

Abstract
Neutrophil migration to sites of infection is the first line of cellular defense. A key event of migration is the maintenance of a polarized morphology, which is characterized by a single leading edge of filamentous actin and a contractile uropod devoid of filamentous actin protrusions. Using a mouse model of high Cdc42 activity, we previously demonstrated the importance of Cdc42 activity in neutrophil migration. However, the specific functions of Cdc42 in this process remain to be understood. Using neutrophils genetically deficient in Cdc42, we show that Cdc42 regulates directed migration by maintaining neutrophil polarity. Although it is known to be activated at the front, Cdc42 suppresses protrusions at the uropod. Interestingly, Cdc42 makes use of the integrin CD11b during this process. Cdc42 determines the redistribution of CD11b at the uropod. In turn, using CD11b-null cells and CD11b crosslinking experiments, we show that CD11b modulates myosin light chain phosphorylation to suppress lateral protrusions. Our results uncover a new mechanism in which Cdc42 regulates the uropod through CD11b signaling to maintain polarity in migrating neutrophils. It also reveals new functions for CD11b in neutrophil polarity.
Introduction
Neutrophils are the first line of cellular defense by moving toward the site of infection. The neutrophil recruitment cascade into tissues is a multistep process that involves elements of tethering and rolling along the endothelium. Subsequent stimulation of neutrophils by inflammatory chemokines induces integrin activation that results in firm adhesion to the endothelium. Neutrophils then migrate along the endothelium before emigration out of the blood vessels.1–3 The recruitment cascade is thus tightly regulated by external cues of adhesion and chemoattractant molecules. Failure to regulate any of these events may lead to abnormal innate immune responses, including immunodeficiency or aberrant inflammatory reactions. Therefore, understanding the molecular mechanisms that control neutrophil migration is of significant therapeutic importance.
Cell migration or chemotaxis involves key processes that enable the cells to interpret small changes in chemoattractant concentration and move persistently toward higher concentration of stimuli. The migration process can be viewed as a cycle of 3 interrelated events: protrusions, polarity, and directional sensing.4,5 The initial event is the accumulation of actin polymers (ie, lamellipodia) that protrudes at one pole of the cells defining the leading edge. At the same time, protrusions are suppressed along the lateral sides of the cells by actomyosin-driven cortical tension to restrict the lamellipodia to the leading edge. This asymmetry characterizes polarity. Although directional sensing promotes the orientation of the leading edge toward the source of the chemoattractant, persistent polarity is necessary to prevent deviation of the cell trajectory. Forward movement is also supported by a series of new integrin-mediated adhesion sites at the leading edge, whereas adhesion is released at the rear of the cells.
Members of the Rho GTPase family are key regulators of cell migration. They cycle between an inactive guanosine diphosphate (GDP)–bound and active guanosine triphosphate (GTP)–bound form that is tightly regulated by 3 families of proteins. Guanine nucleotide exchange factors promote the exchange of GDP for GTP, whereas GTPase-activating proteins (GAPs) accelerate the rate of hydrolysis of GTP. In addition, guanine nucleotide dissociation inhibitors interfere with GTP binding by preventing membrane localization of the protein. Of the 22 Rho GTPases currently known, the best studied Rho GTPases are Rho, Rac, and Cdc42.6 In recent years, it has been established that Rac is the central regulator of lamellipodia at the front.7–10 Conversely, RhoA masters cortical tension by acting at the sides and rear of the cells and by organizing the actomyosin meshwork.11,12 Cdc42 has emerged as a master regulator of cell polarity in numerous cell types. Cdc42 is active toward the front of migrating cells.13 Cdc42 can promote polarity by determining the site where Rac is activated and thus where lamellipodia form.14 Cdc42 can also promote polarity by localizing the microtubule-organizing center and microtubule orientation at the leading edge.15,16 Therefore, the prevailing view is that Cdc42 affects polarity by activating pathways that organize the front of the cells. However, the role of Cdc42 in neutrophil migration remains ill defined with seemingly conflicting results. Cdc42 can be activated at the front by the PAK (p21-activated kinase)–associated Rac guanine nucleotide exchange factor PIXα. Using PIXα-deficient neutrophils, Cdc42 has been proposed to regulate directional sensing and the spatial distribution of filamentous actin (F-actin) at the leading edge.13 In contrast, recent work has suggested that Cdc42 may also regulate pathways that inhibit inappropriate protrusions. In the neutrophil cell line HL-60, abrogation of Cdc42 activity causes the formation of multiple pseudopodia.17,18 Our laboratory showed that neutrophils deficient in Cdc42GAP, a negative regulator of Cdc42, exhibited abnormal lateral protrusions and failed to persistently migrate in one direction.19 These studies were performed in cell lines or using mouse models deficient in regulators of Cdc42, and thus, although very informative, did not directly address the role of Cdc42 in primary neutrophils. Furthermore, details in the mechanisms of Cdc42 functions are not clear.
To understand the precise mechanism by which Cdc42 regulates neutrophil migration, we used mice genetically deficient in Cdc42. We show that Cdc42 regulates polarity in migrating neutrophils. Interestingly, the myeloid integrin CD11b is probably critical during this process. CD11b functioning is defective in Cdc42-null cells, and restoring CD11b functioning in these cells rescues polarity. Furthermore, using CD11b-deficient cells, we show the direct involvement of CD11b in neutrophil polarity. Finally, mechanistic studies indicate that Cdc42 controls polarity by modulating the myosin light chain pathway at the uropod via CD11b. Our study thus emphasizes a new mechanism of Cdc42 in affecting polarity and reinforces the view that Cdc42 can regulate pathways that orchestrate the rear of the cells. Our study also reveals unexpected functions for the integrin CD11b in neutrophil polarity during migration.
Methods
Mouse strains
The conditional Cdc42flox/flox mice were described before.20 These mice were crossed with MxCreTg/+ (MxCreTg/+; Cdc42flox/flox). As controls, we used MxCreTg/+;WT mice. We used a protocol of 3 injections of polyI:polyC (300 μg per mouse, GE Healthcare)8 to induce the deletion of Cdc42 floxed alleles. All mice including the controls were treated with polyI:polyC. Animals used were backcrossed for N = 2 generation. CD11b-deficient animals (Itgamtm1Myd) were purchased from The Jackson Laboratory. All animals were bred in the Cincinnati Children's Research Foundation pathogen-free animal facility. All experimental procedures were approved by the institutional animal committee at the Cincinnati Children's Research Foundation.
Neutrophil isolation
Neutrophils were isolated from bone marrow cells by Percoll gradient as previously described (purity 75% by cytospin).19 Transwell migration assays were performed as previously described.19
Time lapse video microscopy
Time lapse video microscopy was performed in a Zigmond chamber (Neuro Probe) as previously described,19 on glass or on surface coated with fibrinogen (Fg; 25 μg/mL, Sigma-Aldrich). Migration was allowed in gradient of 10μM formyl-methionyl-leucyl-phenylalanine (fMLP; Sigma-Aldrich). Migration was recorded every 5 seconds for 25 minutes with a Zeiss Axiovert 200 microscope at 10×/0.3 NA objective, equipped with ORCA-ER camera (Hamamatsu) and driven by Openlab 5.0.2 software (Improvision). Analysis of cell migration was performed in the motile population that had moved more than 20 μm21 using Openlab. Speed, straightness of migration (distance from origin to total distance covered), and direction of the movement were analyzed. The experiments were performed in 3 independent experiments. Quantification analyses of 70 to 100 cells are from 3 independent videos.
To analyze actin protrusion dynamics, we used enhanced green fluorescent protein (eGFP)-actin cDNA constructed into retrovirus vector (kindly provided by Dr Punam Malik, Cincinnati, OH). MxCreTg/+;WT and MxCreTg/+; Cdc42flox/flox bone marrow cells were transduced with retroviral supernatant eGFP-actin.22 eGFP-actin–transduced cells were injected into lethally irradiated wild-type (WT) C57Bl/6 animals. Five weeks after bone marrow reconstitution, all animals were treated with polyI:polyC to induce floxed-Cdc42 allele deletion. Neutrophils were then isolated and used for experiments. Cells were stimulated with a local source of fMLP (10μM). Fluorescent images were recorded every 30 seconds for 5 minutes with a Zeiss Axiovert 200 microscope at 40× objective NA 0.7.
