

Abstract
AIMS
To determine efficacy of the analgesic flupirtine in the treatment of overactive bladder syndrome in a proof-of-concept study.
METHODS
Double-blind, double-dummy, three-armed comparison of flupirtine extended release (400 mg/day, titrated to 600 mg/day), tolterodine extended release (4 mg/day) and placebo for 12 weeks.
RESULTS
When major elevations of liver enzymes (more than three times the upper normal limit) were detected in several flupirtine-exposed patients, the study was prematurely discontinued. Based on study-end data, hepatotoxicity was detected in 31% of patients receiving flupirtine for ≥6 weeks.
CONCLUSIONS
Unexpected frequent and relevant toxicity can occur when testing an established drug for a new indication.
Keywords: flupirtine, hepatotoxicity, overactive bladder syndrome
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Flupirtine has been on the market for about 30 years in several European countries as an analgesic. This use has not resulted in regulatory action concerning hepatotoxicity.
WHAT THIS STUDY ADDS
When used in a novel indication, hepatotoxicity was frequent with flupirtine, questioning the general assumption that the safety profile in one indication can be extrapolated to other indications.
Introduction
There is growing medical and commercial interest to develop registered drugs for additional indications, with the implicit assumption that the overall pharmacokinetics and safety of that drug are already established, allowing a faster and cheaper drug development for the new indication.
The selective neuronal KCNQ (KV7) potassium channel opener flupirtine [1], [2] has been in clinical use as a centrally acting, non-opioid analgesic in Europe since 1981 [3]. It is undergoing clinical evaluation in fibromyalgia, tinnitus [4] and Creutzfeldt-Jakob disease [5]. Flupirtine is generally considered safe in its licensed indication [3], [6], even upon chronic use [7], with liver enzyme elevations occurring in <0.01% of patients according to the German prescribing information [8]. Based upon promising efficacy data in animal models [9], we have performed a phase II double-blind, randomized proof-of-concept trial with flupirtine in overactive bladder syndrome (OAB) patients. This study was prematurely discontinued owing to frequent serious liver dysfunction, indicating that an established drug is not necessarily safe when used for a new indication.
Patients and methods
In an international (Poland, Germany and Sweden) multicentre trial, OAB patients ≥18 years old were randomized to receive once daily placebo, flupirtine extended release or tolterodine extended release for a planned 12 weeks of double-blind, double-dummy treatment after 2 weeks of a single-blind placebo run-in period (EudraCT 2006-004854-26, http://www.clinicaltrials.gov NCT00439192). Key exclusion criteria were treatment with any OAB medication within 4 weeks prior to study entry, history of liver disease and/or impaired liver function, evidence of significantly impaired renal function or chronic alcohol or drug abuse. The study was in line with the Declaration of Helsinki, adhered to International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use guidelines and had been approved by the responsible ethical committees. Each patient gave written informed consent.
Patients in the flupirtine group received 400 mg/day for 8 weeks, followed by a protocol-specified ‘forced’ titration to 600 mg/day for 4 weeks. Patients in the tolterodine treatment group received 4 mg/day for the duration of the treatment period. It was planned to randomize 110 patients each to flupirtine and placebo treatment and 55 patients to tolterodine treatment. Visits were planned at screening (week −2), at randomization (week 0), after 2, 6 and 8 weeks (forced titration in flupirtine group), after 12 weeks (study end) and 2 weeks after study end. The concentration of flupirtine and its N-acetyl metabolite in plasma was determined by high-performance liquid chromatography using fluorescence detection. The method was validated at Prolytic GmbH (Frankfurt, Germany) and had a lower limit of quantification of 5 ng/ml−1 for both flupirtine and the metabolite when using 250 µl of plasma.
At each visit, treatment-emerging adverse events (AEs) were recorded and encoded at the Medical Dictionary for Regulatory Activities preferred term level. Laboratory tests and vital signs were assessed during each visit while receiving study medication; physical examination and electrocardiograms were performed at visits one and six. All laboratory tests were centrally performed by Laboratorium für Klinische Forschung (Raisdorf, Germany).
