

Abstract
BRCT-containing protein 1 (Brc1) is a multi-BRCT (BRCA1 carboxyl terminus) domain protein in Schizosaccharomyces pombe that is required for resistance to chronic replicative stress, but whether this reflects a repair or replication defect is unknown and the subject of this study. We show that brc1Δ cells are significantly delayed in recovery from replication pausing, though this does not activate a DNA damage checkpoint. DNA repair and recombination protein Rad52 is a homologous recombination protein that loads the Rad51 recombinase at resected double-stranded DNA (dsDNA) breaks and is also recruited to stalled replication forks, where it may stabilize structures through its strand annealing activity. Rad52 is required for the viability of brc1Δ cells, and brc1Δ cells accumulate Rad52 foci late in S phase that are potentiated by replication stress. However, these foci contain the single-stranded DNA (ssDNA) binding protein RPA, but not Rad51 or γH2A. Further, these foci are not associated with increased recombination between repeated sequences, or increased post-replication repair. Thus, these Rad52 foci do not represent sites of recombination. Following the initiation of DNA replication, the induction of these foci by replication stress is suppressed by defects in origin recognition complex (ORC) function, which is accompanied by loss of viability and severe mitotic defects. This suggests that cells lacking Brc1 undergo an ORC-dependent rescue of replication stress, presumably through the firing of dormant origins, and this generates RPA-coated ssDNA and recruits Rad52. However, as Rad51 is not recruited, and the checkpoint effector kinase Chk1 is not activated, these structures must not contain the unprotected primer ends found at sites of DNA damage that are required for recombination and checkpoint activation.
Key words: Brc1, DNA replication, Fission yeast, Recombination
Introduction
During DNA replication, cells are extremely vulnerable to DNA damage. This vulnerability is due to both the genomic lesions themselves, but also to the impediment these lesions cause to the completion of replication. Replication forks that encounter sites of DNA damage stall in the face of these lesions, a process that activates the DNA replication checkpoint to help maintain stalled fork stability, halt the cell cycle, and initiate DNA repair. The majority of replication forks are presumed to retain the full complement of replication machinery in a stable conformation at sites of fork stalling until the impeding lesion is repaired. However, some lesions are not readily resolved and so alternative mechanisms for the completion of replication have evolved. These include the collapse of the stalled replication fork and its repair through homologous recombination (HR), bypass of the DNA lesion through the post-replication repair (PRR) pathway, which includes both error-prone and error-free mechanisms, and the completion of replication through the firing of adjacent origins (Branzei and Foiani, 2008; Branzei and Foiani, 2009).
PRR mediates lesion bypass through a two-step process. Initiation of PRR begins with the mono-ubiquitylation of proliferating cell nuclear antigen (PCNA) on lysine (K)164 by Rad6/Rad18, which enables the recruitment of translesion synthesis (TLS) polymerases that can replicate past the blocking lesion, though in a potentially mutagenic manner. Alternatively, the pathway can be channeled into a second arm, in which the mono-ubiquitylation of PCNA is converted to poly-ubiquitylation with K63 linkages, catalyzed by the Ubc13–Mms2 complex (Lee and Myung, 2008). This arm of the pathway utilizes Rad5 and the HR machinery, specifically Rad55/Rad57, to invade the other nascent strand of DNA and replicate off this template to accomplish error-free lesion bypass (Tapia-Alveal and O'Connell, 2011; Vanoli et al., 2010).
BRCT-containing protein 1 (Brc1) is a six-BRCT (BRCA1 carboxyl terminus) domain protein in Schizosaccharomyces pombe that was identified as a high-copy suppressor of smc6-74, a hypomorphic allele of the essential Smc5–Smc6 complex (Verkade et al., 1999). Although Brc1 is not essential for viability, it is required in strains with compromised Smc5–Smc6 function. The Smc5–Smc6 complex is a large multi-subunit complex composed of the Smc5–Smc6 heterodimer and six non-Smc elements (Nse1–6). Smc5 and Smc6 are members of the structural maintenance of chromosome (SMC) proteins, which include cohesin (Smc1–Smc3) and condensin (Smc2–Smc4). Similar to the cohesin and condensin complexes, Smc5–Smc6 is required for accurate chromosome segregation (Hirano, 2006; Outwin et al., 2009). Although many Smc5–Smc6 mutants show defects in recombinational repair (Ampatzidou et al., 2006; Andrews et al., 2005; Morikawa et al., 2004; Pebernard et al., 2004; Verkade et al., 1999), recent evidence suggests that this defect is due to the recruitment of dysfunctional complexes to lesions, rather than an absolute requirement for Smc5–Smc6 in HR-mediated repair (Tapia-Alveal and O'Connell, 2011). Furthermore, this complex has also been shown to play a role at stably stalled replication forks, where it is required for the recruitment of Rad52, the HR initiating protein, onto chromatin without apparent recruitment of Rad51 (Irmisch et al., 2009). This recruitment could function to prime the loci for recombination, but this is unlikely given the lack of downstream recombination factors. An alternative explanation is that the strand-annealing function of Rad52 (Mortensen et al., 1996; Mortensen et al., 2009) is required for the stabilization of the stalled replication fork structure at the junction of the template and nascent strands.
Genetic epistasis analysis of the suppression of smc6-74 by Brc1 indicates that Brc1 functions in conjunction with the PRR proteins Rhp18 and the TLS polymerases, with the HR machinery, and with multiple structure-specific nucleases (Lee et al., 2007; Sheedy et al., 2005). All of these genes are notable for their function in the processing of stalled and/or collapsed replication forks. Recently, a high frequency of brc1-depleted (brc1Δ) cells was shown to contain Rad52 foci, and the deletion of rad52 is synthetically lethal with brc1Δ (Williams et al., 2010). Such foci are generally equated with active sites of HR, and as all recombination is Rad52-dependent in S. pombe (Doe et al., 2004), the presence of these foci suggests brc1Δ cells contain lesions repaired by HR, or a defect in HR resolution. However, asynchronous (mostly G2) (Forsburg and Nurse, 1991) brc1Δ strains are not sensitive to ionizing radiation (IR) or high-dose ultraviolet C (UV-C) radiation (Verkade et al., 1999), both of which create lesions for HR-dependent repair, but are hypersensitive to radiomimetic drugs that primarily induce DNA damage during replication, such as the alkylating agent methyl methanesulfonate (MMS). Furthermore, this hypersensitivity to replicative damage is only seen upon chronic exposure over several days; transient exposure to these agents does not affect the viability of brc1Δ cells, which is in distinct contrast to genes that act within the DNA repair or checkpoint pathways (Sheedy et al., 2005).
The sensitivity of brc1Δ cells to chronic DNA damage has been interpreted by us and others as a DNA repair defect. However, as damage incurred during S phase also impedes the completion of DNA replication, sensitivity to replicative DNA damage might stem from DNA replication defects, rather then defects in DNA repair. Therefore, we asked whether Brc1 is required for recovery from replication stalling induced by DNA lesions. We show in this paper that, indeed, Brc1 is required for the efficient recovery from a replication arrest, largely through promoting lesion bypass. This process is Rad52-independent, and we show that the Rad52 foci seen in brc1Δ cells do not indicate sites of HR, but could indicate sites of ectopic origin firing in an attempt to overcome inefficient replication re-start.
Results
Brc1 is required for efficient recovery from replication arrest
brc1Δ cells are hypersensitive to chronic exposure to agents that induce replicative DNA damage. Under these conditions, brc1Δ cells die as highly elongated cells that are cell-cycle arrested in a DNA damage checkpoint-dependent manner (Lee et al., 2007; Sheedy et al., 2005). However, unlike DNA repair mutants, brc1Δ cells are not sensitive to DNA damage in G2 (Verkade et al., 1999), nor are they sensitive to acute exposure to the alkylating agent MMS (Sheedy et al., 2005). Therefore, we hypothesized that the sensitivity of brc1Δ cells to DNA damage during S phase might not represent a primary DNA repair defect per se, but rather an inability to efficiently resume DNA replication after a pause induced by collision of the replisome with a lesion in the template. To this end, we studied the response of brc1Δ cells to hydroxyurea (HU), a ribonucleotide reductase inhibitor that stalls DNA replication after origin firing by dNTP depletion. Importantly, in intra-S-phase checkpoint-competent cells, replication forks are stably stalled in HU, and upon HU removal can resume replication once dNTP synthesis resumes. Conversely, replication forks collapse in checkpoint-defective cells, inducing a DNA damage response and replication restart by recombination-dependent mechanisms (Branzei and Foiani, 2009; Irmisch et al., 2009; Kim and Huberman, 2001).
Although brc1Δ cells are sensitive to chronic exposure to HU (Sheedy et al., 2005), this involves the cells cycling slowly through many S phases over several days. We measured the sensitivity of brc1Δ cells to HU over an 8-hour time-course compared with wild-type (checkpoint-competent) and cds1Δ (checkpoint-defective) controls (Fig. 1A). After a 2-hour lag required for asynchronous cells to cycle past S phase, cds1Δ cells precipitously lost viability. Conversely, both wild-type and brc1Δ cells maintained viability over the time-course, showing that the sensitivity of brc1Δ cells to HU requires long-term chronic exposure.
Fig. 1.
