

Abstract
In this study, we expanded regulatory T cells (Tregs) ex vivo from CD4+ CD25+ T cells from cord blood (CB) and CD4+ CD25+ CD127− T cells from adult peripheral blood (APB) and compared the suppressive functions of the newly generated Tregs. The Tregs from CB and APB were expanded either in two cycles with a polyclonal stimulus or in two cycles with an alloantigen stimulus in the first cycle and a polyclonal stimulus in the second cycle. Cell yield after Treg expansion with polyclonal stimulation was greater than that of Tregs expanded with combined alloantigen and polyclonal stimulation. The expanded Tregs expressed high levels of Foxp3, CD39 and cytotoxic T-lymphocyte antigen-4 and low levels of CD127, interleukin-2 and interferon-γ. After two cycles of expansion, the CB Tregs maintained expression of the GARP gene and showed greater suppressive function than APB Tregs. The CB Tregs that were expanded with two cycles of polyclonal stimulation suppressed not only the polyclonal antigen-driven responder T (Tresp) cell proliferation but also the HLA mismatched dendritic cell-driven Tresp cell proliferation. When CB and APB Tregs were expanded with a primary alloantigen stimulus followed by a secondary polyclonal stimulus, the Tregs showed a potent, antigen-specific suppressive capacity. The Tregs expanded with two cycles of polyclonal stimulation from both CB and APB alleviated acute graft-versus-host disease symptoms and prolonged survival in a murine model of graft-versus-host disease. In conclusion, CB Tregs expanded with two cycles of polyclonal stimulation had a stronger immunosuppressive function than APB Tregs. It is feasible to obtain human functional alloantigen-specific Tregs expanded ex vivo from CB and APB in large numbers.
Keywords: acute graft versus host disease, adult peripheral blood, alloantigen stimulation, cord blood, expansion, polyclonal stimulation, regulatory T cells
Introduction
Regulatory T cells (Tregs) play an essential role in modulating the immune system and maintaining peripheral tolerance. Immunotherapy based on Treg infusion (either from in vivo Tregs or ex vivo expanded Tregs) is therefore a potential treatment for many immune disorders. This approach has been successful in animal models of haematopoietic stem cell transplantation,1–3 solid organ transplantation4–6 and autoimmunity.7–9 Because the number of Tregs that can be obtained from donor peripheral blood is limited, freshly isolated Tregs need to be expanded ex vivo to generate a sufficient number of cells for therapeutic applications. Additionally, expanded Tregs have been reported to be more effective therapeutically than primary Tregs.10
It has been shown that naturally occurring human CD4+ CD25+ Tregs can be expanded polyclonally with anti-CD3 and anti-CD28 antibody stimulation in combination with interleukin-2 (IL-2) and/or IL-15.11–14 These polyclonal expansion protocols greatly increase Treg numbers while preserving their suppressive capacity. However, infusion of non-regulatory cells into patients that already suffer from pathological immunological activity should be prevented, as these cells can potentially intensify the disease process, especially when infusing HLA-mismatched Tregs. CD4+ CD127+ conventional T cells are the major contaminating cell type in CliniMACS-isolated CD4+ CD25+ Treg populations from adult peripheral blood (APB). Depletion of CD127+ cells has been found to improve the purity of CD4+ CD25+ FoxP3+ Tregs in CliniMACS-isolated cell populations to approximately 90%,15 and the resulting Treg population showed potent suppressive capacity and high FoxP3 expression. Despite promising results with mouse Tregs, only limited success has been reported in the direct expansion of human alloantigen-specific Tregs with allogeneic antigen-presenting cells. Human peripheral blood mononuclear cells were found to induce modest proliferation of alloreactive CD4+ CD25+ T cells in the presence of exogenous IL-2 plus IL-15. With two cycles of stimulation by alloantigen combined with anti-CD3/CD28 antibodies, an average expansion of 780-fold could be obtained, generating highly suppressive cells consisting of > 90% CD4+ T cells, most of which retained FoxP3 expression.14 Chen et al.16 have reported that human Tregs can be expanded ex vivo with allogeneic B cells. Here, the expanded Tregs expressed very high levels of FoxP3, maintained an anergic phenotype and were potent suppressors capable of inhibiting the alloproliferation of third-party responder T (Tresp) cells at very low Treg to Tresp cell ratios in an alloantigen-specific manner.
As cord blood (CB) is used for haematopoietic stem cell transplantation, the function of CB Tregs is especially important for understanding the low occurrence of graft-versus-host disease (GVHD) in this clinical setting.17,18 Fujimaki et al.19 have found that the suppressive activity of fresh CB CD4+ CD25+ T cells was lacking compared with that found in APB CD4+ CD25+ T cells. However, expansion of the CB CD4+ CD25+ T cells ex vivo resulted in the restoration of suppressive activity levels greater than in those from APB. It remains controversial as to whether Tregs from CB possess better suppressive function than Tregs from APB. We therefore investigated in the current study the suppressive function of ex vivo expanded Tregs from APB and CB with polyclonal or alloantigen stimulation.
Materials and methods
Purification of CD4+ CD25+ Tregs from APB and CB
The APB was obtained from healthy adult donors and CB was obtained from healthy full-term neonates within 24 hr of delivery after written informed consent had been obtained. The APB and CB mononuclear cells were isolated by density gradient centrifugation using Ficoll–Hypaque (Amersham Biosciences, Uppsala, Sweden). CD4+ T cells were purified from CB mononuclear cells using the Dynabeads Untouched Human CD4 T-cell isolation kit (Invitrogen Dynal AS, Oslo, Norway) according to the manufacturer’s instructions. CD4+ CD127− T cells were purified from APB mononuclear cells using monoclonal antibodies directed against CD8 (RPA-T8), CD14 (M5E2), CD16 (3G8), CD56 (B159), CD19 (4G7), CD123 (7G3) and CD127 (hIL-7R-M21) (all from BD Biosciences, San Jose, CA) combined with sheep anti-mouse IgG-coated magnetic beads (Dynal Biotech, Oslo, Norway) according to the manufacturer’s instructions. The CD4+ CD25+ Tregs from CB and CD4+ CD25+ CD127− Tregs from APB were purified from CD4+ T cells using the human CD25 MicroBead II kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. The purity of APB and CB Tregs was > 95% as determined using FACS. The human and mouse B cells were isolated by directly conjugated human or mouse anti-CD19 magnetic microbeads (Miltenyi Biotec). Dendritic cells (DC) were induced from monocytes of peripheral blood mononuclear cells using the adhesion method with 800 U/ml granulocyte–macrophage colony-stimulating factor (R&D Systems, Minneapolis, MN) and 1000 U/ml IL-4 (R&D Systems), and stimulated to maturity with 1μg/ml lipopolysacchairde.
