

Abstract
LSF is a mammalian transcription factor that is rapidly and quantitatively phosphorylated upon growth induction of resting, peripheral human T cells, as assayed by a reduction in its electrophoretic mobility. The DNA-binding activity of LSF in primary T cells is greatly increased after this phosphorylation event [Volker et al., 1997]. We demonstrate here that LSF is also rapidly and quantitatively phosphorylated upon growth induction in NIH 3T3 cells, although its DNA-binding activity is not significantly altered. Three lines of experimentation established that ERK is responsible for phosphorylating LSF upon growth induction in both cell types. First, phosphorylation of LSF by ERK is sufficient to cause the reduced electrophoretic mobility of LSF. Second, the amount of ERK activity correlates with the extent of LSF phosphorylation in both primary human T cells and NIH 3T3 cells. Finally, specific inhibitors of the Ras/Raf/MEK/ERK pathway inhibit LSF modification in vivo. This phosphorylation by ERK is not sufficient for activation of LSF DNA-binding activity, as evidenced both in vitro and in mouse fibroblasts. Nonetheless, activation of ERK is a prerequisite for the substantial increase in LSF DNA-binding activity upon activation of resting T cells, indicating that ERK phosphorylation is necessary but not sufficient for activation of LSF in this cell type.
Keywords: ERK, LSF, T cells, fibroblasts, DNA-binding, phosphorylation
LSF (also known as CP2 [Lim et al., 1992] and LBP-1c [Yoon et al., 1994]) is a ubiquitously expressed mammalian transcription factor [Swendeman et al., 1994] that was originally identified by its ability to bind to and stimulate transcription from the simian virus 40 (SV40) major late promoter [Huang et al., 1990]. LSF is unusual among transcription factors in its ability to bind directly repeated half sites as a homotetramer [Huang et al., 1990; Murata et al., 1998; Shirra and Hansen 1998], or as a tetrameric complex with the highly related LBP-1a/b family member [Yoon et al., 1994] (also named NF2d9 in mouse [Sueyoshi et al., 1995]). However, on a subset of promoters, LSF functions as a heteromeric complex with unrelated partner proteins [Casolaro et al., 2000; Jane et al., 1995; Murata et al., 1998; Romerio et al., 1997; Zhou et al., 2000].
In addition to sites within the SV40 late promoter, LSF/CP2/LBP-1 has been shown to bind and regulate a number of cellular and viral promoters. It binds several promoters regulated at the G0/G1 boundary: the human immunodeficiency virus (HIV) long terminal repeat (LTR) [Jones et al., 1988; Kato et al., 1991; Malim et al., 1989; Wu et al., 1988; Yoon et al., 1994], the human IL-4 promoter [Casolaro et al., 2000], the human c-fos promoter, at a site immediately downstream of the serum response element (R. Misra, H.-C. Huang, M. Greenberg, U. Hansen, unpublished observation) [Volker et al., 1997], and the human ornithine decarboxylase promoter (J. Volker, A. Butler, U. Hansen, unpublished observation). In addition, LSF regulates the thymidylate synthase promoter at the G1/S transition [Powell et al., 2000] and stimulates differentiation-specific promoters, such as those of the murine α-globin gene [Lim et al., 1993], the serum amyloid A3 gene [Bing et al., 1999], and the PAX6 gene [Zheng et al., 2001]. Due to the established regulation of a number of these promoters at the G0/G1 boundary, as well as the coupling of SV40 late gene expression to cell growth, we previously investigated whether LSF DNA-binding activity was modulated by cell growth in human peripheral T cells [Volker et al., 1997]. Indeed, within 15 min of mitogenic stimulation of these cells, the level of LSF-DNA binding activity increased by a factor of five [Volker et al., 1997].
The molecular basis of the enhanced DNA-binding activity of LSF in primary T cells, upon mitogenic signaling, was investigated further. Although the level of LSF protein in the nucleus remained constant throughout this interval, a rapid decrease in the electrophoretic mobility of LSF was observed by Western blot analyses. The modification leading to the altered mobility of LSF was attributed to phosphorylation, with phosphorylation of serine 291 being critical [Volker et al., 1997]. Mitogen activated protein (MAP) kinase, in particular pp42 ERK1, phosphorylated LSF in vitro on this residue, pinpointing ERKs as potential kinases for LSF modification following stimulation of T lymphocytes [Volker et al., 1997]. We therefore hypothesized that ERK phosphorylation of LSF contributed to its enhanced DNA-binding activity in T cells.
The MAP kinases ERK1 and ERK2 represent a central group of signaling kinases that are activated in response to growth stimuli in most cell types (for reviews see [Chang and Karin 2001; English et al., 1999; Hardy and Chaudhri 1997; Marais and Marshall 1996; Su and Karin 1996; Weston et al., 2002]). The best understood mechanism for activation of ERK is via activation of Ras by growth factor receptors or tyrosine kinases. ERK has been implicated in the phosphorylation of a number of transcription factors that are important for expression of genes essential for cell proliferation [Davis 1993; Hill and Treisman 1995; Hunter 1995; Schaeffer and Weber 1999; Vojtek and Cooper 1995]. We therefore explored further the potential connection between ERK activity, LSF phosphorylation, and LSF DNA-binding activity in primary T cells. Specifically, we have now investigated the causative and temporal relationship between ERK activation and the enhancement of LSF-DNA binding activity. We present evidence to support the connection, in that LSF phosphorylation correlates with cellular responses to various mitogens that induce ERK activity, enhanced LSF DNA-binding activity correlates with ERK activation in resting T cells, and activation of ERK is essential for the enhancement of LSF DNA-binding activity exiting G0 in these primary T cells. However, quantitative phosphorylation of LSF by ERK either in vitro or in NIH 3T3 cells does not affect the binding affinity of LSF to DNA. Therefore, the ERK pathway is necessary, although not sufficient, for increased LSF DNA-binding activity in T cells.
METHODS
Cell Culture
Human peripheral blood lymphocytes were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum (JRH Biosciences) at 37°C in 5% CO2 as previously described [Volker et al., 1997]. Erythrocytes were removed from the donor leukopacs immediately after separation on Ficoll-paque (Pharmacia) by incubation in ACK lysis buffer (0.15 M NH4Cl, 1 mM KHCO3, and 0.1 mM EDTA, pH 8.0) for 10 min at room temperature. Lymphocytes were stimulated for the indicated amount of times with phorbol myristate acetate (PMA; Sigma), at concentrations indicated in the figure legends; 2 µg/ml of phytohemagglutinin (PHA; Murex Diagnostics or Sigma); and 0.38 µg/ml of ionomycin (CalBiochem). These human peripheral blood lymphocytes responded identically as highly purified T cells with respect to LSF phosphorylation and DNA binding activities [Volker et al., 1997].
NIH 3T3 fibroblasts were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% calf serum (JRH Biosciences) at 37°C in 10% CO2. Cells were synchronized in G0 by serum deprivation. Briefly, 5 × 105 cells were plated per 100-mm dish in medium containing 10% calf serum. The following day, the medium was replaced with medium containing 0.2% calf serum, in which cells were incubated for 36 h to induce quiescence. Cells were growth stimulated by addition of either 10% calf serum or 10 ng/ml epidermal growth factor (EGF, Gibco BRL). Where indicated, the MEK1/2 inhibitor PD98059 (New England Biolabs) was added two hours prior to mitogenic stimulation of either T or NIH 3T3 cells.
Cell Extracts
Nuclear extracts were isolated as previously described [Volker et al., 1997], using 1 × 108 human peripheral blood lymphocytes or 5 × 10 6 NIH 3T3 cells. SDS total cell lysates were obtained from 2 × 106 NIH 3T3 cells by initially scraping and centrifuging the cells in phosphate-buffered saline solution (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2 PO4). Cells were subsequently resuspended in 2% SDS, 5% β–mercaptoethanol, 6% glycerol, 62.5 mM Tris-HCl, pH 6.8, 0.002% Bromophenol blue and boiled for 10 min. Half of each lysate was analyzed by SDS-PAGE and Western blotting for the migration of LSF.
Western Blotting Analysis
For LSF immunoblots, proteins were separated by 7.5% SDS-PAGE at 4°C for 15 hours at 10 mA and then electrophoretically transferred to nitrocellulose membranes (Schleicher & Schuell). Nitrocellulose membranes were blocked for 1 h in 5% nonfat milk in TBST (150 mM NaCl, 10 mM Tris-HCl, pH 8.0, 0.5% Tween 20). LSF was detected by incubation with a 1/100 dilution of purified α-LSFpep1-1 or α-LSFpep1–2 or a 1/500 dilution of purified α-LSFpep2-2 for 12 h [Volker et al., 1997]. The blots were washed, incubated with goat anti-rabbit antibody conjugated to horseradish peroxidase (HRP; Bio-Rad), and visualized by chemiluminescence as described by the supplier (DuPont NEN). Nitrocellulose blots were stripped for 1 h at 50°C in buffer containing 100 mM β-mercaptoethanol, 2% SDS, and 62.5 mM Tris-HCl, pH 6.7. Stripped blots were rinsed in TBST before blocking and reprobing.