Data analysis was carried out using SAS® version 8.2 (SAS Institute Inc., Cary, NC, USA) and data are reported as means ± SD of n subjects. Upon the decision to discontinue the study prematurely, a specific study-end visit, including laboratory tests, was performed for each patient having had at least one dose of study medication but not yet having completed the study.
Results
207 patients were randomized and received at least one dose of study medication (Figure 1). Demographics, comorbidities and comedications were similar across all three treatment groups (Table 1). Patient compliance based upon pill counts was 77.9%, yielding mean plasma concentrations of flupirtine and its N-acetyl metabolite as measured during exposure to 400 mg/day of 794 ± 329 and 719 ± 310 ng/ml−1 after up to 2 weeks (n = 71) and 788 ± 351 and 682 ± 276 ng/ml−1 after up to 8 weeks of treatment (n = 28); during exposure to 600 mg/day, they were 1248 ± 606 and 1037 ± 404 ng/ml−1, respectively, after up to 12 weeks (n = 12).
Figure 1.
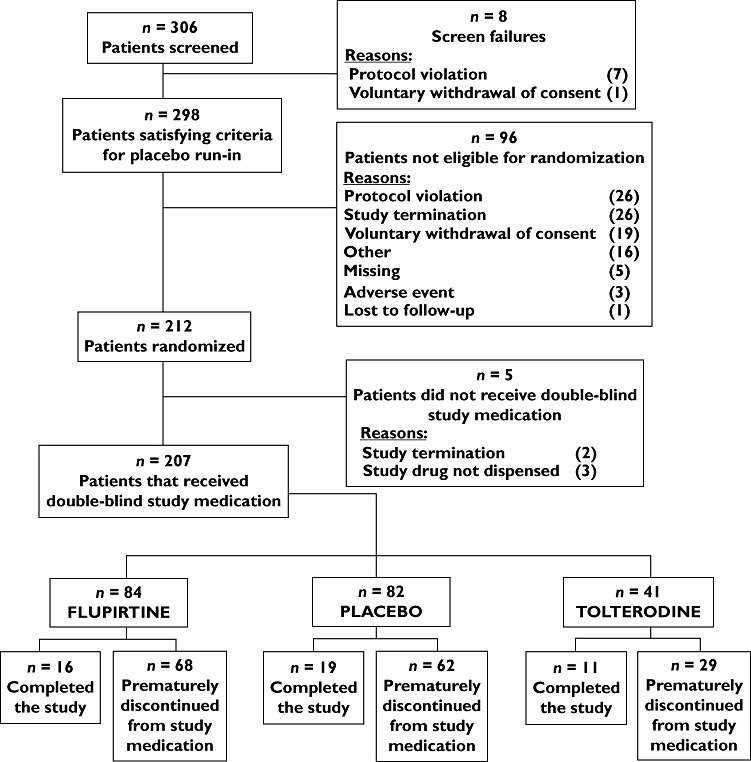
Patient disposition
Table 1.
Demography and baseline characteristics by treatment group
| Placebo | Flupirtine | Tolterodine | Total | |
|---|---|---|---|---|
| n | 82 | 84 | 41 | 207 |
| Age (years) | 60.9 ± 10.8 | 58.9 ± 13.8 | 61.1 ± 12.7 | 60.1 ± 12.4 |
| Female gender | 65 (79.3) | 72 (85.7) | 36 (87.8) | 173 (83.6) |
| Height (cm) | 166 ± 8 | 165 ± 7 | 162 ± 8 | 165 ± 8 |
| Weight (kg) | 81 ± 19 | 78 ± 15 | 75 ± 15 | 78 ± 17 |
| Body mass index (kg/m2) | 29.1 ± 5.8 | 28.6 ± 5.8 | 28.4 ± 5.0 | 28.8 ± 5.6 |
| Duration of overactive bladder syndrome symptoms (months) | 67 ± 65 | 72 ± 84 | 50 ± 39 | 66 ± 70 |
| Most frequent comorbidities | ||||
| Vascular disorders | 33 (40.2) | 24 (28.6) | 13 (31.7) | 70 (33.8) |
| Metabolic disorders | 19 (23.2) | 17 (20.2) | 6 (14.6) | 42 (20.3) |
| Musculoskeletal or connective tissue disorders | 14 (17.1) | 15 (17.9) | 10 (24.4) | 39 (18.8) |
| Concomitant medications | 55 (67.1) | 50 (59.5) | 28 (68.3) | 133 (64.3) |
Data are shown as number of subjects (% of respective group) or as means ± SD. Note that multiple nominations were possible for comorbidities.
