

Abstract
DGCR8 (DiGeorge Critical Region 8) is an essential microRNA (miRNA) processing protein that recognizes primary transcripts of miRNAs (pri-miRNAs) and triggers their cleavage by the Drosha nuclease. We previously found that Fe(III) heme binds and activates DGCR8. Here we report that in HeLa cells, DGCR8 undergoes two proteolytic events that produce two C-terminal fragments called DGCR8C1 and DGCR8C2, respectively. DGCR8C2 accumulates during apoptosis and is generated through cleavage by a caspase. The caspase cleavage site is located in the central loop of the heme-binding domain. Cleavage of DGCR8 by caspase-3 in vitro results in loss of the otherwise tightly bound Fe(III) heme cofactor, dissociation of the N- and C-terminal proteolytic fragments, and inhibition of the pri-miRNA processing activity. These results reveal an intrinsic mechanism in the DGCR8 protein that seems to have evolved for regulating miRNA processing via association with Fe(III) heme and proteolytic cleavage by caspases. Decreased expression of miRNAs has been observed in apoptotic cells, and this change was attributed to caspase-mediated cleavage of a down-stream miRNA processing nuclease Dicer. We suggest that both the Drosha and Dicer cleavage steps of the miRNA maturation pathway may be inhibited in apoptosis and other biological processes where caspases are activated.
Keywords: microRNA biogenesis, RNA-binding protein, Pasha, apoptosis, DiGeorge syndrome, ferric heme
Introduction
MicroRNA (miRNAs) function in diverse biological processes, including apoptosis and developmental timing and patterning.1 Among the >500 human miRNAs, some are directly involved in diseases such as cancers.2 Both altered expression of individual miRNAs and global regulation of miRNAs have been demonstrated in development and in cancers.3, 4 Mature miRNAs are around 22 nt in length. They are processed from long primary transcripts of miRNAs (pri-miRNAs) through sequential cleavages by the Drosha and Dicer nucleases in the nucleus and cytoplasm, respectively.5, 6 The cleavage of pri-miRNAs by Drosha requires its RNA-binding partner DGCR8.7–10 Drosha and DGCR8 copurify with each other and are collectively called the Microprocessor complex. Knockdown of Dgcr8 using RNA interference results in enhanced cellular transformation and tumorigenesis,11 consistent with a link between global down-regulation of miRNAs and cancer development. Knockout of Dgcr8 in embryonic stem cells and in mice results in depletion of nearly all miRNAs.12, 13 Along with ∼30 other nearby genes located in chromosome 22, DGCR8 is heterozygously deleted in DiGeorge syndrome patients.14 A Dgcr8+/− mouse model displays abnormal miRNA biogenesis, and functional deficits in the nervous system similar to the neurological symptoms in DiGeorge syndrome patients.15–17 This observation suggests that the haploinsufficiency of DGCR8 directly contributes to the disease, and a proper expression level of DGCR8 is critical for normal miRNA processing. Consistent with this view, the Microprocessor complex regulates the stability of DGCR8 mRNA by cleaving its miRNA-like hairpins.18, 19
The overall domain structure of the 773-residue DGCR8 protein has been revealed in previous studies. The C-terminal region of DGCR8 contains two double-stranded RNA-binding domains (dsRBDs) and a highly conserved C-terminal tail (CTT). This C-terminal region is sufficient for processing of at least some pri-miRNAs.20–23 The N-terminal region of DGCR8 (residues 1–275) includes a nuclear localization signal.21 The central region of DGCR8 (residues 276–498) is a dimeric heme-binding domain (HBD) and each DGCR8 dimer binds one heme (iron–protoporphorin IX) molecule.22 The N-terminal portion of the HBD contains a dimerization domain that contributes a surface for association with heme.24 Importantly, DGCR8 strongly prefers Fe(III) heme over Fe(II) heme and Fe(III) heme activates DGCR8 for pri-miRNA processing.25 Thus, the central region of DGCR8 appears to serve a regulatory function. The fact that the activity of DGCR8 is regulated by Fe(III) heme is consistent with a relatively recent recognition that heme serves as a signaling molecule for diverse cellular functions.26
In the current study, we show that the DGCR8 protein is regulated through a cleavage by caspases. Caspases are a family of proteases that play essential roles in apoptosis, necrosis, and inflammation.27 Twelve caspases have been identified in humans. They use cysteine residues in their active sites. Caspases recognize specific sequences on the surface of target proteins, cleave polypeptide bonds at the C-terminal side of aspartate residues, and modulate their activity. For example, cleavage of the Forkhead transcription factor FOXO3a by caspase-3-like proteases separates the N-terminal DNA-binding domain of FOXO3a from its C-terminal transactivation domain and inactivates its gene regulation activities.28 Hundreds of cellular proteins have been identified to be cleaved by caspases.29–31 It is interesting to note that caspases often target multiple proteins in common biochemical pathways.30
Here, we report that DGCR8 is cleaved in HeLa cells in two proteolytic events. The approximate locations of the cleavage sites have been mapped. The current study focuses on the cleavage that occurs in a loop in the middle of the HBD and is mediated by caspases. In vitro cleavage of recombinant heme-bound DGCR8 results in loss of heme and separation of the two proteolytic fragments. The caspase-cleavage product of DGCR8 is not active in pri-miRNA processing. The biological implications of our findings are discussed.
Results
In vivo proteolytic cleavage of DGCR8
To address the expression and activity of DGCR8, we transfected human HeLa cells with a plasmid expressing N-flag-DGCR8, a N-terminally flag-tagged DGCR8 [Fig. 1(A)]. SDS-PAGE of whole-cell extracts from these cells and immunoblotting (IB) using an anti-DGCR8 antibody detected full-length N-flag-DGCR8 and two additional bands with lower molecular masses [Fig. 1(B), lanes 2 and 3]. The full-length N-flag-DGCR8 has lower SDS-PAGE mobility, corresponding to ∼110 kDa, than would be expected based on its molecular mass (86 kDa). Lower SDS-PAGE mobility has been generally observed in previously characterized recombinant DGCR8 proteins (see below).22 The two lower-molecular-mass bands, termed DGCR8C1 and DGCR8C2, were likely proteolytic fragments from full-length DGCR8 instead of translation products from alternatively spliced DGCR8 mRNAs, as the expression cassette did not contain introns. IB of these whole-cell extracts with anti-flag antibody detected only N-flag-DGCR8 [Fig. 1(B), lane 6], suggesting that both DGCR8C1 and DGCR8C2 were C-terminus-proximal fragments of DGCR8. The N-terminal fragments originated from these cleavages were not observed, and thus must have been degraded quickly in cells. We named the cleavage sites that are responsible for production of the DGCR8C1 and DGCR8C2 fragments cleavage site 1 (CS1) and 2 (CS2), respectively.
Figure 1.

