

Abstract
The presence and persistence of alloantigen is necessary for graft-specific T-cell-mediated immunity. However, specificity comprises only a single facet of an extremely complex process. Evidence is accruing to suggest that immunogenicity could be manipulated by endogenous ligands released during tissue injury. Stress molecules are significantly up-regulated following transplantation and stimulate conserved receptors on a range of leucocytes, including dendritic cells (DCs). The DCs are essential for co-stimulation and the induction of adaptive immunity. Stress signals can act as an adjuvant leading to DC maturation and activation. DCs stimulated by endogens exhibit enhanced alloantigen presentation, co-stimulation and production of pro-inflammatory cytokines, such as interleukin-1β (IL-1β) and IL-18. Inflammasomes have a major role in IL-1β/IL-18 production and secretion, and can be stimulated by endogens. Importantly, the polarization toward inflammatory T helper type 17 cells as opposed to regulatory T cells is dependent upon, among other factors, IL-1β. This highlights an important differentiation pathway that may be influenced by endogenous signals. Minimizing graft damage and stress expression should hypothetically be advantageous, and we feel that this area warrants further research, and may provide novel treatment modalities with potential clinical benefit.
Keywords: innate immunity, novel biological therapies (anti-cytokines etc.), T cells, toll receptors/toll-like receptors, transplantation
Allograft rejection
Transplantation accompanied by an immunosuppressive regimen, is the established treatment for end-stage organ disease. Engraftment success is principally determined by the extent of the recipient immune response. Both acute and chronic rejection contribute significantly to allograft dysfunction. Acute rejection is typified by a predominantly T-cell-mediated response to the graft, and is observed most frequently in the early post-transplant period (weeks to months). Histological examination of endomyocardial biopsy is the standard diagnostic method, and is graded according to the level of allograft infiltrate and extent of myocyte damage. Acute rejection generally responds well to additional immunosuppressive treatment. Conversely, chronic rejection remains a major limitation to long-term allograft acceptance. Although there are differences in chronic rejection between organs, there is commonly a characteristic fibrosis of the graft and associated vasculature. Chronic rejection typically manifests over a significantly longer period (months to years), and causes a gradual deterioration in graft function. Although the precise mechanisms responsible are not completely understood, it is clear that rejection involves the collective interplay of a milieu of immune processes. Allograft rejection occurs as a direct response to the presence and persistence of alloantigen. Whereas this is an absolute necessity for a graft-specific T-cell-mediated response, specificity comprises only a single facet of an extremely complex immunological network. We feel that additional factors may be more influential than previously thought. Mounting an effective immune response requires a vast array of co-ordinated signalling mechanisms (T helper cell-derived ‘help’, proliferative signals via inflammatory cytokines, cellular diapedesis via chemotactic gradients and adhesion molecule expression) and interactions [MHC–peptide complex ligation to T-cell receptor (TCR), co-stimulation), many of which are inducible in an antigen-independent manner. The current non-self-discrimination paradigm with regard to transplantation relies on the notion that alloantigen is inherently immunogenic. However, evidence is accruing to suggest that several mechanisms in the alloimmune response could be manipulated via the stimulation of conserved pattern recognition receptors (PRRs) by damage-associated molecular patterns (DAMPs) of endogenous origin (see Fig. 1).
Figure 1.
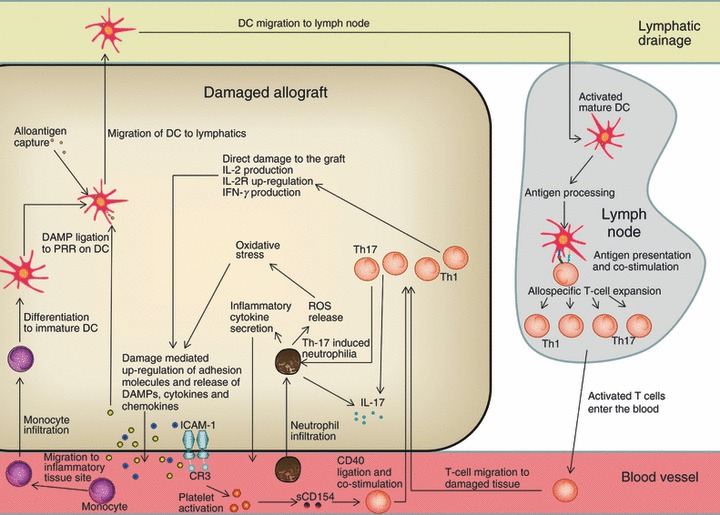
Overview of immune interactions stimulated by damage-associated molecular pattern (DAMP) release. DAMPs released during tissue damage ligate to Toll-like receptors (TLRs)/ nucleotide-binding oligomerization domain-like receptors (NLRs) and instigate the activation of important immune populations, i.e. mature dendritic cells (DCs), which subsequently influence T-cell differentiation [T helper type 1 (Th1), Th2, Th17, regulatory T (Treg)]. Mature DCs will co-stimulate allospecific T cells, inducing interleukin-2 (IL-2) synthesis, CD25 up-regulation and interferon-γ (IFN-γ) production. Stress-induced DAMP release could induce the differentiation of monocytes to DCs, further enhancing the potential for T-cell involvement. Up-regulated intercellular adhesion molecule 1 (ICAM-1) can be bound by CR3, inducing platelet activation and soluble CD154 release, which is able to provide co-stimulation to T cells. IL-2R, IL-2 receptor; PRR, pattern-recognition receptor; ROS, reactive oxygen species.
Donor-derived endogenous stress and the immune response
Experimental data in mice have demonstrated that resting mature dendritic cells (DCs) can be activated in the absence of foreign stimuli, to a level comparable with lipopolysaccharide-stimulated cells (up-regulated antigen presentation and co-stimulation compared with controls).1 These DCs may potentially stimulate naive T cells, prompting an adaptive response. Moreover, stressed/necrotic cells but not healthy or apoptotic cells act as an adjuvant, enabling a primary immune response to ovalbumin in vivo. This illustrates that foreign antigen alone does not initiate a response, highlighting the disparity between antigenicity and immunogenicity.
Innate immunity is important in tissue maintenance, the routine clearance of cellular debris and post-surgical trauma.2–4 Activation can occur as a direct result of DAMPs originating from endogenous tissue and received through PRRs constitutively expressed on a variety of leucocytes [i.e. Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-like receptors (NLRs)]. Immune activation is again demonstrated to occur in the absence of non-self antigens. It is therefore plausible that a similar process could be responsible for innate activation following transplantation.
Early infiltration of the graft is dominated by macrophages, neutrophils and other innate cells, which become activated before, and can substantially influence adaptive cells.5 A milieu of confirmed constitutive/inducible endogenous ligands for PRRs is released in the early post-transplant period in response to the inherent damage and stress caused by the procedure (see Table 1).6–8
Table 1.
