

Abstract
3-Deoxy-D-arabino-heptulosonate 7-phosphate synthase (DAHPS) catalyzes the first step in the biosynthesis of a number of aromatic metabolites. Likely because this reaction is situated at a pivotal biosynthetic gateway, several DAHPS classes distinguished by distinct mechanisms of allosteric regulation have independently evolved. One class of DAHPSs contains a regulatory domain with sequence homology to chorismate mutase—an enzyme further downstream of DAHPS that catalyzes the first committed step in tyrosine/phenylalanine biosynthesis—and is inhibited by chorismate mutase substrate (chorismate) and product (prephenate). Described in this work, structures of the Listeria monocytogenes chorismate/prephenate regulated DAHPS in complex with Mn2+ and Mn2+ + phosphoenolpyruvate reveal an unusual quaternary architecture: DAHPS domains assemble as a tetramer, from either side of which chorismate mutase-like (CML) regulatory domains asymmetrically emerge to form a pair of dimers. This domain organization suggests that chorismate/prephenate binding promotes a stable interaction between the discrete regulatory and catalytic domains and supports a mechanism of allosteric inhibition similar to tyrosine/phenylalanine control of a related DAHPS class. We argue that the structural similarity of chorismate mutase enzyme and CML regulatory domain provides a unique opportunity for the design of a multitarget antibacterial.
Keywords: allostery, 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase, chorismate mutase, Listeria monocytogenes, shikimate pathway, multitarget inhibitor
Introduction
The shikimate pathway is composed of the enzymes that catalyze the first seven reactions in the synthesis of a diverse set of important aromatic compounds.1 Chorismate, the product of the shikimate pathway, is positioned at a biosynthetic branch point from which the paths that lead to specific classes of aromatic metabolites first diverge. Flux into postshikimate pathway branches is mediated by four major chorismate using enzymes: (1) chorismate mutase (CM), which converts chorismate to prephenate in phenylalanine and tyrosine biosynthesis; (2) anthranilate synthase, which converts chorismate and l-glutamine to anthranilate in tryptophan biosynthesis; (3) chorismate lyase, which converts chorismate to 4-hydroxybenzoate in ubiquinone biosynthesis; and (4) aminodeoxychorismate synthase, which converts chorismate and l-glutamine to aminodeoxychorismate in folate biosynthesis (Fig. 1).2
Figure 1.
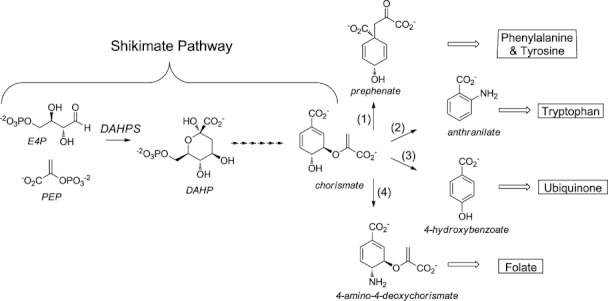
Abridged aromatic metabolite biosynthetic tree. The DAHPS catalyzed reaction and the chorismate branch point at which the paths to specific metabolite classes diverge are emphasized. Major chorismate-using enzymes are (1) CM, (2) anthranilate synthase, (3) chorismate lyase, and (4) aminodeoxychorismate synthase.
3-Deoxy-D-arabino-heptulosonate 7-phosphate synthase (DAHPS) catalyzes the conversion of phosphoenolpyruvate (PEP) and erythrose-4-phosphate to 3-deoxy-D-arabino-heptulosonate 7-phosphate, the first reaction in the shikimate pathway (Fig. 1).3 As the first enzyme in the pathway DAHPS is situated at a pivotal gateway, where pathway input can be efficiently controlled in response to changes in the cellular concentration of pathway outputs. Evolutionary processes have led to at least four disparate mechanisms of conferring DAHPSs allosterically responsive to downstream reaction products.
