

Abstract
d-Tetronitrose is a nitro-containing tetradeoxysugar found attached to the antitumor and antibacterial agent tetrocarcin A. The biosynthesis of this highly unusual sugar in Micromonospora chalcea requires 10 enzymes. The fifth step in the pathway involves the transfer of a methyl group from S-adenosyl-l-methionine (SAM) to the C-3′ carbon of dTDP-3-amino-2,3,6-trideoxy-4-keto-d-glucose. The enzyme responsible for this transformation is referred to as TcaB9. It is a monomeric enzyme with a molecular architecture based around three domains. The N-terminal motif contains a binding site for a structural zinc ion. The middle- and C-terminal domains serve to anchor the SAM and dTDP–sugar ligands, respectively, to the protein, and the active site of TcaB9 is wedged between these two regions. For this investigation, the roles of Tyr 76, His 181, Tyr 222, Glu 224, and His 225, which form the active site of TcaB9, were probed by site-directed mutagenesis, kinetic analyses, and X-ray structural studies. In addition, two ternary complexes of the enzyme with bound S-adenosyl-l-homocysteine and either dTDP-3-amino-2,3,6-trideoxy-4-keto-d-glucose or dTDP-3-amino-2,3,6-trideoxy-d-galactose were determined to 1.5 or 1.6 Å resolution, respectively. Taken together, these investigations highlight the important role of His 225 in methyl transfer. In addition, the structural data suggest that the methylation reaction occurs via retention of configuration about the C-3′ carbon of the sugar.
Keywords: SAM-dependent methyltransferases, tetrocarcin A, d-tetronitrose, enzyme mechanism, unusual sugar biosynthesis, tetradeoxysugar
Introduction
Tetrocarcin A, first identified from a broth culture of Micromonospora chalcea in 1980, belongs to the spirotetronate antibiotic family1 and exhibits both antibiotic and antitumor activities.2 As shown in Scheme 1, it is composed of three structurally distinct moieties: a polycyclic tetronolide core, a monosaccharide referred to as d‐tetronitrose (highlighted in purple), and a tetrasaccharide composed of alternating l‐digitoxose and l‐amicetose residues.3, 4 Interestingly, the antibacterial activities of the tetrocarcins are directly proportional to the number of deoxysugars decorating their aglycone rings.5
Scheme 1.
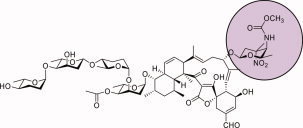
The structure of tetrocarcin A.
The biosynthesis of d-tetronitrose requires 10 enzymes, many of which catalyze very intriguing reactions.4, 6 One example is a C-3′-methyltransferase, which is referred to as TcaB9 and is the focus of this investigation. It catalyzes the fifth step in the biosynthetic pathway, namely the transfer of a methyl group from S-adenosyl-l-methionine (SAM) to the C-3′ position of dTDP-3-amino-2,3,6-trideoxy-4-keto-d-glucose as illustrated in Scheme 2.
Scheme 2.
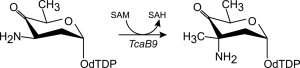
The reaction catalyzed by TcaB9.
The first three-dimensional structure of TcaB9 was recently reported from our laboratory.7 The enzyme was cloned from M. chalcea and was shown to belong to the Class I family of SAM-dependent enzymes.7 A ribbon representation of the monomeric enzyme is presented in Figure 1(a). Quite unexpectedly, the electron density map revealed the presence of a zinc ion coordinated by four cysteine residues near the N-terminus. The molecular architecture of TcaB9 was shown to consist of three domains: an N-terminal region that harbored the zinc-binding site, a middle domain that adopted the canonical “SAM-binding” fold, and a C-terminal domain that appeared to be related to the middle domain by an approximate twofold rotational axis, thus suggesting that TcaB9 arose via gene duplication.
Figure 1.
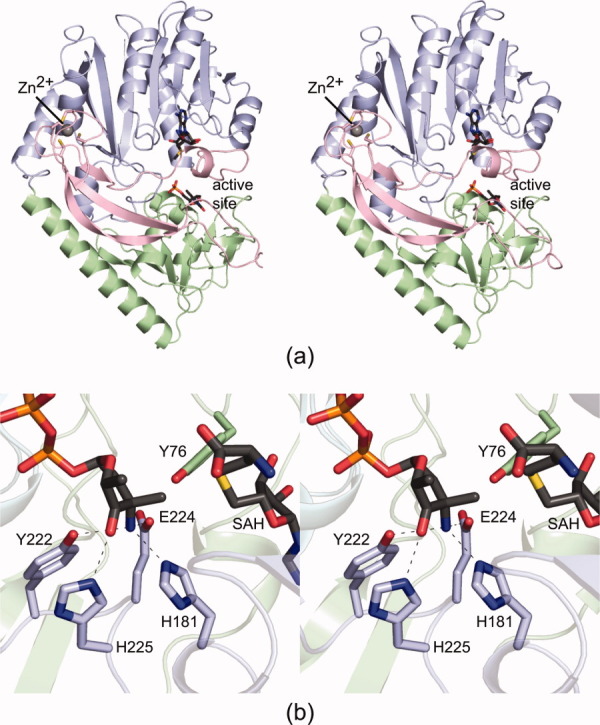
The structure of TcaB9. Shown in (a) is a ribbon representation of the monomeric enzyme in complex with SAM and dTMP. The ligands are drawn in stick representations. The TcaB9 fold can be envisioned in terms of three domains, which are colored in pink, blue, and green. A close-up view of the enzyme active site with bound SAH and dTDP-3-amino-2,3,6-trideoxy-4-keto-3-methyl-d-glucose is presented in (b). The dashed lines indicate potential hydrogen bonding interactions between the protein and the ligands. All figures were prepared with the software package PyMOL.16
In the initial X-ray crystallographic analysis of TcaB9, two ternary complexes were described.7 The first was that of the protein crystallized in the presence of SAM and dTMP, and the second was that of the enzyme with bound S-adenosyl-l-homocysteine (SAH) and the dTDP–sugar product. A close-up view of the active site for the enzyme/SAH/dTDP–sugar model is presented in Figure 1(b). As can be seen, there are three amino acid residues, His 181, Tyr 222, and Glu 224, that lie within hydrogen bonding distance to the dTDP–sugar C-3′ amino group. In addition, the imidazole side chain of His 225 hydrogen bonds to the C-4′ keto moiety of the sugar.