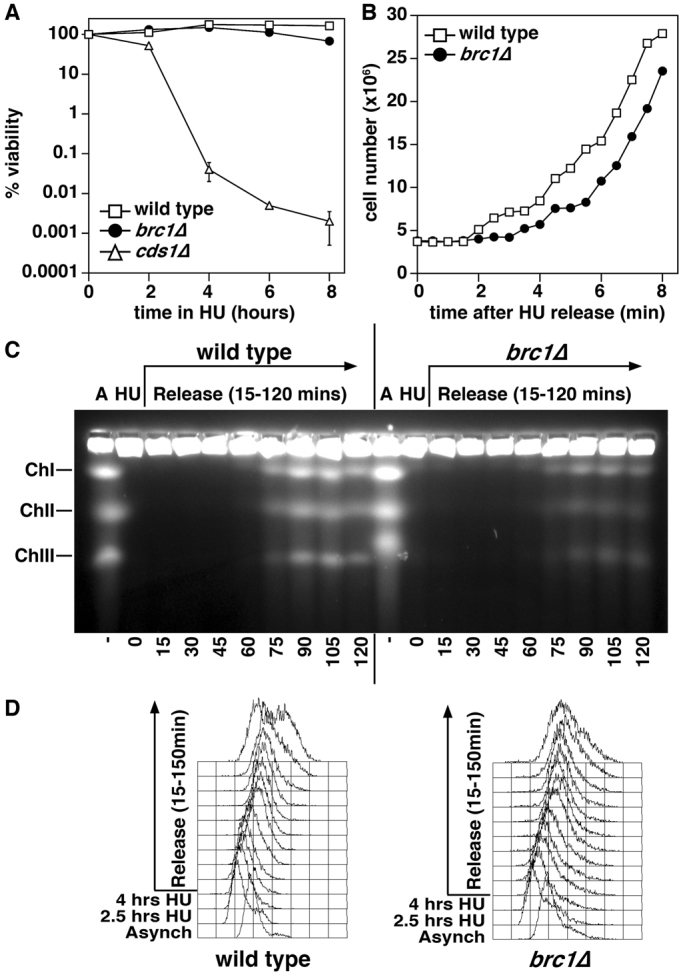
Brc1 is required for efficient recovery from an HU-induced replication arrest. (A) Exponential cultures of wild-type, brc1Δ and cds1Δ cells were treated with 11 mM HU (time 0). At the indicated timepoints, samples were taken, washed free of HU, and viability determined by colony formation on YES plates for 4 days at 30°C. Data are means ± s.d., n=3, and are normalized to untreated cells. Note that brc1Δ cells maintain viability over this acute treatment with HU, whereas cds1Δ cells die rapidly once cycling past the HU arrest point (2 hours). (B) Wild-type and brc1Δ cells were arrested in 11 mM HU for 4 hours at 30°C, washed free of HU and re-inoculated into fresh medium at 30°C. Cell number was determined at the indicated timepoints. Data are means ± s.d., n=3. Note that the first cell number doubling occurs at 3.5 hours for wild type, and 5-hours for brc1Δ. (C) Chromosomes from wild-type and brc1Δ cells were analyzed by pulse field gel electrophoresis (PFGE). Samples were taken from asynchronously growing cultures (A), cultures arrested in 11 mM HU for 4 hours at 30°C (HU), and from cultures washed free of HU and inoculated into fresh medium, with samples taken every 15 minutes for 2 hours (0–120 minutes). Chromosomes with stalled replication forks fail to enter the gel, but do enter upon completion of DNA synthesis, although less efficiently (brc1Δ, 75–90 minutes) as determined by FACS analysis. ChI, ChII and ChIII indicate the position of the three S. pombe chromosomes. The reduced mobility of chromosome III in brc1Δ cells is due to rDNA expansion. (D) FACS analysis of DNA content from the cultures used for PFGE analysis in C.
Next, we measured cell cycle progression on recovery from an HU block for the following 8 hours, corresponding to ~2.5 cell cycles (Fig. 1B). Upon HU removal, the time to cell doubling for wild-type cells was 3.5 hours, compared with 5.0 hours for brc1Δ. Should such a delay occur every cell cycle over a 4-day growth period, the normal time for colony formation, this would result in a 300-fold difference in cell number. For both strains, elongation during the HU arrest results in slightly shorter than normal subsequent cell cycles due to size control, and the rate of cell number increase was only affected in the first cell cycle following HU arrest in brc1Δ cells. Thus, Brc1 is required to efficiently recover from treatment with HU.
We next assayed completion of DNA replication using pulse-field gel electrophoresis (PFGE). In this system, the three S. pombe chromosomes are resolved as discrete bands, but incompletely replicated chromosomes such as those in HU-arrested cells fail to enter the gel, and this was observed for both wild-type and brc1Δ cells (Fig. 1C). The wild-type chromosomes were fully recovered by 75–90 minutes after HU removal, which by fluorescence-activated cell sorting (FACS) corresponds to the completion of bulk DNA replication (Fig. 1D). However, although the timing of the initial appearance of intact chromosomes was similar in brc1Δ, the intensity of the brc1Δ chromosomes re-entering the gel was much lower (Fig. 1C). The simplest explanation for this, given the cell number data (Fig. 1B), is that a substantial proportion of brc1Δ cells are delayed in recovering from the HU arrest. However, other changes to chromosome topology could impact on chromosome resolution, although fragmented chromosomes do enter these gels (Verkade et al., 1999) and catenated chromosomes in top2 mutants do not delay cell cycle progression (Uemura et al., 1987), and thus these are unlikely sources of the inefficient recovery of intact chromosomes. Furthermore, bulk DNA replication appears to be retarded in brc1Δ cells as evidenced by the broader FACS profiles, and by the reduced secondary peak at 150 minutes that is prominent in wild-type cells, which is derived from a second S phase prior to completion of cytokinesis.
From these observations, we conclude that brc1Δ cells inefficiently recover from HU-induced replication arrest, but most cells do eventually recover under conditions of acute HU exposure. Therefore, the sensitivity of brc1Δ cells to chronic HU exposure over several days probably reflects defects from multiple cycles of inefficient recovery.
brc1Δ cells are wild type for checkpoint signaling
Replication arrest in HU activates the intra-S-phase checkpoint and its effector kinase, serine/threonine-protein kinase cds1 (Cds1), to maintain the stability of stalled replication forks. If the replisome becomes uncoupled from the stalled fork (a process known as fork collapse) a DNA damage response is initiated and activates the checkpoint effector kinase serine/threonine-protein kinase chk1 (Chk1). In HU-treated cells lacking both Cds1 and Chk1, this checkpoint relay is short circuited, and cells enter lethal mitoses with incompletely replicated chromosomes (Lindsay et al., 1998; O'Connell and Cimprich, 2005; O'Connell et al., 2000).
Given the importance of checkpoint responses during replication arrest, we crossed brc1Δ to both cds1Δ and chk1Δ cells and also created triple mutant cells. Mitotic defects consistent with checkpoint failure were only seen in HU-treated cells lacking both Cds1 and Chk1, indicating that both checkpoints are operative during the replication arrest in the absence of Brc1 (Fig. 2A). We next assayed Chk1 activation by Rad3-catalyzed phosphorylation, which is evident as a mobility shift on western blots (Latif et al., 2004; Walworth and Bernards, 1996). In wild-type cells, Chk1 was efficiently activated by MMS, but not by HU (Fig. 2B). However, a small but reproducible activation of Chk1 was observed 60 minutes after HU removal. Chk1 activation was similar in brc1Δ cells, but the low level activation observed on HU removal was sustained until 120 minutes (Fig. 2B). We also measured the effect of Chk1 on the viability of brc1Δ cells during acute HU exposure. Consistent with the Chk1 activation data, a small sensitizing effect was observed in chk1Δ brc1Δ cells (Fig. 2C), but this was several orders of magnitude lower than in HU-treated cds1Δ cells (Fig. 1A), or MMS-treated DNA repair mutants (Sheedy et al., 2005). However, given the survival data (Fig. 1A) and lack of increased mitotic defects in brc1Δ chk1Δ cells (Fig. 2A), and the fact that Chk1 activation is binary rather than dose dependent (Latif et al., 2004), Chk1 activation must arise in a minority of cells. Moreover, this suggests that Cds1 activation by HU must be functional in the absence of Brc1, which we assayed using Cds1 immunoprecipitates. HU treatment resulted in a 19-fold increase in activation of Cds1 in wild-type cells, but only a sixfold increase in brc1Δ cells (Fig. 2D). This was also associated with reduced Cds1 protein expression (Fig. 2D), but not lower cds1 mRNA (not shown). Although the significance of this in unclear, the sixfold induction of Cds1 in HU is clearly sufficient to prevent Chk1 activation (Fig. 2B) and mitotic defects in chk1Δ cells (Fig. 2A). Thus, the defect in recovery from replication arrest observed in brc1Δ cells is not accompanied by significant defects in known checkpoint signaling pathways.
Fig. 2.

brc1Δ cells maintain a competent intra-S-phase checkpoint and do not activate the DNA damage checkpoint upon replication stalling. (A) Cell morphology was analyzed in the indicated strains after 4 hours in 0 (control) or 11 mM HU. Cells able to maintain the intra-S checkpoint show elongation after treatment, which was evident in all strains except those lacking both cds1 and chk1. Scale bar: 10 μm. (B) The indicated strains expressing an HA-tagged allele of chk1 were assayed for activation of Chk1 by western blotting following treatment with 0.05% MMS or after release from a 4-hour arrest in HU. Panels show a short (1 minute) and a long (20 minutes) exposure of the same blots. Activation of Chk1 results in a slower migrating species because of phosphorylation of S345 by Rad3, which is prominent upon MMS treatment, but only faint upon HU recovery. (C) The viability of the indicated strains during an HU arrest was assayed over 8 hours. Note that these cells maintain good viability over this timecourse; compare with Fig. 1, but note the scale of the y-axis. (D) Cds1 expression was assayed in wild-type and brc1Δ cells using a C-terminally HA-tagged cds1 allele. Actin was used as a loading control for western blotting, and shows Cds1 expression to be reduced two- to threefold in brc1Δ cells. mRNA levels for Cds1 are not significantly affected in brc1Δ cells (data not shown). The lower panel shows immunoprecipitated Cds1 activity assayed with myelin basic protein (MBP) as a substrate. 32P-incorporation was quantified with a Phosphorimager, and is expressed as arbitrary units.
brc1Δ cells contain Rad52 foci
It has been reported that ~30% of asynchronously growing brc1Δ cells possess Rad52–YFP foci (Williams et al., 2010). These resemble Rad52 foci in wild-type cells following exogenous DNA damage, presumed to mark sites of HR. We confirmed this result (Fig. 3A), although found this surprising because ~70% of a culture of most S. pombe strains, including brc1Δ, are in the G2 phase of the cell cycle (Forsburg and Nurse, 1991), and yet brc1Δ cells are not sensitive to DNA damage in G2 (Sheedy et al., 2005; Verkade et al., 1999). Furthermore, cycling brc1Δ cells do not show Chk1 activation (Fig. 2B), a sensitive marker of DNA damage. Thus, we hypothesized that these foci might not represent sites of DNA damage.
Fig. 3.
