalow W, Sellers EM. Inhibition of human cytochrome P450 2D6 (CYP2D6) by methadone. Br J Clin Pharmacol. 1993;35:30–4. doi: 10.1111/j.1365-2125.1993.tb05666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kerry NL, Somogyi AA, Bochner F, Mikus G. The role of CYP2D6 in primary and secondary oxidative metabolism of dextromethorphan: in vitro studies using human liver microsomes. Br J Clin Pharmacol. 1994;38:243–8. doi: 10.1111/j.1365-2125.1994.tb04348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foster DJR, Somogyi AA, Bochner F. Methadone N-demethylation in human liver microsomes: lack of stereoselectivity and involvement of CYP3A4. Br J Clin Pharmacol. 1999;47:403–12. doi: 10.1046/j.1365-2125.1999.00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang Y, Fang WB, Lin SN, Moody DE. Stereo-selective metabolism of methadone by human liver microsomes and cDNA-expressed cytochrome P450s: a reconciliation. Basic Clin Pharmacol Toxicol. 2011;108:55–62. doi: 10.1111/j.1742-7843.2010.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Somogyi AA, Coller JK. Metabolism of Drugs and Other Xenobiotics. Berlin: Wiley-VCH; 2011. Analgesic and adjuvant drugs. (in press) [Google Scholar]
- 30.Iribarne C, Picart D, Dréano Y, Bail J-P, Berthou F. Involvement of cytochrome P450 3A4 in N-dealkylation of buprenorphine in human liver microsomes. Life Sci. 1997;60:1953–64. doi: 10.1016/s0024-3205(97)00160-4. [DOI] [PubMed] [Google Scholar]
- 31.Rouguieg K, Picard N, Sauvage FL, Gaulier JM, Marquet P. Contribution of the different UDP-glucuronosyltransferase (UGT) isoforms to buprenorphine and norbuprenorphine metabolism and relationship with the main UGT polymorphisms in a bank of human liver microsomes. Drug Metab Dispos. 2010;38:40–5. doi: 10.1124/dmd.109.029546. [DOI] [PubMed] [Google Scholar]
- 32.James HM, Coller JK, Gillis D, Bahnisch J, Sallustio BC, Somogyi AA. A new simple diagnostic assay for the identification of the major CYP2D6 genotypes by DNA sequencing analysis. Int J Clin Pharmacol Ther. 2004;42:719–23. doi: 10.5414/cpp42719. [DOI] [PubMed] [Google Scholar]
- 33.Somogyi AA, Larsen M, Abadi RM, Jittiwutikarn J, Ali R, White JM. Flexible dosing of Tincture of Opium in the management of opioid withdrawal: pharmacokinetics and pharmacodynamics. Br J Clin Pharmacol. 2008;66:640–7. doi: 10.1111/j.1365-2125.2008.03277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirchheiner J, Schmidt H, Tzvetkov M, Keulen J-THA, Lötsch J, Roots I, Brockmöller J. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J. 2007;7:257–65. doi: 10.1038/sj.tpj.6500406. [DOI] [PubMed] [Google Scholar]
- 35.Foster DJ, Morton E, Murtder T, Somogyi A. Stereoselective quantification of methadone and a d(6)-labeled isotopomer using high performance liquid chromatography-atmospheric pressure chemical ionization mass-spectrometry: application to a pharmacokinetic study in a methadone maintained subject. Ther Drug Monit. 2006;28:559–67. doi: 10.1097/00007691-200608000-00012. [DOI] [PubMed] [Google Scholar]
- 36.Newcombe RG. Confidence intervals for an effect size measure based on the Mann-Whitney statistic. Part 1: general issues and tail-area-based methods. Stat Med. 2006;25:543–57. doi: 10.1002/sim.2323. [DOI] [PubMed] [Google Scholar]
- 37.Newcombe RG. Confidence intervals for an effect size measure based on the Mann-Whitney statistic. Part 2: asymptotic methods and evaluation. Stat Med. 2006;25:559–73. doi: 10.1002/sim.2324. [DOI] [PubMed] [Google Scholar]
- 38.Jackson PR, Tucker GT, Lennard MS, Woods HF. Polymorphic drug oxidation: pharmacokinetic basis and comparison of experimental indices. Br J Clin Pharmacol. 1986;22:541–50. doi: 10.1111/j.1365-2125.1986.tb02933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tucker GT, Rostami-Hodjegan A, Jackson PR. Determination of drug-metabolizing enzyme activity in vivo: pharmacokinetic and statistical issues. Xenobiotica. 1998;28:1255–73. doi: 10.1080/004982598238895. [DOI] [PubMed] [Google Scholar]
- 40.Raungrut P, Uchaipichat V, Elliot DJ, Janchawee B, Somogyi AA, Miners JO. In vitro-in vivo extrapolation predicts drug-drug interactions arising from inhibition of codeine glucuronidation by dextropropoxyphene, fluconazole, ketoconazole, and methadone in humans. J Pharmacol Exp Ther. 2010;334:609–18. doi: 10.1124/jpet.110.167916. [DOI] [PubMed] [Google Scholar]