Phosphorylation of Recombinant LSF in vitro
Purified, recombinant histidine-tagged LSF (His-LSF) was obtained as described previously [Volker et al., 1997]. For in vitro kinase assays, 100 ng of activated glutathione S transferase (GST)-ERK2 (Upstate Biotechnology) was incubated with 1 µg His-LSF for 15 min at 30°C in 20 µl buffer containing: 20 mM MOPS, pH 7.2, 25 mM β-glycerophosphate, 25 mM EGTA, 1 mM Na3VO4, 1 mM DTT, 15 mM MgCl2, 5 µg insulin and either 1 mM ATP or 1 mM AMP PNP. After incubation, samples were aliquoted and analyzed either by Western blotting or by EMSA.
Assays for ERK Activation
In gel MAPK assay
Two × 106 NIH 3T3 cells were lysed in 200 µl of buffer containing 0.5% Triton X-100, 20 mM Tris-HCl, pH 7.5, 0.5 mM EGTA, 100 µg/ml aprotinin, 100 µg/ml leupeptin, 2 mM NaF, 2 mM sodium pyrophosphate, 50 mM β-glycerophosphate and 1 mM DTT. Cell lysates were sonicated for 5 sec and centrifuged briefly at 4°C. The supernatant was transferred to a fresh tube and centrifuged for 45 min at 40,000 × g. Final protein concentration in the extracts was 5–10 mg/ml, in a total volume of 100 µl. Twenty µg of each lysate was electrophoresed through a 10% polyacrylamide gel containing 0.5 mg/ml myelin basic protein (Sigma) and the gel was then incubated with [γ-32P] ATP and washed, as described previously [Kameshita and Fujisawa 1989]. Subsequent to scanning (UMAX UC630 scanner) the image of the autoradiograph, the gel bands were quantified using Imagequant software. Kinase activity was determined by the amount of radioactivity at the positions in the gel corresponding to the electrophoretic mobility of ERK1 and ERK2.
Western blotting
For assaying levels of activated ERK, proteins were separated by 7.5% SDS-PAGE and transferred to a nitrocellulose membrane. The blots were incubated for 12 h either with a 1/1000 dilution of phospho-p44/42 MAPK antibody (T202/Y204 E10 monoclonal antibody, New England Bio Labs), which identifies active ERK due to its state of phosphorylation, or with a 1/1000 dilution of ERK1 antibody (C-16, polyclonal antibody, Santa Cruz), which identifies activated ERKs as slower migrating forms of ERK1/2. The blots were washed, incubated with goat anti-rabbit antibody conjugated to horseradish peroxidase (HRP; Bio-Rad), and visualized as described above.
Electrophoretic Mobility Shift Assay (EMSA)
Five µg of T cell nuclear extract or 2.5 µg of NIH 3T3 cell nuclear extract was preincubated at room temperature for 10 min in 1.2% NP-40, 127 mM KCl, 8 mM Tris-HCl, pH 8.0, 20 mM HEPES, pH 7.9, 10% glycerol, 0.2 mM EDTA, 2% polyvinyl alcohol, 1 mM DTT, 10 µg/ml bovine serum albumin, and 0.25 µg of poly[d(I-C)] · poly[d(I-C)], in a final volume of 15 µl. Subsequently, 32 fmoles of 32P-labeled LSF-280 binding site [Huang et al., 1990] were added, and the reactions were incubated for an additional 30 min. For supershift assays, samples were preincubated at room temperature for an additional 10 min with the indicated antibodies prior to addition of labeled DNA. Samples were electrophoresed through a 5% polyacrylamide gel for 2 hr at 11 V/cm in buffer containing 44.5 mM Tris base, 44.5 mM boric acid, and 1 mM EDTA and visualized using a Molecular Dynamics Phosphorimager and quantified, where indicated, using ImageQuant software.
RESULTS
LSF Phosphorylation and Enhanced DNA Binding Activity During T Cell Activation from G0 are Proportional to the Extent of ERK Activation
The relationship between LSF activities and ERK activation was first investigated by examining whether there is a quantitative correlation between the level of ERK activity and the extent of LSF phosphorylation in resting human peripheral T cells stimulated with various combinations of mitogens. Nuclear extracts were prepared to analyze the LSF phosphorylation state by Western blotting. Consistent with our previous study [Volker et al., 1997], PMA, PHA, and ionomycin treatment resulted in quantitative phosphorylation of LSF within 5 min of induction, as indicated by a complete shift in electrophoretic mobility of the protein to a slower migrating form, which is sustained during the 60 min time course (Fig. 1A, compare lane 1 with lanes 2–4). Of note, when normalized for levels of loaded proteins (bottom panel, Fig. 1B), the total amount of LSF was essentially constant over this time period, consistent with our previous observations [Volker et al., 1997]. Similarly, treatment of T cells with the tumor-promoter PMA alone caused rapid, complete phosphorylation of LSF (Fig. 1A, compare lane 1 with lanes 5–6). However, treatment with the lectin PHA alone resulted in a weaker and delayed phosphorylation response, peaking at sixty min and affecting a lesser, final extent of LSF phosphorylation, as indicated by the persistence of the lower, hypophosphorylated LSF species at 5 min and even later, resulting in a doublet of closely migrating bands (Fig. 1A, lanes 7–9).
Fig. 1.
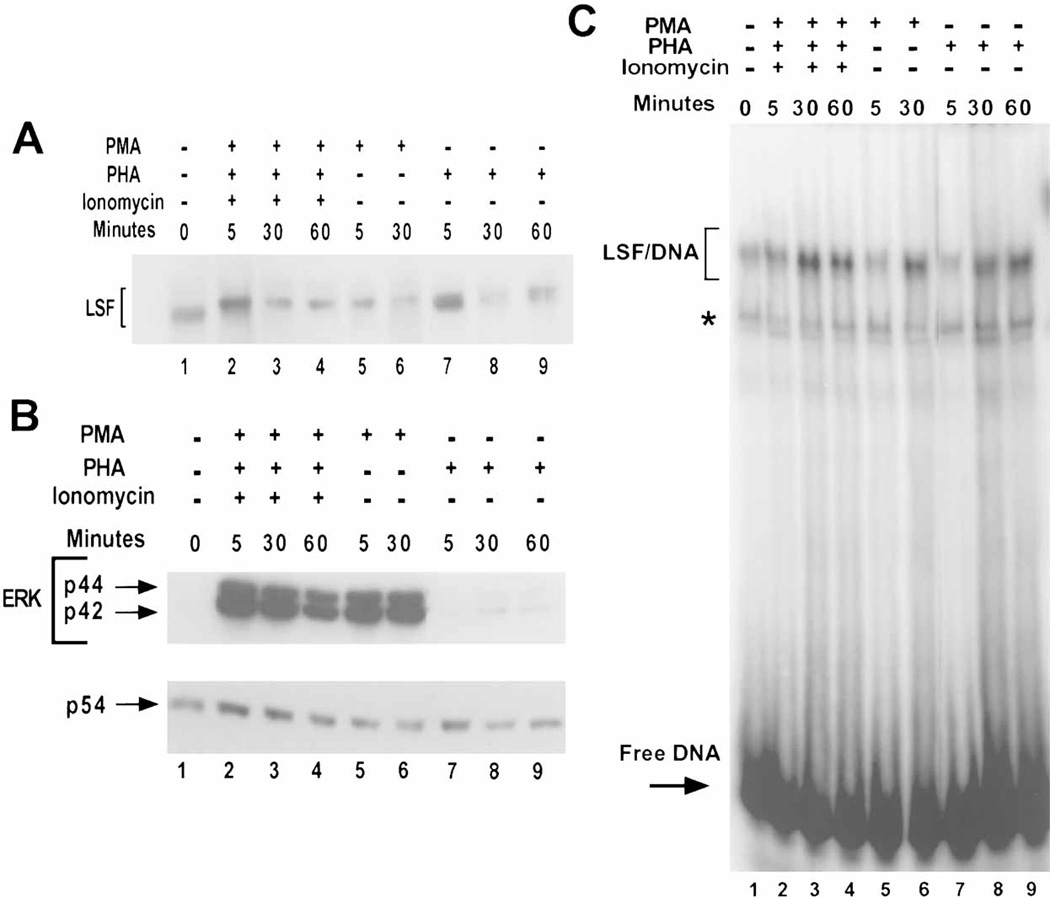
Mitogenic stimulation of human peripheral T cells leads to LSF phosphorylation, increased LSF-DNA binding activity, and ERK activation. A: Western blot analysis of nuclear extracts from either unstimulated or stimulated human peripheral T cells. Approximately 40 µg of extract were loaded per lane. The blot was probed with α-LSF pep1-1 antibody. Lane 1: unstimulated cells, lanes 2–4: cells treated with PHA, ionomycin, and 74 ng/ml of PMA, lanes 5–6: cells treated with 74 ng/ml of PMA, and lanes 7–9: cells treated with PHA. The lengths of time of treatment in min are indicated. Note the slight curvature across the lanes in the relative mobilities of the proteins. B: Kinetics of ERK activation by mitogenic stimuli in T cells. Twenty micrograms of the same samples used in panel A were loaded per lane. The blot was probed with antiphospho-ERK antibody (upper panel) and reprobed with JNK antibody (lower panel) to detect differences in the loading of total protein. Lanes 1–9 are as in panel A. The positions of phosphorylated p44 ERK1, phosphorylated p42 ERK2, and p54 JNK are indicated. Note the slight curvature across the lanes in the relative mobilities of the proteins. C: LSF DNA-binding activity is enhanced in response to mitogenic stimulation in T cells. EMSA of nuclear extracts prepared in a separate experiment from that in panels A and B, either from quiescent cells or from cells stimulated with various mitogens. The positions of migration of LSF/DNA complexes and free DNA are indicated. The asterisk denotes a complex with non-specific DNA-binding proteins. In this experiment, increases in LSF DNA-binding activity were 2.3-fold (lane 3), 1.9-fold (lane 6), and 1.4-fold (lane 9), as compared with lane 1.