One type of DAHPS regulation is exemplified by three Escherichiacoli DAHPS isoenzymes, which each have a distinct regulatory domain inserted into the DAHPS core. This renders one isoenzyme responsive to tyrosine, another to phenylalanine, and another to tryptophan.4 A second type of DAHPS regulation is exemplified by the Mycobacterium tuberculosis enzyme, which is synergistically inhibited by tyrosine and tryptophan binding at discrete allosteric sites.5 Additionally, the M. tuberculosis DAHPS forms a noncovalent complex with CM.6 Apparently, in this complex, CM has a regulatory function, because chorismate has also been reported to inhibit the enzyme.7 A third type of DAHPS regulation (DAHPS-FL) is exemplified by the Thermotoga maritima DAHPS, which contains an N-terminal ferredoxin-like (FL) regulatory domain that dimerizes upon the binding of phenylalanine or tyrosine, adopting a conformation where it blocks substrate access to the active site (Supporting Information Fig. 1).8–10 Finally, a fourth type of DAHPS regulation (DAHPS-CML) is exemplified by the Bacillus subtillis DAHPS, which contains an N-terminal domain with sequence homology to CM (CM-like or CML), that is inhibited by CM substrate and product, chorismate, and prephenate.11–14 Although this domain has residual CM activity, because it is catalytically inefficient and has high affinity for prephenate, it has been argued that the CML domain primarily functions in a regulatory, rather than a catalytic role.14 Interestingly, a similarly functioning C-terminal CML domain is linked to the Porphyromonas gingivalis DAHPS—demonstrating that chorismate/prephenate regulated DAHPSs independently arose at least twice over the course of evolutionary history.14
Here, we report the first crystal structures of a DAHPS-CML, from the Gram-positive pathogen Listeria monocytogenes. These structures reveal a striking domain architecture, in which a pair of CML dimers hang loosely off either side of the DAHPS tetramer and provide clues about the possible mechanism of allosteric inhibition. Based on a structural analysis and comparison to other allosterically regulated DAHPSs, we speculate that inhibitor binding promotes the formation of a specific domain-domain interface. This may inhibit DAHPS activity either by blocking substrate access to the active site or by inducing allosteric changes in the catalytic domain.
Results and Discussion
DAHPS-CML domain architecture
Mn2+ and Mn2+ + PEP bound complexes of the L. monocytogenes DAHPS-CML were determined at a resolution of 1.95 Å in the C2 space group (Table I). The two crystal structures are very similar (RMSD = 0.24 Å over 571 Cα atoms) and contain two molecules within the crystallographic asymmetric unit (Supporting Information Fig. 2). Applying a twofold crystallographic symmetry operator to the contents of the asymmetric unit generates the physiological tetramer. The core of the tetramer is composed of the four catalytic DAHPS domains. Within this core, the individual DAHPS domains alternate in direction, so that diagonally related protomers face the same side of the tetramer. A short ∼ 15 amino acid, N-terminal linker connects each of the DAHPS domains to a CML domain [Fig. 2(A)]. The two CML domains that emerge on the same side of the DAHPS tetramer interact to form a structure very similar to previously characterized CM dimers.6, 15 Because this interaction is mirrored by the CML domains emerging from the opposite side of the tetramer, the biological unit can be described as a catalytic DAHPS tetramer sandwiched by a pair of regulatory CML dimers [Fig. 2(B)].
Table I.
Data Collection and Refinement Statistics
| Holoenzyme | PEP complex | |
|---|---|---|
| Space group | C2 | C2 |
| Unit-cell dimensions | ||
| a, b, c (Å) | a = 111.57 | a = 110.16 |
| b = 111.79 | b = 110.89 | |
| c = 81.05 | c = 81.23 | |
| α, β, γ (°) | α =90.00 | α = 90.00 |
| β = 127.63 | β = 127.21 | |
| γ = 90.00 | γ = 90.00 | |
| Resolution (Å) | 29.04–1.95 (2.00–1.95) | 30.00–1.95 (1.99–1.95) |
| No. reflections | 57343 (3010) | 55389 (3223) |
| Completeness (%) | 99.5 (95.6) | 97.6 (78.4) |
| Redundancy | 6.5 (4.5) | 3.7 (3.3) |
| Rmerge (I) | 0.097 (0.554) | 0.100 (0.555) |
| I/σ (I) | 10.8 (2.7) | 15.7 (2.1) |
| Rwork/Rfree | 0.154/0.198 | 0.175/0.205 |
| No. of atoms | ||
| Protein | 4564 | 4936 |
| Waters | 756 | 352 |
| Mn2+/PEP | 10 | 24 |
| Average B-factors | ||
| Protein | 45.6 | 38.1 |
| Waters | 29.3 | 40.7 |
| Mn2+/PEP | 35.7 | 38.1 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.010 | 0.007 |
| Bond angles (°) | 1.254 | 1.183 |
| Ramachandran map analysis | ||
| Favored regions (%) | 98.8 | 99.2 |
| Disallowed regions (%) | 0 | 0 |
Highest resolution shell in parenthesis.