Prior to the structural analysis of TcaB9, another C-methyltransferase, TylC3, was biochemically characterized in detail.8 This enzyme is required for the biosynthesis of l-mycarose in Streptomyces fradiae. A catalytic mechanism for the enzyme was proposed to occur through the formation of an enediolate intermediate upon abstraction of a proton from the C-3′ carbon. In addition, inversion of the stereochemistry about the C-3′ position was thought to occur.8
Assuming that the methylation reaction catalyzed by TcaB9 also results in inversion of configuration about the C-3′ carbon, the position of the amino group in the dTDP–sugar product represents a good mimic for the location of the proton in the substrate [Fig. 1(b)]. As such, His 181, Tyr 222, and Glu 224 could all possibly function as a general base for abstraction of the C-3′ proton. On the basis of our three-dimensional models for TcaB9, and in light of the previously reported biochemical data on TylC3, a catalytic mechanism was suggested for the enzyme as highlighted in Scheme 3. Accordingly, it was proposed that the imidazole side chain of His 225 functions to stabilize the enolate intermediate that occurs during catalysis, whereas an enzymatic base removes the proton from the C-3′ carbon. The identity of this base could not be determined from the structural data alone. This proposed mechanism also assumed that inversion of configuration about the C-3′ carbon occurs during the reaction.
Scheme 3.
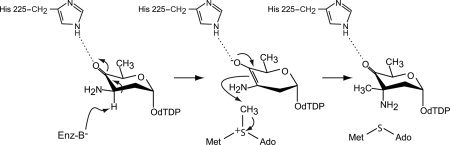
A proposed catalytic mechanism for TcaB9.
Here we report a combined structural and functional analysis to further probe the roles of His 181, Tyr 222, Glu 224, and His 225 in the catalytic mechanism of TcaB9. The positions of these residues are shown in Figure 1(b). For the analyses described here, various site-directed mutant proteins were constructed, their activities tested, and their three-dimensional structures solved to high resolution. In addition, two ternary complexes of the wild-type enzyme with bound SAH and alternative dTDP–sugars were determined. Taken together, these studies provide new insight into the catalytic mechanism of TcaB9 specifically and into the C-3′-methyltransferases in general.
Results and Discussion
Overall structures of the TcaB9 mutant proteins
To ensure that no major conformational changes had occurred upon mutation of specific amino acid residues, the structures of the mutant proteins, H181N, Y222F, E224Q, H225N, and H181N/E224Q, were solved to 1.8 Å resolution or better. The locations of these residues in the TcaB9 active site are displayed in Figure 1(b). X-ray data collection and model refinement statistics are given in Tables I and II, respectively.
Table I.
X-ray Data Collection Statistics
| H181N | Y222F | E224Q | H225N | H181N/E224Q | Y76F | Wild-type/SAH/ dTDP–sugar substrate | Wild-type/SAH/ reduced dTDP–sugar substrate | |
|---|---|---|---|---|---|---|---|---|
| Unit cell dimensions (Å) | a = 100.6 | a = 100.5 | a = 100.4 | a = 102.0 | a = 100.7 | a = 100.8 | a = 100.7 | a = 100.7 |
| b = 114.9 | b = 114.2 | b = 115.0 | b = 114.3 | b = 114.3 | b = 114.4 | b = 114.4 | b = 114.4 | |
| c = 37.8 | c = 37.7 | c = 38.0 | c = 38.0 | c = 37.9 | c = 37.8 | c = 37.7 | c = 37.8 | |
| Resolution limits | 38.0–1.5 (1.6–1.5)a | 38.0–1.4 (1.5–1.4) | 33.0–1.8 (1.9–1.8) | 38.0–1.4 (1.5–1.4) | 38.0–1.5 (1.6–1.5) | 34.0–1.75 (1.85–1.75) | 29.0–1.5 (1.6–1.5) | 38.0–1.6(1.7–1.6) |
| Number of independent reflections | 70194 (11310) | 81798 (13540) | 40896 (5381) | 81683 (13054) | 69471 (11712) | 43793 (6335) | 68740 (11395) | 56697 (8696) |
| Completeness (%) | 97.9 (90.6) | 94.7 (85.2) | 97.6 (94.9) | 93.6 (80.0) | 97.8 (95.3) | 97.0 (92.8) | 96.8 (92.2) | 96.7 (90.6) |
| Redundancy | 4.7 (2.0) | 4.3 (1.7) | 4.0 (2.1) | 4.3 (2.2) | 5.7 (3.0) | 3.4 (1.9) | 4.4 (2.2) | 4.7 (2.4) |
| Avg I/avg σ(I) | 11.2 (2.8) | 10.3 (1.8) | 10.4 (3.4) | 10.5 (2.2) | 10.3 (2.8) | 9.6 (2.7) | 7.4 (2.5) | 11.9 (2.5) |
| Rsym (%)b | 8.4 (29.1) | 7.2 (34.0) | 8.6 (20.9) | 7.6 (32.3) | 7.1 (35.9) | 8.4 (25.5) | 9.9 (32.7) | 7.6 (33.0) |
Statistics for the highest resolution bin.