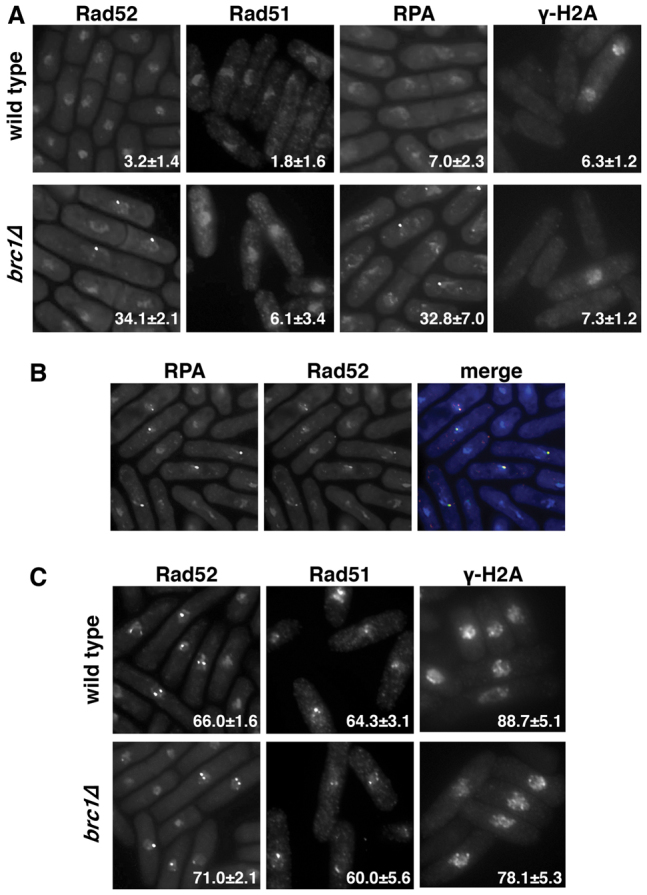
Rad52 foci form in brc1Δ cells. (A) Representative examples of Rad52 (YFP tagged), Rad51 (anti-Rhp51 antibody), RPA (Rad11–GFP) and γH2A (anti-phosphorylated-S139 antibody) foci in asynchronously growing wild-type and brc1Δ strains. The numbers are the mean percentage (± s.d.) of cells showing foci from three counts of 100 cells. (B) RPA and Rad52 foci colocalize in brc1Δ cells. The micrographs show a representative field of asynchronous brc1Δ cells expressing both Rad11–GFP and Rad52–RFP. 83% of Rad52–RFP foci colocalize with Rad11–GFP foci in merged images. (C) Wild-type and brc1Δ cells were treated with 0.05% MMS for 4 hours, and Rad52 (Rad22–YFP), Rad51 (anti-Rhp51) and γH2A (anti-phosphorylated-S139 antibody) foci imaged as in A. 80–90% of MMS-treated cells are positive for these markers.
The best-characterized function for Rad52 at sites of DNA damage is to catalyze the replacement of the high affinity single-stranded DNA (ssDNA) binding protein, replication protein A (RPA), with the recombinase Rad51 (Krogh and Symington, 2004). Thus, we assayed for the presence of RPA and Rad51 foci in wild-type and brc1Δ cells (Fig. 3A). Surprisingly, we found that brc1Δ cells contain high levels of RPA foci, but not Rad51 foci. As a separate chromatin-associated marker for DNA damage, we also stained cells for phosphorylated H2A (γH2A), which was also not elevated in brc1Δ cells (Fig. 3A). Therefore, the Rad52 foci in brc1Δ cells are not markers of sites of DNA damage.
Simultaneous imaging of RPA and Rad52 confirmed that these foci colocalize in asynchronously growing brc1Δ cells (Fig. 3B). Both wild-type and brc1Δ cells were positive for Rad52, Rad51 and γH2A after MMS treatment (Fig. 3C), and thus brc1Δ cells are competent for the recruitment of these proteins to sites of DNA damage. This is in keeping with the fact that brc1Δ cells are HR-proficient and resistant to transient exposure to MMS, whereas repair mutants such as rhp51Δ, which lacks the Rad51 homolog, are not (Sheedy et al., 2005).
Formation of Rad52–RPA foci is enhanced by replication stress
Because Brc1 is required to efficiently recover from replication stalling, we assayed the effect of replication stalling on the formation of Rad52, RPA and Rad51 foci. Wild-type and brc1Δ cells were arrested in early S phase with 11 mM HU, and then released into fresh medium (Fig. 4). In both strains, RPA foci increased upon HU treatment (Fig. 4A,D), consistent with RPA being present at stalled replication forks (O'Connell and Cimprich, 2005). Concomitantly, the frequency of HU-treated brc1Δ cells with Rad52 foci decreased to ~5% (Fig. 4E). Thus, it appears that HU arrests brc1Δ cells at a cell-cycle stage before formation of Rad52 foci. FACS analysis showed that bulk DNA synthesis was completed by ~60–90 minutes after release, which coincided with the return of Rad52 foci, now in a higher proportion (~50%) of brc1Δ cells. These foci persisted after the cells exited mitosis, which is consistent with the observation that they are not associated with a robust checkpoint response (Fig. 2).
Fig. 4.

brc1Δ cells accumulate RPA and Rad52 foci after release from replication stalling. Asynchronous cultures of wild-type and brc1Δ cells were arrested in 11 mM HU for 4 hours at 30°C. Cells were then washed free of HU, and re-inoculated into fresh medium (time 0). Samples were taken at the indicated timepoints to quantify RPA (Rad11–GFP) (A,D), Rad52 (Rad22–YFP) foci (B,E) and Rad51 (anti-Rhp51 antibodies) foci (C,F; all open squares), together with septation indices (closed diamonds). Note that the peak of septation in brc1Δ cells is broadened by the delayed recovery from HU, and that cells expressing Rad11–GFP also show a more modest delay, although synchrony was highly reproducible between individual experiments. Data are means ± s.d. for three populations of 100 cells. FACS profiles of DNA content are shown to the right of the graphs, with bulk DNA synthesis completed by ~60 minutes after release. The >2C DNA content at 150 minutes is due to DNA replication in synchronized cells prior to cytokinesis.
A small proportion of wild-type cells also showed Rad52 foci coincident with completion of replication (Fig. 4B) and the time at which low-level Chk1 activation is seen (Fig. 2B). However, these and the RPA foci were resolved prior to mitosis. Such Rad52 foci have been observed previously in wild-type cells treated transiently with HU, together with a modest activation of Chk1 (Meister et al., 2005) (Fig. 2), suggesting these foci form at sites of DNA damage. In support of this, these timepoints coincided with a small and transient increase in cells with Rad51 foci in both wild-type and brc1Δ cells (Fig. 4C,F). Thus, these are unlikely to be related to the foci seen in asynchronously growing brc1Δ cells.
We also assayed the formation of Rad52 foci in brc1Δ cells synchronized without replication stress by cdc25-22 block-and-release (Fig. 5A). During the G2 arrest imposed by the inactivation of Cdc25, the Rad52 foci were resolved and again reappeared immediately after the peak of septation, which coincides with late S phase (Forsburg and Nurse, 1991). However, unlike the HU synchronized cells, these foci were not as abundant, and ~50% of these were resolved during the next cell cycle. Thus, Rad52 foci are potentiated by replication stress, but occur transiently in the latter portion of an otherwise unperturbed S phase in the absence of Brc1.
Fig. 5.
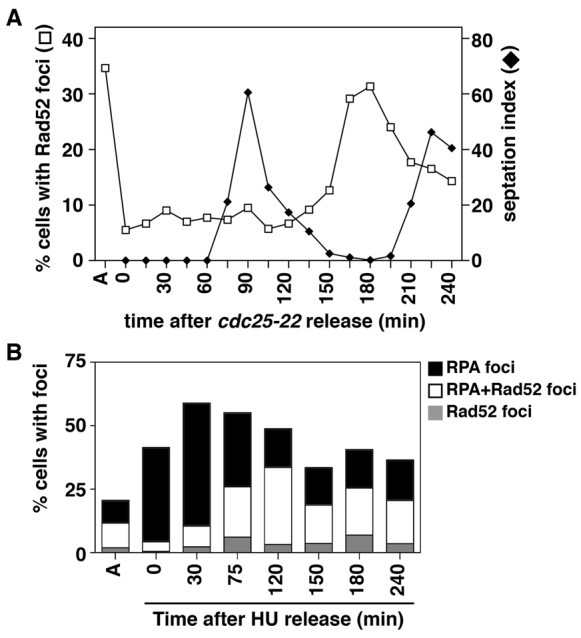
Rad52–RPA foci form without exogenous replication stress. (A) Asynchronous cultures of brc1Δ cells carrying the temperature sensitive cdc25-22 allele and expressing Rad52–YFP were grown to exponential phase at 25°C, and then shifted to 36°C for 4.5 hours to arrest cells late in G2 phase. Cultures were then shifted to 25°C (time 0), and samples taken every 15 minutes to determine the percentage of cells with YFP foci and possessing division septa. Note that the foci present in asynchronously growing brc1Δ cells are resolved during the imposed cell cycle arrest, but reappear upon completion of septation, coincident with completion of S phase. Data are means ± s.d. for three populations of 100 cells. (B) Both Rad52 (Rad22–RFP) and RPA (Rad11–GFP) expressed in the same cells were simultaneously localized in asynchronous brc1Δ cells (as in A) and following a timecourse of release from an arrest in 11 mM HU. Note, the RFP-tagged Rad22 is much dimmer than the YFP-tagged protein used in previous experiments (Coulon et al., 2004), leading to a reduced number of cells with visible Rad52 foci. Nevertheless, the majority of Rad52-positive cells are also positive for colocalizing RPA foci. Data are the means of three populations of 100 cells.
We also repeated the HU block-and-release protocol in brc1Δ cells expressing both Rad11–GFP (RPA) and Rad52–RFP (Fig. 5B). For Rad52, the RFP tag is much dimmer and more quickly bleached than the YFP tag used in the other experiments, and thus we are underestimating the presence of Rad52 foci in this experiment. Nevertheless, this approach enabled us to assess colocalization without risk of ‘bleed through’ of GFP and YFP signals. This confirmed that the majority (83%) of cells with Rad52–RFP foci had colocalizing RPA foci and thus, the foci induced by replication stress, similar to those in asynchronous cultures, are Rad52–RPA foci.
Brc1 facilitates recombination between direct repeats
We next sought to explore Rad51-independent functions for Rad52 as a source of the foci in brc1Δ cells. Perturbations to replication fork progression can induce recombination between repeated sequences. Repeat recombination is Rad52 dependent, but can occur by resection of intervening DNA and repeat annealing in a Rad51-independent process known as single-stranded annealing (SSA) (Ahn et al., 2005). Therefore, a defect in SSA might explain the presence of Rad52 foci that do not contain Rad51.