To determine the consequence of mitogenic stimulation on the activation of p44 ERK2 and p42 ERK1 from these same extracts, the status of these enzymes was assayed by Western blotting, using antibodies specific for the phosphorylated, active forms of the kinases. ERK1 and ERK2 are activated only upon phosphorylation at specific sites by MEK, therefore the degree of phosphorylation of each protein reflects the kinase activity. Activation of the ERKs was most pronounced in T cells stimulated with a mitogenic cocktail (Fig. 1B, compare lane 1 with lanes 2–4), and by far the weakest in T cells stimulated with PHA alone, although activation above the levels observed in resting cells was still evident (Fig. 1B, compare lane 1 with lanes 7–9). The peak in ERK activity generally occurred very rapidly, within five min after mitogenic stimulation, except in PHA-stimulated T cells. In these cells, the peak of ERK activity occurred 30 min after growth stimulation. The weak and delayed activation of ERK in the PHA-induced lysates (Fig. 1B, compare lanes 2–6 to 7–9) corresponded to the delayed and incomplete phosphorylation of LSF induced by PHA alone (Fig. 1A). In conclusion, the levels and temporal expression of activated ERK from these extracts correlate well with the kinetics of appearance and levels of the phosphorylated form of LSF.
In order to assess whether ERK activity in the T cells similarly correlated with increased LSF DNA-binding activity, EMSAs were performed from such extracts. An increase in LSF DNA-binding activity was observed after stimulation of T cells with a combination of PMA, PHA and ionomycin (Fig. 1C, lanes 1–4), consistent with previous results [Volker et al., 1997]. However, PHA treatment alone resulted in a somewhat lesser effect (Fig. 1C, compare lanes 1 and 7–9). In a series of 3–5 sets of such data, stimulation either with PMA alone or with a combination of PMA, PHA, and ionomycin elevated the DNA-binding activity of LSF to similar levels (averaging 3- to 5-fold). However, stimulation with PHA alone consistently resulted in less activation of LSF DNA-binding activity (1.5- to 2-fold). The smaller induction of LSF DNA-binding activity with PHA correlated with the lower extent and slower kinetics of LSF phosphorylation in PHA-induced cells (Fig. 1A).
These results indicated that the signaling pathways induced by either PMA or PHA alone are sufficient not only to cause phosphorylation of LSF, but also to cause an increase in its ability to bind to DNA. However, we note that the kinetics of the increase in LSF DNA-binding were slower than the kinetics of LSF phosphorylation (Fig. 1 and [Volker et al., 1997]). This partial discordance suggests that direct phosphorylation by ERK may not be sufficient for an increase in DNA-binding, and that secondary events impinging directly upon LSF may also be required.
LSF Phosphorylation in NIH 3T3 Cells is not Sufficient for the Increase in DNA-Binding Activity
Activation of the Ras/Raf/MEK/ERK cascade has been characterized in many cell types and species. To explore the general significance of the observed correlation between ERK activation, LSF phosphorylation, and elevated LSF DNA-binding activity, murine NIH 3T3 fibroblasts were used as a model system. NIH 3T3 cells were growth arrested by serum deprivation and then stimulated with either calf serum or EGF to reenter the cell cycle (see Methods). Upon serum deprivation of fibroblasts, LSF is only slowly dephosphorylated, over the course of many days (data not shown). Thus, after 36 h of serum deprivation, as in this experiment, multiple species of LSF are still observed in the serum-deprived fibroblast cell line, both phosphorylated and hypophosphorylated (Fig. 2A, lane 1). This is distinct from the situation in human peripheral T cells, which are in a stable resting state and contain only hypophosphorylated LSF (Fig. 1A). Five min after stimulation with either serum or EGF, Western blot analyses revealed the same rapid phosphorylation of LSF as was observed in T cells, as indicated by the entire population of LSF shifting to the slowest migrating species (Fig. 2A, compare lane 1 with lanes 2 and 5). Furthermore, the serum- or EGF-induced LSF phosphorylation in NIH 3T3 cells was quantitative, as was seen in human peripheral T cells stimulated with PMA, PHA and ionomycin (compare Figs. 1A and 2A). The kinetics and degree of activation of ERK in these cells was determined by an in gel kinase assay. As was the case in T cells, the kinetics of ERK activation were rapid (Fig. 2B, compare lane 1 with lanes 2–4 and 5–7), correlating with the rapid kinetics of LSF phosphorylation following either serum or EGF stimulation of NIH 3T3 cells.
Fig. 2.

LSF is quantitatively phosphorylated without altering DNA-binding activity in NIH 3T3 cells. A: Western blot analysis of LSF in total cell lysates from serum-deprived or mitogenically stimulated NIH 353 cells. Cell lysate from 1 × 106 cells was loaded per lane. The membrane was probed with α-LSFpep1-1 antibody. Lane 1: serum-deprived cells, lanes 2–4: cells treated with EGF, lanes 5–7: cells treated with 10% serum, and lane 8: 250 ng His-LSF as a marker. The lengths of time of treatment are indicated. Note the slight curvature across the lanes in the relative mobilities of the proteins. B: ERK is rapidly activated by mitogenic stimuli in NIH 3T3 cells. An in gel kinase assay was performed on the same samples described in panel A. The mobilities of active p44 and p42 ERKs are indicated. C: Nuclear extracts prepared from serum-deprived cells or from cells stimulated for 5, 30, and 60 min with mitogens were subjected to EMSA. The positions of migration of LSF/DNA complexes, free DNA, and antibody/LSF/DNA complexes (“supershifted LSF”) are indicated. Lane 1: 50 ng His-LSF, lane 2: no added protein, lane 3: untreated cells; lanes 4–6 and 10–12: cells treated with EGF; lanes 7–9: cells treated with 10% serum. Lane 11: incubation with preimmune rabbit serum; lane 12: incubation with polyclonal antibody α-LSFpep1–2 [Volker et al., 1997].
Given the response of LSF to mitogenic stimulation in T cells, we anticipated that LSF DNA-binding activity would similarly increase in growth-stimulated NIH 3T3 cells. To test this hypothesis, LSF activity was measured by EMSA from nuclear extracts of NIH 3T3 cells stimulated to grow with either serum or EGF. Unexpectedly, no increase in LSF DNA-binding activity was observed in these cells (Fig. 2C, compare lane 3 to lanes 4–9). The identity of the LSF-DNA complex in the NIH 3T3 cell extracts was verified by supershifting the complexes with an LSF specific polyclonal antibody (Fig. 2C, lanes 10–12). Thus, in contrast to T lymphocytes, phosphorylation of LSF in NIH 3T3 cells does not lead to enhanced affinity for DNA.