Immunofluorescence
To examine F-actin structures or distribution of phosphorylated myosin light chain (p-MLC), neutrophils (5 × 104) were prestimulated with fMLP in Hanks balanced salt solution, 0.1% bovine serum albumin, 1mM Ca2+, 1mM Mg2+, and on Fg-coated slides for 0 and 10 minutes at 37°C. The cells were fixed, permeabilized with 0.1% Triton X-100, and stained with rhodamine-labeled phalloidin (Invitrogen)19 or with rabbit anti–p-MLC (Cell Signaling Technology) followed by anti–rabbit Alexa 488 (Invitrogen). To examine CD11b distribution at the cell surface, cells were stimulated with fMLP and on fibrinogen and fixed. The cells were first stained with anti-CD45 or anti-CD44 followed by anti–rat Alexa 594, then with biotin-labeled anti–mouse anti-CD11b (clone M1/70), followed by streptavidin Alexa 488 (BD Biosciences). To analyze CD11b clustering in response to fMLP alone, the cells were stimulated with fMLP in water bath at 37°C. The cells were then fixed, allowed to adhere onto slides, and stained for CD11b. To examine intracellular distribution of CD11b, the cells were fixed, permeabilized with Triton, and stained for CD11b.
To block CD11b functions, WT cells were incubated with 10μM anti-CD11b blocking antibody (eBioscience) for 20 minutes at room temperature and then used for experiments. For antibody-mediated crosslinking experiments, we used a method published by the Lowell group.23,24 The slides were coated with 25 μg/mL of streptavidin (Sigma-Aldrich) at 37°C followed by 10 μg/mL of biotin-labeled rat IgG2b–anti-CD11b (BD Biosciences) or isotype control A95-1 at 37°C. The slides were then blocked with 20% fetal bovine serum for 1 hour at 37°C.
To analyze ROCK functions, WT or Cdc42-null neutrophils were preincubated with the ROCK inhibitor Y-27632 (10μM, Calbiochem) for 30 minutes at 37°C and analyzed for F-actin and p-MLC distribution.
Z series of fluorescence images were captured using a Leica DMI6000 or with Zeiss Axiovert 200 fluorescence microscope at 63×/1.3 NA objective, with ORCA-ER C4742-95 camera (Hamamatsu) driven by Openlab software.8,22 Z series were analyzed by deconvolution using Volocity (Improvision). Quantifications were performed using region measurement in Openlab and Volocity and ImageJ software25 (supplemental Methods, available on the Blood website; see the Supplemental Materials link at the top of the online article). At least 30 cells per sample and per experiment were analyzed; at least 3 independent experiments were performed. Quantifications shown in the study are from at least n = 30 cells from 2 independent experiments and representative of all experiments.
Adhesion assays and integrin expression were analyzed as previously described.19 Biochemistry experiments were analyzed as previously described19,26 (supplemental data).
Statistical analysis
All data were analyzed using a 2-tailed t test.
Results
Cdc42 regulates neutrophil migration by maintaining polarity
To examine the role of Cdc42 in neutrophil migration, we extracted neutrophils from mice with a conditional Cdc42 (flox) allele and expressing Cre under an inducible Mx1 promoter (MxCreTg/+;Cdc42flox/flox) and controls (MxCreTg/+;WT),20 previously treated with polyI:polyC. Cdc42 deletion was confirmed by immunoblot (Figure 1A).
Figure 1.

Cdc42 is critical to maintain front/back polarity of neutrophils during migration. (A) Expression of Cdc42 in WT and Cdc42−/− neutrophils analyzed by immunoblot. (B) Neutrophil migration using a Boyden chamber in uniform concentration or in a gradient of 1 μM fMLP in which fMLP is placed only in the lower chamber to measure chemokinesis or chemotaxis, respectively. The histogram represents the number of migrated neutrophils per field (mean ± SD, representative experiment in triplicate of 3 independent experiments). (C) Neutrophil migration using transwells coated with fibrinogen in uniform concentration or in a gradient of 10μM fMLP. The histogram represents the total number of migrated neutrophils recovered from the bottom well (mean ± SD, representative experiment in triplicate of 3 independent experiments). (D) Neutrophil migration was examined by time-lapse video microscopy in a gradient of 10μM fMLP and on surface coated with fibrinogen, in a Zigmond chamber. Representative images (1 minute between each frame) of migrating cells, fMLP concentration increases from left to right. Arrows point to inappropriate protrusions. Cell trajectory analysis: the schema represents the migration trajectory of cells moving up fMLP gradient for 25 minutes. Trajectories were tracked with Volocity software and realigned to the same horizontal axis. The black circle represents the starting position. Cdc42-deficient neutrophils exhibited overall displacement toward the fMLP gradient. Speed (sp, μm/min) and straightness (st) of migration are indicated on the right. Data are mean ± SEM; n = 100. The histogram represents the percentage of cells exhibiting change in direction arising from inappropriate lateral protrusions (mean ± SD; n = 3 independent videos); at least 50 cells per video were analyzed. Images were captured at 37°C using a Zeiss Axiovert 200 microscope at 10× objective, NA 0.3, with ORCA-ER C4742-95 camera driven by Openlab software (supplemental Videos). *Results that are significantly different from WT (P < .05).
Neutrophil migration across the endothelial barrier depends on chemokines and interaction with the endothelium through adhesion molecules, known as integrins. Regulation of neutrophil migration can be context-dependent. We thus examined the role of Cdc42 in neutrophil migration in response to the neutrophil chemokine fMLP and on surface containing or not containing fibrinogen. Fibrinogen is a physiologic ligand of the neutrophil integrin CD11b/CD18 that is relevant in vivo for neutrophil locomotion27 and subsequent inflammation.28 We used Boyden chambers or transwells coated with fibrinogen, both of which depend on integrin activation, to assess random movement (chemokinesis) in uniform concentration of fMLP and directed migration (chemotaxis) in fMLP gradient. Loss of Cdc42 activity resulted in a marked reduction in both chemotaxis and chemokinesis (Figure 1B-C). In the Boyden chamber assay, Cdc42−/− neutrophils exhibited higher chemotactic migration than chemokinesis, which suggests that Cdc42−/− may sense the direction and that the chemotactic defect results from failure in locomotion. To examine the Cdc42-dependent migratory responses in more detail, neutrophil migration was examined using time-lapse videomicroscopy, in fMLP gradient and on fibrinogen surface. WT cells acquired a polarized shape with single pseudopodia facing the fMLP gradient. They produced few lateral protrusions and moved persistently toward fMLP (supplemental Video 1; Figure 1D). Conversely, Cdc42−/− neutrophils exhibited multiple protrusions. Some cells that were able to initiate polarity developed additional lateral protrusions that caused deviation of the cell trajectory (supplemental Video 2; Figure 1D). Subsequently, Cdc42−/− neutrophils failed to migrate persistently in one direction, and the ratio of distance from origin to total distance, or straightness of migration,21 was markedly lower in Cdc42−/− neutrophils than in WT cells. This result indicates that Cdc42−/− neutrophils were unable to maintain polarity of migrating neutrophils. Nonetheless, the speed of migration and tail retraction were comparable between the genotypes. Cdc42−/− neutrophils interpreted the fMLP gradient in that analysis of the cell trajectory, and the positions of the cells at 25 minutes indicated that they migrated up the gradient direction, although with frequent turns (Figure 1D). Therefore, Cdc42 regulates migration by maintaining polarity. As persistent polarity is essential for chemokinesis and chemotactic migration, defective migration of Cdc42−/− neutrophils seen in transwell assays is probably arising from their abnormal polarity.
One feature of polarity is the formation of a lamellipodia made of F-actin at the cell front.4 To examine actin protrusion dynamics, we used neutrophils expressing an eGFP-actin reporter, which has no impact on migration. During migration, eGFP-actin showed persistent polarized distribution at the front of WT neutrophils. In contrast, the eGFP-actin of Cdc42−/− neutrophils was observed both at the front and at the back during the course of the movement (Figure 2). Time-course analysis of F-actin distribution using rhodamine-phalloidin staining confirmed multiple F-actin protrusions around Cdc42−/− cells (supplemental Figure 1). Therefore, Cdc42 limits lateral protrusions and thus regulates migration by maintaining polarity.