Ex vivo expansion of APB and CB Tregs
Cell culture was performed in 96-well round bottom plates with culture medium consisting of RPMI-1640 supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), penicillin (50 U/ml; Sigma, St Louis, MO) and streptomycin (50 μg/ml; Sigma) in a 37°, 95% humidity, 5% CO2 incubator. The APB and CB Tregs were expanded ex vivo either in two cycles with a polyclonal stimulus or in two cycles with an alloantigen stimulus in the first cycle and a polyclonal stimulus in the second cycle (Fig. 1a). Anti-CD3-/anti-CD28-coated microbeads and recombinant human (rh) IL-2 were used for polyclonal stimulation, and allogeneic human B cells or xenogenic mouse B cells, anti-CD28 and rhIL-2 were used for alloantigen expansion. For expansion of APB and CB Tregs with polyclonal stimulation, APB CD4+ CD25+ CD127− T cells and CB CD4+ CD25+ T cells were cultured at 1 × 104 cells per well with 4 × 104 anti-CD3-/anti-CD28-coated microbeads (Invitrogen Dynal AS) and rhIL-2 (200 U/ml) for 6 days. The cells were harvested, washed and rested for 2 days in culture medium with rhIL-2 (20 U/ml). The APB and CB Tregs were then cultured at 2 × 104 cells per well with 4 × 104 anti-CD3-/anti-CD28-coated microbeads and rhIL-2 (200 U/ml) for another 6 days. For expansion of APB and CB Tregs with alloantigen stimulation, APB CD4+ CD25+ CD127− T cells and CB CD4+ CD25+ T cells were cultured at 1 × 104 cells per well with 4 × 104 human or mouse B cells, anti-CD28 (500 ng/ml; BD Biosciences) and rhIL-2 (200 U/ml) for 12 days. The cells were harvested, washed and rested for 2 days in culture medium with rhIL-2 (20 U/ml). Then, the APB and CB Tregs were cultured at 2 × 104 cells per well with 4 × 104 anti-CD3-/anti-CD28-coated microbeads and rhIL-2 (200 U/ml) for another 6 days. Fresh medium containing rhIL-2 (200 U/ml) was added every 2–3 days.
Figure 1.
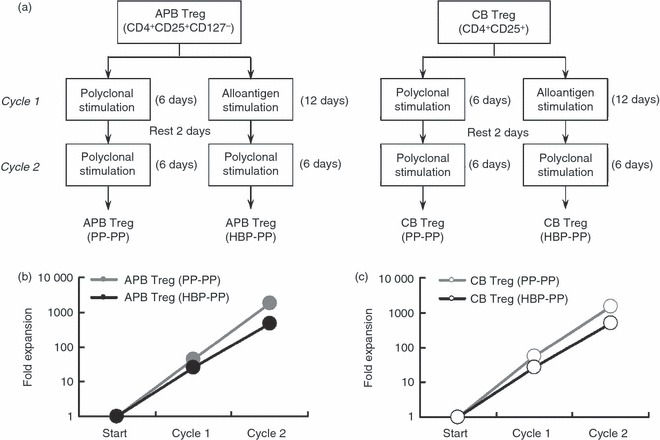
Schematic overview of expansion strategies and regulatory T cell (Treg) expansion after primary and secondary stimulation. (a) Adult peripheral blood (APB) and cord blood (CB) Tregs were expanded in either two cycles with polyclonal stimulation or two cycles with an alloantigen stimulus in the first cycle and a polyclonal stimulus in the second cycle. We therefore generated four types of expanded Tregs: APB Treg (PP-PP), APB Treg (HBP-PP), CB Treg (PP-PP) and CB Treg (HBP-PP). (b, c) APB and CB Tregs were expanded as described above. Treg numbers were determined and compared to initial Treg numbers. Data represent the average expansion ± standard deviation (SD) of six independent experiments.
Immunophenotyping of APB and CB Tregs
The phenotype of APB and CB Tregs was analysed by flow cytometry (FACSCalibur; BD Biosciences). For cell surface staining, the following conjugated monoclonal antibodies were used: CD4(RPA-T4)-fluorescein isothiocyanate (FITC), CD25(M-A251)-phycoerythrin (PE), CD39(TU66)-PE, CD127(hIL-7R-M21)-AlexaFluor647, cytotoxic T-lymphocyte antigen 4 (CTLA4; BN13)-PE, HLA-DR-FITC (all from BD Biosciences) and LAG-3-FITC (R&D). For intracellular staining, Fix and Fix/Perm buffers (eBioscience, San Diego, CA) were used according to the manufacturer’s instructions with FoxP3-PE. Isotype controls were used to set gates. Dead cells were gated out based on their forward-scatter/side-scatter properties. To assess each marker, 1 × 105 cells were incubated with specific antibodies for 20 min at 4° in PBS.
Reverse transcription-PCR analysis of gene expression
The expanded Tregs from either CB or APB were disrupted in TRIzol reagent (Invitrogen), and total cellular RNA was prepared according to the manufacturer’s instructions. Complementary DNA was prepared from equal amounts of total RNA using a reverse transcriptase and the resulting cDNA was used to amplify target genes by reverse transcription–PCR using Taq DNA polymerase (TaKaRa Bio Inc., Otsu, Japan) with β-actin as a reference gene. Primers used were as follows: β-actin: (forward 5′-GTGGGGCGCCCCAGGCACCA-3′, reverse 5′-CTTCCTTAATGTCACGCACGATTTC-3′); IL-2: (forward 5′-AGAATCCCAAACTCACCAGG-3′, reverse 5′-TCAGATCCCTTTAGTTCCAG-3′); interferon-γ: (forward 5′-GCAGGTCATTCAGATGTAGC-3′, reverse 5′-ATGTCTTCCTTGATGGTCTC-3′); IL-10: forward 5′-AACCTGCCTAACATGCTTC-3′, reverse 5′-CAGATCCGATTTTGGAGAC-3′); transforming growth factor-β: (forward 5′-TGGAAACCCACAACGAAATC-3′, reverse 5′-TAAGGCGAAAGCCCTCAAT-3′); GARP (glycoprotein-A repetitions predominant): (forward 5′-TGGTTTGGTGCGGTGAG-3′, reverse 5′-AGCAGGAGA CGGAATGGT-3′).