Mapping in vivo cleavage sites in DGCR8. A: Diagrams of DGCR8 and its expression constructs used in this study. The flag tags are denoted by “F.” B: Immunoblots of whole-cell extracts from HeLa cells. Lane 1, mock transfection. Lanes 2–8, HeLa cells transfected with the plasmids expressing either wild-type (WT) N-flag-DGCR8 or its N-terminal truncations. Extracts were prepared 2 days post-transfection and were analyzed by IB using anti-DGCR8 and anti-flag antibodies after SDS-12% PAGE. The bands of full-length DGCR8 as well as the DGCR8C1 and DGCR8C2 fragments are indicated. C: Same as in (B) except that DGCR8 constructs with two flag tags was used. D: Calibration of molecular masses of DGCR8 and its fragments versus their mobility (Rf) in SDS-12% PAGE, for estimating the molecular masses of the DGCR8C1 and DGCR8C2 fragments with improved accuracy. The mobility of the DGCR8C1 fragment was arbitrarily set to 1. The known molecular masses of the recombinant DGCR8 proteins used for this calibration are: NC9 (29 kDa); NC20-8 (residues 276–589, with a TEV cleavage site between residues 499 and 500; 38 kDa); NC1 (54 kDa); N-flag-DGCR8ΔN239 (62 kDa); and N-flag-DGCR8ΔN199 (67 kDa). E: A schematic drawing indicating locations of CS1 (approximate) and CS2.
To map the CS1 and CS2, we engineered a series of deletion and tagged DGCR8 expression constructs. This strategy was used, instead of mass spectrometry or N-terminal sequencing, because the (ectopic) expression level of DGCR8 was relatively low due to regulation by more than one posttranscriptional mechanisms (Refs.18, 19 and our unpublished data). We expressed two N-terminal truncation constructs of DGCR8 in HeLa cells, including N-flag-DGCR8ΔN119 (lacking N-terminal residues 1–119) and N-flag-DGCR8ΔN159 (lacking N-terminal residues 1–159) [Fig. 1(A)]. Whole-cell extracts of these cells were examined using SDS-12% PAGE and IB with anti-DGCR8 and anti-flag antibodies. The ΔN119 and ΔN159 mutants were clearly cleaved in vivo [Fig. 1(B), lanes 4 and 5]. The DGCR8C1 and DGCR8C2 species generated from these truncations were electrophoretically indistinguishable from those produced from full-length N-flag-DGCR8 and were not detected by the anti-flag antibody [Fig. 1(B), lanes 7 and 8]. These observations suggest that the CS1 and CS2 are located on the C-terminal side of residue 159.
We constructed two additional derivatives of N-flag-DGCR8 that contained a second flag epitope [Fig. 1(A)]. N,C-flag-DGCR8 contained a C-terminal flag and lacked the proteolytically vulnerable residues 752–773, which were found to be readily clipped off during purification of DGCR8 from Escherichia coli extracts (Supporting Information Fig. S1). N,275-flag-DGCR8 contained the 8-residue flag sequence between residues 275 and 276 [Fig. 1(A)]. Since neither proteolytic fragments of N-flag-DGCR8 contains the N-terminal flag tag [Fig. 1(B)], the presence of flag in the cleavage products of a dual-flag construct must be contributed by the second flag epitope. Indeed, the DGCR8C2 fragment generated from N,C-flag-DGCR8 contained the flag epitope and migrated slightly faster than the DGCR8C2 fragment produced from N-flag-DGCR8 as expected due to the deletion of C-terminal 22 residues [Fig. 1(C), lanes 2 and 5]. These observations indicate that DGCR8C2 shares the C-terminus with full-length DGCR8. Much less DGCR8C1 fragment was generated from N,C-flag-DGCR8 than the DGCR8C2 fragment, but was weakly visible on the anti-flag IB, implying that the DGCR8C1 fragment may contain the C-terminus of full-length protein as well. However, we cannot rule out the possibility that DGCR8C1 is generated through two proteolytic cleavages, with one of them located close to the C-terminus of the full-length protein. N,275-flag-DGCR8 was cleaved in vivo to produce a DGCR8C1 that could be recognized by both anti-DGCR8 and anti-flag antibodies, and a DGCR8C2 that is not detected by the anti-flag antibody [Fig. 1(C), lanes 3 and 6]. Together with the findings from the N-terminal truncations, these results indicate that the CS1 is located between residues 160 and 275 of DGCR8 and the CS2 is on the C-terminal side of amino acid 275.
We next measured the electrophoretic mobility of the cleavage fragments to further narrow down the location of these cleavage sites. Because of the lower SDS-PAGE mobility of DGCR8 proteins, we constructed a mobility-versus-mass calibration curve specifically for DGCR8 using various recombinant DGCR8 fragments [Fig. 1(D)]. As expected, logarithm of the mobility (Rf) had a linear relationship with molecular mass. This curve predicted the molecular mass of, for example, the DGCR8276–751 (NC1) fragment to be 55 kDa, close to its actual Mr of 54 kDa. On the basis of this calibration, we predicted molecular masses of the DGCR8C1 and DGCR8C2 fragments to be 57 and 40 kDa, respectively. Given all the evidence described above, the CS1 would be close to residue 275 on its N-terminal side and the CS2 would be around residue 400, in the middle of the HBD [Fig. 1(E)]. These estimates also imply that DGCR8C2 can be derived from either full-length DGCR8 or DGCR8C1.
Apoptosis results in accumulation of the DGCR8C2 fragment
An important class of intracellular proteases is caspases that serve pivotal functions in programmed cell death (apoptosis).27 To test whether the DGCR8 protein is cleaved during apoptosis, we transiently expressed N-flag-DGCR8 in HeLa cells and induced apoptosis through either the intrinsic (internally initiated by damage and stress) pathway using etoposide or the extrinsic (death receptor-mediated) pathway using the tumor necrosis factor alpha (TNFα) and cycloheximide (CHX).27 Etoposide is a chemotherapy drug that inhibits topoisomerase II and causes DNA breakages. TNFα binds to TNF receptor 1 (a death receptor family member) and triggers the formation of two distinct signaling complexes, one regulates inflammation and promotes cell survival, the other initiates pro-apoptotic signaling. CHX sensitizes TNFα-treated cells for apoptosis.32 Whole-cell extracts were prepared and were examined using SDS-PAGE and IB (Fig. 2). IB using an antibody against activated caspase-3 showed a large increase of activated caspase-3 at 24 h in the etoposide-treated cells and at 18 h in the TNFα/CHX-treated cells, confirming the onset of apoptosis (Fig. 2, lanes 4 and 7). DGCR8C2 accumulated during apoptosis, indicative of activation of the protease that is responsible for generating this fragment. In contrast, the levels of full-length N-flag-DGCR8 and the endogenous Drosha did not change much in the etoposide-treated cells, whereas those in the cells treated with TNFα and CHX diminished. The latter observation is likely caused by the inhibition of translation by CHX.
Figure 2.
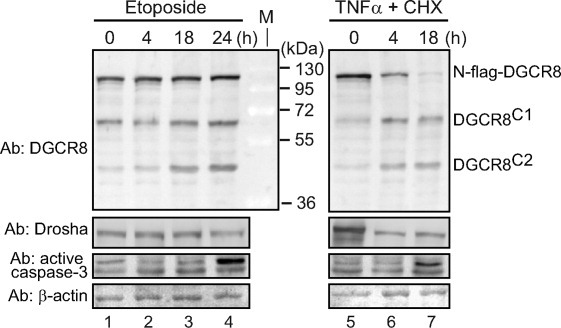
The DGCR8C2 fragment accumulates in apoptotic cells. Immunoblots of whole-cell extracts of HeLa cells that transiently expressed N-flag-DGCR8. Twenty-four hours post-transfection, cells were treated with either etoposide (100 μM) or the combination of TNFα (10 ng/mL) and cycloheximide (20 μg/mL) to induce apoptosis, and extracts were prepared at the indicated time of treatment.