R = (∑|I - I|/ ΣI) × 100.
= (∑|I - I|/ ΣI) × 100.
Table II.
Refinement Statistics
| H181N | Y222F | E224Q | H225N | H181N/E224Q | Y76F | Wild-type/SAH/ dTDP–sugar substrate | Wild-type/SAH/ reduced dTDP–sugar substrate | |
|---|---|---|---|---|---|---|---|---|
| Resolution limits (Å) | 38.0–1.5 | 38.0–1.4 | 33.0–1.8 | 38.0–1.4 | 38.0–1.5 | 34.0–1.75 | 29.0–1.5 | 38.0–1.6 |
| aR-factor (overall)%/no. of reflections | 19.1/70189 | 20.6/81756 | 17.8/40862 | 19.2/81651 | 18.7/69422 | 16.7/43752 | 18.4/68693 | 17.7/56668 |
| R-factor (working)%/no. of reflections | 18.9/66622 | 20.4/77644 | 17.6/38801 | 19.0/77553 | 18.5/65919 | 16.5/41540 | 18.3/65232 | 17.5/53799 |
| R-factor (free)%/no. of reflections | 22.8/3567 | 23.6/4112 | 21.8/2061 | 21.7/4098 | 21.8/3503 | 20.5/2212 | 21.8/3461 | 21.3/2869 |
| Number of protein atoms | 3222 | 3181 | 3178 | 3206 | 3191 | 3186 | 3190 | 3163 |
| Number of heteroatoms | 515 | 541 | 368 | 527 | 488 | 496 | 488 | 463 |
| Average B values | ||||||||
| Protein atoms (Å2) | 11.3 | 13.4 | 16.8 | 15.2 | 17.7 | 14.2 | 13.3 | 14.0 |
| Metal (Å2) | 13.9 | 12.2 | 16.5 | 17.3 | 20.8 | 19.2 | 12.8 | 14.1 |
| Ligands (Å2) | 8.6 | 8.2 | 14.3 | 10.0 | 13.5 | 9.0 | 8.7 | 9.4 |
| Solvent (Å2) | 23.8 | 25.9 | 25.3 | 27.9 | 29.2 | 25.5 | 24.2 | 24.5 |
| Weighted RMS deviations from ideality | ||||||||
| Bond lengths (Å) | 0.010 | 0.010 | 0.010 | 0.010 | 0.010 | 0.010 | 0.010 | 0.010 |
| Bond angles (°) | 2.3 | 2.3 | 2.3 | 2.3 | 2.3 | 2.3 | 2.3 | 2.3 |
| General planes (Å) | 0.009 | 0.009 | 0.009 | 0.009 | 0.009 | 0.009 | 0.009 | 0.009 |
| Ramachandran plot statistics | ||||||||
| Most favored (%) | 92.8 | 92.0 | 92.3 | 92.6 | 92.9 | 92.8 | 93.7 | 92.5 |
| Additionally allowed (%) | 6.6 | 7.4 | 7.4 | 6.8 | 6.8 | 6.6 | 5.7 | 6.9 |
| Generously allowed (%) | 0.6 | 0.6 | 0.3 | 0.6 | 0.3 | 0.6 | 0.6 | 0.6 |
R-factor = (Σ|Fo–Fc|/Σ|Fo|) × 100 where Fo is the observed structure factor amplitude and Fo is the calculated structure factor amplitude.
All mutant protein structures had SAH and dTDP-3-amino-2,3,6-trideoxy-4-keto-3-methyl-d-glucose (dTDP–product) bound in their active sites with the exception of the H181N/E224Q mutant enzyme, which contained only SAH and dTDP. Overall the mutations resulted in no major conformational perturbations in the polypeptide chain backbones of the various proteins. Indeed, the α-carbons for the mutant proteins superimpose upon the wild-type enzyme with root-mean-square deviations ranging from 0.11 to 0.18 Å.
The three residues that lie within hydrogen bonding distance to the dTDP–sugar amino group are His 181, Tyr 222, and Glu 224, all of which could serve as a general base. The substitution of an asparagine residue for a histidine in the H181N mutant protein merely resulted in a water molecule occupying the cavity that was created upon loss of the imidazole side chain. The removal of a hydroxyl group in the Y222F mutant protein had a somewhat more pronounced effect, however. In the structure of the Y222F/SAH/dTDP–sugar complex, the quality of the electron density corresponding to the pyranosyl moiety of the dTDP–sugar was very weak, suggesting that it was adopting multiple conformations. With respect to the E224Q mutant protein, there were relatively minor shifts in the residues lining the active site pocket. The same was true for the H181N/E224Q double mutant protein.
The H225N mutant protein was the only one that demonstrated a marked change in its active site architecture relative to that of the wild-type enzyme. As can be seen in Figure 1(b), His 225 lies within hydrogen bonding distance to the C-4′ keto group of the dTDP–product. In addition, it is positioned within 3.3 Å of the imidazole side chain of His 181 and forms an almost T-shaped stacking interaction with it. A superposition of the active site regions for the wild-type enzyme and the H225N mutant protein is shown in Figure 2. As can be seen, the asparagine residue of the H225N mutant protein rotates away from the C-4′ keto group, and His 181 adjusts in the active site such that it no longer lies within hydrogen bonding distance to the dTDP–sugar amino group.