Suppression assay
Magnetic cell sorting-purified CD4+ CD25− Tresp cells from APB were pulsed with carboxyfluorescein succinimidyl ester (CFSE; 5 μm) and activated with anti-CD3/anti-CD28 antibodies (300 ng/ml) and rhIL-2 (200 U/ml) or HLA mismatched DC (1 × 104) treated with mitomycin (1 μg/ml) for 30 min at 37°. The cells were then cultured at a density of 5 × 104 responders per well in 96-well plates. Third-party Tregs expanded from APB and CB were then added at ratios of 1 : 1, 1 : 4, 1 : 16 and 1 : 64 to the responder cultures in a final volume of 200 μl. Four days later, the cells were collected and analysed by flow cytometry. The results were expressed as the percentage of proliferating cells.
Mice and GVHD model
Wild-type male DBA/2 (H-2Kd) and C57BL/6 (H-2Kb) mice were purchased from the Shanghai Laboratory Animal Centre of the Chinese Academy of Science (Shanghai, China). The DBA/2 (H-2Kd) mice were allowed to reach a minimum weight of 25 g before initiation of host conditioning. All mice, between 6 and 12 weeks of age, were maintained in a specific pathogen-free environment according to the guidelines established by the Animal Care Committee, and received drinking water supplemented with gentamycin. Ten- to twelve-week-old DBA/2 mice (recipients) were given total body lethal irradiation (800 cGy) from a gamma-ray source and after 24 hr, were injected with 3 × 107 bone marrow cells and 1 × 107 spleen cells from C57BL/6 mice (donors) via the tail vein.20
Adoptive transfer of expanded Tregs from APB and CB
As mentioned above, CB and APB Tregs were expanded ex vivo with two cycles of polyclonal stimulation: CB Treg (PP-PP) and APB Treg (PP-PP). The CB Tregs were also expanded with polyclonal stimulation in the first cycle and xenogenic mouse B cells in the second cycle or xenogenic mouse B cells in the first cycle and polyclonal stimulation in the second cycle: CB Treg (PP-MBP) and CB Treg (MBP-PP). In the acute GVHD (aGVHD) mouse model, lethally irradiated recipient DBA/2 (H-2Kd) mice were injected with 3 × 107 bone marrow cells and 1 × 107 spleen cells from donor C57BL/6 mice (H-2Kb). For the adoptive transfer of expanded Tregs, recipient mice received 1 × 106 expanded Tregs from APB and CB via the tail vein immediately after bone marrow cell and splenocyte transplantation. Survival and appearance were monitored daily and body weight was measured weekly. A scoring system that used five clinical variables,21 including weight loss, posture, activity, fur texture and skin integrity (maximum index, 10), was used to assess GVHD.
Statistical analysis
The data are presented as the mean ± standard deviation (SD). All statistical analyses were performed using the Statistica 6.0 statistical software (StatSoft Inc., Tulsa, OK). Statistical differences in animal survival were analysed using the log-rank test. The differences in the groups being compared were considered significant if the P value was < 0·05.
Results
Cell yield after Treg expansion with alloantigen or polyclonal stimulation
Cell yield after Treg expansion with two cycles of polyclonal stimulation was larger than Tregs expanded with combined alloantigen and polyclonal stimulation. The average fold expansions of CB and APB Tregs with two cycles of polyclonal stimulation were 1·5 ± 0·4 × 103 and 1·8 ± 0·5 × 103, respectively (P > 0·05), whereas the average fold expansions of CB and APB Tregs with an alloantigen stimulus followed by a polyclonal stimulus were 5·0 ± 1·1 × 102 and 4·7 ± 1·2 × 102, respectively (P > 0·05). The average fold expansions of CB and APB Tregs with two cycles of polyclonal stimulation were significantly larger than those of CB and APB Tregs with an alloantigen stimulus followed by a polyclonal stimulus (Fig. 1b,c).
Phenotypic characterization of ex vivo expanded Tregs
A potential risk of Treg expansion is the outgrowth of contaminating cell types, such as CD8+ T cells or natural killer cells. In our experiments, we did not find major contaminants, because > 95% of the expanded cells were CD4+ CD25+ T cells (Fig. 2b). The majority of Tregs retained expression of FoxP3 after two cycles of expansion. Although only 65·4% of freshly isolated CB Tregs expressed FoxP3+ CD127− (Fig. 2a), > 90% of expanded CB Tregs expressed FoxP3+ CD127− (Fig. 2b). The FoxP3− CD127+ T cells in the CD4+ CD25+ T cells isolated from CB did not proliferate efficiently after two cycles of expansion. Analysis of CTLA4 surface expression by flow cytometry revealed that ex vivo expanded Tregs expressed more CTLA4 molecules than activated CD4+ CD25− T cells did. Similarly, ex vivo expanded Tregs, but not activated CD4+ CD25− T cells, expressed CD39, which might serve as a marker of the expanded Tregs. The expanded Tregs expressed LAG3 as well as the activated CD4+ CD25− T cells did. Hence, compared with the activated CD4+ CD25− T cells, all the expanded Tregs expressed high levels of FoxP3, CD39 and CTLA4, and low levels of CD127 (Fig. 2b). Only CB Tregs with two cycles of polyclonal stimulation expressed low levels of HLA-DR.
Figure 2.
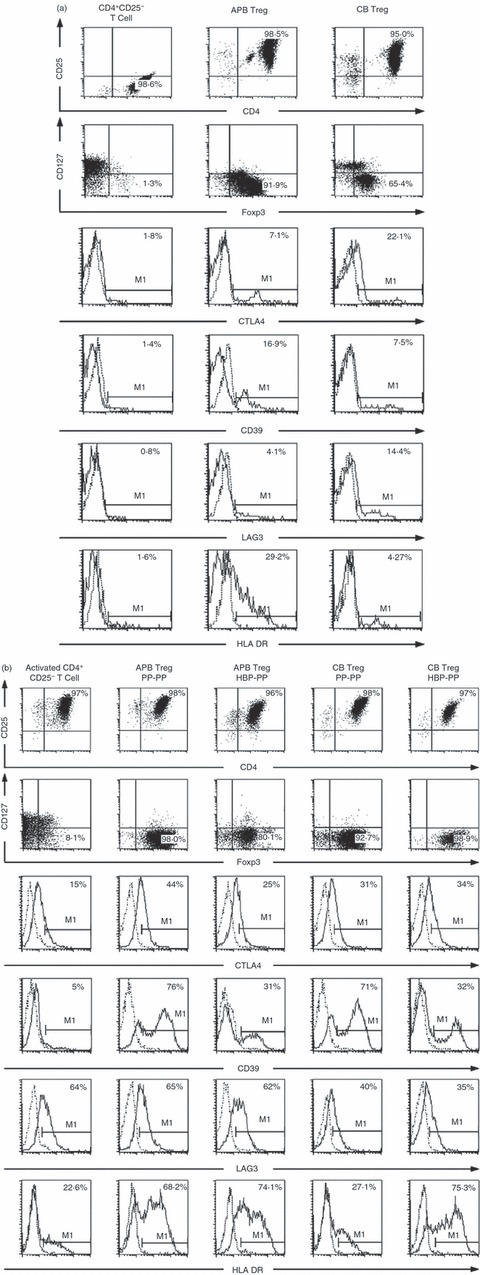
Phenotypic characterization of freshly isolated and expanded regulatory T cells (Tregs). (a) The phenotype of freshly isolated adult peripheral blood (APB) CD4+ CD25− T cells and Tregs from APB and cord blood (CB) was analysed by flow cytometry. (b) CD4+ CD25− T cells were activated with anti-CD3/anti-CD28 microbeads and recombinant human interleukin-2 (rhIL-2) for two cycles. APB and CB Tregs were expanded according to the protocol described in Fig. 1(a). Cell surface expression of CD4/CD25, CD127, CTLA4, CD39, LAG3 and HLA-DR and intracellular expression of FoxP3 were analysed on activated CD4+ CD25− T cells and expanded Tregs. Histograms show the percentage of expression of these markers (bold line) in the gated populations compared with the appropriate isotype control (thin line). Data are representative of three independent experiments.