The DGCR8C1 fragment appears to moderately accumulate during apoptosis, suggesting that the DGCR8C1-generating protease may be activated. In this study, we focus on characterizing the DGCR8C2-producing cleavage. Potential functions of the DGCR8C1 fragment, which appears to be produced by a different protease (see below), will be delineated in a separate study.
DGCR8C2 is generated through cleavage by caspases in vivo and in vitro
Consistent with the accumulation of DGCR8C2 during apoptosis, we found that human DGCR8 sequence near the predicted region of CS2 (around residue 400) contains a potential caspase cleavage site, LDEPD396↓S (↓ is the site of cleavage) [Fig. 3(A)]. DEPD396S (positions P4-P1 and P1′ in the general nomenclature of protease cleavage sites) is highly conserved and is predicted to be cleaved by caspases in general.33 As a hydrophobic residue at the P5 position, Leu392 is predicted to enhance cleavage at Asp396 by caspase-2;34 however, Leu392 is only conserved among mammals. Ser397 should make a favorable P1′ residue in a caspase cleavage site. Furthermore, the sequence in this region is rich in acidic residues and might contain additional caspase cleavage sites. For example, DEKD409↓P is also conserved. However, Asp409 is substituted by a glutamate in frog. Because aspartate at the P1 position is universally required for cleavage by caspases, this substitution is expected to destroy the caspase cleavage site. Charged residues and proline at the P1′ position are poorly tolerated by caspases.33, 35 Thus, Pro410 might make DEKD409P a relatively unfavorable caspase site. Nevertheless, because suboptimal cleavage sites might be compensated by additional interactions between protease and substrates, we cannot rule out Asp409 as a potential cleavage site. The sequences of insect and worm DGCR8 homologs (called Pasha) in this region are also rich in acidic residues, but are too diverge to be aligned with those from humans and other organisms.
Figure 3.
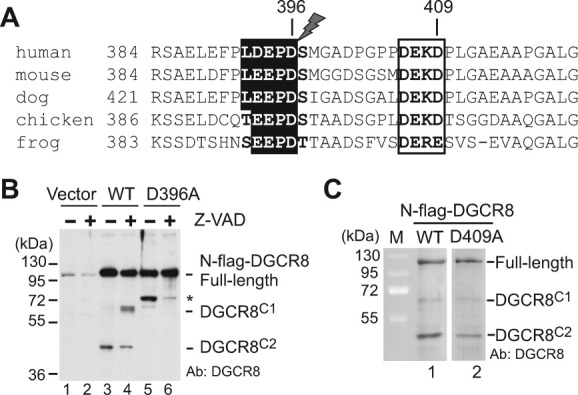
DGCR8 is cleaved by caspases between Asp396 and Ser397 in HeLa cells. A: Sequence alignment of DGCR8 from humans and several other organisms around the cleavage site that produced DGCR8C2. Two potential caspase recognition sites are highlighted. B: Immunoblot of nuclear extracts from HeLa cells transiently expressed N-flag-DGCR8 (wild type and D396A), and treated with and without Z-VAD(Ome)-FMK, a pan-caspase inhibitor. Z-VAD was added to the media to the final concentration of 20 μM 24-h post-transfection, followed by incubation for 24 h. The asterisk indicates a DGCR8 species accumulated when the cleavage at the CS2 site was blocked by the D396A mutation. C: Immunoblot of HeLa nuclear extracts containing transiently expressed N-flag-DGCR8 D409A mutants, as well as the WT. Lanes 1 and 2 are from the same immunoblot.
To examine if DGCR8C2 is produced by caspases, HeLa cells transiently expressing N-flag-DGCR8 were incubated with or without the pan-caspase inhibitor Z-VAD(Ome)-FMK for 24 h, and nuclear extracts were analyzed using SDS-PAGE and IB. Consistent with a previous report showing that DGCR8 is present nearly exclusively in the nucleus,21 we found that nuclear extracts contained majority of full-length and cleaved DGCR8 proteins and showed IB bands similar to those in whole cell extracts [compare Fig. 3(B), lane 3, with Fig. 1(B), lane 2]. Because nuclear extracts contain little cytoplasmic proteins, IB using nuclear extracts has lower background and appears to be more sensitive than that using whole-cell extracts. During the treatment with Z-VAD(Ome)-FMK, the abundance of DGCR8C2 decreased [Fig. 3(B), lanes 3 and 4], indicating that DGCR8C2 is likely generated by caspases. Concomitantly, DGCR8C1 accumulated, suggesting that this fragment is not a direct product of caspase cleavage.
To test if the predicted caspase cleavage sites were used to generate DGCR8C2, we transiently expressed in HeLa cells two single mutants of DGCR8, D396A and D409A, in which the critical aspartate residue in each site was mutated. The D396A mutation blocked the production of DGCR8C1 [Fig. 3(B), lane 5], whereas D409A did not [Fig. 3(C)]. These results suggest that Asp396 is the primary caspase cleavage site on DGCR8. However, we cannot rule out the possibility that Asp409 may be cleaved by caspases in other biological contexts. An additional DGCR8 fragment [marked by “*” in Fig. 3(B)], larger than DGCR8C1 in size, appeared when the cleavage at CS2 was eliminated by the D396A mutation. Z-VAD(Ome)-FMK treatment greatly reduced the intensity of this band [Fig. 3(B), lane 6], suggesting that the “*” fragment may also be generated via caspase cleavage. Because the “*” fragment was not observed in wild-type DGCR8, we did not study it further.
We next validated the cleavage of DGCR8 in vitro. N-terminal His6-tagged NC1 proteins, wild-type and the D396A and D409A mutants, were expressed in E. coli, purified and tested in cleavage assays using recombinant active human caspase-3 (Fig. 4). Subsequent SDS-PAGE analyses showed that the wild-type NC1 was completely cleaved by caspase-3 into two fragments with apparent molecular masses of 41 and 18 kDa, respectively (Fig. 4, lanes 1 and 2). This result agrees well with the C-terminal fragment of 41 kDa and N-terminal fragment of 15 kDa expected from cleavage of His6-NC1 at Asp396. The His6 tag often makes a protein migrate slower than expected for the actual molecular mass, and thus can explain the larger apparent molecular mass of the N-terminal fragment. The D396A mutant failed to be cleaved by caspase-3 and D409A was cleaved just like the wild-type NC1 (Fig. 4, lanes 3–6). Thus, we conclude that caspase-3 cleaves DGCR8 between Asp396 and Ser397 in vitro.
Figure 4.
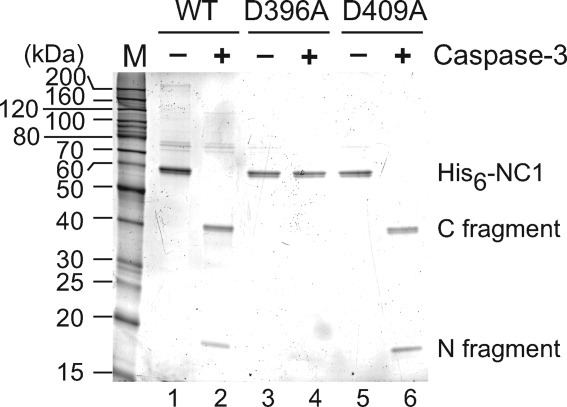
Caspase-3 cleaves recombinant DGCR8 in vitro. Purified His6-NC1 proteins, expressed in E. coli, were incubated with recombinant active caspase-3 at 30°C for 7 h. The cleavage reactions were analyzed using SDS-12% PAGE, and the gel was stained using SYPRO Red.