Endogenous damage-associated molecular patterns (DAMPs), their corresponding receptors on immune cells and the downstream effects of their ligation
| DAMP | Receptor | Function |
|---|---|---|
| Biglycan | TLR2/4 | Increased CXCL13 expression; rapid p38 MAPK, ERK and NF-κB activation; TNF-α and MIP-2 secretion9,10 |
| HMGB1 | RAGE and TLR2/4 | TNF-α, IL-1β, IL-6 and IL-10 secretion; DC maturation |
| Heat-shock protein 60/70 | TLR2/4 | TNF-α, IL-1, IL-6 and IL-12 secretion; DC maturation; C-C chemokine and NO release |
| Fibrinogen | TLR4 | NF-κB activation; TNF-α and IL-1β secretion; MIP-1α, MIP-1β, MIP-2 and MCP-1 secretion |
| Uric acid | TLR2 | Caspase-1 activation and IL-1β/IL-18 release; |
| ATP | P2X7R | Caspase-1 activation and IL-1β/IL-18 release; up-regulated DC co-stimulatory molecules |
DC, dendritic cell; ERK, extracellular signal-regulated kinase; IL-2, interleukin-2; MAPK, mitogen-activated protein kinase, MCP-1, monocytes chemoattractant protein 1; MIP, macrophage inflammatory protein; NF-κB, nuclear factor-κB; TLR, Toll-like receptor; TNF tumour necrosis factor.
The presence of PRR agonists [including High Mobility Group Box 1 (HMGB1), biglycan and heat-shock proteins]11,12 provide a foundation for innate stimulation. PRR ligation can initiate important signalling cascades in an alloantigen-independent process, up-regulating the production of a panoply of pro-inflammatory cytokines (see Table 1) that can influence adaptive immunity and promote acute rejection. Interestingly, stimulated macrophages actually secrete both HMGB1 and biglycan as endogenous pro-inflammatory mediators, thereby escalating the response further and recruiting the involvement of other PRR-expressing cells.9,13
Acute rejection episodes have been demonstrated to be the major risk factor in the development of chronic rejection in solid organ transplantation.14,15 T-cell differentiation to a memory phenotype may enable acute insults to translate into a chronic response. However, chronic rejection manifestation is also significantly influenced by non-antigen-specific factors, such as neutrophilia and oxidative stress, which are responsible for progressive graft damage. Oxidative stress can result from a range of processes, including inflammation and calcineurin inhibitor therapy,16 which disturb the balance between reactive oxygen species (ROS) and natural antioxidants. Innate immune cells, including activated neutrophils, release significant amounts of inducible nitric oxide synthase and NADPH oxidase, contributing to the overwhelming pro-oxidant environment.17 Activation of PRRs as a result of the damage associated with oxidative stress induces ROS production, maintaining the redox imbalance. Oxidative stress and neutrophilia are therefore intrinsically linked in a deleterious cycle which escalates DAMP expression and chronic graft damage, eventually leading to rejection. Interestingly, NADPH oxidase production by neutrophils is able to up-regulate TLR2 and stable intercellular adhesion molecule 1 (ICAM-1) expression on TLR4-stimulated endothelial cells.18 This should allow more efficient adhesion of graft-infiltrating immune cells, and will increase endothelial cell responsiveness to DAMPs.
Conserved endogenous pathways and the local tissue environment
Although both innate and adaptive cells are integral in efficient immunity, the structural components of the tissue also play a vital role. The interplay between tissue and effector cells is strictly regulated to ensure that immune activation occurs only when and where it is necessary, and is adequately specific to minimize collateral tissue damage. Fibroblasts, and epithelial and endothelial cells can release DAMPs, pro-inflammatory or anti-inflammatory cytokines, chemokines and up-regulate adhesion molecules as a signal to immune cells to induce (or cease) a response. For example, fibroblasts significantly express interleukin-6 (IL-6) and allograft inflammatory factor-1 in a model of scleradermatous graft-versus-host-disease, and are therefore of importance in stem cell transplantation.19 Notably, fibroblast activation is elevated during graft rejection and has been associated with significantly increased levels of hyaluronan,20 a confirmed ligand for TLR4. Acute kidney injury induces an increased expression of TLR4 on endothelial cells,21 allowing for co-operation between populations to potentiate the response. The well-documented DAMP HMGB1, which is released during tissue damage and can signal via TLR4, significantly increases adhesion molecule expression on the endothelial surface.21 This is extremely important in the extravasation of leucocytes, hence allowing an efficient immune response. Importantly, the expression of PRRs on structural cells22–24 enables them to receive, as well as act as a source of, DAMP signals. This versatility provides a mechanism whereby stress signals can be efficiently amplified and maintained, so contributing to graft rejection.
Damaged endothelial cells lining graft vasculature release endogenous mediators including IL-1α, IL-1β and IL-18, enhancing immunity in the presence of alloantigen. Importantly, IL-1β and IL-18 production and secretion involves multiple signals (see Fig. 2). Interleukin-1β gene transcription requires PRR stimulation and caspase-1 activation.25 Caspase-1 activation in macrophages requires the assembly of inflammasomes in an NLR-dependent mechanism. The activation requirements of inflammasomes are incompletely defined; however, a number of sources suggest that membrane disruption allowing the release of endogenous molecules is a key mechanism.26,27 The NACHT, LRR and PYD domains-containing protein 3 (NALP3) inflammasome may represent a general sensor for danger and stress signals (which can occur in the absence of allogeneic or microbial stimuli), an assertion that is supported by a range of data.28–31 Inflammasomes have recently been identified as having an important role in translating endogenous DAMP signals (such as uric acid32 and biglycan10,25) into inflammatory responses in a range of diseases.28,30,33,34 Extracellular ATP released via membrane disruption is an extremely important DAMP, and has pluripotent properties dependent upon the relative expression of corresponding receptors in the local environment [including α-1 adrenergic receptor and P2X purinoreceptor 7 (P2X7)].35,36 In several inflammatory diseases, including asthmatic airway inflammation and graft-versus-host diseases, extracellular ATP recognized by P2X7 receptor induces inflammasome activity, including caspase-1 activation, pro-inflammatory cytokine transcription/release and increased expression of co-stimulatory molecules on DCs.37,38 Blockade of this endogenous pathway has reported benefits on clinical outcome (including survival) in recipients.
Figure 2.