To test this possibility, we assayed the frequency of Ade-positive (+) colonies arising by recombination between two ade6 heteroalleles flanking an his3 gene (Fig. 6A) (Osman et al., 2000). In this system, Rad51-independent SSA leads to loss of the his3 marker, whereas conversion of one ade6 allele into a functional gene can occur through gene conversion by mispairing with the sister chromatid and retention of the his3 marker (Doe and Whitby, 2004). Spontaneous recombination frequencies (reported as events per 104 cells) were reduced 19-fold in brc1Δ cells (0.28±0.20) compared with a wild-type strain (5.3±1.67; Fig. 6B).
Fig. 6.
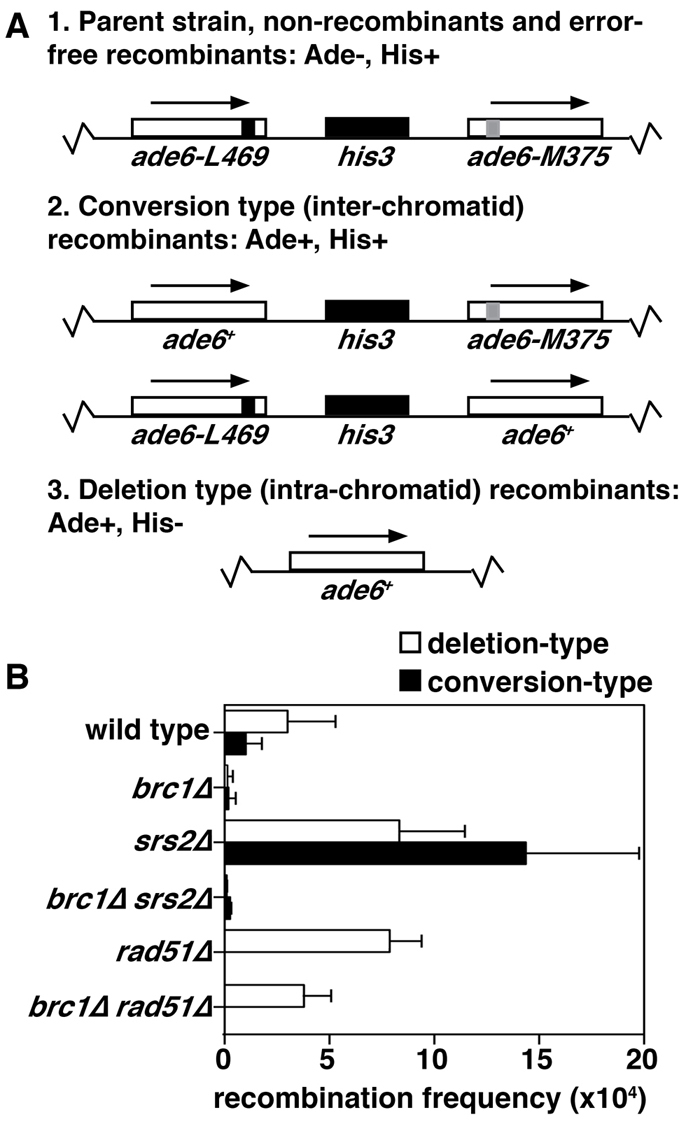
brc1Δ cells do not have increased recombination between direct repeats. (A) Schematic of tandem ade6 heteroalleles used to determine recombination frequencies. Conversion events result in Ade+ His+ progeny, whereas deletion events result in Ade+ His− colonies. (B) Recombination frequencies (per 104 cells) of the following strains: wild type (5.3±1.67), brc1Δ (0.28±0.20), srs2Δ (26.6±5.27), brc1Δ srs2Δ (0.27±0.047), rad51Δ (7.87±1.52), brc1Δ rad51Δ (3.78±1.29). Deletion types (white) and conversion types (black) were determined by replica-plating. Data are means ± s.d.
Srs2 is a DNA helicase that negatively regulates recombination. In S. cerevisiae, this helicase suppresses recombination leading to crossovers through its ability to disrupt Rad51 ssDNA filaments (Ira et al., 2003; Krejci et al., 2003; Veaute et al., 2003). In S. pombe, deletion of srs2 causes elevated rates of spontaneous recombination, presumably through a similar mechanism (Doe and Whitby, 2004). Deletion of brc1 suppressed the hyper-recombination of an srs2Δ strain (Fig. 6B).
One limitation of this assay is that the products of no recombination and those of error-free sister chromatid exchange are the parental chromosome (Ade− His+). In order to determine which of these two possibilities was the case, we utilized the observation that a rhp51Δ strain has elevated recombination events forced through the SSA pathway, which leads to loss of the his3 gene (Doe et al., 2004). rhp51Δ restores deletion-type events to brc1Δ cells (Fig. 6B), suggesting that recombination occurs predominately by error-free sister-chromatid exchange in the absence of brc1. These data show that although Brc1 facilitates SSA between direct repeats, SSA can be successfully completed in the absence of Brc1 and Rad51. Hence, a blockade to recombination between repeated sequences cannot be the source of Rad52–RPA foci in brc1Δ cells.
Rad52–RPA foci are not enriched in ribosomal DNA
Rad52 is also recruited to stalled replication forks that contain RPA, but not detectable Rad51. However, in wild-type cells this can only be assayed by chromatin immunoprecipitation (ChIP), although we considered that in the case of brc1Δ cells an increased recruitment might pass a threshold, leading to microscopically visible foci. This recruitment of Rad52 is most readily demonstrated in the ribosomal DNA (rDNA), presumably because of the high density of replicons (Irmisch et al., 2009). We visualized Rad52–YFP foci in cells expressing mCherry-tagged Gar2, a marker of nucleoli (Gulli et al., 1995), using conventional fluorescence microscopy (Fig. 7A). Only 15% of cells contained a Rad52 focus that colocalized with Gar2, and as these are not confocal images, this is probably an overestimate of colocalization. Thus, the majority of these Rad52–RPA foci are not in the rDNA, and therefore unlikely to arise from perturbations to rDNA recombination.
Fig. 7.
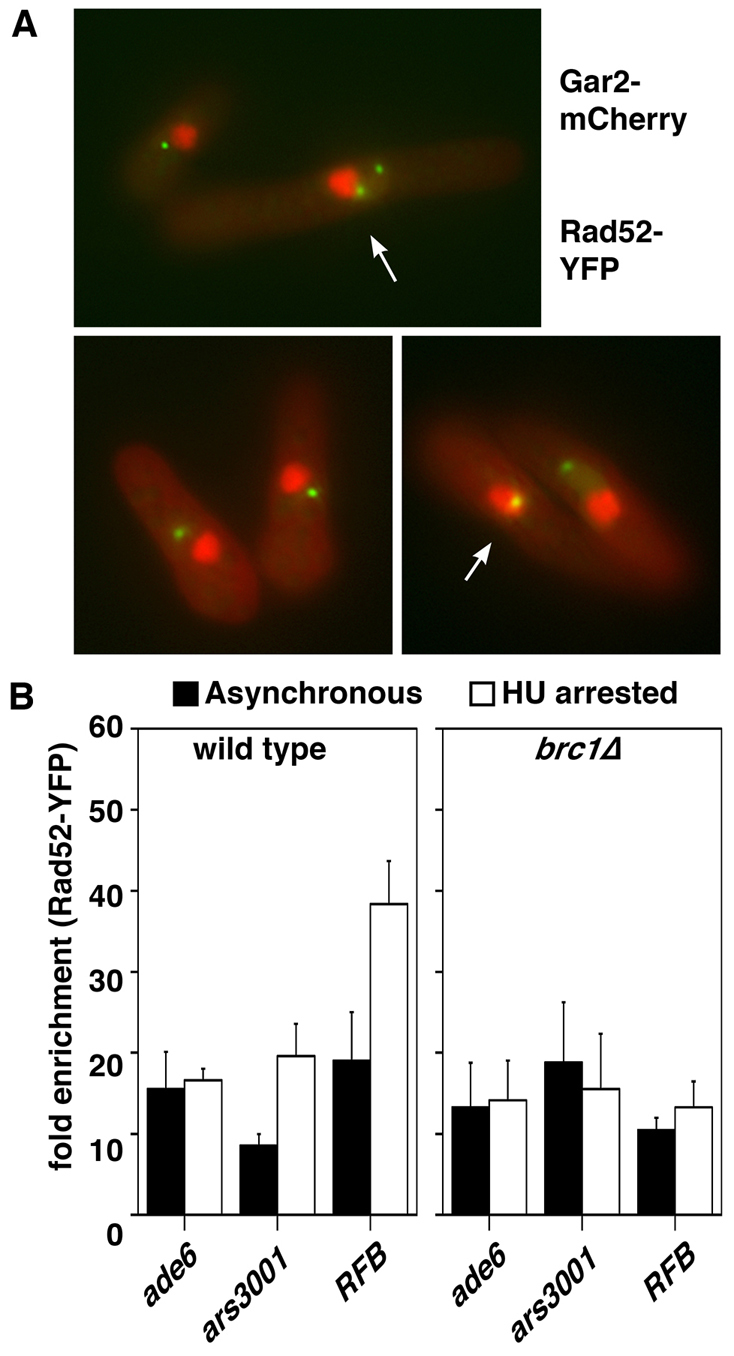
brc1Δ cells do accumulate Rad52 in the rDNA. (A) Rad52–YFP and Gar2–mCherry were simultaneously imaged in brc1Δ cells by conventional fluorescence microscopy. Colocalizing signals (arrowed) were observed in 15% of cells (n=50). (B) Anti-GFP ChIP analysis of the indicated loci from wild-type and brc1Δ cells in a Rad52–YFP background from either asynchronous cultures (solid bars) or following arrest in 11 mM HU (open bars) for 4 hours. Increased rDNA enrichment is evident in wild-type, but not in brc1Δ cells. Data are means ± s.e.m., n=3.
We corroborated these findings using ChIP to analyze recruitment of Rad52 to rDNA (Fig. 7B). As we have previously reported (Irmisch et al., 2009), Rad52 shows strong association with chromatin, but this is further enriched at the replication origin (ars3001) and replication fork barrier within the rDNA in HU-treated cells. However, as seen in smc6-74 mutants (Irmisch et al., 2009), brc1Δ cells are also defective in enrichment of Rad52 at these loci in HU-treated cells, as assayed by ChIP, confirming that the Rad52 foci do not reside in rDNA. Moreover, these two modes of Rad52 recruitment to stably stalled replication forks in the rDNA might contribute to the synthetic lethality of smc6-74 brc1Δ, as well as the high-copy suppression of smc6-74 by brc1 (Verkade et al., 1999).
Brc1 promotes mutagenic lesion bypass
Another Rad52-dependent recombinogenic response to replication stress is the error-free branch of PRR, where replication occurs using the other nascent strand as a template. However, this is preceded by commitment to the error-prone pathway of lesion bypass by mutagenic trans-lesion synthesis (TLS) polymerases, and so is associated with low frequency spontaneous and induced mutagenesis (Kai and Wang, 2003). Thus, we next examined whether PRR was the source of these foci by analyzing the rates of spontaneous and MMS-induced can1 mutations, conferring resistance to canavanine, by fluctuation analysis. MMS both arrests replication and introduces alkylation damage that can be bypassed by the TLS polymerases. brc1Δ cells had spontaneous rates of mutagenesis that were similar to those of wild-type cells, but was induced by MMS by ~2-fold lower than wild type (7.3- versus 4.4-fold; Table 1). Thus, commitment to the use of either arm of PRR is an unlikely source of the Rad52 foci in brc1Δ cells.
Table 1.
Brc1 promotes mutagenesis

Because the effects of brc1Δ on mutagenesis were small, we sought additional links between Brc1 and PRR-induced mutagenesis. Overexpression of Brc1 suppresses the DNA damage sensitivity of Smc5–Smc6 hypomorphs, and brc1Δ is synthetically lethal with these mutants (Lee et al., 2007; Sheedy et al., 2005; Verkade et al., 1999). smc6-74 cells have ~5-fold reduced spontaneous and MMS-induced rates of mutagenesis (Table 1), although this strain is sensitive to transient exposure to MMS and so potentially mutagenized cells might not form colonies. Conversely, wild-type and smc6-74 cells overexpressing Brc1 are not sensitive to transient MMS exposure (Sheedy et al., 2005). Brc1 overexpression in wild-type cells enhanced spontaneous and MMS-induced mutagenesis (63- and 29-fold, respectively). Brc1 overexpression in smc6-74 suppresses sensitivity to MMS but mutagenesis rates were increased by a massive 206-fold (spontaneous) and 407-fold (MMS induced) compared with smc6-74 controls. Deletion of the three TLS polymerases eliminated ~90% of the mutagenesis caused by Brc1 overexpression in the absence of MMS, and the deletion of these polymerases also abolished MMS-induced mutagenesis. These data match the fact that error-prone PRR is required for suppression of smc6-74 by Brc1 overexpression (Sheedy et al., 2005), and corroborates the reduced mutagenesis rates seen in brc1Δ cells. Thus, Brc1 actually promotes the Rad52-independent and error-prone branch of PRR by lesion bypass, and thus the Rad52–RPA foci in brc1Δ cells are not related to increased use of recombinogenic lesion bypass, which requires transient commitment to the error-prone bypass.
Replication recovery in brc1Δ cells is origin recognition complex (ORC) dependent
The foci we have observed contain large amounts of RPA, and therefore ssDNA, and yet do not evoke a DNA damage checkpoint response. Although RPA-bound ssDNA is the template on which checkpoint proteins assemble, studies in Xenopus egg extracts indicate an additional requirement for primer–template junctions within this ssDNA for efficient checkpoint activation (Byun et al., 2005; Lupardus et al., 2002; MacDougall et al., 2007), which presumably are absent at the loci containing Rad52–RPA foci in brc1Δ cells because of the normal checkpoint signaling in these cells (Fig. 2). Thus, although arising in S phase, the ssDNA in brc1Δ cells must emanate from regions of duplex unwinding without priming of DNA synthesis.
We therefore hypothesized that in the absence of Brc1, replication origins that have not fired before the HU arrest might now fire under these conditions of replication stress to overcome the inefficient restart of replication. However, DNA synthesis from these regions must not proceed efficiently, as evidenced by the abundance of RPA and the delayed recovery of brc1Δ cells from an HU arrest. If this hypothesis is correct, then preventing the firing of replication origins after the HU arrest should suppress the formation of the Rad52–RPA foci. To test this notion, we utilized orp1-4, a temperature-sensitive allele of orp1, the S. pombe homolog of ORC1, which encodes the large subunit of the ORC. Because sufficient origins fire in HU-treated cells prior to arrest (Heichinger et al., 2006), orp1-4 cells can be shifted to the non-permissive temperature (36°C) upon HU removal, and DNA synthesis is then completed from the stably stalled origins that fired prior to ORC inactivation. Therefore, the absence of ORC activity in these cells does not manifest until the next cell cycle, by which time the cells are incapable of initiating DNA replication (Grallert and Nurse, 1996).
orp1-4 and orp1-4 brc1Δ cells were arrested in HU at the permissive temperature (25°C), and then released from the HU block at either 25 or 36°C. At both temperatures, orp1-4 reduced the number of brc1Δ cells with Rad52 foci by ~4-fold (Fig. 8) compared with orp1+ cells (Figs 3, 4 and 5). This suggests that the function of the ORC is compromised in orp1-4 even at permissive temperature. Moreover, unlike orp1+ cells (Fig. 4), the HU block-and-release protocol did not induce formation of foci upon recovery from the replication arrest at 25 or 36°C, showing that full ORC activity is required for focus formation in brc1Δ cells. Temperature did not significantly influence the percentage of cells with Rad52 foci in either wild-type or brc1Δ backgrounds (not shown).
Fig. 8.
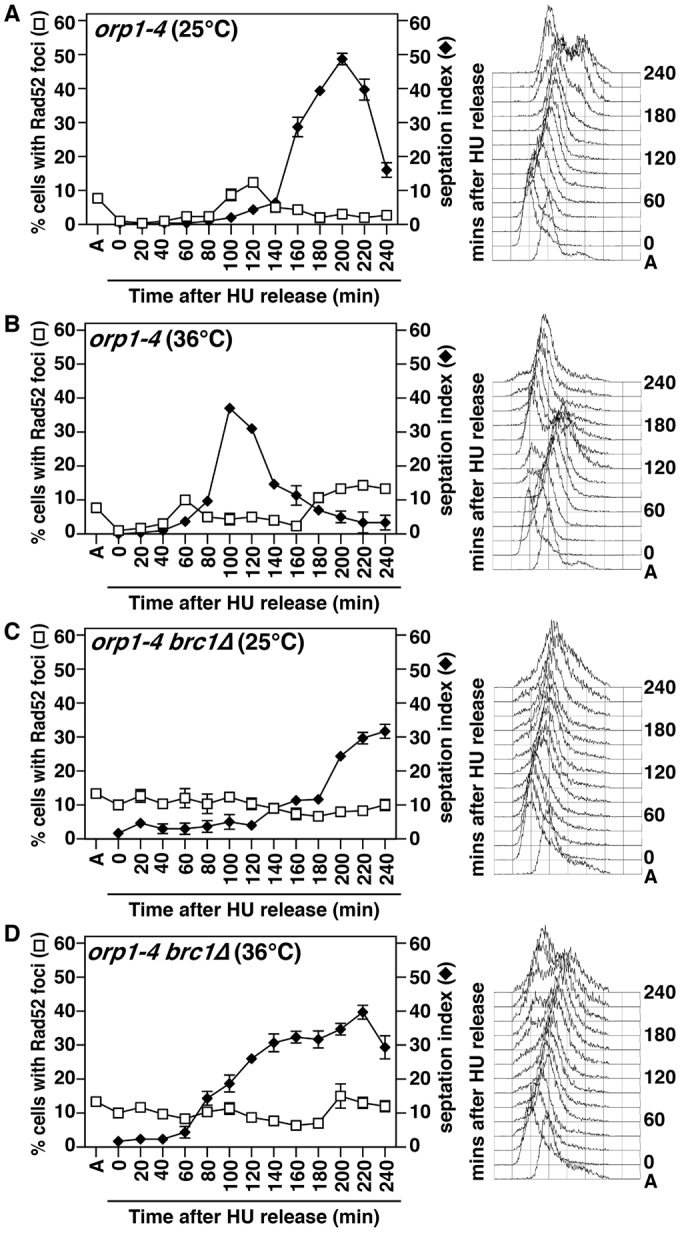
Reduction in ORC function suppresses the formation of Rad52 foci in brc1Δ cells. Asynchronous cultures of the indicated strains expressing Rad52–YFP were arrested in 11 mM HU for 6.5 hours at 25°C. HU was then removed by filtration, and cultures were returned to either 25°C (A,C) or 36°C (B,D). Samples were then taken at 20-minute intervals to determine septation index (closed diamonds) and percentage of cells containing Rad52–YFP foci (open squares). Data are means ± s.d. for three populations of 100 cells. Right panels: samples were also fixed in 70% ethanol and processed for FACS analysis of DNA content.
We also observed that cell cycle progression after HU arrest, assayed by septation index and FACS analysis of DNA content, was delayed in orp1-4 brc1Δ cells. Furthermore, the FACS analysis showed broad DNA profiles with cells containing both more and less DNA than orp1-4 controls (Fig. 8). Such profiles are produced by defects in chromosome segregation, and analysis of mitotic figures by DAPI-staining showed that HU block and release induces severe mitotic failure in orp1-4 brc1Δ cells (Fig. 9A). We then measured the viability of these cells, and not surprisingly, orp1-4 brc1Δ cells were highly sensitive to transient HU exposure at both temperatures, whereas orp1-4 single mutants survived the HU treatment at 25°C, and were dead regardless of HU treatment at 36°C (Fig. 9B). Therefore, the ORC-dependent formation of Rad52 foci in brc1Δ cells upon HU treatment is crucial for recovery from replication arrest. The fact that orp1-4 reduces the formation of foci without affecting viability in untreated cells suggests the formation of these foci is crucial for recovery from replication stress, where the foci persist (Fig. 4), but not during unperturbed S phases where >50% of the foci are rapidly resolved (Fig. 5).
Fig. 9.
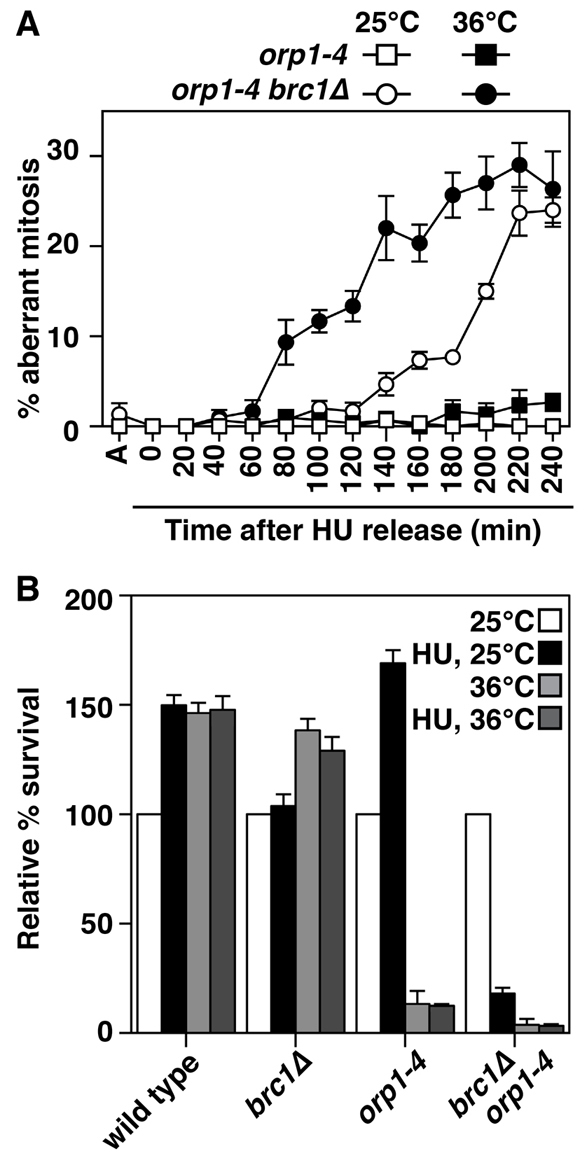
Suppression of replication-stress-induced Rad52 foci formation in brc1Δ cells results in lethal mitoses. (A) The same ethanol fixed cells as in Fig. 8 were stained with DAPI and aberrant mitotic figures were scored by microscopy (DAPI + DIC). Data are means ± s.d. for three populations of 100 cells. (B) The viability of cells grown at 25°C was determined by plating aliquots onto YES medium with plates incubated at 25°C for 5 days. The culture was then shifted to 36°C for 4 hours and viability determined using the same dilutions as for the 25°C culture. Alternatively, cells were arrested in 11 mM HU for 6.5 hours at 25°C, washed by filtration, and then incubated at 25°C or 36°C, followed by viability measurement as for the no HU cultures. Data are normalized to no HU cultures grown at 25°C, and are means ± s.d., n=3.
Discussion
Replicative DNA damage poses two separate problems to the cell: the lesion itself, and the physical barrier it causes to the replicative polymerases, requiring the lesion to be excised or bypassed so completion of DNA replication can occur. If bypassed, the lesion is still present and must be removed by DNA repair mechanisms (Branzei and Foiani, 2009). We have previously attributed the DNA damage sensitivity of brc1Δ cells to a DNA repair defect, which was in keeping with the interactions between Brc1 and the Smc5–Smc6 complex (Lee et al., 2007; Sheedy et al., 2005; Verkade et al., 1999). However, in this study we have separated the blockade to DNA replication from the effects of lesions in the template by utilizing HU-induced dNTP depletion. From these analyses, and from the resistance of brc1Δ cells to acute exposure to DNA damaging agents, we can conclude that Brc1 is required for resumption (or completion) of DNA replication after replication stress. Thus, the sensitivity to chronic exposure to agents such as MMS that elicit replicative DNA damage probably comes from many rounds of inefficient completion of DNA replication, rather than accumulated damage to DNA, as evidenced by the reduced mutagenesis seen in brc1Δ strains. In keeping with this model, DNA repair mutants accumulate markers of DNA damage, such as γH2A and activated Chk1, but brc1Δ cells do not (Figs 2, 3) (Outwin et al., 2009).
Despite the difficulty in completing DNA replication after HU arrest, brc1Δ cells do not show hyperactivation of the intra-S-phase checkpoint, as evidenced by Cds1 activity (Fig. 2). Cds1 is required to maintain stalled forks in a stable configuration that enables their rapid and efficient restart. The mechanism of Cds1 activation requires its recruitment to components at the stalled replisomes. Here, Cds1 interacts with the replisome component Mrc1, which enables the Rad3-catalyzed trans-phosphorylation and subsequent auto-phosphorylation that results in active Cds1 (Xu et al., 2006). brc1Δ cells actually express lower levels of Cds1 protein, but not of its mRNA, as well as displaying lower levels of Cds1 activation. However, as Chk1 is not activated in HU-arrested brc1Δ cells, the reduced Cds1 activity is clearly sufficient to prevent replication fork collapse, which signals a strong DNA damage response to Chk1 (Lindsay et al., 1998). The reason behind the reduced Cds1 expression and activity is not clear, although we note that cells lacking components of the replication fork protection complex are also defective in Cds1 activation (Noguchi et al., 2003). One attractive hypothesis is that the structure of the replication forks is perturbed in the absence of Brc1, and this affects Cds1 stability and activation. However, there are also data suggesting that the activation of Cds1 homologs prevent the firing of other and/or late replication origins (Branzei and Foiani, 2009), and as Brc1 seems to require this to survive replication stress, Cds1 activity might actually be actively suppressed.
This leaves the question of the function of the Rad52 foci in brc1Δ cells. They cannot mark loci that are destined for Rad52-dependent HR-mediated repair because they do not contain Rad51, the downstream mediator of recombination, but instead contain RPA. The presence of Rad52 but absence of Rad51 could explain why brc1Δ is synthetically lethal with rad52Δ (Williams et al., 2010), but not rad51Δ (Sheedy et al., 2005). A previous study using higher concentrations (15–20 mM) of HU did observe high levels of Rad51 and Rad52 foci after a 4-hour treatment (Bailis et al., 2008), suggesting that at these higher concentrations HU affects processes other than dNTP synthesis, or that widespread fork collapse is occurring, and these effects activate HR-mediated repair.
Given the unique nature of the foci, we decided to assess all known potential sources of ssDNA that could involve Rad52 – that is, recombination and replication. The fact that brc1Δ cells are not defective in HR-mediated repair (Verkade et al., 1999), and the absence of a link between Brc1 and error-free template switching, makes recombinational repair an unlikely source of the foci. Moreover, although brc1Δ cells are capable of SSA when forced down this pathway by the absence of Rad51, brc1Δ cells actually show reduced recombination between repeats (Fig. 6). Thus, intermediates of repeat recombination are also an unlikely source of the foci, which is in keeping with their predominately non-nucleolar localization (Fig. 7), where recombination between rDNA repeats modulates rDNA copy number. Hence, we next assayed replication itself as a source of the Rad52–RPA foci.
The fact that the Rad52 foci also contain large RPA foci, suggests there is a substantial amount of ssDNA at these sites. Furthermore, as the foci are not accompanied by strong Chk1 activation (Fig. 2) they are unlikely to contain primer–template junctions necessary for Chk1 activation (MacDougall et al., 2007). Finally, as their formation is ORC dependent (Fig. 8) and required for brc1Δ cells to survive a transient treatment with HU (Fig. 9), they probably originate from localized template unwinding at a sub-set of ORC-resident replication origins, without concomitant origin firing.
Under unstressed conditions, the majority of foci are resolved following bulk DNA replication (Fig. 5). However, after HU treatment, the foci persist and cell cycle progression is delayed (Fig. 4), although the mechanism of delay is not dependent solely on Chk1. As a DNA damage signal is not initiated, one explanation for the persistence of these foci is that replisome components are sequestered at the stably stalled replication forks, and thus replication is not initiated despite template unwinding. In conditions where these foci are substantially reduced (brc1Δ orp1-4), cells exposed to a transient disruption of replication are unable to recover, thus showing that the formation of these foci is necessary to survive a transient arrest in HU (Fig. 9). In addition, the persistence of these foci is induced under these conditions, indicating that HU treatment probably amplifies a condition of replication stress that occurs in the absence of Brc1.
From these observations, we conclude that Brc1 acts during S phase to aid in the completion of replication under conditions of replication stress. A component of this function for Brc1 is engagement of the error-prone branch of PRR. In keeping with this model, studies of the Brc1 homolog in budding yeast, Rtt107, have shown it to play a role in replication re-start mediated through its interaction with Slx4 (Ohouo et al., 2010; Roberts et al., 2006). However, Slx4 is a considerably larger protein in S. cerevisiae than in S. pombe, and the region required for the Slx4–Rtt107 interaction is absent in fission yeast (Coulon et al., 2004). This suggests Brc1 functions differently to Rtt107, however, Brc1-mediated suppression of smc6-74 is dependent on the Slx4-binding nuclease Slx1 (Sheedy et al., 2005), and a related function cannot be ruled out.
If Brc1 is required for replication re-start and functions primarily during S phase, how is it able to suppress the repair defects of smc6-74 in G2? Curiously, PRR seems to function in DNA repair out of the context of collision of the replisome with lesions in the template strand. Rad18-dependent ubiquitylation of PCNA occurs in irradiated G2 cells (Frampton et al., 2006; Karras and Jentsch, 2010), and Rad18 homologs have further been implicated in post-replication recombinational repair (Huang et al., 2009; Szüts et al., 2006). Furthermore, the S. pombe Rad18 homolog Rhp18 is required for DNA damage resistance throughout the cell cycle (Verkade et al., 2001). Thus, the structures that form in smc6-74 cells following DNA damage in G2 might either resemble replication forks, be processed into such structures and/or be resolved in a similar manner. As to whether this occurs in wild-type cells is not clear, although brc1Δ cells show wild-type DNA damage responses outside S phase (Sheedy et al., 2005; Verkade et al., 1999).
Our results also highlight the important finding that Rad52 foci cannot be equated with sites of double-strand break (DSB) repair by HR. Without confirmation that other HR proteins are resident in these foci, this convenient assay can be misleading. By the same argument, the absence of microscopically visible Rad52 foci does not indicate Rad52 is not accumulating at particular loci. For example, we recently showed Smc5–Smc6-dependent recruitment of Rad52 to stably stalled forks by ChIP, but the amount of protein at each locus is insufficient to form a visible focus (Irmisch et al., 2009). Studies in budding yeast have estimated that Rad52 foci at DSBs contain 600–2100 molecules, and can represent clustering of several DSBs into recombination centers (Lisby et al., 2003; Mortensen et al., 2009). Thus, the function of Rad52 is considerably more diverse than simply promoting Rad51 filament formation for HR-dependent repair.
Our studies of Brc1 and the Smc5–Smc6 complex have uncovered two additional roles for Rad52 in replication stress: the recruitment to stalled replication forks (Irmisch et al., 2009), and the formation of large Rad51-HR-independent foci late in S phase after replication stress. It is important to determine what Rad52 is doing under these conditions, and whether these functions extend to Rad52 in human cells. Human Rad52 is not a major initiator of HR, a function that seems to have been replaced by BRCA2, which is not present in the yeasts (Jensen et al., 2010; Kojic et al., 2008; Thorslund and West, 2007; van Veelen et al., 2005). Thus, it will be interesting to see the division of labor between Rad52 and BRCA2 for these processes, and whether these previously unknown functions for Rad52 have ensured its retention in higher eukaryotes despite the presence of BRCA2 to initiate HR.
Materials and Methods
General 975h+ methods
All strains used were derived from the 972h− and 975h+strains. Standard procedures were used for propagation and genetic manipulation (Moreno et al., 1991). For cell counting, cells fixed with 3.7% formaldehyde and washed in 1× PBS were analyzed on a Coulter Z1 particle counter.
For hydroxyurea (HU) sensitivity assays, serial dilutions were plated in triplicate on yeast extract plus supplements (YES) agar plates following treatment for 0–8 hours with 11 mM HU. Colonies were counted and the percentage survival was expressed as a proportion of the untreated controls after 4 days growth at 30°C. cdc25-22 cultures were synchronized by growing cells overnight at 25°C in supplemented EMM2 medium to mid-log phase, followed by incubation at 36°C for 4.5 hours to arrest cells at the G2–M boundary. After return to the permissive (25°C) temperature, these cells proceeded through the cell cycle with a high degree of synchrony, as determined by septation indices determined from three samples of 100 cells. For HU block-and-release, mid-log phase cultures grown in supplemented EMM2 medium at 30°C were treated with 11 mM HU for 4 hours to synchronize cells in early S phase. Cells were then washed by filtration and resuspended in fresh medium and further incubated at 30°C. Samples were taken at appropriate intervals for the described assays. DNA content was determined with a FACS Caliber (Becton Dickinson) as described previously (Calonge et al., 2010). Mutation rates in can1 were calculated using the method of the median from 11 independent cultures as previously described (Lee et al., 2007). Mutants were selected on EMM2 medium with 60 μg/ml canavanine. MMS-induced mutagenesis was performed on cells treated with 0.05% MMS for 60 minutes, with the MMS then inactivated with 5% sodium thiosulphate. PFGE was performed on untreated exponentially growing cells or treated cells at various times after exposure to 11 mM HU as described (Outwin et al., 2009). Recombination frequency was determined by measuring the numbers of Ade-positive colonies arising from strains containing two ade6 heteroalleles flanking a his3 gene (Osman et al., 1996). Frequencies of five to seven colonies were averaged to determine the mean recombination frequency. Error bars indicate standard deviation from the mean.
Chromatin immunoprecipitation (ChIP)
ChIP was performed on 50 ml samples of either asynchronous mid-log phase cultures or cultures treated with 11 mM HU for 4 hours, as previously described (Outwin et al., 2009). Immunoprecipitations were performed with polyclonal anti-GFP antibodies (Invitrogen); real-time PCR reactions were carried out with ChIP primers designed with Primer 3 software, and cycle times calculated as described previously (Outwin et al., 2009).
Microscopy
DNA was visualized with 1 μg/ml 4′,6-diamino-2-phenylindole (DAPI). Data were collected from at least three samples of at least 100 cells. Microscopy was performed on a Nikon E800 microscope with a 100× 1.40 Plan-Apo objective lens. Images were captured on a Spot RT/SE Camera using Spot advanced software, and prepared for publication using Adobe Photoshop. Cells expressing Rad22–YFP were imaged either as live cells, or cells fixed with 70% ethanol. Cells expressing Gar2–mCherry were imaged as live cells. Cells expressing Rad11–GFP were imaged either as live cells, or were fixed with 0.5% formaldehyde. Cells expressing Rad22–RFP were fixed with 0.5% formaldehyde and RFP foci were counted after image capture (because of the quick fading of these foci). Rhp51 was detected by indirect immunofluorescence microscopy as described previously (Lambert et al., 2005) with an anti-Rhp51 antibody (Diagnocine, Hackensack, NJ) used at a 1:400 dilution, and detected with Alexa-Fluor-488-coupled anti-rabbit IgG antibodies at a 1:400 dilution. S129 phosphorylated histone H2A (γH2A) was detected with a phosphorylation-specific antibody (Abcam) at a 1:1500 dilution in cells fixed in 1.6% formaldehyde, 0.2% glutaraldehyde, and processed as for anti-Rhp51 staining.
Western blotting
For detection of epitope-tagged proteins, frozen cells were disrupted with glass beads using a bead beater and extracted into urea lysis buffer (O'Connell et al., 1997). The extract was cleared by centrifugation at 13,000 g for 5 minutes, and the supernatant was boiled in SDS sample buffer. Protein extracts were run on SDS-PAGE gels and transferred to nitrocellulose membrane in 10 mM 3-(cyclohexamino)-1-propanesulfonic acid (pH 11.0) and 10% methanol for 1 hour. Immune complexes were detected with horseradish-peroxidase-linked secondary antibodies (GE Healthcare, Buckinghamshire, UK) followed by chemiluminescence. Chk1 activation was analyzed by western blotting with anti-HA (12CA5) to detect the HA-tagged chk1 allele as described previously (Calonge and O'Connell, 2006), which migrates as a higher molecular mass species following activation.
Cds1 kinase assays
HA-tagged Cds1 was expressed from its endogenous promoter in wild-type and brc1Δ backgrounds. Cultures were grown to mid-log phase and then treated with 0 or 11 mM HU for 4 hours, harvested by centrifugation, and snap frozen in liquid nitrogen. Cell lysates were prepared, as described above, in native lysis buffer [30 mM NaPO4 (pH 7.5), 500 mM NaCl, 50 mM Tris pH 7.4, 50 mM NaF, 10 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride, 0.1% NP-40, 10 mM β-glycerophosphate, 0.5% protease inhibitor cocktail (Sigma), 1 mM dithiothreitol (DTT), 0.1 mM activated sodium orthovanadate, 1.5 mM ρ-NPP, 2 μg/ml pepstatin, 2 μg/ml leupeptin, 2 μg/ml aprotinin, 2 μg/ml E64] and immunoprecipitated using anti-HA (12CA5) antibody. Immunoprecipitates were washed extensively in lysis buffer, and then incubated with 20 μl kinase reaction buffer [20 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1 mM DTT, 75 mM KCl, 100 mM ATP and 5 μCi [γ-32P]ATP, 5 μg myelin basic protein] at 30°C for 20 minutes. The reaction was then run on a 12% SDS-PAGE gel, which was stained and dried. 32P incorporation onto the myelin basic protein was quantified with a Bio-Rad FX Phosphorimager.
Acknowledgements
We are grateful to Paul Russell, Greg Freyer, Fekret Osman, Virginia Zakian and Matthew Whitby for the provision of strains. We also appreciate the critical discussions with Emily Outwin, Claudia Tapia-Alveal, Karen Kuntz and Karen Lee.
Footnotes
Funding
This work was supported by the National Institutes of Health [grant numbers GM087326 and GM088162 to M.J.O.]; Cancer Research UK [grant number C9601/A9484 to J.M.M.]; the Medical Research Council [grant numbers G0901011 and G1001668 to J.M.M.]; and the National Institutes of Health/National Cancer Institute training grant [grant number T32 CA78207 to K.L.B.]. Deposited in PMC for release after 12 months.