The caspase cleavage site is located in a flexible loop dispensable for heme binding
Cleavage sites of proteins are often located in a surface loop, making them accessible to proteases.33 Residues 354–414 of DGCR8, which is located in the central region of the HBD and contains CS2, are predicted by the Predictor of NOn-Regular Secondary Structure program36 to be relatively unstructured. In a previous study, we characterized DGCR8276–412, which contained the dimerization domain and the putative central loop of HBD, using nuclear magnetic resonance (NMR).24 Only ∼50 peaks were observed in the region of the heteronuclear single quantum coherence (HSQC) spectrum that was indicative of ordered three-dimensional structures. These peaks were probably contributed mostly by the similar number of residues in the dimerization domain (residues 298–353), of which we have determined a crystal structure.24 Thus, the residues beyond the dimerization domain in DGCR8276–412 are likely unstructured.
We wondered if the CS2-containing loop is important for heme binding to DGCR8. To address this question, we deleted residues 377–410 in the HBD-His6 expression construct of human DGCR8 (HBDΔ377–410-His6), expressed it in E. coli and purified it following the same procedure as described for the wild-type protein.24 The purified HBDΔ377–410-His6 protein contained Fe(III) heme as indicated by the electronic absorption spectrum (Fig. 5). The absorption peak wavelengths at 368, 450, and 557 nm are nearly identical to those of the wild-type HBD-Fe(III) heme complex.24 The ratio of absorptions at 450 nm (by heme) and 280 nm (contributed primarily by the protein, but influenced by heme) was 0.84, also similar to the ratio of 0.80 for HBD-His6.24 Thus, we conclude that the central loop of DGCR8 HBD is not required for association with heme. Presenting the caspase cleavage site may be a primary function of this loop.
Figure 5.
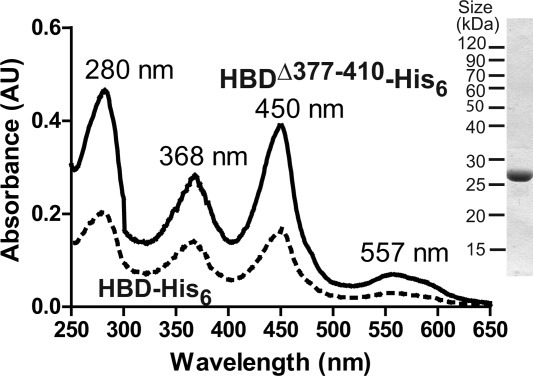
The central loop of the DGCR8 HBD in which the caspase cleavage site resides is dispensable for heme binding. HBDΔ377–410-His6 was overexpressed in E. coli and purified using affinity chromatography followed by SEC. Shown are the electronic absorption spectra of HBDΔ377–410-His6 (solid line) and the wild-type HBD-His6 (dotted line, as previously reported [24]). On the right is a SDS gel image of the purified HBDΔ377–410-His6 protein stained with Coomassie Brilliant Blue.
Caspase cleavage of DGCR8 results in loss of heme and separation of two halves of HBD
We characterized the effects of caspase-mediated cleavage of DGCR8 on its interaction with Fe(III) heme using purified recombinant heme-bound DGCR8276–700 and active recombinant human caspase-3. The DGCR8276–700 contains the intact HBD and two dsRBDs [Fig. 1(A)], and associates with Fe(III) heme in the same manner as that of the NC1 protein.37 This DGCR8 construct does not contain the CTT, which includes an amphipathic helix and appears to increase the tendency of DGCR8 proteins to aggregate.37 Incubation of DGCR8276–700 with recombinant active human caspase-3 resulted in complete digestion of DGCR8276–700 into two fragments with the expected molecular masses (N-terminal fragment ∼14 kDa, C-terminal fragment ∼32 kDa) as indicated by SDS-PAGE [Fig. 6(A)]. Interestingly, the cleavage of DGCR8276–700 by caspase-3 is accompanied by a complete loss of association with heme, indicated by the disappearance of the prominent peaks at 366 and 450 nm in the electronic absorption spectra [Fig. 6(B)]. The absorption of the digested DGCR8276–700 in the 350–500 nm region was not zero and was likely contributed by the released heme molecules.
Figure 6.
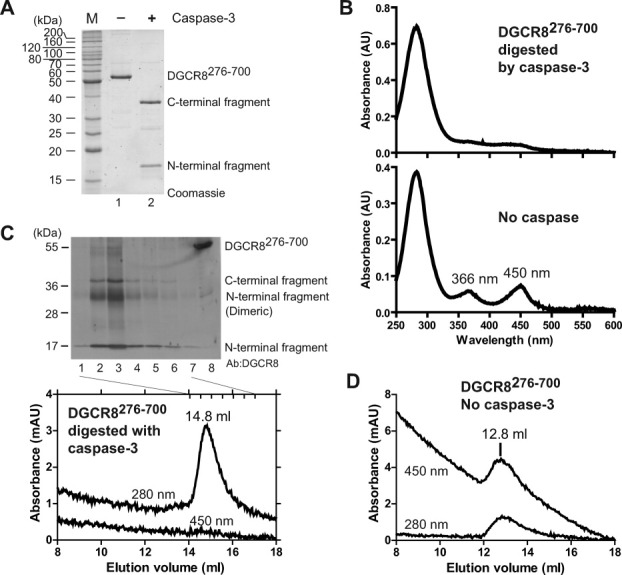
Cleavage of recombinant DGCR8 by caspase-3 results in loss of heme and dissociation of the cleavage fragments. A: DGCR8276–700, produced in E. coli, was incubated with and without active caspase-3 at 30°C for 7 h and the reactions were analyzed using SDS-12% PAGE stained with SYPRO Red. B: Electronic absorption spectra of DGCR8276–700 with and without cleavage by caspase-3. C: SEC analysis of DGCR8276–700 cleaved by caspase-3. Immunoblot of the indicated SEC fractions using anti-DGCR8 antibody is shown above the chromatogram. The centers of the fractions are marked by short vertical bars in the chromatogram. D: SEC of DGCR8276–700 incubated under the same condition as in (C) except without caspase-3.
If heme is dissociated from DGCR8 on caspase-3 cleavage, will the two proteolytic fragments separate from each other? We investigated this question by characterizing the cleaved DGCR8276–700 using size exclusion chromatography (SEC), followed by SDS-PAGE and IB. Both fragments eluted later (14.8 mL) than intact Fe(III) heme-bound DGCR8276–700 dimer (12.8 mL) [Fig. 6(C,D)]. This result indicates that the fragments must have separated from each other. The N-terminal cleavage fragment contains the dimerization domain, has a molecular mass of 28 kDa (twice the mass of the single polypeptide chain) and elutes in a volume similar to that of the 32-kDa C-terminal fragment, which appeared to be monomeric. Our crystal structure of the dimerization domain shows that the dimerization interface is very extensive, and that the two Cys352 residues are right next to each other.24 Hence dimeric species, either covalently linked through a disulfide or non-covalently associated, are sometimes observed on denaturing gels for relatively short dimerization-domain-containing proteins, like reported previously24 and herein. Therefore, we conclude that the caspase cleavage of DGCR8 results in dissociation with Fe(III) heme and separation of the two halves of the HBD.