Phosphorylation of LSF by ERK in vitro does not Enhance its Affinity for DNA
The contrasting responses of LSF DNA-binding activity to mitogenic stimuli in NIH 3T3 versus T cells could be due to many factors, including the presence of secondary, inhibitory modifications on LSF in NIH 3T3 cells. Therefore, we examined directly whether phosphorylation by ERK in vitro would affect the DNA-binding activity of LSF. For this experiment, it was essential to obtain quantitative phosphorylation of LSF. Thus, the degree of modification of LSF was assayed by Western blotting, following incubation of recombinant LSF with a highly active preparation of GST-ERK2, in the presence of either ATP or AMP-PNP, a nonhydrolyzable analog of ATP. ERK phosphorylation of LSF in vitro caused a quantitative decrease in its electrophoretic mobility (Fig. 3A), mimicking the mitogenically-induced modification of LSF in cells. We conclude, therefore, that phosphorylation of LSF by ERK is sufficient to decrease its electrophoretic mobility. In addition, no additional modification(s) on LSF are required for this shift.
Fig. 3.
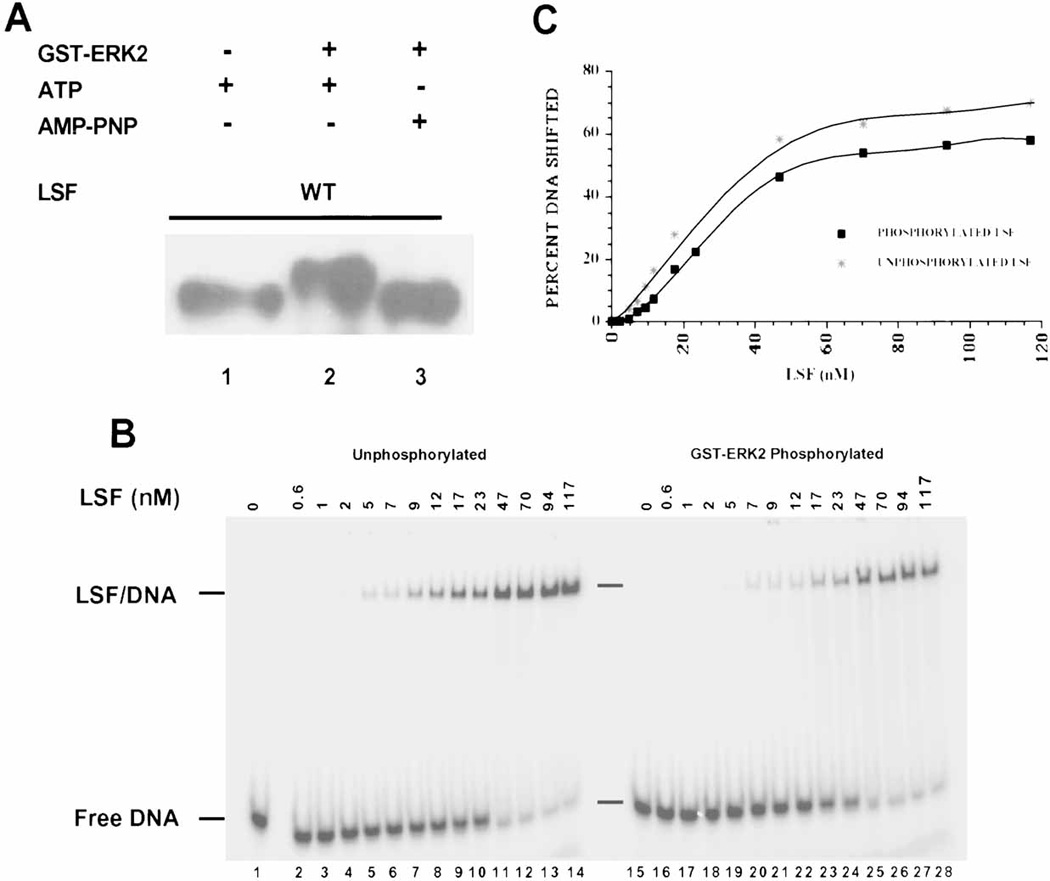
In vitro phosphorylation of LSF by ERK2 does not alter its DNA-binding activity. A: Western blot analysis of LSF phosphorylated by GST-ERK2 in vitro. Lane 1: 1 µg of His-LSF incubated only with ATP, lane 2: 1 µg of His-LSF incubated with activated GST-ERK2 and ATP, lane 3: 1 µg of His-LSF incubated with activated GST-ERK2 and AMP-PNP. The separated products of the kinase reactions were probed with α-LSFpep1-1 antibody. B: Increasing amounts of either unphosphorylated (lanes 1–14) or GST-ERK2 phosphorylated (lanes 15–28) His-LSF were analyzed by EMSA. Positions of free DNA and LSF/DNA complexes and concentrations of LSF monomer in nM are indicated. C: The averaged data of two experiments, including that shown in panel B, are plotted as a function of concentration of LSF monomer in the reaction. DNA binding is expressed as the percentage of DNA that formed a complex with LSF in the reaction. (Note: LSF binds DNA as a homotetramer.)
To test whether such phosphorylation was sufficient to alter LSF DNA-binding activity, we directly compared the affinities of unphosphorylated and phosphorylated LSF for a high affinity DNA-binding site in the SV40 late promoter, the LSF-280 site. The apparent Keq can be determined by titrating a constant amount of DNA with increasing amounts of protein and separating free and bound DNA by EMSA. The concentration of protein resulting in 50% of maximal binding is roughly equal to the Keq, if the concentration of total DNA is much less than the Keq [Ausubel et al., 1997]. LSF/DNA complexes containing either phosphorylated or unphosphorylated LSF did not display any significant differences in their binding for the LSF-280 site (Fig. 3B). In fact, the apparent overall Keq of LSF for the LSF-280 site (approximately 6 nM of LSF tetramer) was not detectably altered with the phosphorylation status of the protein (Fig. 3C). The rates of dissociation of LSF/DNA complexes were also unchanged upon ERK phosphorylation [Volker 1998]. Taken together, these results demonstrate that although phosphorylation of LSF by ERK in vitro is sufficient to alter its electrophoretic mobility, it does not directly alter its DNA-binding affinity. These in vitro data are consistent with the EMSA results obtained from growth-stimulated NIH 3T3 fibroblasts; although LSF is rapidly phosphorylated in these cells, there is no appreciable change in DNA-binding activity.
The MEK 1/2 Inhibitor PD98059 Prevents Complete Phosphorylation of LSF at the G0/G1 Transition both in NIH 3T3 Fibroblasts and T Lymphocytes
Our data have established a strong correlation in both T cells and NIH 3T3 cells between ERK activation and LSF phosphorylation, as monitored by the reduction in the electrophoretic mobility of LSF. Our previous results that general dephosphorylation of purified, mammalian LSF reduced the affinity of LSF for the LSF-280 DNA binding site had led us to hypothesize that phosphorylation by ERK activated LSF DNA-binding activity in T cells [Volker et al., 1997]. However, the differential effects in the two cell types in LSF DNA-binding activity, and the inability of ERK to activate LSF DNA-binding activity directly in vitro raised the question as to whether different signaling pathways might target LSF in the two cell types. In particular, the involvement of ERK signaling in stimulating LSF activity in T cells was brought into question. To resolve these issues, we directly assessed whether ERK actually targeted LSF in both NIH 3T3 cells and T lymphocytes, by use of the MEK 1/2 inhibitor PD98059. PD98059 binds to the inactive form of MEK1 and MEK2, the upstream activators of ERKs, and specifically prevents activation of ERKs [Alessi et al., 1995; Dudley et al., 1995].
Extracts from EGF-treated NIH 3T3 cells were analyzed by Western blotting, with antibody against LSF (Fig. 4A). Unlike T cells that are isolated in the resting state, fibroblast cell lines must be deprived of growth factors in order to enter the resting state. During this starvation period, LSF becomes progressively dephosphorylated, although at a slow rate. Detailed studies in mouse A31 fibroblasts revealed that the slower migrating form of LSF was still significant at 72 hours of starvation, and continued to decrease to minimal levels only after 120 hours of starvation (data not shown). Similarly, in the NIH 3T3 cells, the phosphorylated species of LSF remained evident after the 36 hour starvation period (Fig. 4A, lane 1). As expected (Fig. 2), mitogenic treatment of NIH 3T3 cells with EGF led to rapid phosphorylation of the total LSF population (Fig. 4A, lane 2). The extracts were also analyzed by Western blotting using antibody against all forms of ERK (Fig. 4B), where the activated forms of ERK1 and ERK2 are distinguished by their slower electrophoretic mobility, as compared with the inactive forms of the kinases. Again as expected, mitogenic treatment of NIH 3T3 cells with EGF led to rapid activation of ERK (Fig. 4B, lane 2). Treatment of cells with 25 µM or 50 µM PD98059 prior to stimulation with EGF abolished ERK activation (Fig. 4B, lanes 3–4). The inhibitor also prevented the EGF-induced phosphorylation of LSF, in that the pattern of LSF species remained similar to that in the quiescent cells (Fig. 4A, compare lane 1 with lanes 3–4). Due to the specificity of the MEK 1/2 inhibitor, we can conclude that LSF is either a direct substrate of ERK or a substrate of a downstream kinase in the ERK signaling pathway in NIH 3T3 cells.