Figure 2.

Dynamic analysis of actin distribution using eGFP-actin reporter. Sequential images showing the distribution of eGFP-actin reporter in WT and Cdc42−/− neutrophils in response to a local source of fMLP (10μM, indicated by X) (30 seconds between each frame). Arrows point to actin protrusions. Note that eGFP-actin remains at one pole of the cell facing the source of fMLP in WT but not in Cdc42−/− cells. Each set of images are representative of a set of 10. Scale bar represents 5 μm. Fluorescence images were captured at 37°C using a Zeiss Axiovert 200 fluorescence microscope at 40× objective, NA 0.6, with ORCA-ER C4742-95 camera driven Openlab software. The histograms are speed, straightness of migration, and percentage of cells with multiple protrusions and turns of WT and Cdc42−/− cells expressing eGFP-actin, which were analyzed in a Zigmond chamber assay as in Figure 1 and showed similar results as WT and Cdc42−/− cells (Figure 1).
Cdc42 regulates neutrophil polarity through CD11b
To investigate the mechanism of these events, we examined the role of the integrin CD11b for several reasons. First, our data showed impaired polarity of Cdc42-null neutrophils on adhesion to fibrinogen, a ligand of CD11b; second, we previously suggested that Cdc42GAP inhibits protrusions in a manner dependent on CD11b signaling.19 Furthermore, in migrating cells, the polarized redistribution of integrins and downstream signaling are essential for the regulation of polarity.29 Specifically, in migrating neutrophils, the plasma membrane becomes organized into domains that are different at the front and at the rear (eg, whereas the transmembrane receptor CD45 accumulates at the front, CD44 is concentrated at the rear), and maintaining this organization appears essential for neutrophil polarity.30–32 Interestingly, CD11b is relocalized at the rear, where protrusions are suppressed, and this distribution is essential for adhesion strength and migration.25,33 Thus, we examined the role of Cdc42 in CD11b distribution, in comparison with that of CD45 and CD44, respectively. As previously reported,32 CD45 was enriched in the front of WT polarized neutrophils, whereas CD44 distributed along the lateral sides and in the tail. In agreement with previous studies,33 in WT cells, CD11b was found at the rear, along with CD44 but not with CD45 (Figure 3A). In Cdc42−/− cells, CD11b accumulated at the front, similarly to CD45, or in the tail but was absent along the sides of the cells (Figure 3A). Interestingly, alteration in the pattern of CD44 distribution was also observed in Cdc42−/− cells with significant defect in CD44 staining. Quantification of CD11b and CD44 at the substrate contact surface confirmed impaired CD11b and CD44 distribution at the rear of Cdc42−/− cells compared with WT cells (Figure 3B; supplemental Figure 2). This effect was not the result of differences in integrin expression or endocytosis as CD11b expression at the cell surface, which is up-regulated on chemokine stimulation, was comparable between WT and Cdc42−/− neutrophils both on resting and fMLP-stimulated cells (supplemental Figure 3A). Moreover, intracellular staining of CD11b did not reveal marked differences of intracellular CD11b between the genotypes (supplemental Figure 3B). Expressions of CD18, CD45, and CD44 were similar in each genotype (supplemental Figure 3A).
Figure 3.
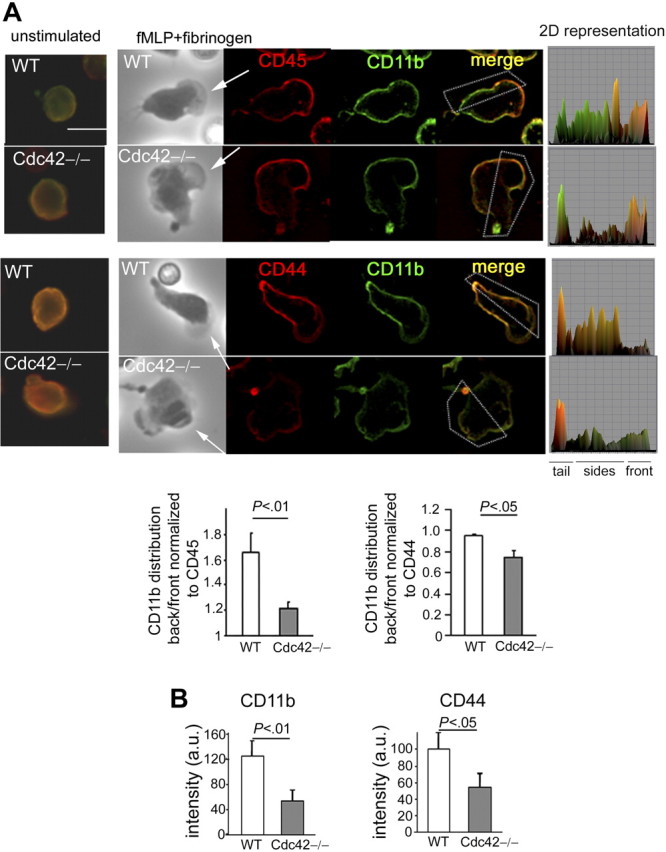
Cdc42 regulates CD11b distribution. (A) WT and Cdc42−/− neutrophils were stimulated or not stimulated with fMLP and on Fg-coated slides for 10 minutes. The cells were fixed and stained with anti-CD45 (in red) or anti-CD44 (in red) and anti-CD11b (in green). The black-and-white pictures are the phase-contrast images. The images are one x-y view of the z-series analyzed by deconvolution in Volocity. Two-dimensional representation of mean intensity of fluorescence of CD11b and CD45 or CD44 of the region of interest indicated by the box, analyzed in ImageJ. Arrows point to the cell front. Histogram is ratio of mean intensity of fluorescence of CD11b along the sides of the cells to the front normalized to CD45 or CD44 (mean ± SD; n = 55). Scale bar represents 5 μm. (B) The average of fluorescence intensity at the substrate contact surface of CD11b and CD44 was measured along the lateral sides of the cells in at least 5 sections per cell (supplemental data). Histogram shows mean ± SEM of all measurements per cell and of n = 20 cells, representative of 3 independent experiments. The slides were mounted with Slowfade Gold antifade reagent. Fluorescence images were captured at room temperature using a Leica DMI6000 fluorescence microscope at 63×/1.3 NA objective, with ORCA-ER C4742-95 camera driven by Openlab software. Scale bar represents 5 μm.
To investigate whether the abnormal CD11b distribution was associated with abnormal CD11b functioning, we examined in detail the regulation of CD11b functions by Cdc42. Like other integrins, CD11b is an adhesion molecule, which is regulated by subsequent inside-out and outside-in signals.34–38 Chemokine stimulation, such as fMLP, initiates an intracellular signaling cascade that leads to conformational changes in CD11b that, in turn, induce CD11b to bind ligand. In response to inside-out signaling, CD11b can also redistribute and form intramembrane clusters that enhance adhesion.25 Subsequently, activated integrins themselves trigger outside-in signals that are important for adhesion and migration.36–38 Conformational changes of the integrin can be assayed with an adhesion assay. Adhesion to immobilized fibrinogen was unchanged in Cdc42−/− neutrophils compared with WT cells (Figure 4B). We next examined CD11b clustering in response to fMLP alone in cell suspension. Interestingly, the formation of CD11b clusters in response to fMLP seen in WT cells39 was significantly reduced in Cdc42−/− cells (Figure 4A), suggesting that Cdc42 regulates CD11b clustering via inside-out signals. One function of integrin clustering, including CD11b, is to strengthen adhesion.25,34,40 The strength of adhesion can be assayed by measuring cell detachment after stimulus removal.41 Indeed, integrin activation is dependent on chemokines, Ca2+, and Mg2+. The strength of integrin-dependent adhesion is estimated, therefore, by the number of remaining attached cells after removal of fMLP, Ca2+, and Mg2+. Cell detachment was higher in Cdc42−/− cells than in WT cells (Figure 4B). These findings suggest that Cdc42 regulates CD11b distribution/clustering and subsequent functioning.
Figure 4.