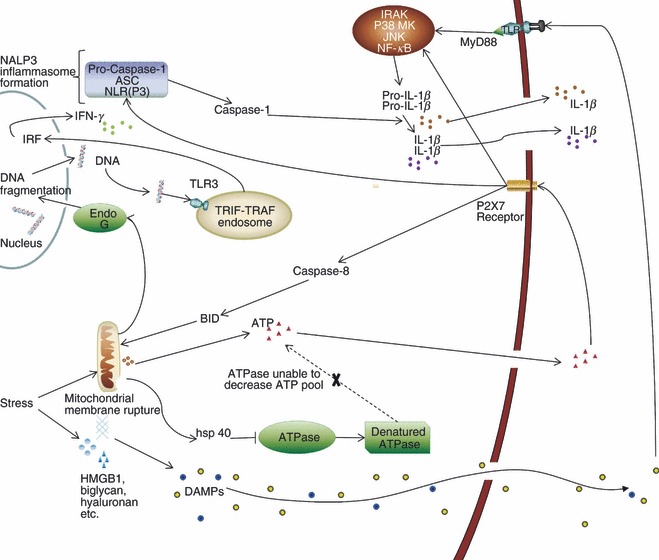
Mitochondrial damage, stress signalling and inflammasome formation. Damage-associated molecular patterns (DAMPs) released during tissue damage ligate to Toll-like receptors (TLRs), which initiate intracellular cascades i.e. interleukin receptor-associated kinase (IRAK), p38 mitogen activated protein kinase (MAPK), Jun N-terminal kinase (JNK) and nuclear factor-κB (NF-κB), leading to pro-interleukin-1β (IL-1β) and pro-IL-18 production. Tissue trauma can also initiate caspase-8/BID-dependent mitochondrial membrane rupture, releasing ATP. ATPase is denatured by heat-shock protein 40 (HSP 40) during stress, contributing to elevated ATP levels during tissue damage. ATP binds to P2X7 purinergic receptor inducing NACHT, LRR and PYD domains-containing protein 3 (NALP3) inflammasome formation and cleavage of pro-caspase-1. ATP-induced caspase-1 cleaves pro-IL-1β and pro-IL-18 to functional IL-1β and IL-18. Mitochondrial disruption also causes endo G-dependent DNA fragmentation. DNA fragments will therefore bind to TLR3 on the TIR-domain-containing adaptor-inducing interferon-β (TRIF) and tumour necrosis factor receptor associated factor (TRAF) endosome, inducing interferon regulatory factor (IRF) signalling and interferon-γ release. ASC, apoptotic speck protein; NLR, nucleotide-binding oligomerization domain-like receptors.
Stimulation of TLR2 and TLR4 alone is sufficient to induce IL-1β release from monocytes, because of their constitutively active form of caspase-1.39 This essentially equates to a potent pro-inflammatory mechanism without the immunological safeguard of a two-signal system. Importantly, TLR2 and TLR4 are pluripotent, with a vast array of endogenous ligands expressed on or released from injured donor tissue, including heat-shock proteins, fibrinogen, HMGB1 and biglycan.12,40 This emphasizes the potential for endogenous signals to play an influential role following transplantation, where the local tissue environment has the capacity to dictate and control subsequent immune responses. The donor endothelium (which represents an early target for recipient T cells infiltrating the graft) can significantly influence T-cell proliferation and differentiation via regulating cytokine secretion.41 This equates to endothelial cell control over the allogeneic inflammatory response. Under inflammatory conditions, the donor endothelium promotes the expansion of helper T cells, pro-inflammatory T helper type 17 (Th17) and regulatory T (Treg) cell populations. The concomitant Th17/Treg expansion has been confirmed elsewhere.42 The reason for such a relationship, other than their (disputed) shared requirement for transforming growth factor-β,42–45 is ill defined considering their vastly different functions. The pro-inflammatory Th17 population is responsible for accelerated graft rejection and vasculopathy in the absence of Th1 cells,46 and is considered to be the major inducer of neutrophilia in transplantation.47 As such, Th17 cells are considered to be an important population in transplantation immunity. The induction of a Th17 response is reliant on, among other factors, IL-1β43,44,48 and IL-6. Mice deficient in IL-1R exhibit significantly impaired Th17 responses.49 As IL-1β production is regulated by conserved PRRs (TLRs/NLRs), and IL-6 expression can be stimulated upon tissue injury, endogenous DAMP signalling could significantly influence this differentiation pathway. Interleukin-1α has an almost identical immunological effect to IL-1β but is expressed in a bioactive form without the need for further processing. Whereas the regulatory step that is characteristic of IL-1β production is absent, IL-1α expression is observed upon tissue damage and is therefore also sensitive to endogenous stimuli.50
Endogenous stress and innate immunity
Neutrophilia in transplantation is an extremely important event, which contributes significantly to chronic rejection. A primary characteristic of neutrophils in the graft is the production of ROS. The direct effect of this will contribute to oxidative stress and DAMP release. Neutrophils are also responsible for much of the early IL-17 production within the graft, before infiltration by Th17 cells.51,52 This process has several implications, including accelerated acute rejection and a reduction in Treg cell expansion, which favours an inflammatory effector environment.53 Although these cells can clearly be detrimental to the graft, early neutrophil extravasation to the site of injury is important for tissue repair. Their involvement in this process is diminished beyond the early phase, whereby macrophages become the main reparative agents. The heterogeneity of macrophages means that, like neutrophils, their precise role is complicated and multifactorial in transplantation. Macrophages express a wide range of PRRs,54 and as such have an extensive ability to survey for damage. Once activated via PRR ligation, macrophages act as non-professional antigen-presenting cells, allowing the induction of a secondary allospecific immune response. These cells also contribute to graft damage directly, via the secretion of ROS, and increase infiltration via the production of inflammatory cytokines (i.e. tumour necrosis factor-α, IL-18 and IL-1β). Consequently, this population is significantly involved in the pathogenesis of both acute and chronic rejection. The factors controlling the balance between inflammatory and immunoregulatory macrophage activity/phenotype requires further research. Theoretically, the polarization towards a reparative, rather than inflammatory, response would minimize direct damage to the graft, reduce DAMP expression and promote quiescence.
Complement co-ordinates and influences multiple facets of the immune response following transplantation. The beauty of the complement system is that it is integrally involved in the recognition of both apoptotic and necrotic cell death, is able to distinguish between the two and prompt innate and adaptive immunity accordingly. Complement receptor-3 (CR3) stimulation can result in phagocytosis of apoptotic cells without an inflammatory response, or induce potent inflammation upon recognition of endogenous DAMPS, such as oleic acid55 and ICAM-1.56 The response therefore appears to be tailored to differentiate between natural and necrotic cell death, regardless of cause. Of particular note for transplantation, the CR3 ligand ICAM-1 is significantly up-regulated during cold ischaemia57 and following endothelial damage.58 ICAM-1 facilitates CR3 activation and so potentially exacerbates the early inflammatory response to the graft. The role of CR3 has been assessed previously,59 and was discovered to be integral to the regulation of IL-12 production, a critical determinant of cell-mediated immunity. The elevated expression of adhesion factors such as P-selectin and ICAMs during non-antigen-dependent stress (i.e. cold ischaemia), initiates an interactive feedback loop with platelets. Platelet activation is sufficient to induce the recruitment and activation of leucocytes and feedback stimulation of endothelial cells, via degranulation and release of an array of growth factors, chemoattractants and adhesion molecules.60 Platelets have also been implicated in the expression of CD154, a co-stimulatory molecule primarily expressed on activated T cells.60 Thrombin-activated platelets also secrete an active, soluble form of CD154. Both platelet-expressed and soluble CD154 induce DC maturation and activation, determined via CD80, CD83, CD86 and HLA-DR expression.61 Therefore endogenous DAMPs indirectly contribute to alloantigen specific immunity via the provision of activating signals to DCs.