During routine safety review, eight patients showed critically high liver enzyme values (more than three times the upper normal limit; mostly alanine aminotransferase, ALT). When these patients were unblinded for safety reasons, seven of them were receiving flupirtine and one placebo. This led to immediate termination of the entire study. At that time point, 19 of 82, 16 of 84 and 11 of 44 patients in the placebo, flupirtine and tolterodine group, respectively, had completed the study as planned. Based on closing documentation, ALT values more than three times the upper normal limit (up to 1034 IU/l) were detected in 14 of 84 flupirtine-exposed patients. Of those, eight also had increased aspartate aminotransferase (AST) up to 1636 IU/l, including one with total bilirubin concentrations of up to 142 µmol/l. Among 82 placebo-exposed patients, one each had a critically high ALT or AST, whereas no critical elevations were seen in the 41 tolterodine-treated patients. Most liver enzyme elevations occurred after 4–6 weeks of treatment. In patients treated for at least 6 weeks with flupirtine, the incidence of liver dysfunction as assessed by abnormal ALT and/or AST was 31%. At the 6 week time point, mean ± SD ALT concentrations were 100 ± 198 (n = 44), 25 ± 11 (n = 47) and 23 ± 9 IU/l (n = 26) in the flupirtine, placebo and tolterodine group, respectively, whereas mean AST concentrations were 84 ± 247, 25 ± 6 and 26 ± 6 IU/l, respectively. Patients experiencing abnormal liver function were asymptomatic (except for fatigue in the patient with high bilirubin), and all recovered fully.
Flupirtine-exposed patients exhibiting liver enzyme elevations did not differ remarkably from those without such alterations in any of the baseline parameters, including comorbidities and comedications, except for four patients with liver enzyme elevations having increased peripheral eosinophil counts, indicating possible drug hypersensitivity. Of note, flupirtine plasma concentrations did not differ significantly or consistently between the two groups (week 2, 831 ± 364 vs. 787 ± 326 ng/ml−1; week 8, 540 ± 404 vs. 829 ± 301 ng/ml−1; and week 12, 1520 ± 978 vs. 1179 ± 518 ng/ml−1).
Other than those liver enzyme elevations, the general safety and tolerability profile of the study drugs was largely as expected. Thus, in the placebo, flupirtine and tolterodine groups, a total of 45 AEs in 18 patients (22.0%), 82 AEs in 40 patients (47.6%) and 46 AEs in 16 patients (39.0%) were observed, respectively. Twenty AEs led to study discontinuation in 13 flupirtine and one placebo patient. Serious AEs were observed in two flupirtine patients (one basal cell carcinoma and one cerebrovasular accident; both leading to study withdrawal) and one in a placebo patient (gastroenteritis; patient continued study as planned); all three serious AEs were judged as not treatment related by the investigator and sponsor. Adverse events leading to study discontinuations were observed in one placebo patient (fatigue), 13 flupirtine patients (five ‘liver function test abnormal’, one ‘hepatic enzyme increased’, one ‘ALT and AST increased’, one ‘rash’, one ‘basal cell carcinoma’, one ‘cerebrovascular accident’, one ‘hypersensitivity, allergic dermatitis’, one ‘abdominal pain, vomiting’ and one ‘pyrexia, pruritus’) and two tolterodine patients (one ‘liver function test abnormal’ and one ‘upper abdominal pain’). Otherwise, no clinically important alterations in biochemical or haematological parameters, urinalysis, electrocardiogram or vital signs on physical examination were noted.