Figure 2.

DAHPS-CML domain architecture. A: Schematic representation of the L. monocytogenes DAHPS-CML domain construction. B: Cartoon representation of DAHPS-CML tetramer highlights the catalytic DAHPS tetramer (blue), the domain linkers (yellow), and the regulatory CML dimers (red). Gray boxes show the interaction between catalytic and regulatory domains and dashed lines trace where the disordered portion of the domain linker may lie. C: Superposition (RMSD = 0.10 Å over 204 Cα atoms) of DAHPS domains from the two molecules within the asymmetric unit reveals nonidentical linker conformations and a related difference in the relative CML domain position. Chain A is colored as in (A), whereas the chain B DAHPS domain is purple and the CML domain is brown. D: The small interface [gray boxes in (B)] formed by the chain A CML domain and a neighboring molecule within the DAHPS tetramer. Key-interacting residues are shown as sticks, and dashed lines trace electrostatic/polar interactions.
Interestingly, the linker that connects DAHPS and CML domains assumes nonidentical conformations in the two molecules within the asymmetric unit. In chain A, the linker adopts a kinked conformation where it interacts with the catalytic tetramer. In chain B, the C-terminal portion of the linker extends away from the DAHPS domain, and the N-terminal portion is disordered. The chain B linker must traverse a greater distance to connect to the CML domain and therefore must adopt a more extended conformation than in chain A [Fig. 2(C)].
Either a cause or effect of the nonidentical linker conformation, the CML regulatory dimer adopts a decidedly asymmetric position relative to the catalytic core [Fig. 2(B,C)]. In this position, α2 and α3 helices on the chain A side of the CML dimer form a small, mostly hydrophobic interface with a pair of α-β connecting loops from a neighboring molecule in the DAHPS tetramer [Fig. 2(B,D)]. Establishment of this interface necessitates the ∼ 10° upward tilt of the CML dimer away from its point of contact with the catalytic domain. This places the chain B linking side of the dimer ∼ 15 Å away from the catalytic core and prevents it from establishing the symmetric interaction on the opposite side of DAHPS tetramer [Fig. 2(B)].
Considering the limited size of the domain–domain interface and the profound regulatory domain positional asymmetry, it is unclear how stable or biologically relevant the observed orientation of the CML dimer is. It is possible that the inhibitor-free CML dimer does not form a single stable interaction with the tetramer core but rather samples an ensemble of low occupancy conformations. In which case, subtle crystal-packing forces may have selected for the observed position over equally relevant alternative conformations.
The catalytic DAHPS domain
The DAHPS-CML catalytic domain adopts a classic TIM barrel (α/β)8 fold and is similar to previously characterized DAHPSs, particularly to the catalytic domain of the T. maritima DAHPS-FL with which it shares ∼ 55% sequence identity [Fig. 3(A)].9, 16, 17 The active site is located on the inside of the C-terminal end of the barrel and is formed by several α–β-connecting loops and two β-strands [Fig. 3(A)]. In the holo structure, the presence of a manganese ion at the active site was confirmed by analysis of the anomalous signal. In the PEP structure, the substrate is adjacent to the manganese ion in a similar position as has been observed in related DAHPSs (Supporting Information Fig. 3).9, 16, 17
Figure 3.

Comparison of DAHPS-CML and DAHPS-FL structures. A: Superposition (RMSD = 0.55 Å over 203 Cα atoms) of DAHPS domains from DAHPS-CML (chain A, colored as in Fig. 2) and the T. maritima DAHPS-FL (the catalytic domain is purple and the regulatory domain is green, PDB code 1RZM), reveals a highly similar catalytic domain tertiary structure and a similar position of unique N-terminal regulatory domains. The PEP is depicted in sphere representation. B: Superposition (RMSD = 0.68 Å over 924 Cα atoms) of DAHPS-CML and DAHPS-FL tetramers reveals that the two DAHPSs also assume similar quaternary structures.