Endogenous stress and the T-cell response – indirect contributions to alloantigen specific immunity
Involvement of T cells is fundamental to rejection. However, T-cell stimulation requires previous innate, non-antigen-dependent input. As the result of MHC restriction and co-stimulation requirements, DCs comprise an integral population that regulates immunity and graft rejection. The result of antigen uptake is determined by the maturation and activation of DCs.62 A vast array of inducible danger signals released by stressed tissues and haematopoietic cells are able to stimulate DC maturation. Joffre et al.63 demonstrated that mature, resting DCs induce the expansion of T-cell populations. This may occur as a mechanism of local surveillance, representing a fundamental immunological safeguard. Expanded cells will be deleted or become anergic without the input of activated DCs as a result of peripheral tolerance mechanisms. However, direct stimulation of surface-expressed PRRs is sufficient to induce full activation.63 Activated DCs undergo phenotypic modifications including the up-regulation of antigen-presenting and co-stimulatory molecules (MHC, CD40, CD80 and CD86) and are able to release IL-12, described as ‘signal 3’.63 Hypothetically such DCs should be functionally capable of priming T cells. A disparity in DC behaviour is therefore observed depending on the signal delivered by the tissue itself (i.e. inducible or constitutive signals), which may be related to the nature and extent of the damage/stress. Endogenous immunoactive ligands are released in abundance as a result of the surgical stress that is inherent in transplantation.6–8 Although important for activation, the presence of endogenous signals in the absence of alloantigen would (hypothetically) not lead to graft rejection. The response will be sustained for the duration of stress, as a result of tolerance to healthy ‘self’ antigens being restored by peripheral mechanisms upon quiescence. Similarly, the presence of alloantigen in an entirely stress-free environment may not lead to graft rejection, because of a lack of DC co-stimulation. According to the danger model,64 such an occurrence would promote T-cell apoptosis, leading to immunological tolerance and graft acceptance.
Pre-transplant aetiology
The deleterious effect of endogenous danger signals in transplantation may be further complicated by pre-transplant disease. Elevated DAMP expression, PRR signalling, and innate cell activation can be observed before transplantation as the result of pre-existing pathology. Consequently, this provides an environment in which a milieu of pro-inflammatory and auxiliary signals is already present, and co-stimulatory molecules are overtly expressed. Introduction of alloantigen may simply provide the antigenic target for the adjuvant-primed response. The nature of the adaptive response means that lymphocyte differentiation into alloantigen-specific memory cells will occur. Importantly, these cells may contribute significantly to episodes of acute, as well as chronic rejection. Therefore, alloantigen provides an antigenic stimulus without pre-existing tolerance. Additionally, pre-transplant infection has been recognized as a significant risk factor in graft rejection. Renal transplant recipients with pre-existing BK polyomavirus infection have a significantly elevated risk of acute and chronic rejection,65 whereas pre-existing cytomegalovirus infection has been implicated as a risk factor for cardiac allograft vasculopathy.66 Furthermore, chronic viral infection in a murine transplantation model has been previously demonstrated to abate tolerance induction and prompt rejection.67 The plasticity of conserved PRRs on immune cells is such that ligation by pathogenic agents or their components (e.g. lipopolysaccharide recognition on TLR4 or cytomegalovirus recognition by TLR3/968) induces downstream signalling cascades that mimic the response to endogenous signals. Moreover, any damage that is caused as a result of infection may enhance the expression of DAMPs on local tissue.
Endogens influencing adaptive memory
Antigen specificity is particularly important for adaptive memory, yet even at this point, endogenous signalling mechanisms remain influential. Lymphocytes express a range of TLRs,69 although the precise role in this setting is uncertain. Cell surface expression on T helper cells only appears to be up-regulated on activated and memory populations, in a TCR-stimulation-dependent manner.69 In this case, TLRs may function as co-stimulatory receptors on memory cells, which could explain their reported reduced dependence on classical co-stimulation pathways.70 Importantly, in the transplant setting, TLRs also (indirectly) modulate Treg cell processes. The Treg cells maintain peripheral tolerance to ‘self’ antigens by suppressing autoreactive T-cell activation, and are activated via TCR ligation, although their effector function is antigen non-specific;71 therefore, Treg cell function requires modulation. Treg-cell-induced immunoregulation can be interrupted via TLR-stimulated DCs.72 This implies that DAMPs could subvert natural suppression to promote an alloresponse. It is interesting to note that Treg cells themselves also express several TLRs.72–74 Their role in this setting is incompletely defined, but may allow Treg cell functions to be directly influenced by the donor tissue environment. Intriguingly, it appears that Treg cell TLR stimulation can inhibit or promote suppression in a variable-dependent manner. Recent studies have yielded disparate results, with TLR2 ligation purported to induce opposite effects with different substrates.75,76 These discrepancies increase the uncertainty surrounding the role of TLRs on Treg cells, necessitating additional research. It is not known whether the abrogation of Treg cell suppression post-transplantation results primarily from TLR stimulation of Treg cells, or through interaction with activated DC. Nevertheless, TLRs clearly play an important role in modulating Treg cell suppressive capacity, and may therefore be central in the overall response.
Reducing graft immunogenicity by modulating endogenous stress expression
Minimizing injury to the graft is the ideal scenario during transplantation. Techniques such as ex vivo treatment of donor organs using non-traumatic perfusion pressures may have some benefit; however, it is inevitable that there will be a certain amount of tissue injury as a result of the procedure. It is therefore necessary to consider methods by which this expression can be minimized or their detrimental interactions inhibited.