Caspase cleavage inhibits the activity of DGCR8
We next tested the pri-miRNA processing activity of the caspase-cleavage product of DGCR8. We designed DGCR8397–751 to approximate the in vivo-produced DGCR8C2 fragment, expressed it in E. coli and characterized it using biochemical assays (Fig. 7). The strategy of using a recombinant protein to mimic the cleavage product helped us to avoid complications from potential cleavage of Drosha by caspases during reconstituted pri-miRNA processing assays. Purified DGCR8397–751 was soluble, most likely to be monomeric, and did not bind heme as indicated by the lack of absorption peaks in the Soret (400–500 nm) region [Fig. 7(A,B)]. The latter result was consistent with the absence of an intact HBD and in particular the residues essential for high-affinity heme binding (Cys352 and Trp329) in both DGCR8397–751 and DGCR8C2. In reconstituted pri-miRNA processing assays, DGCR8397–751 displayed much reduced activity for pri-miR-30a and was inactive for pri-miR-380 [Fig. 7(C,D)]. Increasing the concentration of DGCR8397–751 from 50–100 nM to 200 nM and 400 nM failed to rescue the cleavage of pri-miR-30a and pri-miR-380 by Drosha. Residual levels of activity are often observed for in vitro processing of pri-miR-30a with DGCR8 mutants that are inactive in vivo (M.G, R.S., and F.G, manuscript in preparation). Therefore, we conclude that cleavage of DGCR8 into DGCR8C2 by caspases inhibits its pri-miRNA processing activity.
Figure 7.
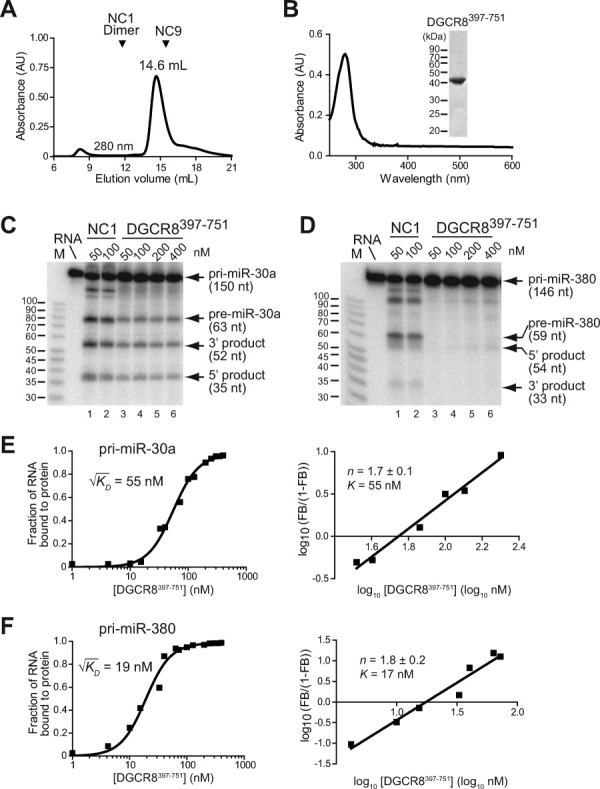
Caspase cleavage of DGCR8 inhibits the pri-miRNA processing activity. A: Size exclusion chromatogram of DGCR8397–751 (41 kDa), produced in E. coli, suggests that it is monomeric. The elution volumes of monomeric NC9 (29 kDa) and heme-bound NC1 dimer (109 kDa) are marked for comparison. B: Electronic absorption spectrum of DGCR8397–751 (the 14.6- mL peak fraction in panel A) lacks peaks in the 350–600 nm region, indicating that DGCR8397–751 does not have any heme bound. A Coomassie-stained SDS gel of this peak fraction is shown in the inset. C and D: The in vitro pri-miRNA processing assays indicate that DGCR8397–751 is inhibited. Pri-miR-30a (C) and pri-miR-380 (D) fragments were uniformly labeled with 32P, and was incubated for 45 min at 37°C with purified recombinant His6-Drosha390–1374 and different concentrations of NC1 (lanes 1–2) or DGCR8397–751 (lanes 3–6). The reactions were analyzed using 15% denaturing PAGE and autoradiography. For convenience of comparison, the indicated concentrations of the dimeric NC1 and monomeric DGCR8397–751 refer to individual polypeptide chains. E and F: Filter binding assays of DGCR8397–751 with 32P-labeled pri-miR-30a (E) and pri-miR-380 (F). The data are best fit with a cooperative dimer model, in which two DGCR8397–751 molecules bind to a pri-miRNA cooperatively. Hill plots of the binding data are shown on the right in each panel. The K [defined as 10(x-intercept)] and Hill constant (n) values are average ± standard deviation of four independent measurements.
We next investigated if the lack of pri-miRNA processing activity of DGCR8397–751 was due to altered interaction with pri-miRNAs using filter binding assays. The results showed that this functionally inhibited DGCR8 protein bound both pri-miR-30a and pri-miR-380 [Fig. 7(E,F)]. The data were best fit with a cooperative dimer model in which two DGCR8397–751 protein molecules bind cooperatively to one RNA molecule. Square roots of the dissociation constants (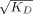 ), which appropriately indicate the protein concentrations at which 50% of the radiolabeled RNA is occupied, are 55 nM for pri-miR-30a and 19 nM for pri-miR-380. These values are similar to that observed for pri-miR-30a with the active Fe(III) heme-bound DGCR8276–751 (NC1) protein (24 nM),22 indicating that the reduced processing activity of DGCR8397–751 is unlikely due to altered affinity for pri-miRNAs.
), which appropriately indicate the protein concentrations at which 50% of the radiolabeled RNA is occupied, are 55 nM for pri-miR-30a and 19 nM for pri-miR-380. These values are similar to that observed for pri-miR-30a with the active Fe(III) heme-bound DGCR8276–751 (NC1) protein (24 nM),22 indicating that the reduced processing activity of DGCR8397–751 is unlikely due to altered affinity for pri-miRNAs.
We previously showed that assembly of DGCR8 to a trimer of dimers on pri-miRNAs is required for triggering cleavage of pri-miRNAs by Drosha.37 Formation of the higher order structure by DGCR8 on pri-miRNAs is indicated by highly cooperative binding: the filter binding data of Fe(III) heme-bound NC1 for pri-miR-30a and pri-miR-21 are best fit with a cooperative trimer model in which three NC1 dimers bind cooperatively with one pri-miRNA molecule, and the Hill coefficients (n) are around 3.22, 37 In contrast, DGCR8397–751 shows reduced cooperativity on binding to pri-miR-30a and pri-miR-380: consistent with the best fit of binding data using a cooperative dimer model, the Hill coefficients are 1.7 ± 0.1 and 1.8 ± 0.2, respectively [Fig. 7(E,F)]. Similar reduction of binding cooperativity without large changes in affinity for pri-miRNAs has been observed for DGCR8 mutants such as C352A and W329A that are deficient in both pri-miRNA processing and heme binding in vitro.22, 24 Altogether, these results suggest that the reduced cooperativity upon binding to a pri-miRNA, likely caused by the failure to add the third DGCR8 molecule to the pri-miRNA, may be the mechanism responsible for inhibition of the pri-miRNA processing activity of DGCR8 through caspase cleavage.
Discussion
Here, we characterize proteolytic cleavages of an essential miRNA processing factor DGCR8 using cultured mammalian cells and biochemical assays. We report that the human DGCR8 protein is cleaved by caspases at a conserved cleavage site in the central loop of the HBD. The caspase-mediated cleavage causes dissociation of Fe(III) heme cofactor from native recombinant DGCR8 protein and inhibition of its pri-miRNA processing activity.
Our study reveals an intrinsic mechanism in the DGCR8 protein for regulating the Drosha processing step of the miRNA maturation pathway. The central loop (containing at least residues 377–410) in the HBD that contains the caspase cleavage site is dispensable for association with heme, suggesting a primary function of presenting the cleavage sites to caspases. Our recent study showed that NC1-Fe(III) heme complex is very stable—no heme dissociation was detected after the complex was incubated with apomyoglobin, a heme scavenger with an extremely high affinity (KD ∼3 × 10−15 M), for 4 days at room temperature.38 Remarkably, the cleavage of NC1 by caspase-3 quickly results in complete dissociation of Fe(III) heme from NC1 within the time frame of the cleavage reaction (7 h), accompanied by physical separation of the two halves of HBD and inhibition of its pri-miRNA processing activity. In another recent study, we found that the activity of apoNC1 (heme-free) dimer is inhibited in the absence of heme; and Fe(III) heme, but not Fe(II) heme, binds the apoNC1 dimer and activates it for pri-miRNA processing.25 Thus, Fe(III) heme-mediated activation and caspase-mediated inhibition appear to be coupled with each other for regulating the DGCR8 protein.
Recent reports showed that Dicer, the RNase III family enzyme responsible for cleavage of pre-miRNAs into miRNA duplexes in the cytoplasm, is cleaved and inactivated by caspases during apoptosis.39–41 Shah and coworkers showed that RNA interference via DNA vector-based expression of small hairpin RNAs (shRNAs) was abrogated in apoptotic cells, and the abundance of natural miRNAs was reduced;40 and these observations were attributed to cleavage and inactivation of Dicer by caspases. In C. elegans, cleavage of DCR-1 (Dicer) by CED-3 (a caspase) destroys the ribonuclease activity, but activates a deoxyribonuclease activity that is responsible for initiating chromosome fragmentation.41 Our findings that DGCR8 is cleaved by caspases in apoptotic cells and that the cleavage inhibits its activity suggest that the Drosha processing step in the miRNA biogenesis pathway may also be blocked during apoptosis. Because DGCR8 functions upstream of Dicer in miRNA maturation, inactivation of DGCR8 likely serves as the first barrier to block miRNA processing. Because caspases are activated in both the nucleus and cytoplasm during apoptosis, both DGCR8 and Dicer may be inhibited due to cleavages by caspases. Thus, our study offers a more complete view of the relationship between miRNA biogenesis and caspase functions.
A number of miRNAs have been shown to be involved in the regulation of apoptosis by targeting apoptotic factors, including caspases, and negatively regulate their expression.2, 42 For example, miR-15 and miR-16a induce apoptosis by targeting BCL2.43 The let-7a miRNA has been shown to down-regulate the expression of caspase-3 in human squamous carcinoma and hepatocellular carcinoma cells and hence suppress therapeutics-induced cancer cell death.44 miR-24a targets caspase-9 and represses apoptosis in developing neural retina.45 Some miRNAs are proapoptotic, others are antiapoptotic. Their effects on apoptosis depend on the cellular context. Regulation of miRNA processing through cleavage and inactivation of DGCR8 and Dicer by caspases suggest that there could be potential feedback and feedforward mechanisms between caspases and miRNA biogenesis, which in turn determine cellular sensitivity to apoptosis signals. Indeed, in glucocorticoid-induced apoptosis of lymphocytes, both decreased expression of miRNA processing factors, including Drosha, DGCR8 and Dicer, and repression of many abundant miRNAs have been observed; and repression of the miRNA processing pathway is important for cell life/death decisions.46 It will be interesting to investigate if DGCR8 and Dicer are cleaved and inactivated by caspases during this process and hence their posttranslational regulation contributes to the decreased miRNA processing.
Caspases also play critical roles in nonapoptotic functions. Caspase-3 activity is important for neurogenesis and synaptic activity.47 Capase-8 is required for embryonic development, for monocyte differentiation and for T cell activation.48 Caspase-2 is involved in DNA damage response, cell cycle regulation, and tumor suppression.49 It would be interesting to examine potential alteration of miRNA biogenesis by these caspases in relevant biological processes.
Materials and Methods
Reagents
A polyclonal antibody to DGCR8 was produced via injecting recombinant DGCR8275–751 in rabbits by Open Biosystems (Huntsville, AL). The untagged DGCR8275–751 fragment (called NC1) was expressed in E. coli and purified as described.22 It contained approximately equal amounts of heme-bound and heme-free forms. Anti-β-actin and anti-flag antibodies, as well as cycloheximide, etoposide and TNFα, were purchased from Sigma (St. Louis, MO). Anti-Drosha and anti-active caspase-3 antibodies were from Cell Signaling Technology (Danvers, MA). Z-VAD(Ome)-FMK was from Alexis Biochemicals (San Diego, CA). Recombinant human caspase-3 was from EMD Chemicals (Gibbstown, NJ).
Plasmids
A plasmid, termed pN-flag-DGCR8, that expressed N-flag-DGCR8 in mammalian cells, was constructed by inserting the PCR-amplified full-length human DGCR8 cDNA sequence into the pCMV-Tag2A vector (Stratagene, La Jolla, CA), between its BamHI and EcoRI sites. The PCR primers were 5′-GCGGATCCCATGGAGACAGATGAGAGCCC-3′ (forward) and 5′-GCTAGAATTCTCAGGGTGCATCTTGCACTGA-3′ (reverse). The BamHI site (GGATCC) in the DGCR8 cDNA sequence was mutated to GGATCA without altering the encoded amino acid sequence. For aims of another study (Gong and Guo, unpublished data), a siRNA target site in the DGCR8 cDNA was also eliminated by codon-synonymous substitutions. The derivatives of pN-flag-DGCR8 were constructed using the standard PCR method. The HBDΔ377–410-His6 expression plasmid was engineered by inserting the coding sequence into the pET-24a(+) vector between the NdeI and NotI sites (EMD Chemicals). The coding sequences of untagged DGCR8276–700 and DGCR8397–751, followed by a stop codon, were inserted into the pET-24a(+) vector between the NdeI and EcoRI sites. The coding sequences of all plasmids were verified via sequencing.