The oligomeric assembly of DAHPS domains, which is quite varied across characterized DAHPSs, is also similar to the DAHPS-FL [Fig. 3(B)].7, 16, 17 The alternating direction of the protomers within the tetramer places the two active sites on one side closer to the CML dimer formed by the other molecules in the tetramer. Interestingly, it was recently shown that the DAHPS-FL regulatory domain is essential for maintaining its tetrameric state.10 Considering the similar quaternary structure and the fact that the extensive CML dimer forming domain–domain interactions occur between tetramer generating chains (Supporting Information Fig. 2), the CML regulatory domain likely stabilizes the tetrameric state of DAHPS-CML.
The regulatory CML domain
The chain A CML domain forms a three-helix bundle that intertwines with the chain B CML domain to generate a symmetric six helix dimer [Fig. 4(A)]. In both chains, the electron density for the seven most N-terminal residues and for the α1–α2 connection is unresolved. However, while only four amino acids (residues 39–43) are disordered in the chain A α1–α2 connection, 21 amino acids (residues 32–53) are not seen in chain B. This difference likely relates to the stable catalytic-regulatory domain interface formed by the chain A side of the dimer. In contrast, as the chain B side of the dimer is further away from the catalytic domain and does not engage in crystal packing, it is less constrained and has ample room to adopt multiple conformations [Fig. 2(B)]. This analysis is supported by trends in the TLS-refined anisotropic displacement parameters, which demonstrate greater conformational heterogeneity on the chain B side of the dimer (Supporting Information Fig. 4). Nevertheless, despite nonidentical linker behavior, unique chain A interaction with the catalytic domain, and greater chain B disorder, the ordered portion of the two CML domains is extremely similar in structure (RMSD = 0.23 Å over 43 Cα atoms).
Figure 4.

Comparison of the regulatory CML and enzymatic CM. A: Superposition (RMSD = 0.94 Å over 81 Cα atoms) of the CML dimer (chain A is shown in red and chain B in brown) to the E. coli CM transition state analog (TSA) complex (the two chains are different shades of blue, PDB code 1ECM), with the TSAs depicted as sticks. B: A different perspective of the superposition in (A) shows a ribbon trace of the main chain. Nonconserved side chains proximal to the TSA are depicted as sticks.
The CML regulatory dimer is most similar to the simplest class of characterized CMs, exemplified by the CM domain from the E. coli P-protein [Fig. 4(A)].15 The most significant distinction from the E. coli enzyme regards the C-terminal α3 helix, which gives way to the linker and consequently is three turns shorter in the CML structure [Fig. 4(A)]. A comparison to the E. coli CM active site reveals that most but not all of the residues are conserved in the regulatory domain. Five charged/polar residues (Arg10, Arg27, Lys38, Glu51, and Gln86) shown to be vital for the E. coli CM function are conserved in the CML domain.18 However, five hydrophobic residues and a serine prominently positioned near the CM active site (Ala32, Val35, Leu55, Ile81, Ser84, and Val85 in the E. coli enzyme) are not conserved in the CML domain (corresponding residues in the L. monocytogenes protein are Val31, Ile39, Met54, Phe79, Gly82, and Leu83) [Fig. 4(B)]. Mutagenesis of four of these residues (Ala32, Val35, Ile81, and Val85) modestly decreased the catalytic efficiency of the E. coli CM.19 Therefore, while the DAHPS-CML structures provide preliminary clues, enough differences between CM and CML dimers persist that follow-up studies will be necessary to pinpoint the source of their differing catalytic and chorismate/prephenate-binding properties.
Model of chorismate/prephenate inhibition of DAHPS-CML
The DAHPS-CML structure provides a new basis for assessing the mechanism of chorismate/prephenate inhibition. Given the discrete nature of catalytic and regulatory domains, the two basic mechanisms by which inhibitor binding might exert allosteric control over DAHPS activity are by (1) directly transmitting inhibitory conformational changes through the domain linker or (2) stabilizing an inhibitory interaction between the catalytic and regulatory domains.