Attempts to reduce the levels of endogenous stress that accompany transplantation have yielded beneficial results in terms of graft survival. Land77 reduced the negative impact associated with ROS-mediated ischaemia–reperfusion (IR) injury, through use of the free radical scavenger, superoxide dismutase. Therapeutic suppression of non-antigen-dependent injury should minimize endogenous ligand release and therefore diminish graft immunogenicity. Further experimental studies with synthetic free radical scavengers, such as MCI-186, have proven similarly beneficial in minimizing graft infiltration and damage.78,79
Sphingosine-1-phosphate (S1P) confers a potent cardioprotective effect via interaction with corresponding sphingosine receptors on lymphocytes. S1P and synthetic homologues (such as FTY720) reduce cell-mediated immunity toward the graft and minimize IR injury.80 However, the cytoprotective role of S1P is disrupted during tissue injury. Sphingosine-1-lyase (S1L) is an inducible stress-activated enzyme that is responsible for irreversibly catabolizing S1P, thereby promoting graft immunogenicity. S1L has been previously identified as a novel therapeutic target for cardiac IR injury, and can be efficaciously inhibited by tetrahydroxybutylimidazole.81 S1L inhibition therapy such as this, or treatment with FTY720, could be potentially beneficial for transplantation by blocking lymphocyte egress.82
The negative association of neutrophilia with chronic rejection83 has increased interest in treatment modalities that may reduce this phenomenon. Azithromycin is an anti-neutrophilic macrolide that has shown some benefit in bronchiolitis obliterans syndrome, particularly in patients with a large initial lavage neutrophil count.84,85 However, depletion of neutrophils may negatively impact tissue repair. A recent report highlighted an increase in IL-10 as an effect of azithromycin on DC; however, an increase in co-stimulatory CD80 expression was also observed.86 The effect of azithromycin on this, and other, important cell types is not well defined, therefore necessitating further research.
The use of anti-oxidants, such as N-acetyl cysteine, has shown some efficacy in reducing primary graft dysfunction following transplantation.87 This is probably because of a protective effect during IR. However, an increase in acute graft-versus-host disease, as well as increased alloantigen-specific T-cell mediated immunity, has been demonstrated in vitro as a result of N-acetyl cysteine treatment.88 This is a particularly detrimental effect that would have potentially devastating consequences in transplantation. Further research is clearly warranted to delineate the cause of this effect, and to ensure that the benefits of this treatment outweigh the risks.
Ethyl pyruvate inhibits HMGB1 release, resulting in diminished inflammatory cytokine production while maintaining IL-10 secretion.89 Ethyl pyruvate treatment also induces protective haem-oxygenase-1 production, which minimizes graft damage during IR injury and acute rejection episodes.90,91 Anti-HMGB1 blockade is similarly effective in murine models in reducing tumour necrosis factor-α and IL-1 production, while improving graft viability.92 These methods may offer clinical benefit, although they do not take into account other expressed/released stress molecules.
Clinical inhibition of other endogenous PRR agonists, such as biglycan and heat-shock proteins, is scarcely reported. However, anti-heat-shock protein 90 treatment efficaciously reduces inflammation and adaptive immunity.93 This may hypothetically be a useful route by which to minimize immune activation following transplantation. Whereas removal of soluble DAMPs such as biglycan from the site of tissue injury should decrease the inflammatory response, monoclonal antibodies may potentially bind detrimentally to important extracellular forms.94
ATP released during tissue injury stimulates neutrophil recruitment and induces the inflammasome assembly and activation necessary for caspase-1 activity. Administration of ATPase markedly diminished neutrophil recruitment following tissue injury.95 P2X7 receptor antagonism also minimizes the inflammatory response to damage, and is therefore of potential therapeutic benefit.96 However, we suggest that the ATP/P2X7 interaction is not the optimal target for treatment. The effect of ATP sequestration/P2X7 blockade on IL-1β/IL-18 secretion will be limited by the constitutively active caspase-1 observed in monocytes.35,39 As IL-1β/IL-18 secretion by monocytes is dependent upon TLR ligation, and TLRs are integral in stress recognition via a milieu of additional ligands, blockade of this pathway may be more successful.
Anti-TLR therapy prevents endogenous stress molecules from binding to the TLR, thereby reducing leucocyte influx, cytokine production and pro-apoptotic signalling. This strategy significantly reduces infarct size and preserves cardiac function during myocardial IR injury.97 This may be a potential therapeutic approach for antagonizing existing stress expression following tissue injury and has proved efficacious in animal models.98 Anti-TLR therapy could be beneficial as an adjunctive to reducing stress expression. Several important endogenous ligands use alternative receptors, such as HMGB1 binding to receptor for advanced glycation end products (RAGE), although the principle remains the same. Pharmacological blockade of RAGE in murine cardiac models significantly delayed acute rejection.99,100
Conclusion
Rejection is a complex, multifactorial process that is not solely reliant upon antigenic disparity between donor and recipient. The influence of DAMPs on proliferation, activation and differentiation of an array of important leucocyte populations has been highlighted herein. Endogenous ligands released during tissue stress and injury play an essential adjuvant role in the initiation of graft rejection. Further research is necessary to assess the implications of treating an endogenous, rather than allogeneic source of rejection.
Disclosures
All authors declare no conflict of interests.