Fig. 4.
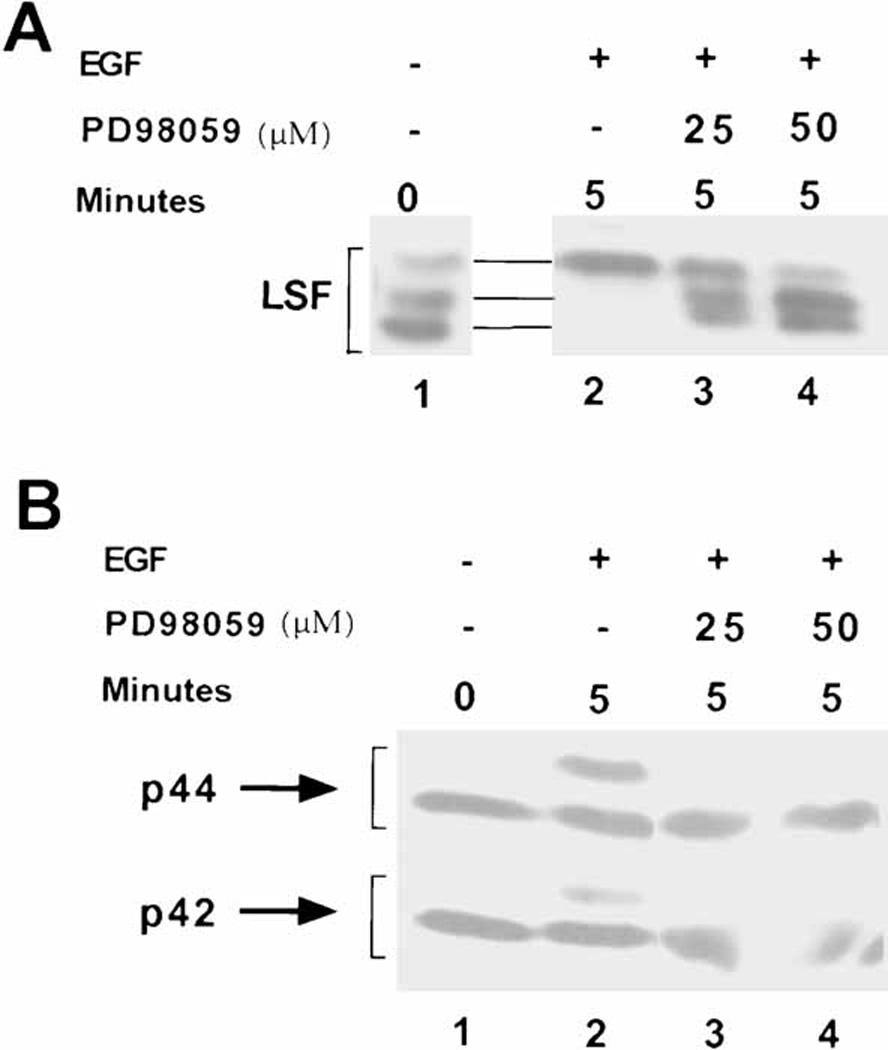
The ERK pathway targets LSF for phosphorylation after mitogenic stimulation of NIH 3T3 cells. A: Western blot analysis of LSF, as described in Fig. 2. Lane 1: extract from serum-deprived NIH 3T3 cells; lanes 2–4: extracts from NIH 3T3 cells stimulated with EGF for 5 min. Lanes 3,4: cells were pre-treated with the MEK1/2 inhibitor PD98059, at the indicated concentrations. Note the slight curvature across the lanes in the relative mobilities of the proteins. B: Western blot analysis of the samples in panel A with polyclonal antibody against ERK1. This antibody recognizes both inactive and active (slower migrating) forms of both ERK1 and ERK2.
In similar experiments with human peripheral T cells, pretreatment of resting cells with MEK inhibitor only delayed, but did not completely abolish LSF phosphorylation after stimulation with PMA (Fig. 5A, compare lanes 1 and 2 with 5 and 6, 9 and 10). [Although curvature in the mobility of samples across the gel upon electrophoresis caused the absolute mobility of LSF in resting cells (lane 1) to appear similar to that in stimulated cells (lane 2), closer examination reveals that the bands in lanes 2–4 comigrate, whereas that in lane 1 migrates faster, consistent with previous experiments.] Similarly, analyses of phosphorylated ERK levels from the same extracts demonstrated that treatment with 50 µM or even 100 µM PD98059 only reduced, but did not abolish the activation of ERK (Fig. 5B, compare lanes 2–4 with 6–8 and 10–12). These correlative data are consistent with ERK activity resulting in LSF phosphorylation.
Fig. 5.
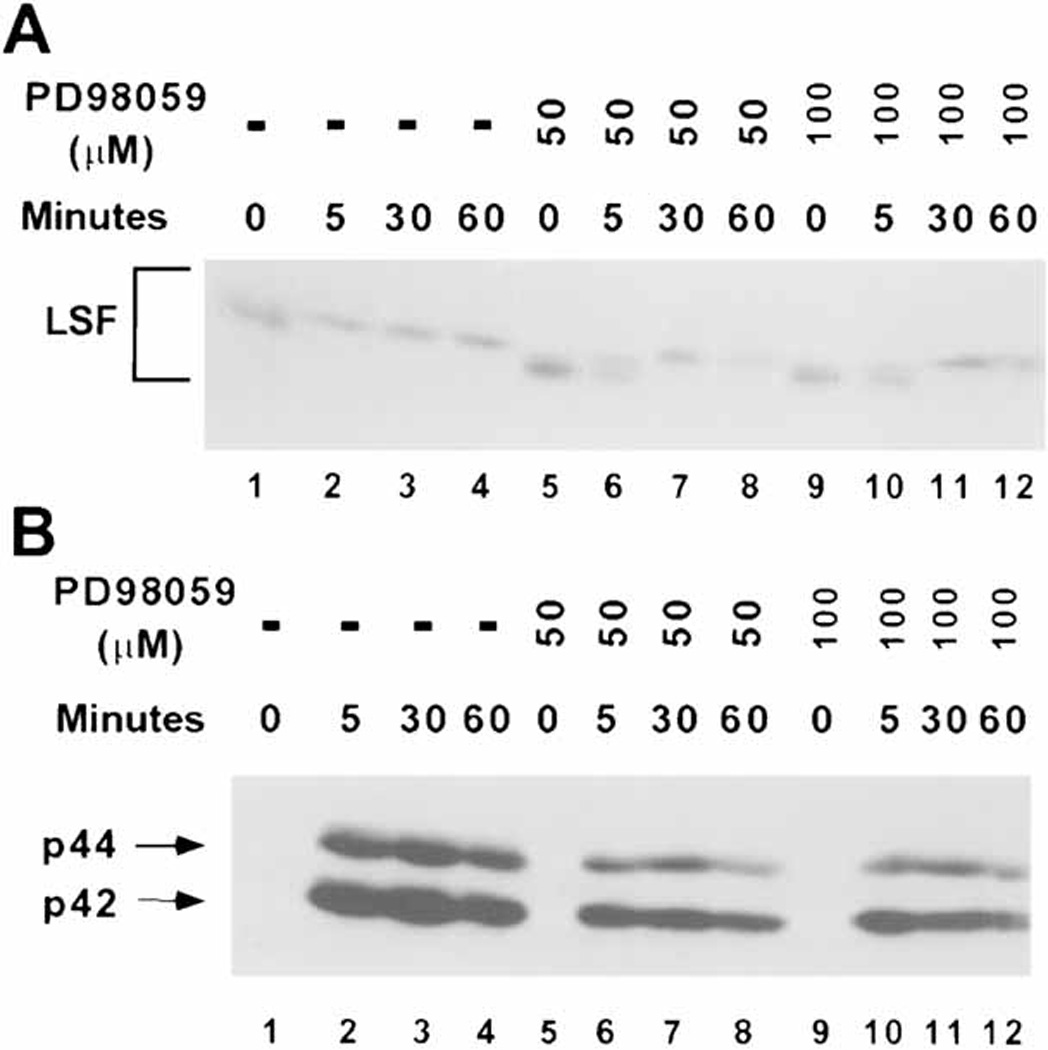
The MEK 1/2 inhibitor PD98059 diminishes the rapid, quantitative phosphorylation of LSF in PMA-induced human peripheral T cells. A: T cells stimulated with 10 ng/ml PMA for the indicated amounts of time were either mock treated (lanes 1–4), or preincubated with 50 µM (lanes 5–8) or 100 µM (lanes 9–12) MEK 1/2 inhibitor. The blot of separated nuclear extracts was probed with α-LSFpep1-1 antibody. The position of LSF is bracketed. Note the curvature across the lanes in the relative mobilities of the proteins. B: The blot from panel A was reprobed with anti-phospho ERK antibody. The positions of phosphorylated p44 and p42 are indicated.