Discussion
Flupirtine has been used as a prescription analgesic in Germany and some other countries for about 30 years, where it has generally been considered to be a safe drug with a low potential for liver enzyme elevations [3], [6], [7], as also reflected in the German prescribing information [8]. Flupirtine was even recommended as a safe alternative for patients experiencing AEs during treatment with nonsteroidal anti-inflammatory drugs [6]. In limited studies with tinnitus patients (24 patients treated with 2 × 100 mg/day for 3 weeks [4]) or Creutzfeldt-Jakob disease patients (13 patients treated with 3–4 × 100 mg/day for up to 10 months [10]), elevated liver enzymes were not reported. Moreover, a single dose of 100 mg was reported not to worsen pre-existing liver disease despite yielding very high drug exposure [11]. Therefore, it was highly surprising that flupirtine treatment of OAB patients in our study was associated with markedly elevated liver enzymes in 31% of patients being exposed to at least 6 weeks of treatment. It was also remarkable that in almost all cases abnormal liver function was asymptomatic. Moreover, some cases of abnormal liver function during flupirtine treatment were accompanied by an increased number of peripheral eosinophils, pyrexia and/or pruritus, indicating a possible immuno-allergic type of hepatotoxicity. A recent histopathological study has also suggested that flupirtine-induced liver injury may have clinical and histological features in line with an immune-mediated toxicity [12].
As the present study was designed as a proof-of-concept for efficacy in OAB patients, we have only limited information elucidating the causes of frequent liver impairment in our cohort. The dosage and formulation in our study was identical to the registered drug in its analgesic indication in Germany [8]. Accordingly, measured plasma concentrations in our study were in the expected range compared with reported values [3] and were similar in flupirtine-exposed patients with and without liver enzyme elevations. Other known factors predisposing to drug-induced hepatotoxicity, such as advanced age or female gender [13], were also similar in our population compared with reported analgesia populations [3], [6], [7]. While many patients using flupirtine for its analgesic indications may take it for only a few days, hepatic damage has not been reported in patients taking it for up to a year [7], [10]. In contrast, a trial published after the premature discontinuation of our study reported elevation of liver enzymes/bilibrubin in almost 3% of patients receiving 300 mg flupirtine for 1 week [14]. A short report from the safety database of the Arzneimittelkommission der deutschen Ärzteschaft published after discontinuation of our study lists 151 cases of flupirtine-associated hepatotoxicity; an incidence estimate was not provided, but the accumulated number of cases is contrasted with >17 million dispensed defined daily doses in 2006 alone [15]. These findings raise the possibility that the high incidence of hepatic toxicity in our study may be related to specific factors in OAB patients, but we have not identified any by comparing our patients with those reported in other flupirtine studies or by comparing patients within our study with and without liver damage. An alternative possibility would be that monitoring during registration studies three decades ago was less strict than today [16] and that postmarketing surveillance since then may have been insufficient [17]. Also, asymptomatic abnormal liver function, as observed in most of our patients, may not be detected in routine clinical practice.
Conclusion
Irrespective of the possible cause of the unexpected hepatotoxicity, our data show that even long-approved drugs may behave quite unexpectedly when used nowadays in different patient populations/indications. Physicians, sponsors and regulators alike should carefully consider this possibility when prescribing, developing or approving existing drugs for novel uses.
Competing Interests
The study was funded by elbion. M.C.M. as co-ordinating investigator had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
M.C.M. has received research support, consultancy or lecturer honoraria in the overactive bladder syndrome field from the following companies: Allergan, AltheRX, Astellas, Bayer and Pfizer. After submission of this manuscript he became an employee of Boehringer Ingelheim.
P.R. has received consultancy or lecturer honoraria in the overactive bladder syndrome field from the following companies: Allergan, Astellas, Pfizer, Pierre Fabre, OCI and ONO.
K.B. acted as Chief Medical Officer for elbion during the conduct and analysis of the study.
The other authors have no competing interests to declare.