Figure 2.
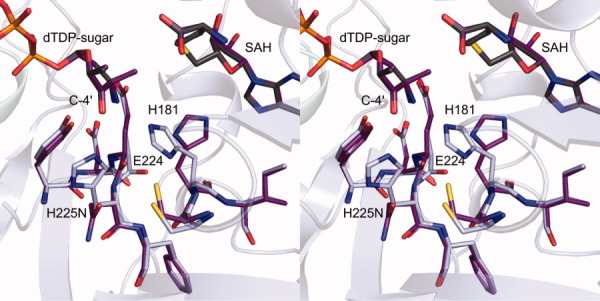
Superposition of the active sites for the wild-type enzyme and the H225N mutant protein. Those amino acid residues highlighted in light blue correspond to the wild-type enzyme, whereas those depicted in purple belong to the H225N mutant protein. The positions of the SAH and dTDP–sugars are virtually identical between the two forms of the enzyme. The conformation adopted by the asparagine in the H225N mutant enzyme creates a steric clash between the backbone carbonyl of Glu 224 and the β-carbon of His 181, which ultimately results in the observed alternative conformation for His 181 in the mutant protein.
As noted in Scheme 3, it is thought that His 225 serves to stabilize the enolate intermediate that occurs during catalysis. To further probe the biochemical role of this residue, activity assays were subsequently conducted on the H225N mutant protein. Strikingly, it exhibited an approximate 36,000-fold decrease in activity as compared to the wild-type enzyme (Table III). Clearly, the loss of the hydrogen bond to the C-4′ keto group upon substitution of a histidine with an asparagine, and the further movement of His 181 away from the amino group of the dTDP–sugar, had a profound effect on catalysis by TcaB9.
Table III.
Specific Activities for Mutant Proteins
| Enzyme | Specific activity (μmol min−1 mg−1) | Fold decrease in activity |
|---|---|---|
| Wild-type | (3.4 ± 0.1) × 10−2 | |
| H181N | (5.6 ± 0.1) × 10−4 | 61 |
| Y222F | (1.0 ± 0.1) × 10−4 | 340 |
| E224Q | (2.6 ± 0.1) × 10−4 | 130 |
| H225N | (9.5 ± 0.8) × 10−7 | 36,000 |
| Y76F | (2.9 ± 0.3) × 10−4 | 120 |
| H181N/E224Q | (1.4 ± 0.1) × 10−6 | 24,000 |
Investigation of the identity of the active site base
The catalytic mechanism originally proposed in Scheme 3 requires an active site base to remove the proton attached to the C-3′ carbon of the dTDP–sugar prior to the methylation event. As noted above, His 181, Tyr 222, and Glu 224 all lie in a position to function as a general base. The catalytic efficiencies of the H181N and E224Q mutant enzymes were mildly perturbed with an observed 61- and 130-fold decrease in kcat/Km relative to the wild-type enzyme, respectively. As noted in Table IV, this effect was primarily due to the reduction in the value of the kcat (wild-type kcat = 1.70 ± 0.05 min−1, H181N kcat = 0.044 ± 0.002 min−1, E224Q kcat = 0.011 ± 0.001 min−1) as the apparent Km values for the dTDP–sugars were similar (wild-type Km = 4.3 ± 0.2 μM, H181N Km = 6.8 ± 1.0 μM, E224Q Km = 3.7 ± 0.2 μM).
Table IV.
Kinetic Parameters for the Wild-type and the Y76F, H181N, and E224Q Mutant Proteins
| Wild-type | Y76F | H181N | E224Q | |
|---|---|---|---|---|
| Vmax (μM min−1) | 0.37 ± 0.01 | 0.0052 ± 0.0002 | 0.019 ± 0.001 | 0.0049 ± 0.0001 |
| Km for dTDP–sugar (μM) | 4.3 ± 0.2 | 4.6 ± 0.2 | 6.8 ± 1.0 | 3.7 ± 0.2 |
| Km for SAM (μM) | 180 ± 30 | 240 ± 30 | 160 ± 20 | 95 ± 20 |
| kcat (min−1) | 1.70 ± 0.05 | 0.012 ± 0.001 | 0.044 ± 0.002 | 0.011 ± 0.001 |
| kcat/Km for dTDP–sugar (M−1 min−1) | 4.0 × 105 | 2.6 × 103 | 6.5 × 103 | 3.0 × 103 |
| kcat/Km for SAM (M−1 min−1) | 9.4 × 103 | 5.0 × 101 | 2.8 × 102 | 1.2 × 102 |
Abbreviations: dTMP, thymidine monophosphate; dTDP, thymidine diphosphate; ESI, electrospray ionization; HEPES, N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid; HEPPS, N-2-hydroxyethylpiperazine-N′-3-propanesulfonic acid; HPLC, high-performance liquid chromatography; NADPH, nicotinamide adenine dinucleotide phosphate.
These data suggest that neither His 181 nor Glu 224, alone, can act as the general base. To test if these two residues act in concert, the specific activity of the H181N/E224Q double mutant enzyme was determined. A 24,000-fold reduction in the specific activity was observed. This decrease in activity compared to the wild-type enzyme or to the individual mutant proteins is significant and suggests that these residues in combination play an important role in the catalytic mechanism of TcaB9. The low catalytic activity of the H181N/E224Q was incompatible with the discontinuous assay employed in our investigation, and thus it was not possible to determine its kcat and Km parameters. As a consequence, the roles of these two residues in substrate binding versus catalysis remain unclear.