Cdc42 regulates CD11b functioning. (A) Neutrophils were stimulated or not stimulated with fMLP in suspension at 37°C. The reaction was stopped by adding paraformaldehyde, and the cells were stained for CD11b. The arrows indicate small clusters and patches of CD11b in WT cells. CD11b clusters were analyzed in ImageJ software. A 3-dimensional representation of fluorescence intensity, which was generated in ImageJ, is shown. Histogram represents the number of CD11b clusters per cell (mean ± SD; n = 40 cells from 3 independent experiments). Scale bar represents 5 μm. The slides were mounted with Slowfade Gold antifade reagent. Fluorescence images were captured at room temperature using a Leica DMIRB fluorescence microscope at 63× objective, NA 1.3, with ORCA-ER C4742-95 camera driven and analyzed with Openlab software. (B) Adhesion and deadhesion of neutrophils. Neutrophils, stimulated with fMLP in the presence of Ca2+ and Mg2+, were allowed to adhere to fibrinogen at the indicated time. The nonadherent fraction was removed. The wells were carefully washed with phosphate-buffered saline, and the adherent fraction was immediately enumerated at the light microscope. Deadhesion: 30 minutes after adhesion to fibrinogen, the nonadherent fraction was removed and fMLP was replaced with phosphate-buffered saline without Ca2+ and Mg2+. The remaining adherent fraction was enumerated at the light microscope 10 minutes after fMLP removal. The histograms represent the number of adherent cells per field (mean ± SD; n = 3 independent experiments).
To determine whether abnormal CD11b functioning is relevant to neutrophil polarity, we first used CD11b−/− neutrophils. Because CD11b−/− neutrophils cannot adhere to fibrinogen (not shown), we analyzed polarity of CD11b−/− cells on glass, which allows adhesion although with little spreading. Whereas WT cells developed polarity with single lamellipodia, CD11b−/− neutrophils exhibited multiple leading edges of F-actin, despite a lack of spreading (Figure 5A). This indicated that CD11b is required to limit protrusions. In comparison, Cdc42−/− cells plated on glass also showed significant increased F-actin protrusions (Figure 5A). To further investigate the role of CD11b of cells plated on fibrinogen, we used a functional blocking anti-CD11b antibody, classically used to block CD11b functions.42 Contrary to the use of CD11b-null cells, this method has the advantage to maintain CD11b expression while altering its function. It still preserves enough adhesion to fibrinogen to allow examination of the consequences of impaired CD11b functions on cellular polarity of cells plated on fibrinogen substrate. In WT neutrophils treated with anti-CD11b, such as like Cdc42−/− neutrophils, CD11b was no longer clustered to the uropod (supplemental Figure 4); instead, it remained diffuse in the cell. Anti-CD11b treatment was associated with a significant increase in cell spreading, as seen in Cdc42−/− cells. Importantly, anti-CD11b treatment led to the formation of multiple F-actin protrusions (Figure 5B). Therefore, CD11b plays a role in neutrophil F-actin polarity. Of note, the seemingly opposite effect of CD11b−/−, Cdc42−/− cells, and cells treated with anti-CD11b on cell spreading may be explained by differences in CD11b expression and potentially by diversity in integrin functions. Indeed, contrary to CD11b−/− cells, Cdc42−/− neutrophils, or cells treated with anti-CD11b maintained CD11b expression and some static adhesion via CD11b although weaker than WT cells. Integrins play important roles in both spreading and contraction but using independent mechanisms.43 Neutrophils spread during the first minute of stimulation and then acquire an elongated shape and narrow lamellipodia, suggesting an initial phase of spreading (which is not impaired in Cdc42−/− cells) followed by contraction, as shown in supplemental Figure 1. The fact that CD11b-null neutrophils do no spread whereas cells treated with anti-CD11b, in which CD11b functions are not totally abrogated, exhibited increased spreading suggests that, like other integrins,43 CD11b may be involved in the initial phase of cell spreading on stimulation and then during contraction.
Figure 5.
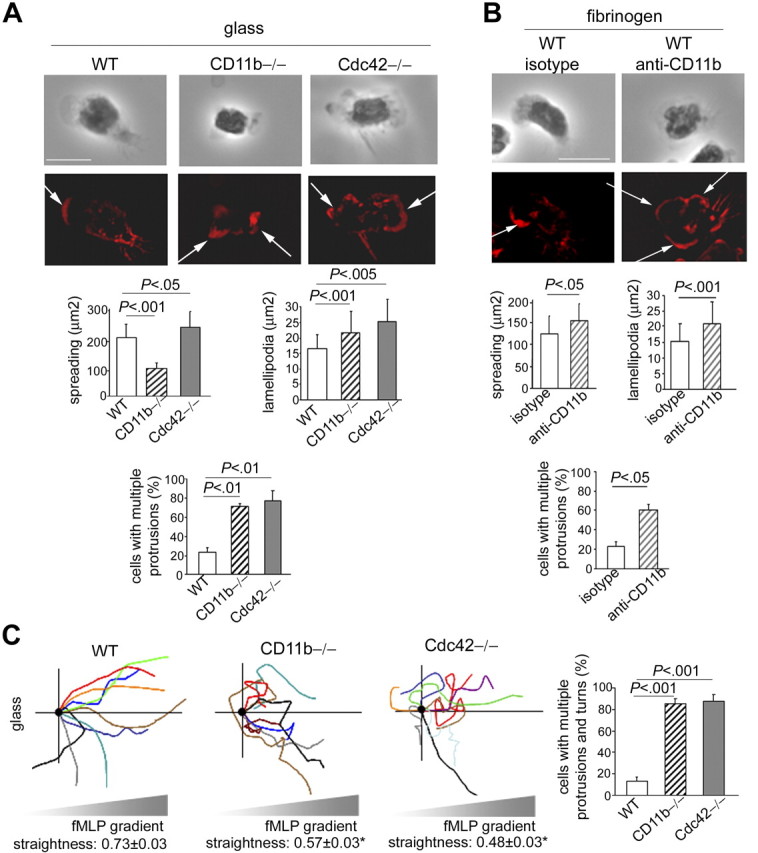
CD11b is critical for polarity of neutrophils during migration. (A) F-actin (rhodamine-phalloidin) analysis of WT, CD11b−/−, and Cdc42−/− neutrophils 10 minutes after stimulation with fMLP and on glass. The black-and-white pictures are the phase-contrast images. The images are one x-y view of the z-series analyzed by deconvolution in Volocity. Histogram represents total surface area and area of lamellipodia (mean ± SD of n = 30 cells) and number of cells with more than one protrusion (mean ± SD; 3 independent experiments). Arrows indicate F-actin protrusions. Scale bar represents 5 μm. (B) WT cells were treated with functional anti-CD11b blocking antibody or isotype control for 20 minutes at room temperature and stimulated with fMLP and on fibrinogen-coated slides in the presence of antibody. The cells were analyzed for F-actin structures with rhodamine-phalloidin (mean ± SD; n = 30 from 3 independent experiments). The slides were mounted with Slowfade Gold antifade reagent. Z series of fluorescence images were captured using a Leica DMIRB or Leica DMI6000 fluorescence microscope at 63×/1.3 NA objective, with ORCA-ER C4742-95 camera driven by Openlab software and analyzed by deconvolution with Volocity software. (C) Migration of cells analyzed by time lapse video microscopy in a Zigmond chamber in gradient of fMLP on glass, as in Figure 1. Schema of cell trajectory is shown. Straightness of migration is indicated as mean ± SEM. *Results that are significantly different from WT (P < .001). Histogram represents percentage of cells that developed lateral protrusions during the course of migration and changed direction (mean ± SD; n = 3 independent videos). Only cells that had moved more than 20 μm were analyzed. Images were captured at 37°C using a Zeiss Axiovert 200 microscope at 10×/0.3 NA objective, with ORCA-ER C4742-95 camera driven by Openlab software.
Cell migration trajectory up fMLP gradient was next examined. CD11b−/− cells extended protrusions back and forth, probably because of abnormal adhesion. These cells showed very little locomotion as previously described.27 Interestingly, the cells that successfully covered more than 50 μm failed to persistently migrate up fMLP gradient. Although their unstable adhesion may contribute to this migratory response, these cells did also develop multiple protrusions simultaneously, which caused changes in direction, as seen in Cdc42-null cells (supplemental Videos 3-5; Figure 5C). This was also true for WT cells treated with anti-CD11b antibody and plated on fibrinogen (data not shown). Therefore, CD11b plays a role in persistent polarity of migrating cells. Collectively, these findings suggest that modulation of CD11b functioning is relevant to neutrophil polarity.
Finally, to determine whether the regulation of neutrophil polarity by Cdc42 is CD11b-dependent, we examined polarity of Cdc42−/− cells in which CD11b functioning had been restored. We reasoned that if Cdc42 regulates CD11b clusters and subsequent functioning and that this regulation is important for polarity, then restoration of CD11b functioning in Cdc42−/− cells should rescue polarity. To restore CD11b functioning, we used the integrin crosslinking method, which is a well-established physiologic way to enforce integrin clustering and subsequent signaling.23,24 This consists of plating the cells on surface coated with anti-CD11b. This treatment crosslinks integrin molecules at the plasma membrane, thereby activating downstream integrin signals. CD11b crosslinking in Cdc42−/− cells rescued the strength of adhesion to WT levels, indicating that CD11b functioning was rescued under these experimental conditions (Figure 6A). Remarkably, Cdc42−/− cells plated on an anti-CD11b antibody-coated surface acquired single lamellipodia at their leading edge similarly to WT cells, indicating rescue of polarity (Figure 6B). As expected, cells plated on control surfaces coated with isotype-matched antibody show poor polarity. We next examined migration in a Zigmond chamber in fMLP gradient and on anti-CD11b–coated surface. Under these conditions, the migration speed was reduced to 3 μm/min because of the strength of adhesion, in a similar manner between the genotypes. Cdc42−/− cells persistently migrated up the fMLP gradient on anti-CD11b in contrast to Fg surface (supplemental Videos 6-7; Figure 6C). Cdc42−/− cells exhibited protrusions only at the leading edge during the course of migration and maintain the direction, although not to the level of WT cells, indicating that strong adhesion partially rescued persistent migration of Cdc42-null cells. Whereas F-actin staining, which was examined at one time point of stimulation, indicated rescue of cells with single leading edge, video microscopy revealed partial rescue of migration, suggesting the involvement of additional mechanism(s) that are needed during chemotaxis in fMLP gradient. These findings indicate that the polarity defect in Cdc42−/− cells arises secondary to defective CD11b functioning. Together, these results suggest that the loss of CD11b functioning of Cdc42−/− cells is a cause of their loss of polarity.
Figure 6.

Cdc42 regulates neutrophil polarity via CD11b. (A) Deadhesion. WT and Cdc42−/− neutrophils were stimulated with fMLP and on slides coated with anti-CD11b to enforce CD11b activation. The stimulus was removed 30 minutes after adhesion, and the remaining adherent fraction was enumerated 10 minutes after stimulus removal at the light microscope (mean ± SD; n = 3 independent experiments, as in Figure 3). (B) WT and Cdc42−/− neutrophils were stimulated with fMLP and on Fg-coated slides or on slides coated with anti-CD11b to enforce CD11b activation, compared with isotype control. The cells were then fixed and stained with rhodamine-phalloidin (in red) to analyze F-actin structures. The black-and-white pictures represent the phase-contrast images. The images are one x-y view of the z-series analyzed by deconvolution in Volocity. Histograms are number of cells with more than one protrusion (percentage, mean ± SD; n = 3 independent experiments); at least 30 cells per experiment were analyzed. Surface area of F-actin (square microns) and total cell spreading (square microns) are shown as mean ± SD of at least n = 30 cells from at least 2 independent experiments. Arrows point to actin protrusions. Scale bar represents 5 μm. The slides were mounted with Slowfade Gold antifade reagent. Z series of fluorescence images were captured at room temperature using a Leica DMI6000 fluorescence microscope at 63×/1.3 NA objective, with ORCA-ER C4742-95 camera driven by Openlab software and analyzed by deconvolution with Volocity software. (C) Migration responses of cells plated on anti-CD11b–coated surface was analyzed in a Zigmond chamber in fMLP gradient as in Figure 1. The schema represents the migration trajectory of cells moving up fMLP gradient for 25 minutes. Straightness of migration is indicated as mean ± SEM; n = 70 cells from 3 independent videos. Histogram represents percentage of cells with change in direction arising from inappropriate lateral protrusions (mean ± SD; n = 3 independent videos).
Cdc42 recruits myosin light chain activity at the lateral sides of migrating neutrophils via CD11b
The suppression of membrane protrusions requires the assembly of myosin cytoskeleton,4,5,44 which is regulated by RhoA signaling in neutrophils.12 RhoA induces myosin filament reorganization through the activation of its effector ROCK, which in turn phosphorylates the myosin light chain (MLC).12,45 Interestingly, Cdc42 has been shown to control the activity of RhoA in HL-60 cells.18 Because RhoA activity can be modulated by integrins,46,47 we investigated the possibility that Cdc42 controls polarity by regulating myosin cytoskeleton assembly via CD11b. We first confirmed that CD11b can activate the RhoA/MLC pathway and examined RhoA and MLC activities on CD11b crosslinking. CD11b crosslinking promotes the activation of RhoA and MLC compared with unstimulated cells (Figure 7A). We next examined the role of Cdc42 and CD11b on MLC phosphorylation on stimulation on fibrinogen. MLC phosphorylation was significantly decreased in Cdc42−/− cells compared with WT cells (Figure 7B). To examine the role of CD11b in MLC activation, we induced loss of CD11b functions with an antibody against CD11b because the extremely low adhesion characteristics of CD11b−/− cells did not permit p-MLC analysis by immunoblot. MLC activation was significantly reduced in WT cells treated with anti-CD11b (Figure 7B). Therefore, MLC activation depends on intact functioning of Cdc42 and CD11b. Interestingly, p-MLC was restored to WT levels in Cdc42−/− cells plated on anti-CD11b–coated surface (Figure 7B). The activation of MLC by Cdc42 can thus be modulated by CD11b. We did not detect significant change in the level of RhoA activity in total cell lysates of Cdc42−/− cells plated on fibrinogen compared with WT, by the pulldown assay. However, the sensitivity of the method may not effectively reveal differences.26 Nevertheless, the fact that the ROCK inhibitor Y-27632 abrogated MLC activity (Figure 7B) suggested that MLC activation by Cdc42/CD11b axis may be RhoA/ROCK-dependent.
Figure 7.
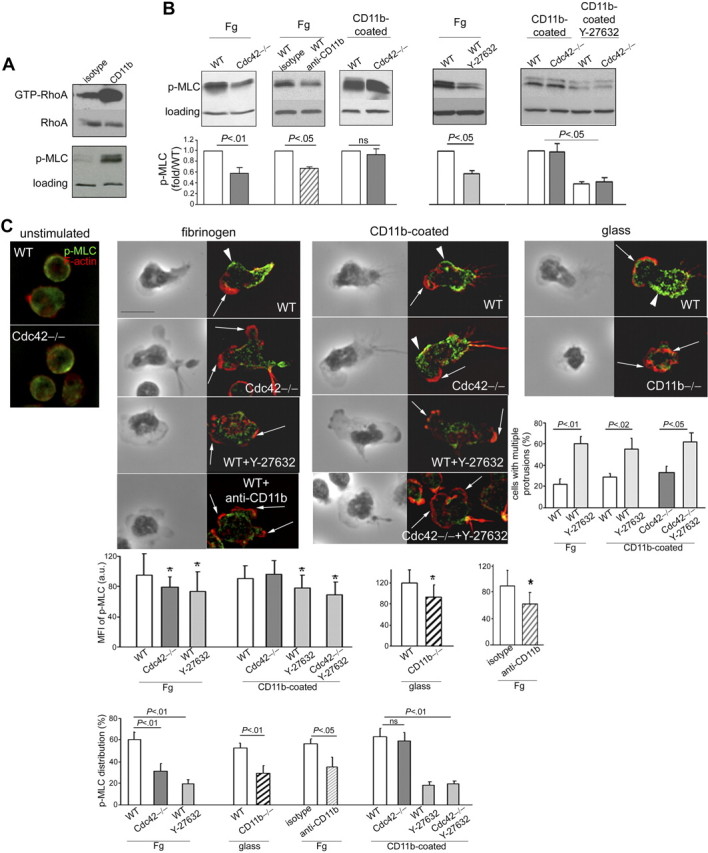
Cdc42 suppresses actin through CD11b-induced p-MLC signaling. (A) WT cells were crosslinked with CD11b or isotype controls for 20 minutes and were analyzed for RhoA activity using the pulldown assay with Rhotekine beads. Total RhoA of cell lysates is used as loading control. The cells were also analyzed for phosphorylated MLC at Ser19 and total p38MAPK as loading control (one blot representative of 3 independent experiments). (B) WT and Cdc42−/− neutrophils with or without Y-27632 treatment were stimulated with fMLP and seeded on fibrinogen or on an anti-CD11b–coated plate for 10 minutes. In addition, WT cells treated with functional anti-CD11b blocking antibody were stimulated with fMLP and on Fg-coated slides. The cells were lysed on plate and analyzed for p-MLC. Histograms represent densitometry analysis compared with WT after normalization to loading (mean ± SD; n = 3 independent experiments). Of note, the experiments with and without Y-27632 were performed independently such that the absolute level of p-MLC is not comparable. (C) Cells, treated or not with Y-27632, were stimulated or not on fMLP and on Fg or glass or anti-CD11b–coated slides for 10 minutes. The cells were stained with anti-p-MLC (in green) and rhodamine-phalloidin (in red). In addition, WT cells treated with anti-CD11b or isotype were plated on fibrinogen and analyzed for F-actin and p-MLC distribution. These later experiments were performed separately. WT cells treated with isotype are not different from nontreated WT cells, and only one image of WT control is shown. The black-and-white pictures represent the phase-contrast images. The images colored in red and green are one x-y view of the z-series analyzed by deconvolution in Volocity. Percentage of cells with p-MLC staining along the lateral sides of the cells is enumerated (mean ± SD; n = 3 independent experiments). The average intensity of fluorescence per area of p-MLC along the sides of the cells was analyzed in Openlab, and the data are shown as histogram (mean ± SD of n = 30-50 cells, from at least 2 independent experiments). Percentage of cells with more than one protrusion (mean ± SD; n = 3 independent experiments); at least 30 cells per experiments were analyzed. *Results that are significantly different from WT (P < .05). Arrows point to actin protrusions. Arrowheads point to p-MLC lateral distribution. Scale bar represents 5 μm. The slides were mounted with Slowfade Gold antifade reagent. Z series of fluorescence images were captured at room temperature using a Leica DMI6000 fluorescence microscope at 63×/1.3 NA objective, with ORCA-ER C4742-95 camera driven by Openlab software and analyzed by deconvolution with Volocity software.
MLC-induced myosin assembly occurs at the uropod and along the sides of the cells.2,4 We next examined the cellular distribution of p-MLC using immunofluorescence staining with an antibody against p-MLC.12 Consistently, in WT cells, p-MLC staining was localized at the back and along the lateral sides of WT cells after stimulation with fMLP and on fibrinogen substrate (Figure 7C). However, in Cdc42−/− neutrophils, p-MLC staining was weak on the sides of the cells (Figure 7C). CD11b−/− cells plated on glass or WT cells treated with anti-CD11b plated on fibrinogen also showed weak lateral p-MLC staining (Figure 7C). In contrast, p-MLC staining was restored to WT levels in Cdc42−/− cells plated on anti-CD11b–coated surface (Figure 7C). Consistent with the biochemistry results, Y-27632–treated cells showed impaired p-MLC signal along their lateral sides either on fibrinogen or on CD11b-coated plates. Interestingly, these cells exhibited multiple protrusions (Figure 7C).12 Together, these results suggest that Cdc42 controls polarity via CD11b-induced ROCK/MLC myosin contraction.
Discussion
Our study provides evidence that Cdc42 is required to maintain polarity during neutrophil migration and describes a pathway linking Cdc42 to integrin activation during this process. Using neutrophils genetically deficient in Cdc42, we show that Cdc42 regulates CD11b distribution and functioning. In turn, CD11b appears to be necessary for proper MLC-based contractility to suppress inappropriate protrusions. Therefore, our study uncovers a new mechanism regulated by Cdc42 that is critical in maintaining neutrophil polarity. Furthermore, it reveals an unexpected role for CD11b in polarity during neutrophil migration.
An important aspect of the regulation of polarity is to determine where protrusions and contractions are formed. This can be achieved by controlling the local activation of divergent signals that are segregated at the front and at the back.4 In many studies, Cdc42 has been suggested to control polarity by determining where lamellipodia form, either through Rac activity and/or via the reorientation of the microtubule-organizing center and Golgi apparatus toward the leading edge.14–16,48 Our study clearly shows that Cdc42 is essential for neutrophil polarity, but it reveals that Cdc42 controls this function by regulating signals outside the leading edge (eg, contractile signals at the back). This is in agreement with previous studies in the neutrophilic cell line HL-6017,18 and in cells deficient in the negative regulator of Cdc42, Cdc42GAP.19 Although associated with increased GTP-bound Cdc42, Cdc42GAP deficiency caused increased membrane protrusions.19 Depending on the cellular context, it is thought that “too much” or “too little” protein activity leads to defective downstream signaling and thereby loss of function of the protein. These observations highlight the importance of having a tight control of the cycle GTP-bound and GDP-bound Cdc42 for proper Rho GTPase functioning. On the other hand, Cdc42GAP deficiency, but not Cdc42 deficiency, caused increased velocity of migration.19 The load of GTP-bound versus GDP-bound Cdc42 may quantitatively modulate chemokinesis. Alternatively, we cannot fully exclude that Cdc42GAP regulates some aspect of migration independently of Cdc42 activity. Other studies in neutrophils deficient in a Cdc42-positive regulator PIXα, reported that Cdc42 controls directional sensing and determines where F-actin is polymerized at the front.13 Although we cannot exclude such a role for Cdc42, our study did not detect a defect in directional sensing in that Cdc42-deficient cells were able to move toward higher fMLP concentration, although with frequent turns. These variations may reflect an agonist dependency of some Cdc42 functions. Indeed, fMLP, but not C5a, which was used in the study in PIXα-deficient neutrophils,13 reportedly recruits neutrophils via β2 integrins.49 However, these previous studies lack specificity regarding Cdc42 functions. Our new data using Cdc42-null neutrophils do provide direct evidence that Cdc42 is required to maintain polarity of migrating neutrophils by regulating contractile signals at the uropod.
Our study suggests a new mechanism of neutrophil polarity and links Cdc42 to integrin signaling during this process. The interplay of Rho GTPases and integrins in the regulation of polarity was previously described in fibroblasts. Rac can recruit high-affinity αvβ3 receptor at the lamellipodia.50 The integrin α4 binds to paxillin and recruits an adenosine diphosphate-ribosylation factor-GAP that leads to the inhibition of Rac activity at the back of the cells, thereby restricting Rac-mediated protrusions to the front.29 This indicates that integrins are not just attachment molecules but also serve a critical role in polarity. Our study indicates that Cdc42 uses CD11b to limit protrusions, which suggests that integrin-mediated adhesion is also critical for neutrophil polarity. We show defective polarity of CD11b-null cells or WT cells treated with an antibody against CD11b. Furthermore, although Cdc42-null cells have defective CD11b functioning, restoration of CD11b functions using antibody crosslinking rescued polarity and partially rescued chemotaxis. The partial rescue of chemotaxis may indicate that an additional mechanism may play an important role in Cdc42 polarity, either by acting upstream or downstream or by acting in parallel. Nevertheless, these findings reveal an unexpected role for CD11b in polarity. CD11b plays a fundamental role during neutrophil migration both in vitro and in vivo.27,42,51 CD11b regulates locomotion on endothelium in which the cells migrate on endothelium to distant emigration sites.27 As CD11b-null cells showed dramatic reduction in speed, the prevailing view has been that CD11b contributes to pulling the cell forward. However, analyzing the trajectory of CD11b-null neutrophils that traveled at least for a short distance in gradient of fMLP clearly indicates defective polarity as the cells developed multiple protrusions that induced deviation of their trajectory. A similar pattern of movement was observed in wild-type monocytes treated with an antibody against β2 integrin.2 Therefore, CD11b may play several functions during migration. These functions may result from specific location of the integrin. CD11b can be found in multiple cellular domains, eg, granule-like compartments and at the leading edge52 and at the uropod.25,33 The plasma membrane at the uropod is specifically enriched in several adhesion molecules, including ICAM-1, ICAM-2, and CD44,32 and this organization is essential for neutrophil polarity.30,31 Interestingly, CD11b redistribution at the uropod with CD44 is Cdc42-dependent. Therefore, there may be a CD11b-dependent process that is regulated by Cdc42 and that specifically promotes polarity.
Our study suggests that the mechanism by which the Cdc42-CD11b axis limits membrane protrusions involves the ROCK/MLC pathway leading to myosin contraction at the trailing edge. In neutrophils, the assembly of myosin II filaments depends on RhoA signaling, which acts through the activation of its effector ROCK and then MLC phosphorylation.12,45 A study by Van Keymeulen et al, performed in HL-60 cells, previously showed that Cdc42 inhibition causes increased protrusions.18 Although of unknown mechanism, this phenotype was associated with decreased RhoA activity, as assessed by fluorescence resonance energy transfer analysis, thus suggesting that, although Cdc42 is activated at the front, it stabilizes polarity by regulating signals at the back.18 Although we did not detect any change in the activity of RhoA in Cdc42-deficient cells using the pulldown assay, we show that Cdc42 regulates myosin contraction at the uropod. Furthermore, Cdc42 controls the local distribution of CD11b; CD11b, in turn, acts as a signaling molecule by modulating the MLC pathway at the trailing edge in a ROCK-dependent manner. These observations suggest that CD11b emerges as a secondary signal induced by Cdc42 to limit protrusions at the uropod. Because CD11b can modulate RhoA activity, Cdc42 may well use CD11b to determine where RhoA is active in the cells. A detailed analysis of the distribution of RhoA activity in these cells will require fluorescence resonance energy transfer analysis.18 Finally, because Cdc42 has been shown to control the distribution of Rac,14 we cannot fully exclude an additional effect on Rac-induced protrusions in our model.
It will be interesting to analyze in detail how Cdc42 controls CD11b distribution in polarized neutrophils. One possibility is that Cdc42 does this by controlling the compartmentalization of the plasma membrane. During neutrophil polarization, the plasma membrane becomes organized into discrete domains with different lipid and protein compositions that are important for polarity.30–32 Lipid rafts have been implicated in the regulation of integrin clustering by providing platform for adaptor and signaling molecules.53 In favor of this, we show that the distribution of both CD11b and the rear marker CD44 is impaired in Cdc42-deficient cells and CD44 is known to distribute at the rear into lipid raft-like domains.32
In conclusion, our study supports a link between Cdc42 and CD11b-mediated adhesion in neutrophil polarity. Mutations that impair expression or activation of all forms of β2 integrins, including CD11b/CD18, are the cause of major neutrophil-associated immunodeficiencies, the leukocytes adhesion deficiencies.54 Cdc42 may thus serve a critical role in neutrophil-mediated innate immunity and may represent a molecular target for novel therapeutic modalities of innate immunity.
Supplementary Material
Acknowledgments
The authors thank Dr Jose Cancelas (Division of Experimental Hematology, Cincinnati Children's Hospital Medical Center) for important comments on the manuscript, Shelli Homan and Christina Sexton for animal husbandry, the Comprehensive Mouse and Cancer Core at Cincinnati Children's Hospital, and Jeff Bailey and Victoria Summey for bone marrow transplantation.
This work was supported by an American Society of Hematology Scholar Award (M.-D.F.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: K.S. performed research; Y.Z. contributed vital new reagents by providing the Cdc42 knockout mouse and key advice in research design and data analysis, and edited the manuscript; and M.-D.F. directed the program research, designed and performed research, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Marie-Dominique Filippi, Division of Experimental Hematology and Cancer Biology, S7.605, Cincinnati Children's Research Foundation, 3333 Burnet Ave, Cincinnati, OH 45229; e-mail: Marie-Dominique.Filippi@cchmc.org.
References
- 1.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 2.Schenkel AR, Mamdouh Z, Muller WA. Locomotion of monocytes on endothelium is a critical step during extravasation. Nat Immunol. 2004;5(4):393–400. doi: 10.1038/ni1051. [DOI] [PubMed] [Google Scholar]
- 3.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7(9):678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 4.Ridley AJ, Schwartz MA, Burridge K, et al. Cell migration: integrating signals from front to back. Science. 2003;302(5651):1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 5.Van Haastert PJ, Devreotes PN. Chemotaxis: signalling the way forward. Nat Rev Mol Cell Biol. 2004;5(48):626–634. doi: 10.1038/nrm1435. [DOI] [PubMed] [Google Scholar]
- 6.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 7.Glogauer M, Marchal CC, Zhu F, et al. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol. 2003;170(11):5652–5657. doi: 10.4049/jimmunol.170.11.5652. [DOI] [PubMed] [Google Scholar]
- 8.Gu Y, Filippi MD, Cancelas JA, et al. Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science. 2003;302(5644):445–449. doi: 10.1126/science.1088485. [DOI] [PubMed] [Google Scholar]
- 9.Roberts AW, Kim C, Zhen L, et al. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity. 1999;10(2):183–196. doi: 10.1016/s1074-7613(00)80019-9. [DOI] [PubMed] [Google Scholar]
- 10.Weiner OD, Servant G, Welch MD, Mitchison TJ, Sedat JW, Bourne HR. Spatial control of actin polymerization during neutrophil chemotaxis. Nat Cell Biol. 1999;1(2):75–81. doi: 10.1038/10042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong K, Pertz O, Hahn K, Bourne H. Neutrophil polarization: spatiotemporal dynamics of RhoA activity support a self-organizing mechanism. Proc Natl Acad Sci U S A. 2006;103(10):3639–3644. doi: 10.1073/pnas.0600092103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J, Wang F, Van Keymeulen A, et al. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell. 2003;114(2):201–214. doi: 10.1016/s0092-8674(03)00555-5. [DOI] [PubMed] [Google Scholar]
- 13.Li Z, Hannigan M, Mo Z, et al. Directional sensing requires G beta gamma-mediated PAK1 and PIX alpha-dependent activation of Cdc42. Cell. 2003;114(2):215–227. doi: 10.1016/s0092-8674(03)00559-2. [DOI] [PubMed] [Google Scholar]
- 14.Cau J, Hall A. Cdc42 controls the polarity of the actin and microtubule cytoskeletons through two distinct signal transduction pathways. J Cell Sci. 2005;118(pt 12):2579–2587. doi: 10.1242/jcs.02385. [DOI] [PubMed] [Google Scholar]
- 15.Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 2001;106(4):489–498. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- 16.Fukata M, Watanabe T, Noritake J, et al. Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell. 2002;109(7):873–885. doi: 10.1016/s0092-8674(02)00800-0. [DOI] [PubMed] [Google Scholar]
- 17.Srinivasan S, Wang F, Glavas S, et al. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J Cell Biol. 2003;160(3):375–385. doi: 10.1083/jcb.200208179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Keymeulen A, Wong K, Knight ZA, et al. To stabilize neutrophil polarity, PIP3 and Cdc42 augment RhoA activity at the back as well as signals at the front. J Cell Biol. 2006;174(3):437–445. doi: 10.1083/jcb.200604113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szczur K, Xu H, Atkinson S, Zheng Y, Filippi MD. Rho GTPase CDC42 regulates directionality and random movement via distinct MAPK pathways in neutrophils. Blood. 2006;108(13):4205–4213. doi: 10.1182/blood-2006-03-013789. [DOI] [PubMed] [Google Scholar]
- 20.Yang L, Wang L, Geiger H, Cancelas JA, Mo J, Zheng Y. Rho GTPase Cdc42 coordinates hematopoietic stem cell quiescence and niche interaction in the bone marrow. Proc Natl Acad Sci U S A. 2007;104(12):5091–5096. doi: 10.1073/pnas.0610819104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferguson GJ, Milne L, Kulkarni S, et al. PI(3)Kgamma has an important context-dependent role in neutrophil chemokinesis. Nat Cell Biol. 2007;9(1):86–91. doi: 10.1038/ncb1517. [DOI] [PubMed] [Google Scholar]
- 22.Filippi MD, Harris CE, Meller J, Gu Y, Zheng Y, Williams DA. Localization of Rac2 via the C terminus and aspartic acid 150 specifies superoxide generation, actin polarity and chemotaxis in neutrophils. Nat Immunol. 2004;5(3):744–751. doi: 10.1038/ni1081. [DOI] [PubMed] [Google Scholar]
- 23.Lowell CA, Fumagalli L, Berton G. Deficiency of Src family kinases p59/61hck and p58c-fgr results in defective adhesion-dependent neutrophil functions. J Cell Biol. 1996;133(4):895–910. doi: 10.1083/jcb.133.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mocsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. Syk is required for integrin signaling in neutrophils. Immunity. 2002;16(4):547–558. doi: 10.1016/s1074-7613(02)00303-5. [DOI] [PubMed] [Google Scholar]
- 25.Zhang H, Schaff UY, Green CE, et al. Impaired integrin-dependent function in Wiskott-Aldrich syndrome protein-deficient murine and human neutrophils. Immunity. 2006;25(2):285–295. doi: 10.1016/j.immuni.2006.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Filippi MD, Szczur K, Harris CE, Berclaz PY. Rho GTPase Rac1 is critical for neutrophil migration into the lung. Blood. 2007;109(7):1257–1264. doi: 10.1182/blood-2006-04-017731. [DOI] [PubMed] [Google Scholar]
- 27.Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. 2006;203(12):2569–2575. doi: 10.1084/jem.20060925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flick MJ, LaJeunesse CM, Talmage KE, et al. Fibrin(ogen) exacerbates inflammatory joint disease through a mechanism linked to the integrin alphaMbeta2 binding motif. J Clin Invest. 2007;117(11):3224–3235. doi: 10.1172/JCI30134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishiya N, Kiosses WB, Han J, Ginsberg MH. An alpha4 integrin-paxillin-Arf-GAP complex restricts Rac activation to the leading edge of migrating cells. Nat Cell Biol. 2005;7(4):343–352. doi: 10.1038/ncb1234. [DOI] [PubMed] [Google Scholar]
- 30.Bodin S, Welch MD. Plasma membrane organization is essential for balancing competing pseudopod- and uropod-promoting signals during neutrophil polarization and migration. Mol Biol Cell. 2005;16(12):5773–5783. doi: 10.1091/mbc.E05-04-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pierini LM, Eddy RJ, Fuortes M, Seveau S, Casulo C, Maxfield FR. Membrane lipid organization is critical for human neutrophil polarization. J Biol Chem. 2003;278(12):10831–10841. doi: 10.1074/jbc.M212386200. [DOI] [PubMed] [Google Scholar]
- 32.Seveau S, Eddy RJ, Maxfield FR, Pierini LM. Cytoskeleton-dependent membrane domain segregation during neutrophil polarization. Mol Biol Cell. 2001;12(11):3550–3562. doi: 10.1091/mbc.12.11.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kindzelskii AL, Laska ZO, Todd RF, 3rd, Petty HR. Urokinase-type plasminogen activator receptor reversibly dissociates from complement receptor type 3 (alpha M beta 2′ CD11b/CD18) during neutrophil polarization. J Immunol. 1996;156(1):297–309. [PubMed] [Google Scholar]
- 34.Takagi J, Springer TA. Integrin activation and structural rearrangement. Immunol Rev. 2002;186:141–163. doi: 10.1034/j.1600-065x.2002.18613.x. [DOI] [PubMed] [Google Scholar]
- 35.Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nat Rev Immunol. 2005;5(7):546–559. doi: 10.1038/nri1646. [DOI] [PubMed] [Google Scholar]
- 36.Ginsberg MH, Partridge A, Shattil SJ. Integrin regulation. Curr Opin Cell Biol. 2005;17(5):509–516. doi: 10.1016/j.ceb.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 37.Lowell CA. Src-family kinases: rheostats of immune cell signaling. Mol Immunol. 2004;41(6):631–643. doi: 10.1016/j.molimm.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 38.Williams MA, Solomkin JS. Integrin-mediated signaling in human neutrophil functioning. J Leukoc Biol. 1999;65(6):725–736. doi: 10.1002/jlb.65.6.725. [DOI] [PubMed] [Google Scholar]
- 39.Patcha V, Wigren J, Winberg ME, Rasmusson B, Li J, Sarndahl E. Differential inside-out activation of beta2-integrins by leukotriene B4 and fMLP in human neutrophils. Exp Cell Res. 2004;300(2):308–319. doi: 10.1016/j.yexcr.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 40.Hato T, Pampori N, Shattil SJ. Complementary roles for receptor clustering and conformational change in the adhesive and signaling functions of integrin alphaIIb beta3. J Cell Biol. 1998;141(7):1685–1695. doi: 10.1083/jcb.141.7.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson SI, Hotchin NA, Nash GB. Role of the cytoskeleton in rapid activation of CD11b/CD18 function and its subsequent downregulation in neutrophils. J Cell Sci. 2000;113(15):2737–2745. doi: 10.1242/jcs.113.15.2737. [DOI] [PubMed] [Google Scholar]
- 42.Ding ZM, Babensee JE, Simon SI, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol. 1999;163(9):5029–5038. [PubMed] [Google Scholar]
- 43.Flevaris P, Stojanovic A, Gong H, Chishti A, Welch E, Du X. A molecular switch that controls cell spreading and retraction. J Cell Biol. 2007;179(3):553–565. doi: 10.1083/jcb.200703185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Affolter M, Weijer CJ. Signaling to cytoskeletal dynamics during chemotaxis. Dev Cell. 2005;9(1):19–34. doi: 10.1016/j.devcel.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 45.Worthylake RA, Burridge K. RhoA and ROCK promote migration by limiting membrane protrusions. J Biol Chem. 2003;278(15):13578–13584. doi: 10.1074/jbc.M211584200. [DOI] [PubMed] [Google Scholar]
- 46.Dib K, Melander F, Andersson T. Role of p190RhoGAP in beta 2 integrin regulation of RhoA in human neutrophils. J Immunol. 2001;166(10):6311–6322. doi: 10.4049/jimmunol.166.10.6311. [DOI] [PubMed] [Google Scholar]
- 47.Janiak A, Zemskov EA, Belkin AM. Cell surface transglutaminase promotes RhoA activation via integrin clustering and suppression of the Src-p190RhoGAP signaling pathway. Mol Biol Cell. 2006;17(4):1606–1619. doi: 10.1091/mbc.E05-06-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Etienne-Manneville S, Hall A. Cdc42 regulates GSK-3beta and adenomatous polyposis coli to control cell polarity. Nature. 2003;421(6924):753–756. doi: 10.1038/nature01423. [DOI] [PubMed] [Google Scholar]
- 49.Carrigan SO, Pink DB, Stadnyk AW. Neutrophil transepithelial migration in response to the chemoattractant fMLP but not C5a is phospholipase D-dependent and related to the use of CD11b/CD18. J Leukoc Biol. 2007;82(6):1575–1584. doi: 10.1189/jlb.0806528. [DOI] [PubMed] [Google Scholar]
- 50.Kiosses WB, Shattil SJ, Pampori N, Schwartz MA. Rac recruits high-affinity integrin alphavbeta3 to lamellipodia in endothelial cell migration. Nat Cell Biol. 2001;3(43):316–320. doi: 10.1038/35060120. [DOI] [PubMed] [Google Scholar]
- 51.Powner DJ, Pettitt TR, Anderson R, Nash GB, Wakelam MJ. Stable adhesion and migration of human neutrophils requires phospholipase D-mediated activation of the integrin CD11b/CD18. Mol Immunol. 2007;44(412):3211–3221. doi: 10.1016/j.molimm.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 52.Francis JW, Todd RF, 3rd, Boxer LA, Petty HR. Sequential expression of cell surface C3bi receptors during neutrophil locomotion. J Cell Physiol. 1989;140(3):519–523. doi: 10.1002/jcp.1041400317. [DOI] [PubMed] [Google Scholar]
- 53.Leitinger B, Hogg N. The involvement of lipid rafts in the regulation of integrin function. J Cell Sci. 2002;115(5):963–972. doi: 10.1242/jcs.115.5.963. [DOI] [PubMed] [Google Scholar]
- 54.Anderson DC, Springer TA. Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu Rev Med. 1987;38:175–194. doi: 10.1146/annurev.me.38.020187.001135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}