REFERENCES
- 1.Mackie AR, Byron KL. Cardiovascular KCNQ (Kv7) potassium channels: physiological regulators and new targets for therapeutic intervention. Mol Pharmacol. 2008;74:1171–9. doi: 10.1124/mol.108.049825. [DOI] [PubMed] [Google Scholar]
- 2.Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156:1185–95. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Devulder J. Fluirtine in pain management. Pharmacological properties and clinical use. CNS Drugs. 2010;24:867–81. doi: 10.2165/11536230-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Salembier L, de Ridder D, van de Heyning PH. The use of flupirtine in treatment of tinnitus. Acta Otolaryngol. 2006;126:93–5. doi: 10.1080/03655230600895242. [DOI] [PubMed] [Google Scholar]
- 5.Appleby BS, Lyketsos CG. Rapidly progressive dementias and the treatment of human prior diseases. Expert Opin Pharmacother. 2011;12:1–12. doi: 10.1517/14656566.2010.514903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Treudler R, Pohle K, Simon JC. Flupirtine is a safe alternative drug in patients with hypersensitivity to NSAIDs. Eur J Clin Pharmacol. 2011;67:961–3. doi: 10.1007/s00228-011-1022-7. [DOI] [PubMed] [Google Scholar]
- 7.Herrmann WM, Hiersemenzel R, Aigner M, Lobisch M, Riethmüller-Winzen H, Michel I. Die Langzeitverträglichkeit von Flupirtin. Offene multizentrische Studie über ein Jahr. Fortschr Med. 1993;111:266–70. [PubMed] [Google Scholar]
- 8.German Prescribing Information. Katadolon S long. 2011. Available at http://www.fachinfo.de (last accesssed 23 November 2011)
- 9.Michel MC, Radziszeswski P, Falconer C, Marschall-Kehrel D, Rundfeldt C, Vanhoutte F. The centrally acting ion channel modulator flupirtine improves bladder function in animal models and patients with overactive bladder syndrome. 2008. Available at http://www.icsoffice.org/Abstracts/Publish/46/000406pdf (last accesssed 13 May 2011)
- 10.Otto M, Cepek L, Ratzka P, Doehlinger S, Boekhoff I, Wiltfang J, Irle E, Pergande G, Ellers-Lenz B, Windl O, Kretzschmar HA, Poser S, Prange H. Efficacy of flupirtine on cognitive function in patients with CJD. A double-blind study. Neurology. 2004;62:714–8. doi: 10.1212/01.wnl.0000113764.35026.ef. [DOI] [PubMed] [Google Scholar]
- 11.Powell-Jackson P, Williams R. Use of flupirtine maleate as an analgesic in patients with liver disease. Br J Clin Pract. 1985;39:63–6. [PubMed] [Google Scholar]
- 12.Puls F, Agne C, Klein F, Koch M, Rifai K, Manns MP. Pathology of flupirtine-induced liver injury: a histological and clinical study of six cases. Virchows Archiv A. 2011;458:709–16. doi: 10.1007/s00428-011-1087-9. [DOI] [PubMed] [Google Scholar]
- 13.Navarro VJ, Senior JR. Drug-related hepatotoxicity. N Engl J Med. 2006;354:731–9. doi: 10.1056/NEJMra052270. [DOI] [PubMed] [Google Scholar]
- 14.Li C, Ni J, Wang Z, Li M, Gasparic M, Terhaag B, Überall MA. Analgesic efficacy and tolerability of flupirtine vs. tramadol in patients with subacute low back pain: a double-blind multicentre-trial. Curr Med Res Opin. 2008;24:3523–30. doi: 10.1185/03007990802579769. [DOI] [PubMed] [Google Scholar]
- 15.Arzneimittelkommission der deutschen Ärzteschaft. Leberschäden unter Flupirtin. Dtsch Arztebl. 2007;104:A3200. [Google Scholar]
- 16.U.S.Department of Health and Human Services. Guidance for industry. Drug-induced liver injury: premarketing clinical evaluation. 2009. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM174090pdf (last accesssed 3 October 2011)
- 17.European Commission. The rules governing medicinal products in the European Union Volume 9A. 2008. Available at http://ec.europa.eu/health/files/eudralex/vol-9/pdf/vol9a_09-2008_enpdf (last accesssed 3 October 2011)