The relatively mild decrease (340-fold) in catalytic activity for the Y222F mutant enzyme indicates that Tyr 222 does not function as an active site base. Rather, the kinetic and structural data suggest that it functions to correctly orient the dTDP–sugar into the active site. It may also act as a selector for deoxysugars that have been dehydrated at the C-2′ carbon position.
Structures of TcaB9 complexed with SAH and either the dTDP–substrate or a dTDP–substrate analog
The original catalytic mechanism proposed for TcaB9 was based on the crystal structure of the wild-type enzyme in complex with SAH and its dTDP–sugar product.7 Clearly, the model represents a molecular snapshot after catalysis. But what is the active site conformation prior to catalysis, and how is the dTDP–sugar substrate accommodated? Given the inability to confidently identify an active site base on the basis of the enzyme/SAH/product complex structure and the above-described activity assays, we subsequently determined the molecular architecture of TcaB9 in the presence of SAH and its natural dTDP–sugar substrate. The structure, which represents an abortive complex, was solved to 1.5-Å resolution. A close-up view of the electron density for the dTDP–sugar substrate is presented in Figure 3(a). As can be seen, the density is unambiguous and shows that the amino group attached to the C-3′ carbon of the dTDP–sugar substrate lies in the axial position. Specifically, the S-configuration is observed about the C-3′ carbon.
Figure 3.
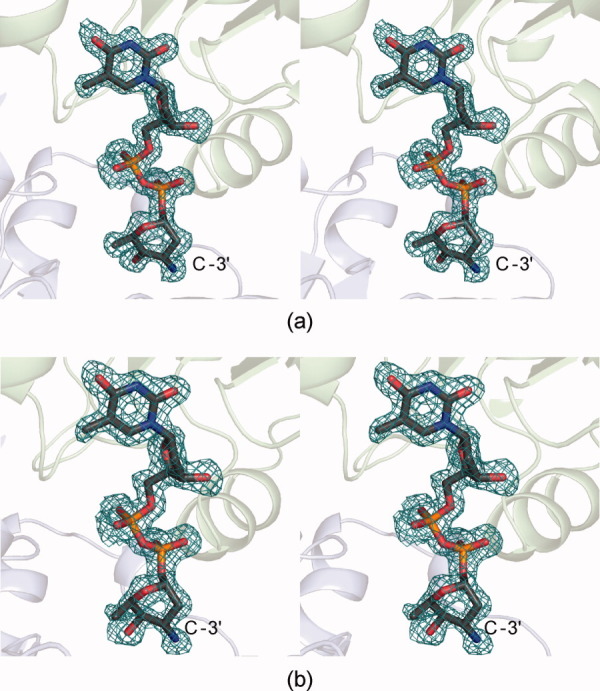
Electron density maps. The electron density shown in (a) corresponds to the TcaB9 substrate. The omit map, contoured at 2σ, was calculated with coefficients of the form (Fo–Fc) where Fo was the native structure factor amplitude, and Fc was the calculated structure factor amplitude. Shown in (b) is the observed electron density for the dTDP-substrate analog in which the C-4′ keto group has been reduced to a hydroxyl moiety.
To confirm that the observed stereochemistry was not an artifact of some type of reaction occurring in the crystalline lattice due to the binding of an activated dTDP–sugar, the substrate analog dTDP-3-amino-2,3,6-trideoxy-d-galactose was subsequently synthesized. This sugar, which is reduced at the C-4′ position, was soaked into crystals of wild-type TcaB9 grown in the presence of SAH, and the structure of the ternary complex was solved to 1.6-Å resolution. Again, the electron density for the sugar was unambiguous [Fig. 3(b)], and it showed the C-3′ amino group pointing toward His 181, Tyr 222, and Glu 224. Overall, there were minimal conformational changes in the TcaB9 polypeptide chain backbone with either bound substrate or substrate analog as compared to the wild-type TcaB9 product complex. Specifically the α-carbons for the TcaB9/SAH/dTDP–substrate or the TcaB9/SAH/dTDP–substrate analog complexes superimpose upon those of the wild-type enzyme with root-mean square deviations of 0.11 or 0.10 Å, respectively.
The catalytic mechanism of TcaB9 originally put forth assumed that inversion of stereochemistry about the C-3′ position of the substrate occurred during catalysis.7 The mechanism was proposed under the assumption that the dTDP–sugar substrate binds in the active site with its C-3′ amino group in the equatorial position as previously suggested.4, 6, 9 The structures of the abortive complexes determined in this investigation, however, and the previously described structure of the TcaB9/SAH/dTDP–product complex, show that the stereochemistry around the C-3′ position is the same for the substrate, the substrate analog, and the product sugar. As a consequence, His 181 and Glu 224 are not in the proper position to abstract the C-3′ proton. If, however, the sugar pucker is different in the true Michaelis complex with bound SAM and dTDP–sugar substrate, then possibly they might function in concert to remove the proton.
Given the structures of the TcaB9/SAH/dTDP–substrate and the TcaB9/SAH/dTDP–substrate analog determined in this investigation, we searched for another residue in the TcaB9 active site that might function as a general base. The only other possible candidate was Tyr 76, which is located in the active site such that its hydroxyl group is oriented between the sulfur of SAH and the C-3′ carbon of the sugar [Fig. 1(b)]. Specifically, in the product complex, the hydroxyl group of Tyr 76 is located at 3.2 Å from the methyl group attached to the C-3′ carbon. The Y76F mutant protein was constructed, and its three-dimensional structure was shown to be nearly identical to that of the wild-type enzyme. The catalytic efficiency of the Y76F mutant enzyme was mildly perturbed with an observed 150-fold decrease in kcat/Km relative to the wild-type enzyme. As noted in Table IV, this effect was primarily due to the reduction in the value of the kcat (wild-type kcat = 1.70 ± 0.05 min−1, Y76F kcat = 0.012 ± 0.001 min−1) as the apparent Km value for the dTDP–sugar was similar (wild-type Km = 4.3 ± 0.2 μM, Y76F Km = 4.6 ± 0.2 μM). These data suggest that Tyr 76 does not act as the general base.