Because the domain linker is a significant distance from the active sites, it is difficult to envision how inhibitor binding could directly transmit conformational changes through the linker (Fig. 3). Furthermore, several circumstantial factors argue against the linker assuming a direct inhibitory role. First, the linker has a lower level of sequence conservation than catalytic or regulatory domains in representative DAHPS-CMLs (Supporting Information Fig. 5). If the linker functioned in a finely tuned role, then a higher level of sequence conservation would be expected. Second, a model involving linker transmitted inhibition would require that N- and C-terminal DAHP-CMLs (discussed in the Introduction), which by definition have differently positioned linkers, operate by distinct mechanisms. Although this possibility cannot be conclusively ruled out, a common mechanism of N- and C-terminal DAHPS-CML inhibition provides a more parsimonious explanation for the origin and persistence of the two DAHPS-CML subtypes. Finally, considering the nearly identical tertiary and quaternary structure of the DAHPS-CML and DAHPS-FL catalytic domains and the superficially similar behavior of their inhibitor-free regulatory domains (Fig. 2), the two DAHPS classes might reasonably be expected to employ related mechanisms of inhibition. As such, the fact that DAHPS-FL inhibition is characterized by a regulatory domain positional change that creates a novel domain–domain interface (Supporting Information Fig. 1) argues for a similar mechanism—and against a linker transmitting model—of DAHPS-CML inhibtion.9, 10
For these reasons, it is more likely that inhibitor binding causes conformational changes that promote a direct interaction between regulatory and catalytic domains. According to this model, in the absence of inhibitor, the CML dimers are loosely associated with the DAHPS tetramer, either adopting the conformation observed in our crystal structures or an ensemble of low occupancy positions (Fig. 5, top panel). Chorismate or prephenate binding would be postulated to induce conformational changes in the CML domain that stabilize a specific interaction between catalytic and regulatory domains. Although at present it is not possible to definitively determine the borders of such an interface, the radius of possible interactions is limited by the length of the domain linker. The most probable scenario has the CML domain interacting with the sizable cleft that contains the two active sites of neighboring molecules in the tetramer. This rearrangement would place the CML dimers in a similar position as the inhibitor bound DAHPS-FL regulatory domains (Supporting Information Fig. 1) and could inhibit activity either by directly blocking substrate access to the active site or by inducing conformational changes in the catalytic domain (Fig. 5, bottom panel). Because the linker only functions to tether the CML domain, keeping it close to the catalytic domain, this model permits the CML domain to be linked to either DAHPS terminus with minimal consequence to regulatory function.
Figure 5.
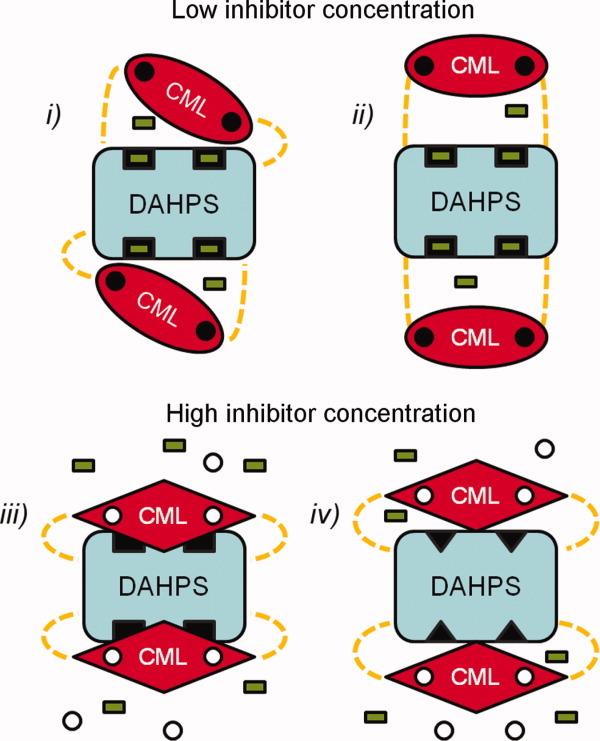
Model of DAHPS-CML inhibition. The DAHPS-CML catalytic tetramer (blue), regulatory dimers (red), domain linkers (dashed yellow), active sites (black rectangles), substrate (green rectangles), and inhibitor (white circles) are schematically depicted. In the inhibitor-free state, the active sites are accessible for substrate binding as the regulatory CML dimers either (i) assume the position observed in crystal structures or (ii) do not stably interact with the catalytic tetramer. Inhibitor binding results in a conformational change within the CML domain that stabilizes a catalytic-regulatory domain–domain interface. In the inhibited state, the CML dimer either (iii) blocks substrate access to the active site or (iv) induces conformational changes in the DAHPS tetramer that impair catalysis (black triangles). Note that for the sake of simplicity, a symmetric effect of inhibitor binding at both the CML dimer-binding sites is depicted. In actuality, a more complex cooperative mechanism of inhibition may occur.