References
- Ahn J. S., Osman F., Whitby M. C. (2005). Replication fork blockage by RTS1 at an ectopic site promotes recombination in fission yeast. EMBO J. 24, 2011-2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ampatzidou E., Irmisch A., O'Connell M. J., Murray J. M. (2006). Smc5/6 is required for repair at collapsed replication forks. Mol. Cell. Biol. 26, 9387-9401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews E. A., Palecek J., Sergeant J., Taylor E., Lehmann A. R., Watts F. Z. (2005). Nse2, a component of the Smc5-6 complex, is a SUMO ligase required for the response to DNA damage. Mol. Cell. Biol. 25, 185-196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailis J. M., Luche D. D., Hunter T., Forsburg S. L. (2008). Minichromosome maintenance proteins interact with checkpoint and recombination proteins to promote S-phase genome stability. Mol. Cell. Biol. 28, 1724-1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D., Foiani M. (2008). Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 9, 297-308 [DOI] [PubMed] [Google Scholar]
- Branzei D., Foiani M. (2009). The checkpoint response to replication stress. DNA Repair (Amst.) 8, 1038-1046 [DOI] [PubMed] [Google Scholar]
- Byun T. S., Pacek M., Yee M. C., Walter J. C., Cimprich K. A. (2005). Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 19, 1040-1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calonge T. M., O'Connell M. J. (2006). Antagonism of Chk1 signaling in the G2 DNA damage checkpoint by dominant alleles of Cdr1. Genetics 174, 113-123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calonge T. M., Eshaghi M., Liu J., Ronai Z., O'Connell M. J. (2010). Transformation/transcription domain-associated protein (TRRAP)-mediated regulation of Wee1. Genetics 185, 81-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon S., Gaillard P. H., Chahwan C., McDonald W. H., Yates J. R., 3rd, Russell P. (2004). Slx1-Slx4 are subunits of a structure-specific endonuclease that maintains ribosomal DNA in fission yeast. Mol. Biol. Cell 15, 71-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe C. L., Whitby M. C. (2004). The involvement of Srs2 in post-replication repair and homologous recombination in fission yeast. Nucleic Acids Res. 32, 1480-1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe C. L., Osman F., Dixon J., Whitby M. C. (2004). DNA repair by a Rad22-Mus81-dependent pathway that is independent of Rhp51. Nucleic Acids Res. 32, 5570-5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsburg S. L., Nurse P. (1991). Cell cycle regulation in the yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe. Annu. Rev. Cell Biol. 7, 227-256 [DOI] [PubMed] [Google Scholar]
- Frampton J., Irmisch A., Green C. M., Neiss A., Trickey M., Ulrich H. D., Furuya K., Watts F. Z., Carr A. M., Lehmann A. R. (2006). Postreplication repair and PCNA modification in Schizosaccharomyces pombe. Mol. Biol. Cell 17, 2976-2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grallert B., Nurse P. (1996). The ORC1 homolog orp1 in fission yeast plays a key role in regulating onset of S phase. Genes Dev. 10, 2644-2654 [DOI] [PubMed] [Google Scholar]
- Gulli M. P., Girard J. P., Zabetakis D., Lapeyre B., Melese T., Caizergues-Ferrer M. (1995). gar2 is a nucleolar protein from Schizosaccharomyces pombe required for 18S rRNA and 40S ribosomal subunit accumulation. Nucleic Acids Res. 23, 1912-1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heichinger C., Penkett C. J., Bähler J., Nurse P. (2006). Genome-wide characterization of fission yeast DNA replication origins. EMBO J. 25, 5171-5179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T. (2006). At the heart of the chromosome: SMC proteins in action. Nat. Rev. Mol. Cell Biol. 7, 311-322 [DOI] [PubMed] [Google Scholar]
- Huang J., Huen M. S., Kim H., Leung C. C., Glover J. N., Yu X., Chen J. (2009). RAD18 transmits DNA damage signalling to elicit homologous recombination repair. Nat. Cell Biol. 11, 592-603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G., Malkova A., Liberi G., Foiani M., Haber J. E. (2003). Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 115, 401-411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmisch A., Ampatzidou E., Mizuno K., O'Connell M. J., Murray J. M. (2009). Smc5/6 maintains stalled replication forks in a recombination-competent conformation. EMBO J. 28, 144-155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen R. B., Carreira A., Kowalczykowski S. C. (2010). Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467, 678-683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai M., Wang T. S. (2003). Checkpoint responses to replication stalling: inducing tolerance and preventing mutagenesis. Mutat. Res. 532, 59-73 [DOI] [PubMed] [Google Scholar]
- Karras G. I., Jentsch S. (2010). The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 141, 255-267 [DOI] [PubMed] [Google Scholar]
- Kim S. M., Huberman J. A. (2001). Regulation of replication timing in fission yeast. EMBO J. 20, 6115-6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojic M., Mao N., Zhou Q., Lisby M., Holloman W. K. (2008). Compensatory role for Rad52 during recombinational repair in Ustilago maydis. Mol. Microbiol. 67, 1156-1168 [DOI] [PubMed] [Google Scholar]
- Krejci L., Van Komen S., Li Y., Villemain J., Reddy M. S., Klein H., Ellenberger T., Sung P. (2003). DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423, 305-309 [DOI] [PubMed] [Google Scholar]
- Krogh B. O., Symington L. S. (2004). Recombination proteins in yeast. Annu. Rev. Genet. 38, 233-271 [DOI] [PubMed] [Google Scholar]
- Lambert S., Watson A., Sheedy D. M., Martin B., Carr A. M. (2005). Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell 121, 689-702 [DOI] [PubMed] [Google Scholar]
- Latif C., den Elzen N. R., O'Connell M. J. (2004). DNA damage checkpoint maintenance through sustained Chk1 activity. J. Cell Sci. 117, 3489-3498 [DOI] [PubMed] [Google Scholar]
- Lee K. M., Nizza S., Hayes T., Bass K. L., Irmisch A., Murray J. M., O'Connell M. J. (2007). Brc1-mediated rescue of Smc5/6 deficiency: requirement for multiple nucleases and a novel Rad18 function. Genetics 175, 1585-1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. Y., Myung K. (2008). PCNA modifications for regulation of post-replication repair pathways. Mol. Cells 26, 5-11 [PMC free article] [PubMed] [Google Scholar]
- Lindsay H. D., Griffiths D. J. F., Edwards R. J., Christensen P. U., Murray J. M., Osman F., Walworth N., Carr A. M. (1998). S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev. 12, 382-395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M., Mortensen U. H., Rothstein R. (2003). Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat. Cell Biol. 5, 572-577 [DOI] [PubMed] [Google Scholar]
- Lupardus P. J., Byun T., Yee M. C., Hekmat-Nejad M., Cimprich K. A. (2002). A requirement for replication in activation of the ATR-dependent DNA damage checkpoint. Genes Dev. 16, 2327-2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDougall C. A., Byun T. S., Van C., Yee M. C., Cimprich K. A. (2007). The structural determinants of checkpoint activation. Genes Dev. 21, 898-903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister P., Taddei A., Vernis L., Poidevin M., Gasser S. M., Baldacci G. (2005). Temporal separation of replication and recombination requires the intra-S checkpoint. J. Cell Biol. 168, 537-544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno S., Klar A., Nurse P. (1991). Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194, 795-823 [DOI] [PubMed] [Google Scholar]
- Morikawa H., Morishita T., Kawane S., Iwasaki H., Carr A. M., Shinagawa H. (2004). Rad62 protein functionally and physically associates with the smc5/smc6 protein complex and is required for chromosome integrity and recombination repair in fission yeast. Mol. Cell. Biol. 24, 9401-9413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen U. H., Bendixen C., Sunjevaric I., Rothstein R. (1996). DNA strand annealing is promoted by the yeast Rad52 protein. Proc. Natl. Acad. Sci. USA 93, 10729-10734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen U. H., Lisby M., Rothstein R. (2009). Rad52. Curr. Biol. 19, R676-R677 [DOI] [PubMed] [Google Scholar]
- Noguchi E., Noguchi C., Du L. L., Russell P. (2003). Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol. Cell. Biol. 23, 7861-7874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell M. J., Cimprich K. A. (2005). G2 damage checkpoints: what is the turn-on? J. Cell Sci. 118, 1-6 [DOI] [PubMed] [Google Scholar]
- O'Connell M. J., Raleigh J. M., Verkade H. M., Nurse P. (1997). Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J. 16, 545-554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell M. J., Walworth N. C., Carr A. M. (2000). The G2-phase DNA-damage checkpoint. Trends Cell Biol. 10, 296-303 [DOI] [PubMed] [Google Scholar]
- Ohouo P. Y., Bastos de Oliveira F. M., Almeida B. S., Smolka M. B. (2010). DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol. Cell 39, 300-306 [DOI] [PubMed] [Google Scholar]
- Osman F., Fortunato E. A., Subramani S. (1996). Double-strand break-induced mitotic intrachromosomal recombination in the fission yeast Schizosaccharomyces pombe. Genetics 142, 341-357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman F., Adriance M., McCready S. (2000). The genetic control of spontaneous and UV-induced mitotic intrachromosomal recombination in the fission yeast Schizosaccharomyces pombe. Curr. Genet. 38, 113-125 [DOI] [PubMed] [Google Scholar]
- Outwin E. A., Irmisch A., Murray J. M., O'Connell M. J. (2009). Smc5-Smc6-dependent removal of cohesin from mitotic chromosomes. Mol. Cell. Biol. 29, 4363-4375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pebernard S., McDonald W. H., Pavlova Y., Yates J. R., 3rd, Boddy M. N. (2004). Nse1, Nse2, and a novel subunit of the Smc5-Smc6 complex, Nse3, play a crucial role in meiosis. Mol. Biol. Cell 15, 4866-4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts T. M., Kobor M. S., Bastin-Shanower S. A., Ii M., Horte S. A., Gin J. W., Emili A., Rine J., Brill S. J., Brown G. W. (2006). Slx4 regulates DNA damage checkpoint-dependent phosphorylation of the BRCT domain protein Rtt107/Esc4. Mol. Biol. Cell 17, 539-548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedy D. M., Dimitrova D., Rankin J. K., Bass K. L., Lee K. M., Tapia-Alveal C., Harvey S. H., Murray J. M., O'Connell M. J. (2005). Brc1-mediated DNA repair and damage tolerance. Genetics 171, 457-468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szüts D., Simpson L. J., Kabani S., Yamazoe M., Sale J. E. (2006). Role for RAD18 in homologous recombination in DT40 cells. Mol. Cell. Biol. 26, 8032-8041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-Alveal C., O'Connell M. J. (2011). Nse1-dependent recruitment of Smc5/6 to lesion-containing loci contributes to the repair defects of mutant complexes. Mol. Biol. Cell 22, 4669-4682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorslund T., West S. C. (2007). BRCA2: a universal recombinase regulator. Oncogene 26, 7720-7730 [DOI] [PubMed] [Google Scholar]
- Uemura T., Ohkura H., Adachi Y., Morino K., Shiozaki K., Yanagida M. (1987). DNA topoisomerase II is required for condensation and separation of mitotic chromosomes in S. pombe. Cell 50, 917-925 [DOI] [PubMed] [Google Scholar]
- van Veelen L. R., Essers J., van de Rakt M. W., Odijk H., Pastink A., Zdzienicka M. Z., Paulusma C. C., Kanaar R. (2005). Ionizing radiation-induced foci formation of mammalian Rad51 and Rad54 depends on the Rad51 paralogs, but not on Rad52. Mutat. Res. 574, 34-49 [DOI] [PubMed] [Google Scholar]
- Vanoli F., Fumasoni M., Szakal B., Maloisel L., Branzei D. (2010). Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genet. 6, e1001205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veaute X., Jeusset J., Soustelle C., Kowalczykowski S. C., Le Cam E., Fabre F. (2003). The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423, 309-312 [DOI] [PubMed] [Google Scholar]
- Verkade H. M., Bugg S. J., Lindsay H. D., Carr A. M., O'Connell M. J. (1999). Rad18 is required for DNA repair and checkpoint responses in fission yeast. Mol. Biol. Cell 10, 2905-2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkade H. M., Teli T., Laursen L. V., Murray J. M., O'Connell M. J. (2001). A homologue of the Rad18 postreplication repair gene is required for DNA damage responses throughout the fission yeast cell cycle. Mol. Genet. Genomics 265, 993-1003 [DOI] [PubMed] [Google Scholar]
- Walworth N. C., Bernards R. (1996). rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science 271, 353-356 [DOI] [PubMed] [Google Scholar]
- Williams J. S., Williams R. S., Dovey C. L., Guenther G., Tainer J. A., Russell P. (2010). gammaH2A binds Brc1 to maintain genome integrity during S-phase. EMBO J. 29, 1136-1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. J., Davenport M., Kelly T. J. (2006). Two-stage mechanism for activation of the DNA replication checkpoint kinase Cds1 in fission yeast. Genes Dev. 20, 990-1003 [DOI] [PMC free article] [PubMed] [Google Scholar]