The catalytic mechanism of TcaB9 revisited
The TcaB9 catalyzed methylation of dTDP‐3‐amino‐2,3,6‐trideoxy‐4‐keto‐d‐glucose was proposed to proceed through inversion of configuration about the C‐3′ carbon (Scheme 3). To initiate catalysis, it was thought that an active site base removes the proton on the C‐3′ carbon of the dTDP–sugar substrate thereby generating an enolate intermediate. In the product complex of TcaB9, His 225 was shown to be situated at 2.8 Å from the C‐4′ keto oxygen of the sugar, and it was postulated to stabilize the enolate. Upon collapse of the enolate, the C‐3′ carbon of the sugar attacks the methyl group of SAM generating dTDP‐3‐amino‐2,3,6‐trideoxy‐4‐keto‐3‐methyl‐d‐glucose and SAH.7 Indeed, the kinetic and structural data presented here suggest that His 225 serves an important role in methyl transfer.
The issue of what residue(s) functions as the general base is more complicated, however. Indeed, the structures of the ternary complexes determined in this investigation were especially enlightening because they demonstrated a different stereochemistry of the substrate at the C-3′ carbon than that observed by Chen et al. in the biosynthesis of dTDP-l-epivancosamine.9 On the basis of these complexes, the proposed mechanism for TcaB9 must be modified. Accordingly, we suggest that methylation occurs via retention of configuration about the C-3′ carbon, thus reflecting the stereochemistry of the substrate and the product observed in our X-ray crystallographic models (Scheme 4). The active site base required to remove the proton from the C-3′ carbon of the dTDP–sugar, however, is still unidentified.
Scheme 4.
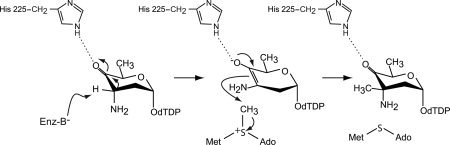
A revised catalytic mechanism for TcaB9.
Materials and Methods
Cloning, site-directed mutagenesis, protein expression, and purification of TcaB9
The gene encoding TcaB9 was previously cloned from M. chalcea to produce the TcaB9-pET28JT expression plasmid used throughout this investigation.7 Six mutant proteins were created via site-directed mutagenesis following the QuikChange® protocol (Stratagene). Incorporation of the desired mutation into each plasmid was confirmed by DNA sequence analysis. Mutant proteins were expressed and purified as previously reported for the wild-type enzyme.7 All proteins were dialyzed against 20 mM HEPES (pH 7.5) and 100 mM NaCl and concentrated to 18–35 mg mL−1 on the basis of a calculated extinction coefficient of 0.68 (mg/mL)−1 cm−1 at 280 nm.
Enzymatic synthesis of the TcaB9 substrate
To synthesize dTDP-3-amino-2,3,6-trideoxy-4-keto-d-glucose, a reaction mixture containing 50 mM HEPES (pH 7.5), 50 mM NaCl, 50 mMl-glutamate, 1 mM dTDP-d-glucose, 0.1 mM pyridoxal 5′-phosphate, 1 mg mL−1 of Escherichia coli RmlB (a 4,6-dehydratase), 0.2 mg mL−1 of Amycolatopsis orientalis EvaA (a 2,3-dehydratase), and 5 mg mL−1M. chalcea TcaB8 (an aminotransferase) was incubated at 37°C for 1 h and quenched via filtration to remove the enzymes. The filtrate was diluted 1:30 with water and the dTDP–sugar was purified via an ÄKTA Purifier HPLC system equipped with a 1 mL Resource Q anion exchange column and using a 5 mL linear gradient from 0 to 250 mM ammonium acetate (pH 6.0). Fractions containing the dTDP–sugar substrate were pooled and concentrated via lyophilization. The sugar concentration was determined by measuring the absorbance of dTDP at 267 nm (ε = 9650 M−1 cm−1), and the sugar purity was assessed via HPLC monitoring at 267 nm. All enzymes required for the dTDP–sugar substrate synthesis were cloned and purified in the laboratory.
Enzymatic synthesis of a TcaB9 substrate analog
To synthesize dTDP-3-amino-2,3,6-trideoxy-d-galactose, a reaction mixture containing 50 mM HEPPS (pH 8.0), 100 mM NaCl, 20 mMl-glutamate, 4 mM dTDP-glucose, 3 mM NADPH, 0.1 mM pyridoxal 5′-phosphate, 0.5 mg mL−1 of E. coli RmlB, 0.1 mg mL−1 of A. orientalis EvaA, 2 mg mL−1 of M. chalcea TcaB8, and 0.5 mg mL−1Streptomyces achromogenes RubN6 (a C-4′ ketoreductase) was incubated at 30°C for 2.5 h and quenched via filtration to remove the enzymes. The filtrate was diluted 1:40 with water and the dTDP–sugar was purified via an ÄKTA Purifier HPLC system equipped with a 6 mL Resource Q anion exchange column and using a 27 mL linear gradient from 0 to 180 mM ammonium acetate (pH 4.0). Fractions containing the dTDP–sugar were pooled and lyophilized to dryness. The final product was subjected to ESI mass spectrometry for analysis. The sugar concentration was determined by measuring the absorbance of dTDP at 267 nm (ε = 9650 M−1 cm−1), and the sugar purity was assessed via HPLC monitoring at 267 nm. Again, all enzymes required for the dTDP–sugar analog synthesis were cloned and purified in the laboratory.