To clarify the mechanism of allosteric inhibition, cocrystallization and crystal soaking experiments with chorismate and prephenate were attempted. Although these experiments failed to produce a structure with unambiguously bound inhibitor, prephenate soaks resulted in a considerable reduction in the level of electron density for the CML domain position observed in the inhibitor-free structures and a concordant increase in the level of uninterruptable density in the cleft directly above the DAHPS active sites (data not shown). Although the apparent mixture of CML conformational states made it impossible to accurately model the domain in any conformation, this effect of prephenate is generally supportive of the inhibitor induced CML positional change predicted by our model.
Implications of the DAHPS-CML structure for multi-target drug discovery
Because they are essential for a number of bacteria but absent from humans, the enzymes involved in amino acid biosynthesis have long been considered attractive targets for the development of novel antibiotics.20, 21 DAHPS-CMLs are the sole annotated DAHPSs within several bacterial pathogens (most notably, L. monocytogenes, Staphylococcus aureus, and Bacillus anthracis) and therefore represent a potential drug target in a class of medically relevant organisms. Consistent with the notion that the CML domain primarily functions in a regulatory role, these DAHPS-CML-containing pathogens also have a separate gene that encodes a CM enzyme—which presumably serves the organisms' primary CM needs.
It has been argued that the DAHPS-CML likely arose through a gene duplication and fusion to DAHPS, followed by adaptation over evolutionary time.14 What had been a functional CM domain was in this way coopted for the purpose of aromatic metabolite regulation. Although this strategy of module swapping and repurposing likely represented an efficient use of available genetic resources to evolve a new functionality, it may have introduced a vulnerability that can be exploited in a rational drug discovery campaign.
The theoretical benefits of a multitarget antibacterial therapy—to which bacteria should develop resistance to at a substantially slower rate—are well explored.22, 23 Another consideration, a downside of competitive enzyme inhibition, concerns the continued function of upstream enzymes, which can allow substrate of the inhibited enzyme to accumulate to a concentration which overcomes inhibition.24 Because it would prevent substrate accumulation for downstream targets, a case can be made that an inhibitor with multiple targets in a single pathway would be particularly advantageous. Nonetheless, despite a well-established rationale, there are few examples of successful directed multitarget discovery campaigns, due in large part to the inherent challenges in optimizing an inhibitor to efficiently bind multiple disparate sites.
Previous studies have shown that the CM enzyme and CML regulatory domain have similar small molecule binding and catalytic properties.13, 14 The findings presented here demonstrate that CM and CML domain also have a highly conserved structure. Considering these similarities, an inhibitor (particularly a CM transition state analog) might be expected to favorably interact with both CM enzyme and CML regulatory domain. In light of the relative functions of the CM enzyme versus the CML regulatory domain, such a compound could simultaneously act as a competitive inhibitor of CM and a noncompetitive inhibitor of DAHPS, thereby intervening at two points along the phenylalanine/tyrosine biosynthetic pathway. The existence of two enzymes in an essential biosynthetic pathway with nearly identical binding and catalytic properties despite distinct functionalities provides a rare opportunity for multitarget drug discovery, in which a reasonable expectation of a multitarget effect can be maintained while optimizing an inhibitor to a single binding site.
Materials and Methods
Cloning and protein expression
The annotated L. monocytogenes aroA gene was amplified from L. monocytogenes EGD-e genomic DNA by PCR, cloned in the pMCSG7 expression vector, and transformed into BL21 (DE3) E. coli cells using previously described methods.25 Cells were grown for 4 h at 37°C. At which point, the temperature was reduced to 25°C and protein overexpression induced by the addition of isopropyl-1-thio-d-galactopyranoside to a final concentration of 0.5 mM. Following overnight growth, cells were harvested by centrifugation, resuspended in a buffer containing 10 mM Tris–HCl (pH 8.3), 500 mM NaCl, 10% glycerol, and 5 mM β-mercaptoethanol and lysed by sonication. Following centrifugation, the soluble fraction of the resulting cell lysate was purified by Ni-NTA affinity chromatography. DAHPS-CML protein was step-eluted with 500 mM imidazole and collected in a buffer containing 10 mM Tris–HCl (pH 8.3), 500 mM sodium chloride, 5 mM β-mercaptoethanol, and 0.5 mM manganese chloride.