To obtain a more definitive result, we examined the effects of the MEK inhibitor upon stimulation of T cells with the weaker mitogen PHA, which only barely activates ERK, in stark contrast to the robust activation by PMA (Fig. 1B). In contrast to PMA induction, incubation of T cells with PD98059 in the presence of PHA was able to inhibit fully the limited activation of ERK (Fig. 6B, compare lanes 1–4 with 5–8). [Note that, due to the extremely low levels of phosphorylated ERK, the major band apparent in the blot, labeled with an asterisk, is a non-specific cross-reacting protein.] The partial PHA-induced phosphorylation of LSF was also eliminated by the presence of the inhibitor (Fig. 6A, compare lanes 1–4 with 5–8). Therefore, we can conclude that in T cells, as in NIH 3T3 cells, LSF phosphorylation that results directly from mitogenic stimulation is dependent on ERK signaling.
Fig. 6.

The MEK 1/2 inhibitor PD98059 prevents both phosphorylation of LSF and activation of ERK activity in PHA-stimulated human peripheral T cells. A: T cells stimulated for the indicated amounts of time with PHA were either mock treated (lanes 1–4) or preincubated with 50 µM PD98059 (lanes 5–8). The blot of separated nuclear extracts (40 µg per lane) was probed with α-LSFpep1–2 antibody. The position of LSF is bracketed. Note the slight curvature across the lanes in the relative mobilities of the proteins. B: The blot from panel A was reprobed with anti-phospho ERK antibody. The positions of phosphorylated p44 and p42 are indicated. The asterisk denotes a non-specific, cross-reacting band.
The MEK Inhibitor PD98059 Prevents Activation of LSF DNA-Binding Activity upon Mitogen Induction of T Cells
To address whether the in vivo phosphorylation of LSF by ERK in T cells is necessary for the enhancement of LSF DNA-binding activity, extracts of T cells stimulated with PHA in the presence or absence of MEK1/2 inhibitor were analyzed by EMSA. Consistent with our previous results (Fig. 1C), LSF DNA binding activity increased 2-fold within 30 to 60 min after stimulation with PHA (Fig. 7A, lanes 1–4). Levels of PD98059 sufficient to completely block the PHA-induced phosphorylation of LSF also blocked this increase in LSF DNA-binding activity (Fig. 7A, compare lane 4 with lanes 8 and 12), indicating that activation of ERK is necessary for stimulating LSF DNA-binding in T cells in vivo. The maximal increase in LSF-DNA binding activity varied between six different batches of T cells (from different human donors), ranging from fifteen to sixty min after mitogenic stimulation with PHA (compare Figs. 7A and 7B). Regardless of the time of the maximum response, however, the MEK inhibitor reduced LSF-DNA binding activity by 80% ± 30%. Variability in the degree of inhibition was related to the level of the increase in DNA binding for the given batch of primary T cells.
Fig. 7.
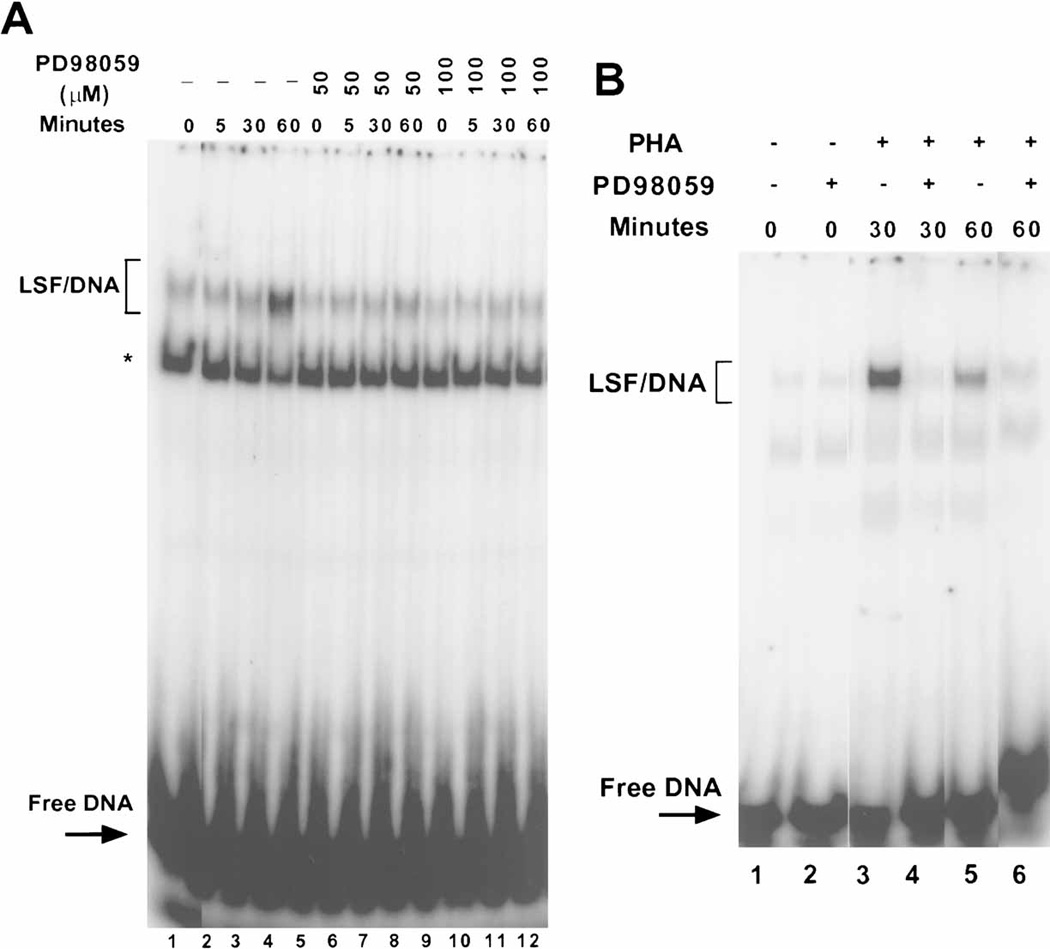
MEK 1/2 inhibitor prevents activation of LSF DNA-binding activity in human peripheral T cells. A: Nuclear extracts from T cells stimulated with PHA, which were either mock treated (lanes 1–4), or preincubated with 50 or 100 µM PD98059 (lanes 5–8 or 9–12, respectively), were subjected to EMSA. The positions of migration of LSF/DNA complexes and of free DNA are indicated. The asterisk denotes a non-specific DNA-binding protein. B: A different donor set of T cells from that in panel A were either preincubated with 100 µM MEK 1/2 inhibitor (lanes 2,4,6) or mock treated (lanes 1,3,5), followed by stimulation with PHA for the indicated periods of time. Extracts were analyzed as in panel A.
DISCUSSION
Phosphorylation of LSF by ERK
The following lines of evidence support a physiological role for ERK in phosphorylation of LSF in diverse cell types. First, LSF is phosphorylated by ERK in vitro on the same residues that are phosphorylated in vivo, with serine 291 being the major site of phosphorylation [Volker et al., 1997]. Second, the shift in electrophoretic mobility of LSF upon growth induction in vivo is mimicked by phosphorylation of LSF by ERK in vitro (Fig. 3). Moreover, phosphorylation at serine 291 in vivo is necessary for this altered mobility of LSF [Volker et al., 1997]. Third, stimulation of human peripheral T lymphocytes or NIH 3T3 fibroblasts by a variety of mitogens that stimulate the ERK pathway results in phosphorylation of LSF with kinetics similar to the kinetics of ERK activation (Figs. 1 and 2). This effect is not restricted to these two cell types, having been observed in a number of fibroblast cell lines, including mouse (NIH3T6; A31), rat (Rat-1; Rat-2), and monkey (COS), after stimulation with epidermal growth factor, platelet derived growth factor, insulin, or high levels of serum (Z. Pagon and U. Hansen, unpublished observations). Finally, EGF-induced phosphorylation of LSF in NIH 3T3 cells was abolished by the specific MEK 1/2 inhibitor PD98059. Consistent with these data, EGF induction of LSF phosphorylation in NIH 3T3 cells is blocked by a dominant-negative form of Ras, which also prevents activation of the Raf/MEK/ERK signaling pathway (Z. Pagon, U. Hansen, and G. Cooper; unpublished observation). The MEK 1/2 inhibitor also reduced phosphorylation of LSF in PMA-stimulated and PHA-stimulated human peripheral T cells, to the extent that it effectively inhibited ERK activation under these conditions. PD98059 is a highly specific inhibitor for MEK 1/2, not impacting the activities of Raf kinase, phosphatidylinositol 3-kinase, p38 MAP kinase, JNK, MKK4, pp70 S6 kinase, or many other kinases [Alessi et al., 1995; Dudley et al., 1995]. The high degree of selectivity for ERK inhibition achieved by treatment of cells with PD98059 provides strong evidence for a direct role of ERK activation in LSF phosphorylation. Taken together, these data demonstrate that LSF is a direct downstream target of ERK signaling upon growth induction of resting cells.