Structural analyses of the TcaB9 mutant proteins and the wild-type protein ternary complexes
Crystallization conditions were surveyed with an enzyme solution containing 15 mg mL−1 of protein, 20 mM HEPES (pH 7.5), 100 mM NaCl, 10 mM dTMP, and 5 mM SAH. The hanging drop method of vapor diffusion was employed with a sparse matrix screen developed in the laboratory. X-ray diffraction quality crystals of the Y76F, H181N, Y222F, E224Q, and H181N/E224Q mutant proteins were obtained by mixing in a 1:1 ratio the enzyme/dTMP/SAH mixture with a precipitant solution composed of 1.2–1.6M sodium/potassium phosphate and 100 mM HEPPS (pH 8.0). Crystals of the H225N mutant protein were grown in the presence of 100 mM HEPES (pH 7.5) rather than HEPPS.
To obtain ternary complexes of the mutant proteins with SAH and the dTDP–sugar product (dTDP-3-amino-2,3,6-trideoxy-4-keto-3-methyl-d-glucose), crystals grown in the presence of SAH and dTMP were transferred to a synthetic mother liquor containing 1.6M sodium/potassium phosphate, 100 mM HEPPS (pH 8.0), 5 mM SAH, and 100 mM NaCl for 4 h to remove dTMP. Next, they were transferred to a synthetic mother liquor containing 1.6M sodium/potassium phosphate, 100 mM HEPPS (pH 8.0), 5 mM SAH, 100 mM NaCl, and 15 mM dTDP-3-amino-2,3,6-trideoxy-4-keto-3-methyl-d-glucose and allowed to soak for 24 h. Finally, the various crystals were placed in a cryo-protectant solution containing 1.95M sodium/potassium phosphate, 100 mM HEPPS (pH 8.0), 3 mM SAH, 100 mM NaCl, 15 mM dTDP-3-amino-2,3,6-trideoxy-4-keto-3-methyl-d-glucose, and 15% ethylene glycol. The dTDP–sugar product was synthesized as previously described.10
For preparation of the wild-type enzyme crystals containing SAH and either the natural dTDP–sugar substrate or the dTDP–sugar substrate analog, crystals were first prepared in an identical manner to that described above for the mutant proteins. These crystals were then soaked in a synthetic mother containing 1.6M sodium/potassium phosphate, 100 mM HEPPS (pH 8.0), 5 mM SAH, and 100 mM NaCl for 4 h to remove the dTMP ligand. They were subsequently transferred to a synthetic mother liquor containing 1.6M sodium/potassium phosphate, 100 mM HEPPS (pH 8.0), 5 mM SAH, 100 mM NaCl, and either 0.5 mM dTDP-3-amino-2,3,6-trideoxy-4-keto-d-glucose (substrate) or 20 mM dTDP-3-amino-2,3,6-trideoxy-d-galactose (substrate analog) and allowed to soak for 24 h. The cryo-protectant conditions were identical to those described above for the mutant proteins except for the replacement of 15 mM dTDP-3-amino-2,3,6-trideoxy-4-keto-3-methyl-d-glucose with either 0.5 mM dTDP-3-amino-2,3,6-trideoxy-4-keto-d-glucose or 20 mM dTDP-3-amino-2,3,6-trideoxy-d-galactose. All of the crystals belonged to the space group P21212 with one polypeptide chain per asymmetric unit.
High-resolution X-ray data sets were collected at 100 K with a Bruker AXS Platinum 135 CCD detector equipped with Montel optics and controlled by the Proteum software suite (Bruker AXS Inc.). All data sets were processed with SAINT version 7.06A (Bruker AXS) and internally scaled with SADABS version 2005/1 (Bruker AXS Inc.). Relevant X-ray data collection statistics are listed in Table I.
The structures of the various TcaB9 complexes were determined via molecular replacement with the software package PHASER11 using as the search probe the previously determined model of the wild-type enzyme in a complex with SAH and its dTDP–sugar product.7 All of the structures were subjected to alternate cycles of manual model building with Coot12 and refinement with Refmac.13 Relevant refinement statistics are given in Table II. The Ramachandran plot statistics presented in Table II were calculated with PROCHECK.14
Activity assays
Specific activities were determined for the wild-type and all mutant proteins in triplicate via a discontinuous HPLC as described below. For each enzyme, a 0.7 mL reaction contained 25 mM HEPPS (pH 8.0), 60 μM dTDP–sugar, 2 mM SAM, and an appropriate amount of the enzyme. Wild-type enzyme, Y76F, H181N, and E224Q mutant proteins were assayed as described below. For the H181N/E224Q, H225N, and Y222F mutant enzymes, 100 μL aliquots were quenched at intervals appropriate for each enzyme. Relative activities are listed in Table III.