Protein crystallization and data collection
Immediately following purification, DAHPS-CML protein was concentrated to 7.5 mg/mL and crystallized by the sitting-drop vapor diffusion method, using a 1:1 ratio of protein to reservoir. For cocrystallization experiments, 2 mM PEP was added to the protein mixture. Sizable crystals were observed within weeks of the initial screening experiments. Crystals were flash-frozen directly from drops containing: 0.2 M magnesium acetate, 0.1 M sodium cacodylate pH 6.5, 20% (w/v) PEG 8000 (holo crystal), 0.1 M PCB buffer pH 8.0, and 25% (w/v) PEG 1500 (PEP crystal). Diffraction data were collected at 100 K at the Life Sciences-Collaborative Access Team at the Advanced Photon Source, Argonne National Laboratory, Argonne, IL.
Structure determination and refinement
Data were processed using HKL-3000 for indexing, integration, and scaling.26 Phases were obtained by molecular replacement in Phaser,27 using the Thermotoga maritima DAHPS structure (PDB code 1RZM) as the DAHPS domain search model and the Thermus Thermophilus CML structure (PDB code 2D8D) as the CML domain search model. Structures were refined with Refmac.28 Models were displayed in Coot29 and manually corrected based on electron density maps. Twelve TLS groups, identified by the TLSMD webserver,30 were used for final rounds of TLS refinement. Final atomic coordinates were deposited in the PDB and assigned codes 3NVT (holo structure) and 3TFC (PEP bound structure). All structure figures were prepared using PyMOL Molecular Graphics System, Version 1.3 (Schrödinger, LLC).
Acknowledgments
We thank Dr. Scott Peterson and Dr. Keehwan Kwon for providing the aroA expression clone.
Glossary
Abbreviations
- CM
chorismate mutase
- CML
chorismate mutase-like regulatory domain
- DAHPS
3-deoxy-D-arabino-heptulosonate 7-phosphate synthase
- FL
ferredoxin-like
- PDB
Protein Data Bank
- PEP
phosphoenolpyruvate
- RMSD
root mean square deviation
- TSA
transition state analog
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Bentley R. The shikimate pathway—a metabolic tree with many branches. Crit Rev Biochem Mol Biol. 1990;25:307–384. doi: 10.3109/10409239009090615. [DOI] [PubMed] [Google Scholar]
- 2.Dosselaere F, Vanderleyden J. A metabolic node in action: chorismate-utilizing enzymes in microorganisms. Crit Rev Microbiol. 2001;27:75–131. doi: 10.1080/20014091096710. [DOI] [PubMed] [Google Scholar]
- 3.Herrmann KM, Poling MD. The synthesis of 3-deoxyheptulosonic acid 7-phosphate. J Biol Chem. 1975;250:6817–6821. [PubMed] [Google Scholar]
- 4.Byng GS, Johnson JL, Whitaker RJ, Gherna RL, Jensen RA. The evolutionary pattern of aromatic amino acid biosynthesis and the emerging phylogeny of pseudomonad bacteria. J Mol Evol. 1983;19:272–282. doi: 10.1007/BF02099974. [DOI] [PubMed] [Google Scholar]
- 5.Webby CJ, Jiao W, Hutton RD, Blackmore NJ, Baker HM, Baker EN, Jameson GB, Parker EJ. Synergistic allostery, a sophisticated regulatory network for the control of aromatic amino acid biosynthesis in Mycobacterium tuberculosis. J Biol Chem. 2010;285:30567–30576. doi: 10.1074/jbc.M110.111856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sasso S, Okvist M, Roderer K, Gamper M, Codoni G, Krengel U, Kast P. Structure and function of a complex between chorismate mutase and DAHP synthase: efficiency boost for the junior partner. EMBO J. 2009;28:2128–2142. doi: 10.1038/emboj.2009.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Webby CJ, Baker HM, Lott JS, Baker EN, Parker EJ. The structure of 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase from Mycobacterium tuberculosis reveals a common catalytic scaffold and ancestry for type I and type II enzymes. J Mol Biol. 2005;354:927–939. doi: 10.1016/j.jmb.2005.09.093. [DOI] [PubMed] [Google Scholar]
- 8.Wu J, Howe DL, Woodard RW. Thermotoga maritima 3-deoxy-d-arabino-heptulosonate 7-phosphate (DAHP) synthase: the ancestral eubacterial DAHP synthase? J Biol Chem. 2003;278:27525–27531. doi: 10.1074/jbc.M304631200. [DOI] [PubMed] [Google Scholar]
- 9.Shumilin IA, Bauerle R, Wu J, Woodard RW, Kretsinger RH. Crystal structure of the reaction complex of 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase from Thermotoga maritima refines the catalytic mechanism and indicates a new mechanism of allosteric regulation. J Mol Biol. 2004;341:455–466. doi: 10.1016/j.jmb.2004.05.077. [DOI] [PubMed] [Google Scholar]