Cell cultures and transfection
Human HeLa cell line was grown in DMEM medium (Invitrogen, Carlsbad, CA), 5% fetal bovine serum (Omega Scientific, Tarzana, CA) and 5% newborn calf serum (Omega Scientific) (complete medium) in 5% CO2 at 37°C. Before transfection, ∼1 × 106 HeLa cells were seeded in 6-well plates in complete medium. Transient transfections were carried out using Lipofectamine 2000 (Invitrogen), according to manufacturers' protocols. Plasmids were purified using the HiSpeed Maxi Plasmid Kit (Qiagen, Valencia, CA).
Immunoblotting
HeLa cells were washed twice with PBS and collected via centrifugation at 4°C. To prepare whole cell extracts, ∼1 × 106 cells were resuspended in 0.1 mL of a lysis buffer containing 0.1M KCl, 5 mM EDTA, 20 mM Tris-HCl (pH 8.0), 1× Halt Protease Inhibitor Cocktail (Thermo Scientific, Rockford, IL). The cells were sonicated with 15 1-s pulses at 29% amplitude with 2-s breaks, using a 750-Watt VibraCell ultrasonic processor (Sonics, Newton, CT). The extracts were centrifuged at 16,000g for 15 min at 4°C, and the supernatants were used as whole-cell extracts. Nuclear extracts used were prepared using a previously described micropreparation protocol.50 The PageRuler Plus prestained protein ladder (Fermentas, Glen Burnie, MD) was used as molecular mass markers. The samples were mixed with equal volume of 2 × SDS-sample loading buffer (1% SDS, 10% glycerol, 0.04% bromophenol blue, 20 mM DTT, 0.1M Tris-HCl, pH 6.8) and were analyzed using SDS-12% PAGE followed by electroblotting to Immobilon-P membrane (Millipore, Billerica, MA). The membrane was incubated for 60 min in PBST [PBS with 0.1% (v/v) Tween-20] containing 5% (w/v) nonfat milk to block nonspecific binding, followed by incubation overnight with primary antibodies diluted 1:5000 (rabbit anti-DGCR8), 1:40,000 (mouse anti-β-actin), 1:1000 (mouse anti-flag), 1:1000 (rabbit anti-Drosha), or 1:10,000 (rabbit anti-active caspase-3). Membranes were washed three times in PBST, followed by incubation for 2 h at room temperature with goat anti-rabbit antibodies conjugated with horseradish peroxidase (HRP; Promega, Madison, WI) (1:30,000 dilution) or anti-mouse antibodies conjugated with HRP (Novagen, Madison, WI) (various dilutions were used). The membrane was washed thoroughly in PBST, and the ECL Plus Western Blotting Detection System (GE Healthcare, Piscataway, NJ) protocol was then used according to the manufacturer's instructions. The images were recorded using chemiluminescence and autoradiography films, or using chemifluorescence and Typhoon 9410 Variable Mode Imager (GE Healthcare). Bands were quantified using the program Quantity One (version 4.4.1, Bio-Rad, Hercules, CA). Statistical analyses, curve fitting, and plotting were performed using Prism (version 4.03, GraphPad, La Jolla, CA).
Expression and Purification of HBDΔ377–410-His6, DGCR8276–700 and DGCR8397–751
Human HBDΔ377–410-His6 was expressed and purified using the same procedure as that described for HBD-His6.24 Briefly, HBDΔ377–410-His6 was overexpressed in E. coli BL21(DE3) CodonPlus cells (Stratagene) with δ-aminolevulinic acid, a precursor of heme biosynthesis, added to 1 mM at the time of induction. HBDΔ377–410-His6 was purified using Ni affinity chromatography followed by size exclusion chromatography.
Expression in E. coli and purification of DGCR8276–700 and DGCR8397–751 were carried out using procedures previously described for DGCR8276–751 (NC1).22
NC1, NC9 (DGCR8499–751), and NC20-8 were used as DGCR8-specific molecular mass markers. NC20-8 (38 kDa) contained residues 275–589 of DGCR8, with a TEV protease cleavage site inserted between residues 499 and 500. Expression in E. coli and purification of NC1, NC20-8, and NC9 proteins were performed using procedures described previously for NC1.22
Electronic absorption spectroscopy
The spectra were recorded using a Beckman DU500 absorption spectrophotometer.
In vitro cleavage of DGCR8 by caspase-3
Approximately 9 μg of DGCR8276–700 dimer was incubated with 2000 units of recombinant active caspase-3 at 30°C for 7 h. The assay buffer contained 100 mM NaCl, 10 mM DTT, 1 mM EDTA, 10% (v/v) glycerol, 0.1% (w/v) CHAPS, 50 mM HEPES pH 7.4. The cleavage reactions were analyzed using SDS-12% PAGE. The gels were stained using SYPRO Red (Cambrex, Rockland, ME) and were scanned using Typhoon 9410 Variable Mode Imager.
In vitro pri-miRNA binding and pri-miRNA processing assays
These assays were performed as previously described.22
Acknowledgments
The authors thank A. Varshavsky for extensive discussion and comments, D. Black, E. De Robertis, G. Weinmaster and J. Wohlschlegel for advice.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 4.Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006;20:2202–2207. doi: 10.1101/gad.1444406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 6.Faller M, Guo F. MicroRNA biogenesis: there's more than one way to skin a cat. Biochim Biophys Acta. 2008;1779:663–667. doi: 10.1016/j.bbagrm.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- 8.Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. The Microprocessor complex mediates the genesis of MicroRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 9.Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landthaler M, Yalcin A, Tuschl T. The human DiGeorge syndrome critical region gene 8 and its D. melanogaster homolog are required for miRNA biogenesis. Curr Biol. 2004;14:2162–2167. doi: 10.1016/j.cub.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 11.Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–677. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet. 2007;39:380–385. doi: 10.1038/ng1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yi R, Pasolli HA, Landthaler M, Hafner M, Ojo T, Sheridan R, Sander C, O'Carroll D, Stoffel M, Tuschl T, Fuchs E. DGCR8-dependent microRNA biogenesis is essential for skin development. Proc Natl Acad Sci USA. 2009;106:498–502. doi: 10.1073/pnas.0810766105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shiohama A, Sasaki T, Noda S, Minoshima S, Shimizu N. Molecular cloning and expression analysis of a novel gene DGCR8 located in the DiGeorge syndrome chromosomal region. Biochem Biophys Res Commun. 2003;304:184–190. doi: 10.1016/s0006-291x(03)00554-0. [DOI] [PubMed] [Google Scholar]
- 15.Stark KL, Xu B, Bagchi A, Lai WS, Liu H, Hsu R, Wan X, Pavlidis P, Mills AA, Karayiorgou M, Gogos JA. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat Genet. 2008;40:751–760. doi: 10.1038/ng.138. [DOI] [PubMed] [Google Scholar]