Enhancement of LSF DNA-Binding Activity in T Cells: T Cell-Specific Protein-Protein Interactions, or Additional Signaling Pathways Targeting LSF
ERK activity is also necessary for enhanced LSF DNA-binding activity in T lymphocytes, as evidenced by the ability of the MEK 1/2 inhibitor to decrease the elevation in LSF DNA-binding activity from resting cells stimulated with PHA. However, the increase in LSF DNA-binding activity cannot be the sole consequence of ERK phosphorylation, for the following reasons. First, the kinetics of ERK activation are not coincident with the kinetics of increased LSF DNA-binding activity, there being a delay between ERK activation and the increase in binding activity. Second, although ERK is strongly activated in EGF- or serum-stimulated NIH 3T3 cells, there is minimal if any change in the levels of LSF-DNA binding activity upon growth induction in these cells. Finally, and most conclusively, phosphorylation of LSF by ERK in vitro does not increase the affinity of LSF for DNA. The most likely hypothesis to explain these data is that a second factor/signal is required, in addition to activation of ERK, for the growth-induced DNA-binding increase. Given no shift in the mobility of the protein/DNA complex using a high affinity LSF DNA-binding site, we infer that the second signal directly impacts either LSF or a factor that inhibits LSF DNA-binding activity [Bruni et al., 2002]. This signaling component must be T cell-specific in response to cellular growth stimuli.
This phenomenon is distinct from that described in Jurkat cells with the binding site for LSF in the IL-4 promoter. In T cell lines, such as Jurkat cells, the G0 state is no longer achievable, IL-4 is expressed constitutively at baseline levels [Casolaro et al., 2000], and LSF is also constitutively phosphorylated on S291 (J. Volker and U. Hansen, unpublished observations). Furthermore, the general LSF DNA-binding activity to a consensus LSF DNA-binding site remains unchanged even upon stimulation of the Jurkat cells [Casolaro et al., 2000], which is consistent with DNA-binding activity inherent in LSF already being maximized. Once this T cell line is stimulated, however, the protein/DNA complex on the IL-4 site migrates more slowly than that of the LSF homotetramer, due apparently to the modification of an LSF/CP2 partner protein [Casolaro et al., 2000]. This additional modification appears to boost activation of expression from the IL-4 promoter above the constitutive level [Casolaro et al., 2000].
What other potential signaling cascades might target LSF in primary T cells? All of the mitogenic signals that affect LSF phosphorylation and DNA binding activity in T cells activate not only ERK, but also JNK and p38 MAP kinases, pp70 S6 kinase, RSK family members, and a large number of kinases through calcium mobilization [Hardy and Chaudhri 1997]. The JNK and p38 pathways were initially attractive candidates for this second signaling pathway, as both JNK and p38 phosphorylate LSF in vitro on similar and additional sites as ERK [Volker 1998]. Additionally, unlike the kinetics of ERK activation, the kinetics of JNK and p38 induction in PMA or PHA stimulated T cells are coincident with the increase in LSF DNA-binding activity [Volker 1998]. However, treatment of T cells with 40 µM of the inhibitor SB 202190, which inhibits both JNK and p38 at this concentration [Chen et al., 1998], did not alter the degree of LSF DNA-binding activity in T cells (Z. Pagon and U. Hansen, unpublished observations). Moreover, co-stimulation of NIH 3T3 cells with serum and anisomycin, a potent activator of the JNK and p38 pathways, did not result in increased LSF DNA-binding activity [Volker 1998]. Therefore, these MAP kinase signaling pathways cannot account for the induction of LSF DNA-binding activity in T cells.
There are two consensus phosphorylation sites in LSF capable of being phosphorylated by RSK family members, pp70 S6 kinase, and the CaM kinases [Hill and Treisman 1995; Songyang et al., 1996]. These sites contain target threonine residues located in the C-terminus of the protein. Although we have previously demonstrated that LSF was primarily phosphorylated on serine residues within 30 min after growth stimulation in T cells [Volker et al., 1997], it is conceivable that threonine phosphorylation of LSF could be maximal only somewhat later. The timing of the increase in LSF DNA binding activity, which trails LSF phosphorylation by ERK, varies with different batches of T cells, peaking in a range from 15 min to 60 min after mitogenic stimulation. Therefore, delayed phosphorylation at sites other than ERK target sites may promote binding of LSF to DNA.
Biological Relevance of LSF Phosphorylation by ERK and Other Potential Kinases
LSF/CP2/LBP-1 is ubiquitous, and the phosphorylation of LSF by ERK may generally either directly influence its transcriptional potential, or may prime the protein for recognition by subsequent signaling events during the cell cycle. In this regard, we have demonstrated that LSF is critical for progression of cells through the cell cycle, with a specific requirement for the transcription factor for progression through S phase [Powell et al., 2000]. However, given the specific enhancement of LSF DNA-binding activity upon mitogenic stimulation of T cells, we anticipate that LSF will regulate genes that are subjected to rapid induction in this specific cell type. As indicated above, IL-4 is one likely candidate.
Recently, mice lacking CP2/LSF were generated [Ramamurthy et al., 2001]. Examination of T cell function in these mice revealed no apparent defects. However, Ramamurthy, et al demonstrated that the mouse paralog gene, NF2d9, was expressed in all the same tissues as was CP2/LSF, could interact with similar proteins as could CP2/LSF, and could bind the same DNA sequences. Thus, they hypothesized that the paralog was compensating for the loss of the CP2/LSF gene. In this regard, the NF2d9/LBP-1a proteins in both mouse and human include sequences similar to the phosphorylation sites for ERK (primarily S291) in CP2/LSF. Thus, it is likely that this entire family of proteins can be regulated similarly by the ERK pathway, supporting the notion that NF2d9/LBP-1a would have similar biological roles in T cells to those described here.
LSF can not only form homotetramers to bind DNA, but can also heterooligomerize with other proteins in order to generate novel DNA-binding or regulatory activities [Jane et al., 1995; Romerio et al., 1997; Sueyoshi et al., 1995; Zhou et al., 2000]. Of particular relevance to the studies described above, LSF oligomerizes with the transcription factor YY1 to repress HIV viral replication [Romerio et al., 1997]. Specifically, LSF is required for recruitment of YY1 to the LSF binding site in the HIV LTR, thus mediating the synergistic repression of HIV transcription by the two factors [Coull et al., 2000]. Due to this repressive activity, the LSF/YY1 complex is hypothesized to play a role in latency of HIV. In contrast to what has been observed for LSF homotetramers (Fig. 3), phosphorylation of LSF in vitro diminishes the effective binding of the LSF/YY1 complex to the HIV binding site (D. Margolis, personal communication); phosphorylation may interfere with the stability of the LSF/YY1 complex or its interaction with DNA. This result suggests that ERK phosphorylation of LSF in vivo would relieve transcriptional repression. One can therefore imagine that LSF phosphorylation status may serve as an integrator of a variety of extracellular signals at the HIV LTR.
ACKNOWLEDGMENTS
We thank Huseyin Aktas, Hong Cai, Ulisses Lopes, David Hartley, and members of the Hansen laboratory for helpful suggestions and discussions. We thank James Deshler for comments on the manuscript.
Grant sponsors: NIH, Grant numbers: R01 CA81157 (UH) and R01 CA18689 (GMC); and Sandoz/DFCI Drug Discovery Program (UH).
Zrinka Pagon was supported by the Zlatko and Joyce Balokovic Fellowship, the Professor Mary Ellen Avery Fellowship, and the Infectious Diseases Society of America Connaught Laboratories Fellowship.