- 10.Cross PJ, Dobson RC, Patchett ML, Parker EJ. Tyrosine latching of a regulatory gate affords allosteric control of aromatic amino acid biosynthesis. J Biol Chem. 2011;286:10216–10224. doi: 10.1074/jbc.M110.209924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nester EW, Lorence JH, Nasser DS. An enzyme aggregate involved in the biosynthesis of aromatic amino acids in Bacillus subtilis. Its possible function in feedback regulation. Biochemistry. 1967;6:1553–1563. doi: 10.1021/bi00857a042. [DOI] [PubMed] [Google Scholar]
- 12.Huang L, Nakatsukasa M, Nester E. Regulation of aromatic amino acid biosynthesis in Bacillus subtilis 168. Purification, characterization, and subunit structure of the bifunctional enzyme 3-deoxy-d-arabinoheptulosonate 7-phosphate synthetase-chorismate mutase. J Biol Chem. 1974;249:4467–4472. [PubMed] [Google Scholar]
- 13.Wu J, Sheflyan GY, Woodard RW. Bacillus subtilis 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase revisited: resolution of two long-standing enigmas. Biochem J. 2005;390:583–590. doi: 10.1042/BJ20050294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu J, Woodard RW. New insights into the evolutionary links relating to the 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase subfamilies. J Biol Chem. 2006;281:4042–4048. doi: 10.1074/jbc.M512223200. [DOI] [PubMed] [Google Scholar]
- 15.Lee AY, Karplus PA, Ganem B, Clardy J. Atomic-structure of the buried catalytic pocket of Escherichia coli chorismate mutase. Journal of the American Chemical Society. 1995;117:3627–3628. [Google Scholar]
- 16.Webby CJ, Patchett ML, Parker EJ. Characterization of a recombinant type II 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase from Helicobacter pylori. Biochem J. 2005;390:223–230. doi: 10.1042/BJ20050259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shumilin IA, Kretsinger RH, Bauerle RH. Crystal structure of phenylalanine-regulated 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase from Escherichia coli. Structure. 1999;7:865–875. doi: 10.1016/s0969-2126(99)80109-9. [DOI] [PubMed] [Google Scholar]
- 18.Liu DR, Cload ST, Pastor RM, Schultz PG. Analysis of active site residues in Escherichia coli chorismate mutase by site-directed mutagenesis. Journal of the American Chemical Society. 1996;118:1789–1790. [Google Scholar]
- 19.Lassila JK, Keeffe JR, Kast P, Mayo SL. Exhaustive mutagenesis of six secondary active-site residues in Escherichia coli chorismate mutase shows the importance of hydrophobic side chains and a helix N-capping position for stability and catalysis. Biochemistry. 2007;46:6883–6891. doi: 10.1021/bi700215x. [DOI] [PubMed] [Google Scholar]
- 20.Coggins JR, Abell C, Evan LB, Frederickson M, Robinson DA, Roszak AW, Lapthorn AP. Experiences with the Shikimate-pathway enzymes as targets for rational drug design. Biochemical Society Transactions. 2003;31:548–552. doi: 10.1042/bst0310548. [DOI] [PubMed] [Google Scholar]
- 21.Benowitz AB, Hoover, Jennifer L, Payne, David J. Antibacterial Drug Discovery in the Age of Resistance. Microbe. 2010;5:390–396. [Google Scholar]
- 22.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 23.Brotz-Oesterhelt H, Brunner NA. How many modes of action should an antibiotic have? Curr Opin Pharmacol. 2008;8:564–573. doi: 10.1016/j.coph.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 24.Copeland RA. Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods Biochem Anal. 2005;46:1–265. [PubMed] [Google Scholar]
- 25.Light SH, Minasov G, Shuvalova L, Duban ME, Caffrey M, Anderson WF, Lavie A. Insights into the mechanism of type I dehydroquinate dehydratases from structures of reaction intermediates. J Biol Chem. 2011;286:3531–3539. doi: 10.1074/jbc.M110.192831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 27.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallographica Section D-Biological Crystallography. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 28.Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallographica Section D-Biological Crystallography. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- 29.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica Section D-Biological Crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.