- 16.Fenelon K, Mukai J, Xu B, Hsu PK, Drew LJ, Karayiorgou M, Fischbach GD, Macdermott AB, Gogos JA. Deficiency of Dgcr8, a gene disrupted by the 22q11.2 microdeletion, results in altered short-term plasticity in the prefrontal cortex. Proc Natl Acad Sci USA. 2011;108:4447–4452. doi: 10.1073/pnas.1101219108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schofield CM, Hsu R, Barker AJ, Gertz CC, Blelloch R, Ullian EM. Monoallelic deletion of the microRNA biogenesis gene Dgcr8 produces deficits in the development of excitatory synaptic transmission in the prefrontal cortex. Neural Dev. 2011;6:11. doi: 10.1186/1749-8104-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han J, Pedersen JS, Kwon SC, Belair CD, Kim YK, Yeom KH, Yang WY, Haussler D, Blelloch R, Kim VN. Posttranscriptional crossregulation between Drosha and DGCR8. Cell. 2009;136:75–84. doi: 10.1016/j.cell.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Triboulet R, Chang HM, Lapierre RJ, Gregory RI. Post-transcriptional control of DGCR8 expression by the Microprocessor. RNA. 2009;15:1005–1011. doi: 10.1261/rna.1591709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 21.Yeom KH, Lee Y, Han J, Suh MR, Kim VN. Characterization of DGCR8/Pasha, the essential cofactor for Drosha in primary miRNA processing. Nucleic Acids Res. 2006;34:4622–4629. doi: 10.1093/nar/gkl458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faller M, Matsunaga M, Yin S, Loo JA, Guo F. Heme is involved in microRNA processing. Nat Struct Mol Biol. 2007;14:23–29. doi: 10.1038/nsmb1182. [DOI] [PubMed] [Google Scholar]
- 23.Sohn SY, Bae WJ, Kim JJ, Yeom KH, Kim VN, Cho Y. Crystal structure of human DGCR8 core. Nat Struct Mol Biol. 2007;14:847–853. doi: 10.1038/nsmb1294. [DOI] [PubMed] [Google Scholar]
- 24.Senturia R, Faller M, Yin S, Loo JA, Cascio D, Sawaya MR, Hwang D, Clubb RT, Guo F. Structure of the dimerization domain of DiGeorge Critical Region 8. Protein Sci. 2010;19:1354–1365. doi: 10.1002/pro.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barr I, Smith AT, Chen Y, Senturia R, Burstyn JN, Guo F. Ferric, not ferrous, heme activates RNA-binding protein DGCR8 for primary microRNA processing. Proc Natl Acad Sci USA. 2012;109:1919–1924. doi: 10.1073/pnas.1114514109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mense SM, Zhang L. Heme: a versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res. 2006;16:681–692. doi: 10.1038/sj.cr.7310086. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27:6194–6206. doi: 10.1038/onc.2008.297. [DOI] [PubMed] [Google Scholar]
- 28.Charvet C, Alberti I, Luciano F, Jacquel A, Bernard A, Auberger P, Deckert M. Proteolytic regulation of Forkhead transcription factor FOXO3a by caspase-3-like proteases. Oncogene. 2003;22:4557–4568. doi: 10.1038/sj.onc.1206778. [DOI] [PubMed] [Google Scholar]
- 29.Luthi AU, Martin SJ. The CASBAH: a searchable database of caspase substrates. Cell Death Differ. 2007;14:641–650. doi: 10.1038/sj.cdd.4402103. [DOI] [PubMed] [Google Scholar]
- 30.Mahrus S, Trinidad JC, Barkan DT, Sali A, Burlingame AL, Wells JA. Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell. 2008;134:866–876. doi: 10.1016/j.cell.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dix MM, Simon GM, Cravatt BF. Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell. 2008;134:679–691. doi: 10.1016/j.cell.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nolop KB, Ryan US. Enhancement of tumor necrosis factor-induced endothelial cell injury by cycloheximide. Am J Physiol. 1990;259:L123–129. doi: 10.1152/ajplung.1990.259.2.L123. [DOI] [PubMed] [Google Scholar]
- 33.Timmer JC, Salvesen GS. Caspase substrates. Cell Death Differ. 2007;14:66–72. doi: 10.1038/sj.cdd.4402059. [DOI] [PubMed] [Google Scholar]
- 34.Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW. Substrate specificities of caspase family proteases. J Biol Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- 35.Stennicke HR, Renatus M, Meldal M, Salvesen GS. Internally quenched fluorescent peptide substrates disclose the subsite preferences of human caspases 1, 3, 6, 7 and 8. Biochem J. 2000;350:563–568. [PMC free article] [PubMed] [Google Scholar]
- 36.Rost B, Yachdav G, Liu J. The PredictProtein server. Nucleic Acids Res. 2004;32:W321–W326. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Faller M, Toso D, Matsunaga M, Atanasov I, Senturia R, Chen Y, Zhou ZH, Guo F. DGCR8 recognizes primary transcripts of microRNAs through highly cooperative binding and formation of higher-order structures. RNA. 2010;16:1570–1583. doi: 10.1261/rna.2111310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barr I, Smith AT, Senturia R, Chen Y, Scheidemantle BD, Burstyn JN, Guo F. DiGeorge Critical Region 8 (DGCR8) is a double-cysteine-ligated heme protein. J Biol Chem. 2011;286:16716–16725. doi: 10.1074/jbc.M110.180844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matskevich AA, Moelling K. Stimuli-dependent cleavage of Dicer during apoptosis. Biochem J. 2008;412:527–534. doi: 10.1042/BJ20071461. [DOI] [PubMed] [Google Scholar]
- 40.Ghodgaonkar MM, Shah RG, Kandan-Kulangara F, Affar EB, Qi HH, Wiemer E, Shah GM. Abrogation of DNA vector-based RNAi during apoptosis in mammalian cells due to caspase-mediated cleavage and inactivation of Dicer-1. Cell Death Differ. 2009;16:858–868. doi: 10.1038/cdd.2009.15. [DOI] [PubMed] [Google Scholar]
- 41.Nakagawa A, Shi Y, Kage-Nakadai E, Mitani S, Xue D. Caspase-dependent conversion of Dicer ribonuclease into a death-promoting deoxyribonuclease. Science. 2010;328:327–334. doi: 10.1126/science.1182374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lima RT, Busacca S, Almeida GM, Gaudino G, Fennell DA, Vasconcelos MH. MicroRNA regulation of core apoptosis pathways in cancer. Eur J Cancer. 2011;47:163–174. doi: 10.1016/j.ejca.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 43.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsang WP, Kwok TT. Let-7a microRNA suppresses therapeutics-induced cancer cell death by targeting caspase-3. Apoptosis. 2008;13:1215–1222. doi: 10.1007/s10495-008-0256-z. [DOI] [PubMed] [Google Scholar]
- 45.Walker JC, Harland RM. microRNA-24a is required to repress apoptosis in the developing neural retina. Genes Dev. 2009;23:1046–1051. doi: 10.1101/gad.1777709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith LK, Shah RR, Cidlowski JA. Glucocorticoids modulate microRNA expression and processing during lymphocyte apoptosis. J Biol Chem. 2010;285:36698–36708. doi: 10.1074/jbc.M110.162123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.D'Amelio M, Cavallucci V, Cecconi F. Neuronal caspase-3 signaling: not only cell death. Cell Death Differ. 2010;17:1104–1114. doi: 10.1038/cdd.2009.180. [DOI] [PubMed] [Google Scholar]
- 48.Maelfait J, Beyaert R. Non-apoptotic functions of caspase-8. Biochem Pharmacol. 2008;76:1365–1373. doi: 10.1016/j.bcp.2008.07.034. [DOI] [PubMed] [Google Scholar]
- 49.Kumar S. Caspase 2 in apoptosis, the DNA damage response and tumour suppression: enigma no more? Nat Rev Cancer. 2009;9:897–903. doi: 10.1038/nrc2745. [DOI] [PubMed] [Google Scholar]
- 50.Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.