Tregs retained anergic properties after expansion
One of the hallmarks of Tregs is their anergic (hyporesponsive) behaviour ex vivo. Tregs retained their anergic state after expansion irrespective of the strategy employed, whereas activated CD4+ CD25− T cells were not anergic. Indeed, all the expanded Tregs did not proliferate upon stimulation in the absence of exogenous T-cell growth factors (Fig. 3g). All the Tregs showed low gene expression of IL-2 and interferon-γ when compared with activated CD4+ CD25− T cells (Fig. 3c,d). However, there was no significant difference in the gene expression levels of IL-10 and transforming growth factor-β between the expanded Tregs and activated CD4+ CD25− T cells (Fig. 3e,f). After two cycles of expansion, the CB Tregs maintained the expression of the GARP gene, whereas the APB Tregs lost GARP expression (Fig. 3b).
Figure 3.
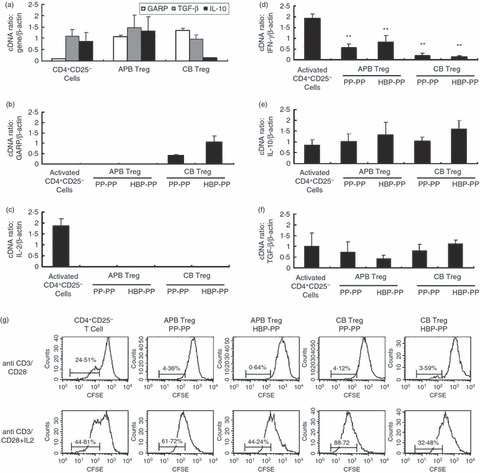
Reverse transcription-PCR analysis of GARP and cytokine gene expression in expanded regulatory T cells (Tregs) and stimulation assay to analyse T-cell anergy. Adult peripheral blood (APB) and cord blood (CB) Tregs were expanded according to the protocol described in Fig. 1(a). Total RNA was isolated, reverse-transcribed and used for quantitative analysis. (a) Gene expression of GARP, transforming growth factor-β (TGF-β) and interleukin-10 (IL-10) of freshly isolated APB CD4+ CD25− T cells and Tregs from APB and CB was displayed as relative expression normalized to β-actin. Gene expression of GARP (b), IL-2 (c), interferon-γ (IFN-γ) (d), IL-10 (e) and TGF-β (f) of activated CD4+ CD25− T cells and expanded Tregs from APB and CB was displayed as relative expression normalized to β-actin. Values are given as the mean ± standard deviation (SD) of three to five independent experiments. **P < 0·01 compared with activated CD4+ CD25− T cells. (g) CFSE-labelled CD4+ CD25− T cells and expanded Tregs from APB and CB were stimulated with anti-CD3/anti-CD28 antibodies in the absence or presence of exogenous IL-2. Proliferation was determined by CFSE dilution on Day 4.
CB Tregs expanded with polyclonal stimulation showed potent suppressive capacity
We used third-party Tregs prepared from HLA-unrelated donors in the suppression experiments based on previous reports.16 The limited availability of Tregs from a single donor or cord blood source could constitute a major barrier to the development of Treg cell-based therapy in humans. This barrier, however, might be readily circumvented if third-party Tregs could be used in place of donor or recipient Tregs. Although APB Tregs showed greater ex vivo expansion with two cycles of polyclonal stimulation than CB Tregs, we found that the CB Tregs had greater suppressive capacity than the APB Tregs. The CB and APB Tregs showed > 80% and > 40% suppression, respectively, of CD4+ CD25− Tresp cell proliferation upon stimulation with anti-CD3/anti-CD28 antibodies and rhIL-2 at Treg : Tresp ratios of 1 : 1 and > 50% and > 20% suppression, respectively, at Treg : Tresp ratios of 1 : 4 (Fig. 4). The CB Tregs even showed > 30% suppression in an HLA-mismatched DC-driven Tresp cell proliferation with Treg : Tresp ratios of 1 : 16 (Fig. 4), but APB Tregs showed little suppression at the same Treg : Tresp ratios. Hence, CB Tregs expanded with polyclonal stimulation suppressed the proliferation of not only the polyclonal antigen-driven Tresp cells but also the HLA-mismatched DC-driven Tresp cells.
Figure 4.
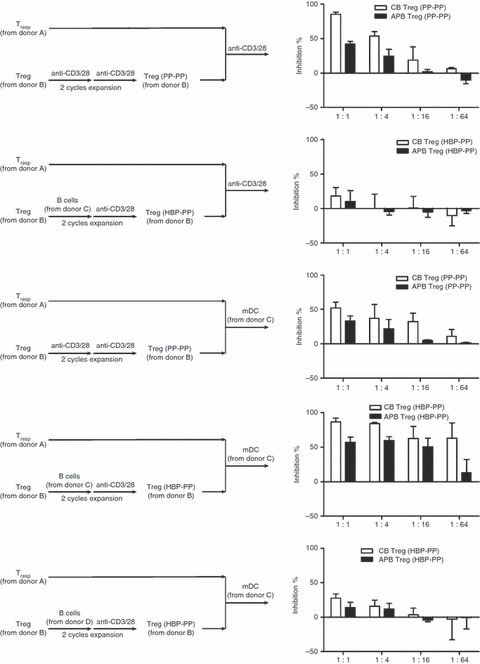
Suppressive capacity of regulatory T cells (Tregs) expanded from adult peripheral blood (APB) and cord blood (CB). The APB and CB Tregs were expanded according to the protocol described in Fig. 1(a). Progressive CFSE dilution was used as a read-out of responder cell proliferation. CFSE-labelled CD4+ CD25− responder T (Tresp) cells were stimulated with anti-CD3/anti-CD28 antibodies and recombinant human interleukin-2 (rhIL-2) or HLA mismatched dendritic cells presenting the target alloantigen (same as or different form the B cells used for Treg expansion). Third-party expanded Tregs from APB and CB were added to these cultures at the indicated Treg : Tresp ratios (1 : 1, 1 : 4, 1 : 16 and 1 : 64). Proliferation was determined by CFSE dilution of Tresp cells on Day 4. The inhibition rate was calculated as followed: (1 – percentage of proliferated Tresp with expanded Treg/percentage of proliferated Tresp with no Treg) ×100%. Data represent the average inhibition rate of three independent experiments.
Tregs expanded by an alloantigen stimulus acquired antigen specificity
The main feature of alloantigen-specific Tregs is their strong suppressive capacity when stimulated by target antigens. Our data showed that highly antigen-specific Tregs were generated when APB and CB Tregs were expanded ex vivo with a primary alloantigen stimulus followed by a secondary polyclonal stimulus. The CB and APB Tregs expanded with allogeneic human B cells showed very high suppressive activities of > 80% and > 60%, respectively, with Treg : Tresp ratios of 1 : 4 in a target antigen-driven Tresp cell proliferation, in this case B cells to expand Tregs and DCs to stimulate Tresp cells were from the same blood donor (Fig. 4). However, these Tregs showed little suppression (about 10%) in mismatched alloantigen-driven Tresp cell proliferation, in this case B cells to expand Tregs and DCs to stimulate Tresp cells were from different blood donors, and hardly any suppression of the proliferation of CD4+ CD25− Tresp cells when stimulated with anti-CD3/anti-CD28 antibodies and rhIL-2 at the same Treg : Tresp ratios. The CB and APB Tregs expanded with polyclonal stimulation showed low suppressive activity with the same Treg : Tresp ratios in an alloantigen-driven Tresp cell proliferation (> 30% and > 20% suppression, respectively, Fig. 4). When Tregs were expanded with an alloantigen stimulus followed by a polyclonal stimulus, they expanded efficiently and showed a highly potent, strictly antigen-specific suppressive capacity. As expected, two cycles of polyclonal stimulation yielded high numbers of Tregs. These cells were suppressive, but no enrichment of alloantigen-specific Tregs had taken place. The CB and APB Tregs expanded by an alloantigen stimulus lost their capacity to potently inhibit the alloreactivity elicited by non-specific alloantigens but remained highly potent in inhibiting the alloproliferation of responder cells elicited by specific alloantigens. The alloantigen specificity demonstrated by B-cell-expanded Tregs was not determined by the HLA haplotypes of the Tregs but was induced and determined by the haplotype of the B cells used to expand them.
Ex vivo expanded Tregs prevented aGVHD in mice
The in vivo activity of ex vivo expanded human APB and CB Tregs was further evaluated in a mouse aGVHD model. The mice with aGVHD showed serious aGVHD symptoms, such as hunched back, diarrhoea and weight loss, and the mice usually died within 18 days (Fig. 5a). When co-transferred with CB or APB expanded Tregs, the ex vivo expanded Tregs significantly enhanced the survival of the aGVHD mice (Fig. 5a). When CB and APB Tregs expanded with two cycles of polyclonal stimulation were transferred to the aGVHD mice, 87·5% and 16·7% of the mice, respectively, survived for at least 63 days. The CB Tregs expanded with polyclonal stimulation followed by xenogenic mouse B-cell stimulation or with xenogenic mouse B-cell stimulation followed by polyclonal stimulation did not show better suppressive function than polyclonal stimulation alone as expected (Fig. 5a).
Figure 5.
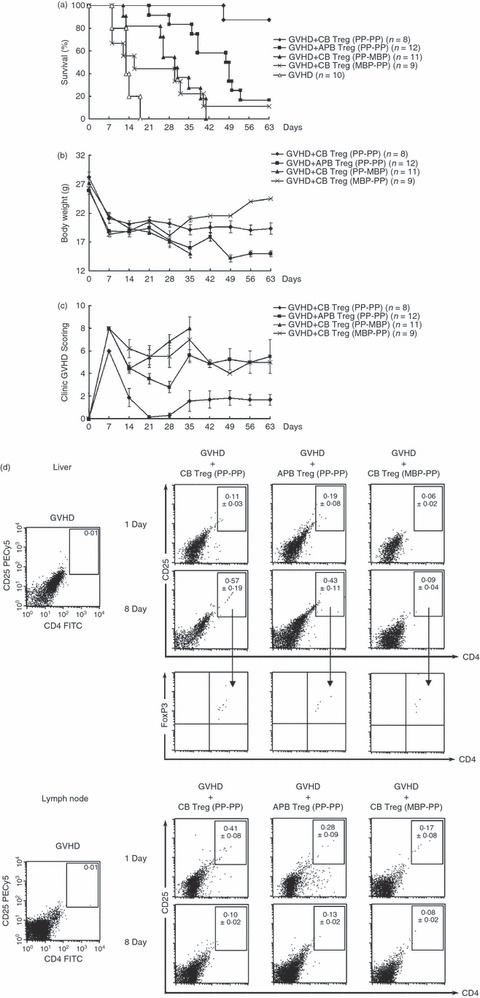
Adoptive transfer of ex vivo expanded regulatory T cells (Tregs) from cord blood (CB) and adult peripheral blood (APB) into mice with acute graft-versus-host disease (aGVHD). CB and APB Tregs were expanded ex vivo with two cycles of polyclonal stimulation: CB Treg (PP-PP) and APB Treg (PP-PP). CB Tregs were also expanded with polyclonal stimulation in the first cycle and xenogenic mouse B-cell stimulation in the second cycle or with xenogenic mouse B-cell stimulation in the first cycle and polyclonal stimulation in the second cycle: CB Treg (PP-MBP) and CB Treg (MBP-PP). In the aGVHD mouse model, 1 × 106 of the variously expanded Tregs were adoptively transferred into lethally irradiated recipient DBA/2 (H-2Kd) mice after injection with 3 × 107 bone marrow cells and 1 × 107 spleen cells from donor C57BL/6 mice (H-2Kb). The survival (a), body weight (b) and clinical GVHD score (c) were assessed. The GVHD score and body weight were determined from the surviving mice. (d) The mononuclear cells from the liver and lymph node of aGVHD mice were isolated by density gradient centrifugation using Ficoll–Hypaque on Day 1 or Day 8 after adoptive transfer of ex vivo expanded Tregs. The human molecules of CD4, CD25 and FoxP3 were determined by flow cytometry. Data represent the average percentage of human CD4+ CD25+ T cells ± standard deviation (SD) of six independent experiments.