References
- 1.Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249–55. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- 2.Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol. 2008;3:99–126. doi: 10.1146/annurev.pathmechdis.3.121806.151456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raoof M, Zhang Q, Itagaki K, Hauser CJ. Mitochondrial peptides are potent immune activators that activate human neutrophils via FPR-1. J Trauma. 2010;68:1328–32. doi: 10.1097/TA.0b013e3181dcd28d. discussion 1332–24. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He H, Stone JR, Perkins DL. Analysis of robust innate immune response after transplantation in the absence of adaptive immunity. Transplantation. 2002;73:853–61. doi: 10.1097/00007890-200203270-00005. [DOI] [PubMed] [Google Scholar]
- 6.Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–21. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- 7.Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. 2001;13:114–9. doi: 10.1016/s0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- 8.Pockley AG. Heat shock proteins, anti-heat shock protein reactivity and allograft rejection. Transplantation. 2001;71:1503–7. doi: 10.1097/00007890-200106150-00001. [DOI] [PubMed] [Google Scholar]
- 9.Schaefer L, Babelova A, Kiss E, et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest. 2005;115:2223–33. doi: 10.1172/JCI23755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moreth K, Brodbeck R, Babelova A, et al. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J Clin Invest. 2010;120:4251–72. doi: 10.1172/JCI42213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 12.Wang S, Schmaderer C, Kiss E, et al. Recipient Toll-like receptors contribute to chronic graft dysfunction by both MyD88- and TRIF-dependent signaling. Dis Model Mech. 2010;3:92–103. doi: 10.1242/dmm.003533. [DOI] [PubMed] [Google Scholar]
- 13.Chen G, Li J, Ochani M, et al. Bacterial endotoxin stimulates macrophages to release HMGB1 partly through CD14- and TNF-dependent mechanisms. J Leukoc Biol. 2004;76:994–1001. doi: 10.1189/jlb.0404242. [DOI] [PubMed] [Google Scholar]
- 14.Tejani A, Ho PL, Emmett L, Stablein DM. Reduction in acute rejections decreases chronic rejection graft failure in children: a report of the North American Pediatric Renal Transplant Cooperative Study (NAPRTCS) Am J Transplant. 2002;2:142–7. doi: 10.1034/j.1600-6143.2002.020205.x. [DOI] [PubMed] [Google Scholar]
- 15.Matas AJ, Humar A, Payne WD, Gillingham KJ, Dunn DL, Sutherland DE, Najarian JS. Decreased acute rejection in kidney transplant recipients is associated with decreased chronic rejection. Ann Surg. 1999;230:493–8. doi: 10.1097/00000658-199910000-00005. discussion 498–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong F, Lee J, Song JW, Lee SJ, Ahn H, Cho JJ, Ha J, Kim SS. Cyclosporin A blocks muscle differentiation by inducing oxidative stress and inhibiting the peptidyl-prolyl-cis-trans isomerase activity of cyclophilin A: cyclophilin A protects myoblasts from cyclosporin A-induced cytotoxicity. FASEB J. 2002;16:1633–5. doi: 10.1096/fj.02-0060fje. [DOI] [PubMed] [Google Scholar]
- 17.Bansal S, Siddarth M, Chawla D, Banerjee BD, Madhu SV, Tripathi AK. Advanced glycation end products enhance reactive oxygen and nitrogen species generation in neutrophils in vitro. Mol Cell Biochem. 2012;361:289–96. doi: 10.1007/s11010-011-1114-9. [DOI] [PubMed] [Google Scholar]
- 18.Fan J, Frey RS, Malik AB. TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. J Clin Invest. 2003;112:1234–43. doi: 10.1172/JCI18696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamamoto A, Ashihara E, Nakagawa Y, et al. Allograft inflammatory factor-1 is overexpressed and induces fibroblast chemotaxis in the skin of sclerodermatous GVHD in a murine model. Immunol Lett. 2011;135:144–50. doi: 10.1016/j.imlet.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 20.Hellkvist J, Tufveson G, Gerdin B, Johnsson C. Characterization of fibroblasts from rejecting tissue: the hyaluronan production is increased. Transplantation. 2002;74:1672–7. doi: 10.1097/00007890-200212270-00004. [DOI] [PubMed] [Google Scholar]
- 21.Chen J, John R, Richardson JA, et al. Toll-like receptor 4 regulates early endothelial activation during ischemic acute kidney injury. Kidney Int. 2011;79:288–99. doi: 10.1038/ki.2010.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang S, Lee SP, Kim KE, Kim HZ, Memet S, Koh GY. Toll-like receptor 4 in lymphatic endothelial cells contributes to LPS-induced lymphangiogenesis by chemotactic recruitment of macrophages. Blood. 2009;113:2605–13. doi: 10.1182/blood-2008-07-166934. [DOI] [PubMed] [Google Scholar]
- 23.Neal MD, Leaphart C, Levy R, et al. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J Immunol. 2006;176:3070–9. doi: 10.4049/jimmunol.176.5.3070. [DOI] [PubMed] [Google Scholar]
- 24.Eisinger K, Bauer S, Schaffler A, Walter R, Neumann E, Buechler C, Muller-Ladner U, Frommer KW. Chemerin induces CCL2 and TLR4 in synovial fibroblasts of patients with rheumatoid arthritis and osteoarthritis. Exp Mol Pathol. 2011;92:90–6. doi: 10.1016/j.yexmp.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Babelova A, Moreth K, Tsalastra-Greul W, et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem. 2009;284:24035–48. doi: 10.1074/jbc.M109.014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–32. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 27.Ogura Y, Sutterwala FS, Flavell RA. The inflammasome: first line of the immune response to cell stress. Cell. 2006;126:659–62. doi: 10.1016/j.cell.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 28.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 29.Williams CD, Antoine DJ, Shaw PJ, et al. Role of the Nalp3 inflammasome in acetaminophen-induced sterile inflammation and liver injury. Toxicol Appl Pharmacol. 2011;252:289–97. doi: 10.1016/j.taap.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 32.Gasse P, Riteau N, Charron S, et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med. 2009;179:903–13. doi: 10.1164/rccm.200808-1274OC. [DOI] [PubMed] [Google Scholar]
- 33.Rajamaki K, Lappalainen J, Oorni K, Valimaki E, Matikainen S, Kovanen PT, Eklund KK. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS ONE. 2010;5:e11765. doi: 10.1371/journal.pone.0011765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghaemi-Oskouie F, Shi Y. The role of uric acid as an endogenous danger signal in immunity and inflammation. Curr Rheumatol Rep. 2011;13:160–6. doi: 10.1007/s11926-011-0162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1β and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A. 2008;105:8067–72. doi: 10.1073/pnas.0709684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eckle T, Krahn T, Grenz A, et al. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation. 2007;115:1581–90. doi: 10.1161/CIRCULATIONAHA.106.669697. [DOI] [PubMed] [Google Scholar]
- 37.Wilhelm K, Ganesan J, Muller T, et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med. 2010;16:1434–8. doi: 10.1038/nm.2242. [DOI] [PubMed] [Google Scholar]