REFERENCES
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen- activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Bing Z, Reddy SAG, Ren Y, Qin J, Liao WS-L. Purification and characterization of the serum amyloid A3 enhancer factor. J Biol Chem. 1999;274:24649–24656. doi: 10.1074/jbc.274.35.24649. [DOI] [PubMed] [Google Scholar]
- Bruni P, Minopoli G, Brancaccio T, Napolitano M, Faraonio R, Zambrano N, Hansen U, Russo T. Fe65, a ligand of the Alzheimer's β-amyloid precursor protein, blocks cell cycle progression by downregulating thymidylate synthase expression. J Biol Chem. 2002;277:35481–35488. doi: 10.1074/jbc.M205227200. [DOI] [PubMed] [Google Scholar]
- Casolaro V, Keane-Myers AM, Swendeman SL, Steindler C, Zhong F, Sheffery M, Georas SN, Ono SJ. Identification and characterization of a critical CP2-binding element in the human interleukin-4 promoter. J Biol Chem. 2000;275:36605–36611. doi: 10.1074/jbc.M007086200. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Chen CY, Del Gatto-Konczak F, Wu Z, Karin M. Stabilization of interleukin-2 mRNA by the c-Jun NH2-terminal kinase pathway. Science. 1998;280:1945–1949. doi: 10.1126/science.280.5371.1945. [DOI] [PubMed] [Google Scholar]
- Coull JJ, Romerio F, Sun J-M, Volker JL, Galvin KM, Davie JR, Shi Y, Hansen U, Margolis DM. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol. 2000;74:6790–6799. doi: 10.1128/jvi.74.15.6790-6799.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]
- Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English J, Pearson G, Wilsbacher J, Swantek J, Karandikar M, Xu S, Cobb MH. New insights into the control of MAP kinase pathways. Exp Cell Res. 1999;253:255–270. doi: 10.1006/excr.1999.4687. [DOI] [PubMed] [Google Scholar]
- Hardy K, Chaudhri G. Activation and signal transduction via mitogen-activated protein (MAP) kinases in T lymphocytes. Immunol Cell Biol. 1997;75:528–545. doi: 10.1038/icb.1997.84. [DOI] [PubMed] [Google Scholar]
- Hill CS, Treisman R. Transcriptional regulation by extracellular signals: mechanisms and specificity. Cell. 1995;80:199–211. doi: 10.1016/0092-8674(95)90403-4. [DOI] [PubMed] [Google Scholar]
- Huang HC, Sundseth R, Hansen U. Transcription factor LSF binds two variant bipartite sites within the SV40 late promoter. Genes Dev. 1990;4:287–298. doi: 10.1101/gad.4.2.287. [DOI] [PubMed] [Google Scholar]
- Hunter T. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- Jane SM, Nienhuis AW, Cunningham JM. Hemoglobin switching in man and chicken is mediated by a heteromeric complex between the ubiquitous transcription factor CP2 and a developmentally specific protein. EMBO J. 1995;14:97–105. doi: 10.1002/j.1460-2075.1995.tb06979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Luciw PA, Duchange N. Structural arrangements of transcription control domains within the 5'- untranslated leader regions of the HIV-1 and HIV-2 promoters. Genes Dev. 1988;2:1101–1114. doi: 10.1101/gad.2.9.1101. [DOI] [PubMed] [Google Scholar]
- Kameshita I, Fujisawa H. A sensitive method for detection of calmodulin-dependent protein kinase II activity in sodium dodecyl sulfate-polyacrylamide gel. Anal Biochem. 1989;183:139–143. doi: 10.1016/0003-2697(89)90181-4. [DOI] [PubMed] [Google Scholar]
- Kato H, Horikoshi M, Roeder RG. Repression of HIV-1 transcription by a cellular protein. Science. 1991;251:1476–1479. doi: 10.1126/science.2006421. [DOI] [PubMed] [Google Scholar]
- Lim LC, Fang L, Swendeman SL, Sheffery M. Characterization of the molecularly cloned muring α-globin transcription factor CP2. J Biol Chem. 1993;268:18008–18017. [PubMed] [Google Scholar]
- Lim LC, Swendeman SL, Sheffery M. Molecular cloning of the α-globin transcription factor CP2. Mol Cell Biol. 1992;12:828–835. doi: 10.1128/mcb.12.2.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim MH, Fenrick R, Ballard DW, Hauber J, Bohnlein E, Cullen BR. Functional characterization of a complex protein-DNA-binding domain located within the human immunodeficiency virus type 1 long terminal repeat leader region. J Virol. 1989;63:3213–3219. doi: 10.1128/jvi.63.8.3213-3219.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais R, Marshall CJ. Control of the ERK MAP kinase cascade by Ras and Raf. Cancer Surveys. 1996;27:101–125. [PubMed] [Google Scholar]
- Murata T, Nitta M, Yasuda K. Transcription factor CP2 is essential for lens-specific expression of the chicken αA-crystallin gene. Genes Cells. 1998;3:443–457. doi: 10.1046/j.1365-2443.1998.00204.x. [DOI] [PubMed] [Google Scholar]
- Powell CMH, Rudge TL, Zhu Q, Johnson LF, Hansen U. Inhibition of the mammalian transcription factor LSF induces S phase-dependent apoptosis by downregulating thymidylate synthase expression. EMBO J. 2000;19:4665–4675. doi: 10.1093/emboj/19.17.4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamurthy L, Barbour V, Tuckfield A, Clouston DR, Topham D, Cunningham JM, Jane SM. Targeted disruption of the CP2 gene, a member of the NTF family of transcription factors. J Biol Chem. 2001;276:7836–7842. doi: 10.1074/jbc.M004351200. [DOI] [PubMed] [Google Scholar]
- Romerio F, Gabriel MN, Margolis DM. Repression of human immunodeficiency virus type 1 through the novel cooperation of human factors YY1 and LSF. J Virol. 1997;71:9375–9382. doi: 10.1128/jvi.71.12.9375-9382.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer HJ, Weber MJ. Mitogen-activated protein kinases: Specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirra MK, Hansen U. LSF and NTF-1 share a conserved DNA recognition motif yet require different oligomerization states to form a stable protein-DNA complex. J Biol Chem. 1998;273:19260–19268. doi: 10.1074/jbc.273.30.19260. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Lu KP, Kwon YT, Tsai LH, Filhol O, Cochet C, Brickey DA, Soderling TR, Bartleson C, Graves DJ, DeMaggio AJ, Hoekstra MF, Blenis J, Hunter T, Cantley LC. A structural basis for substrate specificities of protein Ser/Thr kinases: primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol Cell Biol. 1996;16:6486–6493. doi: 10.1128/mcb.16.11.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su B, Karin M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr Opin Immunol. 1996;8:402–411. doi: 10.1016/s0952-7915(96)80131-2. [DOI] [PubMed] [Google Scholar]
- Sueyoshi T, Kobayashi R, Nishio K, Aida K, Moore R, Wada T, Handa H, Negishi M. A nuclear factor (NF2d9) that binds to the male-specific P450 (Cyp 2d-9) gene in mouse liver. Mol Cell Biol. 1995;15:4158–4166. doi: 10.1128/mcb.15.8.4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swendeman SL, Speilholz C, Jenkins NA, Gilbert DJ, Copeland NG, Sheffery M. Characterization of the genomic structure, chromosomal location, promoter, and developmental expression of the α-globin transcription factor CP2. J Biol Chem. 1994;269:11663–11671. [PubMed] [Google Scholar]
- Vojtek AB, Cooper JA. Rho family members: activators of MAP kinase cascades. Cell. 1995;82:527–529. doi: 10.1016/0092-8674(95)90023-3. [DOI] [PubMed] [Google Scholar]
- Volker JL. Regulation of the mammalian transcription factor LSF at the G0/G1 boundary. Boston, MA: Harvard University; 1998. [Google Scholar]
- Volker JL, Rameh LE, Zhu Q, DeCaprio J, Hansen U. Mitogenic stimulation of resting T cells causes rapid phosphorylation of the transcription factor LSF and increased DNA-binding activity. Genes Dev. 1997;11:1435–1446. doi: 10.1101/gad.11.11.1435. [DOI] [PubMed] [Google Scholar]
- Weston CR, Lambright DG, Davis RJ. MAP kinase signaling specificity. Science. 2002;296:2345–2347. doi: 10.1126/science.1073344. [DOI] [PubMed] [Google Scholar]
- Wu FK, Garcia JA, Harrich D, Gaynor RB. Purification of the human immunodeficiency virus type 1 enhancer and TAR binding proteins EBP-1 and UBP-1. EMBO J. 1988;7:2117–2130. doi: 10.1002/j.1460-2075.1988.tb03051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JB, Li G, Roeder RG. Characterization of a family of related cellular transcription factors which can modulate human immunodeficiency virus type 1 transcription in vitro. Mol Cell Biol. 1994;14:1776–1785. doi: 10.1128/mcb.14.3.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JB, Zhou Y-H, Maity T, Liao WS-L, Saunders GF. Activation of the human PAX6 gene through the exon 1 enhancer by transcription factors SEF and Sp1. Nucl Acids Res. 2001;29:4070–4078. doi: 10.1093/nar/29.19.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Clouston DR, Wang X, Cerruti L, Cunningham JM, Jane SM. Induction of fetal globin gene expression by a novel erythroid factor, NF-E4. Mol Cell Biol. 2000;20:7662–7672. doi: 10.1128/mcb.20.20.7662-7672.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]