We also compared the capacity of ex vivo expanded CB and APB Tregs to ameliorate the clinical symptoms of aGVHD by measuring five clinical variables of aGVHD. The mean GVHD scores of the aGVHD mice given APB Tregs expanded with two cycles of polyclonal stimulation were significantly greater than the scores of mice given CB Tregs expanded with two cycles of polyclonal stimulation (Fig. 5c; P < 0·05). The aGVHD mice given APB Tregs exhibited hair loss, hunched back, swollen face and diarrhoea. Similar results were observed in aGVHD mice given CB Tregs expanded with polyclonal stimulation followed by xenogenic mouse B-cell stimulation or with xenogenic mouse B-cell stimulation followed by polyclonal stimulation. The aGVHD mice given APB Tregs expanded with two cycles of polyclonal stimulation lost more weight than the mice given CB Tregs with two cycles of polyclonal stimulation (Fig. 5b; P < 0·01).
These results indicate that the adoptive transfer of ex vivo expanded human Tregs alleviates the symptoms of aGVHD and markedly prolongs survival time in mice with aGVHD and that CB Tregs expanded with two cycles of polyclonal stimulation have better immunosuppressive function than APB Tregs.
Ex vivo expanded human Tregs survived and expanded in the liver of aGVHD mice
Human CD4+ CD25+ T cells could be detected in the liver and lymph node of aGVHD mice on Day 8 after adoptive transfer of ex vivo expanded Tregs and all the human CD4+ CD25+ T cells in the mice expressed the FoxP3 molecule (Fig. 5d). The percentage of human CD4+ CD25+ T cells increased from 0·11 ± 0·03% on Day 1 to 0·57 ± 0·19% on Day 8 in the liver after adoptive transfer of CB Tregs expanded with two cycles of polyclonal stimulation. The CB Tregs expanded in response to mouse B cells (MBP-PP) did not persist in the liver. In the lymph nodes, the three populations tested (CB Treg PP-PP, APB Treg PP-PP and CB Treg MBP-PP) diminished their frequency from Day 1 to Day 8. We detected a small amount of human CD4+ CD25+ T cells in the spleen and bone marrow of aGVHD mice on Day 1 after adoptive transfer of ex vivo expanded Tregs (data not shown).
Discussion
High numbers of Tregs are needed for effective Treg immunotherapy in humans to facilitate tolerance in patients with autoimmune disorders or after transplantation. Ex vivo Treg expansion with polyclonal stimulation has been shown to be an effective method for obtaining these high numbers.22,23 In the current study, the average fold expansion of CB and APB Tregs with two cycles of polyclonal stimulation was 1·5 × 103 and 1·8 × 103, respectively. A 2-day rest period between the two proliferation cycles of CB Tregs can prevent the apoptosis of CB Tregs induced by excessive activation. In contrast, APB Tregs need not rest between the two proliferation cycles, but the rest period has no adverse effects on the expansion of the APB Tregs. Hence, we can obtain about 1 × 109 to 4 × 109 Tregs from one unit of CB or APB, which will meet the needs of clinical application. However, there are some drawbacks regarding the clinical application of polyclonally expanded Tregs. First, because this type of activation generates Tregs with a broad range of specificities, the infused polyclonal Tregs may suppress immune responses other than the target response, thereby increasing the risk for opportunistic infections and tumour growth. Second, because of the low percentage of Tregs within a polyclonal cell pool that is specific for a given target antigen, large numbers of Tregs need to be infused. Although the average fold expansions of CB and APB Tregs expanded with an alloantigen stimulus followed by a polyclonal stimulus were 5·0 × 102 and 4·7 × 102, respectively, which were significantly less than that with polyclonal stimulation, these Tregs had a potent, strictly antigen-specific suppressive capacity that was greater than Tregs expanded with polyclonal stimulation. If Treg populations were expanded for more than two expansion cycles, we observed a loss of suppressive capacity and high cell death. Because primary human B cells can efficiently expand allogeneic Tregs but cannot activate non-Treg CD4+ cells,16 we used human allogeneic B cells as the alloantigen stimulus to expand the CB and APB Tregs and obtained a high purity of expanded cells. These cells may be especially beneficial to patients receiving an HLA mismatched haematopoietic stem cell transplant, where this type of alloantigen-reactivity is important in graft versus host pathology.
The high level of expression of FoxP3 in all of the ex vivo expanded Tregs suggested that these cells maintained the properties of Tregs. The expanded Tregs also expressed high levels of CD39 and CTLA4 and had low gene expression of IL-2 and interferon-γ, which was in contrast to activated CD4+ CD25− T cells. CD39 is the dominant ectoenzyme in the immune system that hydrolyses ATP or ADP to AMP and is expressed by B cells, DC, all mouse Tregs, and about 50% of human Tregs.24 Freshly isolated Tregs do not hydrolyse ATP, but activated Tregs can mediate hydrolysis. ATP can up-regulate CD86 expression on DC. Pre-exposure of Tregs to ATP-containing medium has been found to reduce ATP-driven DC maturation. In addition to FoxP3, CD39 might serve as another Treg marker of ex vivo expanded Tregs. Another cell surface antigen that may play a role in Treg suppression of DC function is LAG-3 (CD223), a CD4 homologue that binds MHC class II molecules with very high affinity. Binding of LAG-3 to MHC class II molecules expressed by immature DC induces an immunoreceptor tyrosine-based inhibitory motif-mediated inhibitory signal that suppresses DC maturation and immunostimulatory capacity.25 Although the expanded Tregs partially expressed LAG-3, the activated CD4+ CD25− T cells also expressed LAG-3 (Fig. 2).
To reduce the contaminating CD4+ CD127+ conventional T cells within the population of APB Tregs, CD127+ cells should be depleted. In this study, we expanded CB Tregs ex vivo from CD4+ CD25+ T cells with either polyclonal or alloantigen stimulation, and the expanded CB Tregs demonstrated similar phenotypic and functional characteristics as APB Tregs expanded from CD4+ CD25+ CD127− T cells. FoxP3− CD127+ T cells in the CD4+ CD25+ T cells isolated from CB did not proliferate efficiently after two cycles of expansion. Clinical grade [Good Manufacturing Practice, (GMP)] CD4+ CD25+ Treg isolation by a magnetic-bead-based method is now feasible using the CliniMACS system.26,27 Hence, the procedure of CB Treg expansion we used fits with the currently available clinical grade isolation tools with the objective of facilitating easy translation into clinical practice. Clearly, with regard to purity of the starting population, magnetic bead isolation of CD4+ CD25+ T cells is inferior to FACS sorting or further CD127 depletion, but these latter methods are not readily available for GMP purposes.
Our study showed that CB Tregs had better immunosuppressive function than APB Tregs, and this might be the result of the expression of GARP by CB Tregs, as APB Tregs lost GARP expression after expansion. GARP (or LRRC32) is an orphan toll-like receptor composed of leucine-rich repeats. GARP mRNA is almost undetectable in freshly isolated CD4+ T cells (both natural Treg cells and non-Treg cells) and is rapidly up-regulated after T-cell receptor-mediated activation, but only in natural Treg cells.28–30 GARP mRNA then gradually decreases with time. This is the first study in which we found that CB Tregs continued to express the GARP gene after expansion for 14 days. The CB Tregs expanded with polyclonal stimulation followed by xenogenic mouse B-cell stimulation or with xenogenic mouse B-cell stimulation followed by polyclonal stimulation did not show better suppressive function than polyclonal stimulation alone as we expected (Fig. 5a). Because CB Tregs expanded ex vivo with mouse B-cell stimulation (MBP-PP) scarcely survived in the liver or lymph nodes of the DBA/2 hosts (Fig. 5d), this may be the reason for the worse performance obtained with their transfer in the suppression of murine aGVHD. As the frequency of CB Tregs expanded with polyclonal stimulation was clearly higher, the ex vivo selection of CB Tregs in response to mouse B cells may have altered their repertoire and made them unable to expand in the xenogeneic host. The expansion/survival of human T cells after transfer to control DBA/2 mice, not injected with responder C57BL/6 spleen cells might clarify this point and may be addressed in our further studies.
Cao et al.31 demonstrated that the infusion of ex vivo expanded human Tregs prevented lethal xenogenic GVHD in a human peripheral blood lymphocyte–NOD/SCID mouse model, suggesting the potential of ex vivo expanded human CD4+ CD25+ Foxp3+ Tregs as a therapeutic approach for GVHD. In this study, we found that the Tregs from CB and APB expanded with two cycles of polyclonal stimulation alleviated GVHD symptoms and prolonged survival after MHC mismatched allogeneic bone marrow transplantation in a murine model. The aGvHD mouse model we used can reflect the real situation of bone marrow transplantation, so the experiment results will be used to guide the clinical application of the third-party human Tregs in bone marrow transplantation. In the aGVHD murine model, all aGVHD mice died within 18 days. When the CB and APB expanded Tregs were transferred into aGVHD mice, 87·5% and 16·7% of the mice survived for at least 63 days, respectively. We also found that only CB Tregs with two cycles of polyclonal stimulation expressed low levels of HLA-DR (Fig. 2b). Baecher-Allan et al.32 demonstrated that HLA-DR expression on human CD4+ CD25high T cells identified a functionally distinct population of Tregs that induced early contact-dependent suppression that was associated with high FoxP3 expression. In striking contrast, HLA-DR− CD4+ CD25high Treg induced early IL-4 and IL-10 secretion and a late FoxP3-associated contact-dependent suppression. Ashley and Baecher-Allan33 showed strong T-cell receptor stimulation markedly increased the expression of GzmB in all dividing responder CD4 T cells and mitigated the suppression by HLA-DR+ Tregs. The HLA-DR+ Treg suppressive activity re-emerged if GzmB was neutralized. Responder cells actively killed HLA-DR+ effector Tregs by producing GzmB in response to strong T-cell receptor stimulation. Because we used third-party Tregs in the suppression experiments and adoptive transferred human Tregs in the mouse model, the high expression of HLA-DR of ex vivo expanded Tregs may stimulate the proliferation of Tresp at lower Treg : Tresp ratios (Fig. 4) and be easily rejected by immune cells of mice in vivo (Fig. 5d). The suppressive mechanisms of the expanded human Tregs require further evaluation.
In summary, this study demonstrated that CD4+ CD25+ T cells from CB and CD4+ CD25+ CD127− T cells from APB were efficiently expanded ex vivo with polyclonal or alloantigen stimulation. The expanded Tregs from CB and APB remained anergic and displayed potent suppressive functions. The CB Tregs expanded with two cycles of polyclonal stimulation had better immunosuppressive function than APB Tregs expanded by either method. Adoptive transfer of ex vivo expanded human Tregs alleviated the symptoms of aGVHD and markedly prolonged survival time in aGVHD mice. As it is feasible to obtain human functional alloantigen-specific Tregs from CB and APB in large numbers, the ex vivo expansion protocol described here will very probably increase the success of clinical Treg-based immunotherapy.
Acknowledgments
This study was supported by a grant (09140903000) from the Science and Technology Commission of Shanghai Municipality. We would like to express our gratitude to the Department of Animal Science at the Shanghai Jiaotong University School of Medicine for the laboratory animal husbandry.
Disclosure
The authors declare no potential conflict of interest, including all relevant financial interests in any company or institution that might benefit from this publication.
References
- 1.Zhao D, Zhang C, Yi T, et al. In vivo-activated CD103+CD4+ regulatory T cells ameliorate ongoing chronic graft-versus-host disease. Blood. 2008;112:2129–38. doi: 10.1182/blood-2008-02-140277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joffre O, Gorsse N, Romagnoli P, et al. Induction of antigen-specific tolerance to bone marrow allografts with CD4+CD25+ T lymphocytes. Blood. 2004;103:4216–21. doi: 10.1182/blood-2004-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trenado A, Charlotte F, Fisson S, et al. Recipient-type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus- leukemia. J Clin Invest. 2003;112:1688–96. doi: 10.1172/JCI17702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bushell A, Jones E, Gallimore A, et al. The generation of CD25+CD4+ regulatory T cells that prevent allograft rejection does not compromise immunity to a viral pathogen. J Immunol. 2005;174:3290–7. doi: 10.4049/jimmunol.174.6.3290. [DOI] [PubMed] [Google Scholar]
- 5.Golshayan D, Jiang S, Tsang J, et al. In vitro expanded donor alloantigen-specific CD4+CD25+ regulatory T cells promote experimental transplantation tolerance. Blood. 2006;6:1879–82. doi: 10.1182/blood-2006-05-025460. [DOI] [PubMed] [Google Scholar]
- 6.Joffre O, Santolaria T, Calise D, et al. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat Med. 2008;14:88–92. doi: 10.1038/nm1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tarbell KV, Petit L, Zuo X, et al. Dendritic cell expanded, islet-specific CD4+CD25+ CD62L+ regulatory T cells restore normoglycemia in diabetic NOD mice. J Exp Med. 2007;204:191–201. doi: 10.1084/jem.20061631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang Q, Henriksen KJ, Bi M, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–65. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masteller EL, Warner MR, Tang Q, et al. Expansion of functional endogenous antigen-specific CD4+CD25+ regulatory T cells from nonobese diabetic mice. J Immunol. 2005;175:3053–9. doi: 10.4049/jimmunol.175.5.3053. [DOI] [PubMed] [Google Scholar]
- 10.Chai JG, Coe D, Chen D, et al. In vitro expansion improves in vivo regulation by CD4+CD25+ regulatory T cells. J Immunol. 2008;180:858–69. doi: 10.4049/jimmunol.180.2.858. [DOI] [PubMed] [Google Scholar]
- 11.Karakhanova S, Munder M, Schneider M, et al. Highly efficient expansion of human CD4+CD25+ regulatory T cells for cellular immunotherapy in patients with graft-versus-host disease. J Immunother. 2006;29:336–49. doi: 10.1097/01.cji.0000203080.43235.9e. [DOI] [PubMed] [Google Scholar]
- 12.Kreijveld E, Koenen HJ, Hilbrands LB, et al. Ex vivo expansion of human CD4+CD25high regulatory T cells from transplant recipients permits functional analysis of small blood samples. J Immunol Methods. 2006;314:103–13. doi: 10.1016/j.jim.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 13.Trzonkowski P, Szarynska M, Mysliwska J, et al. Ex vivo expansion of CD4+CD25+ T regulatory cells for immunosuppressive therapy. Cytometry A. 2009;75:175–88. doi: 10.1002/cyto.a.20659. [DOI] [PubMed] [Google Scholar]
- 14.Peters JH, Hilbrands LB, Koenen HJ, et al. Ex vivo generation of human alloantigen-specific regulatory T cells from CD4posCD25high T cells for immunotherapy. PLoS One. 2008;3:e2233. doi: 10.1371/journal.pone.0002233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peters JH, Preijers FW, Woestenenk R, et al. Clinical grade Treg: GMP isolation, improvement of purity by CD127pos depletion, Treg expansion, and Treg cryopreservation. PLoS One. 2008;3:e3161. doi: 10.1371/journal.pone.0003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen LC, Delgado JC, Jensen PE, et al. Direct expansion of human allospecific FoxP3+CD4+ regulatory T cells with allogeneic B cells for therapeutic application. J Immunol. 2009;183:4094–102. doi: 10.4049/jimmunol.0901081. [DOI] [PubMed] [Google Scholar]
- 17.Hess AD. Modulation of graft-versus-host disease: role of regulatory T lymphocytes. Biol Blood Marrow Transplant. 2006;1(Suppl 2):13–21. doi: 10.1016/j.bbmt.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Hoffmann P, Edinger M. CD4+CD25+ regulatory T cells and graft-versus-host disease. Semin Hematol. 2006;43:62–9. doi: 10.1053/j.seminhematol.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 19.Fujimaki W, Takahashi N, Ohnuma K, et al. Comparative study of regulatory T cell function of human CD25+CD4+ T cells from thymocytes, cord blood, and adult peripheral blood. Clin Dev Immunol. 2008;2008:305859. doi: 10.1155/2008/305859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang J, Gao L, Liu Y, et al. Adoptive therapy by transfusing expanded donor murine natural killer T cells can suppress acute graft-versus-host-disease in allogeneic bone marrow transplantation. Transfusion. 2010;50:407–17. doi: 10.1111/j.1537-2995.2009.02395.x. [DOI] [PubMed] [Google Scholar]
- 21.Cooke KR, Kobzik L, Martin TR, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88:3230–9. [PubMed] [Google Scholar]
- 22.Riley JL, June CH, Blazar BR. Human T regulatory cell therapy: take a billion or so and call me in the morning. Immunity. 2009;30:656–65. doi: 10.1016/j.immuni.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Godfrey WR, Ge YG, Spoden DJ, et al. In vitro expanded human CD4+CD25+ T-regulatory cells can markedly inhibit allogeneic dendritic cell-stimulated MLR cultures. Blood. 2004;104:453–61. doi: 10.1182/blood-2004-01-0151. [DOI] [PubMed] [Google Scholar]
- 24.Borsellino G, Kleinewietfeld M, Di Mitri D, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–32. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- 25.Liang BT, Workman C, Lee J, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol. 2008;180:5916–26. doi: 10.4049/jimmunol.180.9.5916. [DOI] [PubMed] [Google Scholar]
- 26.Hoffmann P, Boeld TJ, Eder R, et al. Isolation of CD4+CD25+ regulatory T cells for clinical trials. Biol Blood Marrow Transplant. 2006;12:267–74. doi: 10.1016/j.bbmt.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Wichlan DG, Roddam PL, Eldridge P, et al. Efficient and reproducible large-scale isolation of human CD4+CD25+ regulatory T cells with potent suppressor activity. J Immunol Methods. 2006;315:27–36. doi: 10.1016/j.jim.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 28.Tran DQ, Andersson J, Wang R, et al. GARP (LRRC32) is essential for the surface expression of latent TGF-β on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci USA. 2009;106:13445–50. doi: 10.1073/pnas.0901944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang R, Wan Q, Kozhaya L, et al. Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PLoS One. 2008;3:e2705. doi: 10.1371/journal.pone.0002705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Probst-Kepper M, Geffers R, Kroger A, et al. GARP: a key receptor controlling FOXP3 in human regulatory T cells. J Cell Mol Med. 2009;13:3343–57. doi: 10.1111/j.1582-4934.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao T, Soto A, Zhou W, et al. Ex vivo expanded human CD4+CD25+Foxp3+ regulatory T cells prevent lethal xenogenic graft versus host disease (GVHD) Cell Immunol. 2009;258:65–71. doi: 10.1016/j.cellimm.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 32.Baecher-Allan C, Wolf E, Hafler DA. MHC class II expression identifies functionally distinct human regulatory T cells. J Immunol. 2006;176:4622–31. doi: 10.4049/jimmunol.176.8.4622. [DOI] [PubMed] [Google Scholar]
- 33.Ashley CW, Baecher-Allan C. Cutting Edge: Responder T cells regulate human DR+ effector regulatory T cell activity via granzyme B. J Immunol. 2009;183:4843–7. doi: 10.4049/jimmunol.0900845. [DOI] [PMC free article] [PubMed] [Google Scholar]