- 38.Idzko M, Hammad H, van Nimwegen M, et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med. 2007;13:913–9. doi: 10.1038/nm1617. [DOI] [PubMed] [Google Scholar]
- 39.Netea MG, Nold-Petry CA, Nold MF, et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1β in monocytes and macrophages. Blood. 2009;113:2324–35. doi: 10.1182/blood-2008-03-146720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol. 2004;76:514–9. doi: 10.1189/jlb.0304127. [DOI] [PubMed] [Google Scholar]
- 41.Taflin C, Favier B, Baudhuin J, et al. Human endothelial cells generate Th17 and regulatory T cells under inflammatory conditions. Proc Natl Acad Sci U S A. 2011;108:2891–6. doi: 10.1073/pnas.1011811108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benghiat FS, Charbonnier LM, Vokaer B, De Wilde V, Le Moine A. Interleukin 17-producing T helper cells in alloimmunity. Transplant Rev (Orlando) 2009;23:11–8. doi: 10.1016/j.trre.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 43.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–9. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 44.Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, Soumelis V. A critical function for transforming growth factor-β, interleukin 23 and proinflammatory cytokines in driving and modulating human TH-17 responses. Nat Immunol. 2008;9:650–7. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 45.Manel N, Unutmaz D, Littman DR. The differentiation of human TH-17 cells requires transforming growth factor-β and induction of the nuclear receptor RORγt. Nat Immunol. 2008;9:641–9. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan X, Paez-Cortez J, Schmitt-Knosalla I, et al. A novel role of CD4 Th17 cells in mediating cardiac allograft rejection and vasculopathy. J Exp Med. 2008;205:3133–44. doi: 10.1084/jem.20081937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vanaudenaerde BM, De Vleeschauwer SI, Vos R, et al. The role of the IL23/IL17 axis in bronchiolitis obliterans syndrome after lung transplantation. Am J Transplant. 2008;8:1911–20. doi: 10.1111/j.1600-6143.2008.02321.x. [DOI] [PubMed] [Google Scholar]
- 48.Wilson NJ, Boniface K, Chan JR, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–7. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 49.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–91. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rao DA, Tracey KJ, Pober JS. IL-1α and IL-1β are endogenous mediators linking cell injury to the adaptive alloimmune response. J Immunol. 2007;179:6536–46. doi: 10.4049/jimmunol.179.10.6536. [DOI] [PubMed] [Google Scholar]
- 51.Min SI, Ha J, Park CG, Won JK, Park YJ, Min SK, Kim SJ. Sequential evolution of IL-17 responses in the early period of allograft rejection. Exp Mol Med. 2009;41:707–16. doi: 10.3858/emm.2009.41.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li L, Huang L, Vergis AL, et al. IL-17 produced by neutrophils regulates IFN-γ-mediated neutrophil migration in mouse kidney ischemia–reperfusion injury. J Clin Invest. 2010;120:331–42. doi: 10.1172/JCI38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Itoh S, Kimura N, Axtell RC, et al. Interleukin-17 accelerates allograft rejection by suppressing regulatory T cell expansion. Circulation. 2011;124(11 Suppl):S187–96. doi: 10.1161/CIRCULATIONAHA.110.014852. [DOI] [PubMed] [Google Scholar]
- 54.Hennessy EJ, Parker AE, O’Neill LA. Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov. 2010;9:293–307. doi: 10.1038/nrd3203. [DOI] [PubMed] [Google Scholar]
- 55.Mastrangelo AM, Jeitner TM, Eaton JW. Oleic acid increases cell surface expression and activity of CD11b on human neutrophils. J Immunol. 1998;161:4268–75. [PubMed] [Google Scholar]
- 56.Smith CW, Marlin SD, Rothlein R, Toman C, Anderson DC. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J Clin Invest. 1989;83:2008–17. doi: 10.1172/JCI114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lange V, Renner A, Sagstetter MR, Lazariotou M, Harms H, Gummert JF, Leyh RG, Elert O. Heterotopic rat heart transplantation (Lewis to F344): early ICAM-1 expression after 8 hours of cold ischemia. J Heart Lung Transplant. 2008;27:1031–5. doi: 10.1016/j.healun.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 58.Jaakkola K, Jalkanen S, Kaunismaki K, et al. Vascular adhesion protein-1, intercellular adhesion molecule-1 and P-selectin mediate leukocyte binding to ischemic heart in humans. J Am Coll Cardiol. 2000;36:122–9. doi: 10.1016/s0735-1097(00)00706-3. [DOI] [PubMed] [Google Scholar]
- 59.Marth T, Kelsall BL. Regulation of interleukin-12 by complement receptor 3 signaling. J Exp Med. 1997;185:1987–95. doi: 10.1084/jem.185.11.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elzey BD, Sprague DL, Ratliff TL. The emerging role of platelets in adaptive immunity. Cell Immunol. 2005;238:1–9. doi: 10.1016/j.cellimm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 61.Kaneider NC, Kaser A, Tilg H, Ricevuti G, Wiedermann CJ. CD40 ligand-dependent maturation of human monocyte-derived dendritic cells by activated platelets. Int J Immunopathol Pharmacol. 2003;16:225–31. doi: 10.1177/039463200301600307. [DOI] [PubMed] [Google Scholar]
- 62.Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440:808–12. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 63.Joffre O, Nolte MA, Sporri R, Reis e Sousa C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev. 2009;227:234–47. doi: 10.1111/j.1600-065X.2008.00718.x. [DOI] [PubMed] [Google Scholar]
- 64.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 65.Hayat A, Mukhopadhyay R, Radhika S, Sachdeva MS, Nada R, Joshi K, Sakhuja V, Jha V. Adverse impact of pretransplant polyoma virus infection on renal allograft function. Nephrology (Carlton) 2008;13:157–63. doi: 10.1111/j.1440-1797.2007.00861.x. [DOI] [PubMed] [Google Scholar]
- 66.Hussain T, Burch M, Fenton MJ, Whitmore PM, Rees P, Elliott M, Aurora P. Positive pretransplantation cytomegalovirus serology is a risk factor for cardiac allograft vasculopathy in children. Circulation. 2007;115:1798–805. doi: 10.1161/CIRCULATIONAHA.106.627570. [DOI] [PubMed] [Google Scholar]
- 67.Williams MA, Onami TM, Adams AB, Durham MM, Pearson TC, Ahmed R, Larsen CP. Cutting edge: persistent viral infection prevents tolerance induction and escapes immune control following CD28/CD40 blockade-based regimen. J Immunol. 2002;169:5387–91. doi: 10.4049/jimmunol.169.10.5387. [DOI] [PubMed] [Google Scholar]
- 68.Delale T, Paquin A, Asselin-Paturel C, et al. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-α release and initiation of immune responses in vivo. J Immunol. 2005;175:6723–32. doi: 10.4049/jimmunol.175.10.6723. [DOI] [PubMed] [Google Scholar]
- 69.Xu D, Komai-Koma M, Liew FY. Expression and function of Toll-like receptor on T cells. Cell Immunol. 2005;233:85–9. doi: 10.1016/j.cellimm.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 70.Yang J, Brook MO, Carvalho-Gaspar M, et al. Allograft rejection mediated by memory T cells is resistant to regulation. Proc Natl Acad Sci U S A. 2007;104:19954–9. doi: 10.1073/pnas.0704397104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thornton AM, Shevach EM. Suppressor effector function of CD4+ CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000;164:183–90. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- 72.Sutmuller RP, Morgan ME, Netea MG, Grauer O, Adema GJ. Toll-like receptors on regulatory T cells: expanding immune regulation. Trends Immunol. 2006;27:387–93. doi: 10.1016/j.it.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 73.Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197:403–11. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu G, Zhao Y. Toll-like receptors and immune regulation: their direct and indirect modulation on regulatory CD4+ CD25+ T cells. Immunology. 2007;122:149–56. doi: 10.1111/j.1365-2567.2007.02651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zanin-Zhorov A, Cahalon L, Tal G, Margalit R, Lider O, Cohen IR. Heat shock protein 60 enhances CD4+ CD25+ regulatory T cell function via innate TLR2 signaling. J Clin Invest. 2006;116:2022–32. doi: 10.1172/JCI28423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 76.Sutmuller RP, den Brok MH, Kramer M, et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485–94. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Land WG. The role of postischemic reperfusion injury and other nonantigen-dependent inflammatory pathways in transplantation. Transplantation. 2005;79:505–14. doi: 10.1097/01.tp.0000153160.82975.86. [DOI] [PubMed] [Google Scholar]
- 78.Akao T, Takeyoshi I, Totsuka O, et al. Effect of the free radical scavenger MCI-186 on pulmonary ischemia–reperfusion injury in dogs. J Heart Lung Transplant. 2006;25:965–71. doi: 10.1016/j.healun.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 79.Suzuki F, Hashikura Y, Ise H, Ishida A, Nakayama J, Takahashi M, Miyagawa S, Ikeda U. MCI-186 (edaravone), a free radical scavenger, attenuates hepatic warm ischemia–reperfusion injury in rats. Transpl Int. 2005;18:844–53. doi: 10.1111/j.1432-2277.2005.00094.x. [DOI] [PubMed] [Google Scholar]
- 80.Fuller TF, Hoff U, Kong L, et al. Cytoprotective actions of FTY720 modulate severe preservation reperfusion injury in rat renal transplants. Transplantation. 2010;89:402–8. doi: 10.1097/TP.0b013e3181caa499. [DOI] [PubMed] [Google Scholar]
- 81.Bandhuvula P, Honbo N, Wang GY, et al. S1P lyase (SPL): a novel therapeutic target for ischemia/reperfusion injury of the heart. Am J Physiol Heart Circ Physiol. 2011;300:H1753–61. doi: 10.1152/ajpheart.00946.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kappos L, Antel J, Comi G, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355:1124–40. doi: 10.1056/NEJMoa052643. [DOI] [PubMed] [Google Scholar]
- 83.Devouassoux G, Drouet C, Pin I, Brambilla C, Brambilla E, Colle PE, Pison C. Alveolar neutrophilia is a predictor for the bronchiolitis obliterans syndrome, and increases with degree of severity. Transpl Immunol. 2002;10:303–10. doi: 10.1016/s0966-3274(02)00074-6. [DOI] [PubMed] [Google Scholar]
- 84.Vos R, Vanaudenaerde BM, Ottevaere A, et al. Long-term azithromycin therapy for bronchiolitis obliterans syndrome: divide and conquer? J Heart Lung Transplant. 2010;29:1358–68. doi: 10.1016/j.healun.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 85.Federica M, Nadia S, Monica M, Alessandro C, Tiberio O, Francesco B, Mario V, Maria FA. Clinical and immunological evaluation of 12-month azithromycin therapy in chronic lung allograft rejection. Clin Transplant. 2011;25:E381–9. doi: 10.1111/j.1399-0012.2011.01435.x. [DOI] [PubMed] [Google Scholar]
- 86.Sugiyama K, Shirai R, Mukae H, et al. Differing effects of clarithromycin and azithromycin on cytokine production by murine dendritic cells. Clin Exp Immunol. 2007;147:540–6. doi: 10.1111/j.1365-2249.2007.03299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Inci I, Erne B, Arni S, et al. Prevention of primary graft dysfunction in lung transplantation by N-acetylcysteine after prolonged cold ischemia. J Heart Lung Transplant. 2010;29:1293–301. doi: 10.1016/j.healun.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 88.Karlsson H, Nava S, Remberger M, Hassan Z, Hassan M, Ringden O. N-acetyl-l-cysteine increases acute graft-versus-host disease and promotes T-cell-mediated immunity in vitro. Eur J Immunol. 2011;41:1143–53. doi: 10.1002/eji.201040589. [DOI] [PubMed] [Google Scholar]
- 89.Dave SH, Tilstra JS, Matsuoka K, et al. Ethyl pyruvate decreases HMGB1 release and ameliorates murine colitis. J Leukoc Biol. 2009;86:633–43. doi: 10.1189/jlb.1008662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kotsch K, Francuski M, Pascher A, et al. Improved long-term graft survival after HO-1 induction in brain-dead donors. Am J Transplant. 2006;6:477–86. doi: 10.1111/j.1600-6143.2005.01208.x. [DOI] [PubMed] [Google Scholar]
- 91.Baan C, Peeters A, Lemos F, et al. Fundamental role for HO-1 in the self-protection of renal allografts. Am J Transplant. 2004;4:811–8. doi: 10.1111/j.1600-6143.2004.00420.x. [DOI] [PubMed] [Google Scholar]
- 92.Gao Q, Ma LL, Gao X, Yan W, Williams P, Yin DP. TLR4 mediates early graft failure after intraportal islet transplantation. Am J Transplant. 2010;10:1588–96. doi: 10.1111/j.1600-6143.2010.03151.x. [DOI] [PubMed] [Google Scholar]
- 93.Yun TJ, Harning EK, Giza K, et al. EC144, a synthetic inhibitor of heat shock protein 90, blocks innate and adaptive immune responses in models of inflammation and autoimmunity. J Immunol. 2011;186:563–75. doi: 10.4049/jimmunol.1000222. [DOI] [PubMed] [Google Scholar]
- 94.Polgar A, Falus A, Koo E, et al. Elevated levels of synovial fluid antibodies reactive with the small proteoglycans biglycan and decorin in patients with rheumatoid arthritis or other joint diseases. Rheumatology (Oxford) 2003;42:522–7. doi: 10.1093/rheumatology/keg168. [DOI] [PubMed] [Google Scholar]
- 95.McDonald B, Pittman K, Menezes GB, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–6. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 96.Peng W, Cotrina ML, Han X, et al. Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc Natl Acad Sci U S A. 2009;106:12489–93. doi: 10.1073/pnas.0902531106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Arslan F, Smeets MB, O’Neill LA, et al. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation. 2010;121:80–90. doi: 10.1161/CIRCULATIONAHA.109.880187. [DOI] [PubMed] [Google Scholar]
- 98.Shimamoto A, Chong AJ, Yada M, et al. Inhibition of Toll-like receptor 4 with eritoran attenuates myocardial ischemia–reperfusion injury. Circulation. 2006;114(1 Suppl):I270–4. doi: 10.1161/CIRCULATIONAHA.105.000901. [DOI] [PubMed] [Google Scholar]
- 99.Moser B, Szabolcs MJ, Ankersmit HJ, Lu Y, Qu W, Weinberg A, Herold KC, Schmidt AM. Blockade of RAGE suppresses alloimmune reactions in vitro and delays allograft rejection in murine heart transplantation. Am J Transplant. 2007;7:293–302. doi: 10.1111/j.1600-6143.2006.01617.x. [DOI] [PubMed] [Google Scholar]
- 100.Huang Y, Yin H, Han J, et al. Extracellular HMGB1 functions as an innate immune-mediator implicated in murine cardiac allograft acute rejection. Am J Transplant. 2007;7:799–808. doi: 10.1111/j.1600-6143.2007.01734.x. [DOI] [PubMed] [Google Scholar]