Measurement of the kinetic constants for the wild-type enzyme and the Y76F, H181N, and E224Q site-directed mutant proteins
The kinetic constants for the wild-type and the Y76F, H181N, and E224Q mutant proteins were determined following the formation of SAH via a discontinuous HPLC assay using an ÄKTA HPLC system equipped with a 1 mL Resource S cation exchange column. Each reaction was conducted at 23°C and initiated by the addition of enzyme. At each time point, 100 μL aliquots were quenched by the addition of HCl to a final concentration of 0.55M. Afterward, 40 μL of CCl4 were added to the samples, which were then vigorously mixed and centrifuged for 1 min to precipitate the protein. An 80-μL sample of the aqueous phase was diluted 1:28 with water, and 2 mL were loaded onto the Resource S column for analysis. SAH and SAM were separated using a 20 mL linear gradient from 1 to 320 mM HCl with the SAH peak eluting at 260 mM HCl. The area of the peak corresponding to SAH was determined and then correlated to a concentration based on a calibration curve. Initial velocities for each reaction were determined by plotting the SAH concentration versus time and were fit to the Michaelis–Menten equation v = VA/(K + A). The kinetic constants were calculated using non-linear regression,15 where K was the Michaelis constant for the varied substrate and V was the maximum velocity.
For the wild-type enzyme, fourteen 0.7 mL reactions were run, and the initial velocities with either the dTDP–sugar (2–60 μM) or SAM (0.075–2 mM) as the varied substrates were determined at fixed, saturating concentrations of the other substrate. Each reaction mixture also contained 25 mM HEPPS (pH 8.0) and 0.22 μM enzyme. For each reaction, 100 μL aliquots were quenched at time zero and 2 min intervals for 10 min.
For the Y76F, H181N, and E224Q mutant proteins, similar experiments were conducted. Specifically, for each mutant protein, nine 0.7 mL reactions were run, and the initial velocities with either the dTDP–sugar (2–60 μM) or SAM (0.09–2.0 mM) as the varied substrates were determined at fixed, saturating concentrations of the other substrate. Each reaction mixture also contained 25 mM HEPPS (pH 8.0) and 0.44 μM enzyme. For each reaction, 100 μL aliquots were quenched at time zero and 1 h intervals for 5 h. The kinetic parameters are presented in Table IV.
Acknowledgments
The authors thank Dr. James B. Thoden, Ms. Rachel L. Kubiak, and Professor Grover L. Waldrop for helpful discussions. X-ray coordinates have been deposited in the Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (accession nos. 4E2W, 4E2X, 4E2Y, 4E2Z, 4E30, 4E31, 4E32, 4E33).
References
- 1.Tomita F, Tamaoki T, Shirahata K, Kasai M, Morimoto M, Ohkubo S, Mineura K, Ishii S. Novel antitumor antibiotics, tetrocarcins. J Antibiot. 1980;33:668–670. doi: 10.7164/antibiotics.33.668. [DOI] [PubMed] [Google Scholar]
- 2.Morimoto M, Fukui M, Ohkubo S, Tamaoki T, Tomita F. Tetrocarcins, new antitumor antibiotics. 3. Antitumor activity of tetrocarcin A. J Antibiot. 1982;35:1033–1037. doi: 10.7164/antibiotics.35.1033. [DOI] [PubMed] [Google Scholar]
- 3.Tamaoki T, Tomita F. Biosynthesis of tetrocarcin. Incorporation of 14C- and 13C-labeled compounds into tetrocarcin. J Antibiot. 1983;36:595–598. doi: 10.7164/antibiotics.36.595. [DOI] [PubMed] [Google Scholar]
- 4.Fang J, Zhang Y, Huang L, Jia X, Zhang Q, Zhang X, Tang G, Liu W. Cloning and characterization of the tetrocarcin A gene cluster from Micromonospora chalcea NRRL 11289 reveals a highly conserved strategy for tetronate biosynthesis in spirotetronate antibiotics. J Bacteriol. 2008;190:6014–6025. doi: 10.1128/JB.00533-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tamaoki T, Kasai M, Shirahata K, Tomita F. Tetrocarcins E1, E2, F and F-1, new antibiotics. Fermentation, isolation and characterization. J Antibiot. 1982;35:979–984. doi: 10.7164/antibiotics.35.979. [DOI] [PubMed] [Google Scholar]
- 6.Zhang H, White-Phillip JA, Melancon CE, 3rd, Kwon HJ, Yu WL, Liu HW. Elucidation of the kijanimicin gene cluster: insights into the biosynthesis of spirotetronate antibiotics and nitrosugars. J Am Chem Soc. 2007;129:14670–14683. doi: 10.1021/ja0744854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bruender NA, Thoden JB, Kaur M, Avey MK, Holden HM. Molecular architecture of a C-3′-methyltransferase involved in the biosynthesis of D-tetronitrose. Biochemistry. 2010;49:5891–5898. doi: 10.1021/bi100782b. [DOI] [PubMed] [Google Scholar]
- 8.Chen H, Zhao Z, Hallis TM, Guo Z, Liu HH. Insights into the branched-chain formation of mycarose: methylation catalyzed by an (S)-adenosylmethionine-dependent methyltransferase. Angew Chem Int Ed Engl. 2001;40:607–610. doi: 10.1002/1521-3773(20010202)40:3<607::AID-ANIE607>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 9.Chen H, Thomas MG, Hubbard BK, Losey HC, Walsh CT, Burkart MD. Deoxysugars in glycopeptide antibiotics: enzymatic synthesis of TDP-L-epivancosamine in chloroeremomycin biosynthesis. Proc Natl Acad Sci USA. 2000;97:11942–11947. doi: 10.1073/pnas.210395097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruender NA, Thoden JB, Holden HM. X-ray structure of KijD3, a key enzyme involved in the biosynthesis of D-kijanose. Biochemistry. 2010;49:3517–3524. doi: 10.1021/bi100318v. [DOI] [PubMed] [Google Scholar]
- 11.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 13.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 14.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 15.Cleland WW. Statistical analysis of enzyme kinetic data. Methods Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 16.DeLano WL. 2002. The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA, The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA.