

Abstract
Given the importance of protein–protein interactions for nearly all biological processes, the design of protein affinity reagents for use in research, diagnosis or therapy is an important endeavor. Engineered proteins would ideally have high specificities for their intended targets, but achieving interaction specificity by design can be challenging. There are two major approaches to protein design or redesign. Most commonly, proteins and peptides are engineered using experimental library screening and/or in vitro evolution. An alternative approach involves using protein structure and computational modeling to rationally choose sequences predicted to have desirable properties. Computational design has successfully produced novel proteins with enhanced stability, desired interactions and enzymatic function. Here we review the strengths and limitations of experimental library screening and computational structure-based design, giving examples where these methods have been applied to designing protein interaction specificity. We highlight recent studies that demonstrate strategies for combining computational modeling with library screening. The computational methods provide focused libraries predicted to be enriched in sequences with the properties of interest. Such integrated approaches represent a promising way to increase the efficiency of protein design and to engineer complex functionality such as interaction specificity.
Keywords: protein design, library screening, computational modeling, protein interaction specificity
Introduction
DNA stores the information needed for life. One of the ways this one-dimensional information generates functional complexity is by encoding proteins that participate in elaborately structured interaction networks. Proteins interact with one another to form macromolecular assemblies, machines and cellular scaffolds, to relay signals, and to catalyze biochemical reactions. Correct function and information processing require correct physical interactions, indicating a strong evolutionary pressure for protein association to occur selectively. Indeed, the highly specific nature of protein–protein interactions has been demonstrated in vitro and in vivo1 in numerous studies. Recent efforts to measure protein–protein interactions comprehensively in selected organisms have provided a picture of specificity on a large scale.2–4 And assays of protein–protein interactions in isolation of their native environment, especially studies done using purified proteins, have demonstrated that interaction specificity can be encoded within proteins themselves.5,6
A key question for protein biochemists is how protein–protein interaction specificity is achieved. It is not surprising that proteins with very different sequences can fold into different structures that have distinct interaction properties. However, many recent studies have revealed that proteins (or protein domains/motifs) highly similar in sequence and/or structure can participate in different interactions. These observations indicate that diverse interaction profiles can be evolved from a common protein sequence family/structural fold. A major implication is that interaction networks can be evolved with increasing complexity without the need to reinvent components from scratch.7 Examples of recurring structures with distinct specificities include modular domains such as the PDZ,6,8–10 Src homology 2 (SH2),11,12 and Src homology 3 (SH3)13,14 families that are present in many cell signaling proteins, the coiled-coil motifs of different bZIP transcription factors,5 the Bcl-2 proteins involved in apoptosis,15 and cell-adhesion molecules such as the Drosophila protein Dscam16 (Fig. 1). Dscam represents a particularly interesting case from an evolutionary perspective. Dscam consists of 10 immunoglobulin-like domains, three of which are variable and play important roles in homodimerization. Each of these three variable domains is encoded by an exon block, and mutually exclusive splicing at each block gives rise to more than 10,000 distinct isoforms. It was shown that each variable domain is largely specific for interaction with itself, and the combined action of the three domains results in high binding specificity of the full-length Dscam,16 a key property for neurons to distinguish self from nonself (self-avoidance) in development. Evolutionary analysis suggested that each exon block was evolved by exon duplication followed by sequence divergence,17 illustrating how selective pressure exerted by the need to maintain self-avoidance can help shape the remarkable homo-specificity of the Dscam family.
Figure 1.
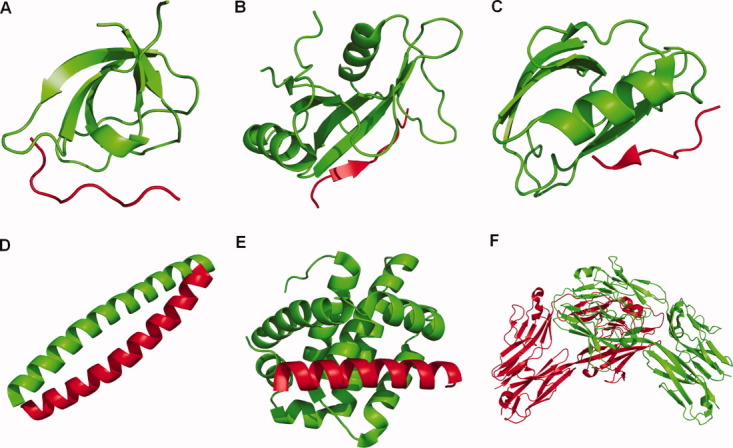
Examples of different types of protein–protein interactions where the protein fold is conserved but the specificity can be varied. A representative complex is shown for each class of interaction: A: Complex between SH3 domain from the Abl tyrosine kinase (green) and a proline-rich peptide (red). (PDB ID: 1ABO).169 B: Complex between SH2 domain from the SAP protein (green) and a phosphotyrosine peptide (red) (PDB ID: 1D4W).170 C: Complex between Erbin PDZ domain (green) and the C-terminal tail of the ErbB2 receptor (red) (PDB ID: 1MFG).171 D: Complex between the bZIP coiled-coil motifs of FOS (green) and JUN (red) (PDB ID: 1FOS).172 E: Complex between anti-apoptotic protein Mcl-1 (green) and the BH3 region of Bim (red) (PDB ID: 2PQK).173 F: Complex of a homodimer formed by the N-terminal domain of a particular Dscam isoform (PDB ID: 2V5M).174 Figure generated using PyMol (Delano Scientific).
Interestingly, solved structures of proteins with similar sequences but distinct interactions have revealed that often the same binding interface is utilized, and differences in binding preferences can be attributed to local differences in structures.18–26 Some natural proteins share a similar protein fold yet differ in their interaction properties through use of different conformations for loops linking helices and/or strands that define a basic scaffold. A good example is the SH2 family, which interacts with peptides that contain a phosphorylated tyrosine (pTyr).11,12 Specificity of SH2 domains interacting with different pTyr peptides is crucial for correctly transmitting signals from protein tyrosine kinases to downstream pathways. Three main classes of SH2 domains recognize peptides with different sequence signatures C-terminal to pTyr. Loops flanking the binding interface confer selectivity toward these 3 types of peptides by opening or blocking binding pockets for the P+2, P+3 or P+4 residues.19,20 The SH3 family also utilizes different loop conformations at the binding interface to provide specificity toward different peptides that are rich in prolines.19 Antibodies provide another example of using loops to confer different binding properties,23 sharing a common immunoglobulin scaffold but using variation in 6 surface loops, the complementarity-determining regions (CDR), to achieve exquisite specificities for antigens.
Although changes in local structures such as loops present a convenient way to change interaction properties, examples of more subtle sequence/structural features providing specificity abound in nature as well. One example is the interaction between colicin endonucleases (DNases) and immunity (Im) proteins. Colicins are stress-induced bacterial bacteriocins. Toxicity of colicins against their own producing cells is neutralized by interaction with cognate Im proteins, so high interaction specificity is critical. A crystal structure of a noncognate complex between DNase ColE9 and Im2 was solved recently and comparison was made to the structure of the cognate complex between DNase ColE9 and Im9.24 The backbone and side-chain packing at the core of the two interfaces was highly similar. However, the presence of unfavorable polar/charged residue burial and suboptimal hydrogen bonding patterns weakened interaction significantly for the noncognate complex. For bZIP coiled coils, structural and mutational analyses have revealed specificity features defined by particular patterns of hydrophobic packing, hydrogen bonding, and electrostatic interactions between different side-chain pairs.25 Interaction can be encoded through combinations of these features, without any significant change in backbone structure. For the SH2 and SH3 domains discussed in the previous paragraph, it has also been shown that structural features similar to the ones described above are important to further fine-tune the selectivity obtained from loops.12,14,19
Examining strategies used by nature to achieve interaction specificity offers the exciting possibility that we might learn to mimic or devise new strategies to design selective binding. In fact, many attempts have been made to change interaction specificity for the different protein systems described above by altering loops or simply introducing one or a few amino acid mutations at binding interfaces.27–30 Given the importance of protein–protein interactions, an ability to design protein–protein interaction specificity could find many applications in the study of cell biology.31 Proteins have already been redesigned to create dominant negatives and potential therapeutics specific for a target,32–34 to generate obligate heterodimers,35,36 to test the functional significance of the many different interactions of an original protein,37,38 and to create novel interactions to rewire cell signaling in synthetic biology applications.39,40 In addition to these applications, evaluating design successes and failures can help advance our understanding of how protein sequence influences interaction specificity.
Traditionally, researchers have attempted design using general knowledge obtained from structural and mutational analysis of protein–protein interactions. For example, the importance of hydrogen bonding, favorable electrostatics, and shape complementarity are well known.41 Nonetheless, this approach often fails to capture the subtlety of how sequence influences interaction specificity. Experimental alanine scanning42,43 and hydrophile scanning44 have been used to generate proteins/peptides with novel interaction specificities, but the chemical diversity accessible by such approaches is limited. Recent technologies, both computational and experimental, have helped revolutionize the field of protein design. Below we first introduce computational protein design and its application to designing protein–protein interaction specificity. We then survey the field of experimental library screening, with particular focus on how the combination of computational protein design with screening could become a powerful approach moving forward.
Computational protein design
Computational protein design posits that because protein sequence determines protein function, it should be possible to develop a quantitative understanding of the relation between sequence and function.45–47 In the context of a protein–protein interaction,48,49 function refers to the free energy change (ΔΔG) of a protein interacting with its partner. If such an energy (or “score”) could be computed, one could perform computational instead of experimental searches to identify sequences with desired properties. But capturing the relationship between sequence and function in a score and searching through the vast sequence/structure space are both daunting tasks.
Scoring functions
Different types of scoring models have been developed to compute energy from sequence. Among them, physics-based structural modeling is the most general.50 In this approach, the structure of a protein complex is predicted from its sequence, and an energy is computed from the structure that accounts for terms such as van der Waals interactions and Coulomb electrostatics.51 Solvation of the protein is often approximated using a polar and a nonpolar component to avoid the computational cost of treating water molecules explicitly.52 The polar component addresses the screening of electrostatic interactions within the protein and the energetic cost of burying and thus desolvating charged or polar groups. It is often computed using a continuum electrostatics model.53–55 The nonpolar component approximates other effects resulting from the structure and interactions of water, which are frequently calculated using terms that depend on solvent accessible surface area.56–60 The accuracy of physics-based models is limited by imperfect knowledge of the correct protein structure and the many approximations made in scoring.
In contrast to physics-based models, statistical potentials estimate energies using the frequencies of different interactions in known protein structures. Such potentials usually approximate the energy as a sum of terms describing interactions between pairs of residues61 or atoms62,63 in the structure being evaluated, although attempts to capture higher order interactions have been reported.64,65 The score for a particular residue–residue or atom–atom interaction is based on the observed number of such contacts in known structures, corrected by the expected number of random encounters for the same residue or atom pair. A contact can be defined simply by distance,62,63 but more sophisticated potentials take into account other information such as orientation66,67 or the environment of the contact as well.68 One advantage of statistical potentials is their speed compared with physics-based structural models. In addition, current physics-based models do not always accurately describe the geometry of interactions in known structures. Examples include certain packing preferences among hydrophobic sidechains69 and the angle distribution of hydrogen bonds.70 On the other hand, statistical potentials make approximations in converting observed statistics into energies.71 Statistical potentials have been used in multiple prediction and design problems72 including modeling of protein–protein interactions.73,74 Potentials that include terms from both physics-based models and statistical potentials have also been developed (e.g., Rosetta75) and have had success in many applications.
Design objectives
After choosing a scoring function, one must define the objective(s) for which a designed sequence will be optimized. One option is to minimize the interaction energy (ΔΔG), calculated by subtracting the predicted energy of each protein partner modeled in its unbound form from the energy of the modeled complex. Approximations such as a rigid–body docking or even less formal definitions are often employed, due to the difficulty of modeling the unbound reference states when experimental structures are not available.76,77
When designing protein–protein interaction specificity, it is necessary to consider interactions with one or more undesired proteins, in addition to that with the target. Various objectives have been considered, ranging from simple differences in the energy between two complexes, for example, S = Edesired_complex – Eundesired_complex, to statistical mechanical expressions that capture more states.78–80 It is tempting to pick designed proteins that are predicted to be highly specific, that is to have a large predicted energy difference between desired and undesired states. However, it is known that tradeoffs can exist between affinity for the target and specificity against undesired proteins.78,79,81 Focusing only on widening the specificity gap can therefore create designed proteins that are specific but bind the target weakly. Depending on the application, it can be beneficial to explore a range of different designs with different tradeoffs in affinity and specificity. Examples of this are presented below.
Search in structure and sequence space
Guided by a scoring function and seeking to optimize one or more design objectives, the next step is to search for sequences in an immense combinatorial space. Because evaluating a sequence requires determining the optimal structure of the protein, this search is performed in both sequence and structure space. When redesigning protein–protein interactions, if a crystal structure of the complex being redesigned is available, it is often assumed that the backbone conformation of the redesigned complex will remain invariant as the sequence is changed. Such a fixed-backbone approach considers only structural degrees of freedoms for the side chains. Optimization of side-chain conformations can be simplified by sampling from a predefined rotamer library.82 Energy minimization can be used to relieve serious steric clashes that stem from artifacts in discretizing the side-chain conformers.
Although success has been reported for many design applications using a fixed-backbone approach, it is clear that even a small number of mutations can sometimes lead to significant variations in backbone geometry.83,84 Designing on a fixed backbone therefore risks the elimination of viable sequences. When designing specificity, an unfavorable interaction modeled for an undesired partner on a fixed backbone might not represent the lowest energy conformation. Different approaches have been proposed to introduce backbone flexibility on a local or global scale.85–91 For example, Fu et al. demonstrated that designing on an ensemble of helices generated using normal mode analysis produced binders of the protein Bcl-xL with more diverse sequences.88 Smith et al. found that incorporation of “backrub sampling,” a method inspired by examining small, local structural variations within the PDB, improved performance in predicting binding profiles for different PDZ domains.90
Both deterministic algorithms (e.g., dead end elimination, A* and integer linear programming) or stochastic ones (e.g., Monte Carlo, FASTER and genetic algorithms) can be used for optimization in structure/sequence space.92–97 Deterministic algorithms are powerful but can be computationally slow or memory intensive and can present problems when the scoring function contains a nonpairwise decomposable term such as continuum electrostatics, or when backbone flexibility is treated explicitly. Strategies have been presented to partially overcome such difficulties.98–100 In contrast to deterministic algorithms, stochastic algorithms might not converge on the optimal solution, but they can be more robust in accommodating different formulations/objectives and are sometimes the only viable option when a search space is too large for deterministic methods.97 Heuristics have been presented to manage the search problem. For example, search using a fast but less accurate pair-wise decomposable scoring function can be performed to narrow down a sequence space, and a more sophisticated scoring function can then be used for evaluation.101 Recently, Grigoryan et al. proposed a novel framework, CLASSY, in which a technique called cluster expansion is first used to approximate a structure-based scoring function as a linear sequence-based scoring function102; the optimization algorithm integer linear programming (ILP) can then be run for optimization in sequence space only.79 In addition to dramatically reducing the time spent evaluating sequences during the optimization, the ILP formulation allows the incorporation of multiple linear constraints, making it ideal for exploring different tradeoffs in specificity design.
Application to designing protein–protein interaction specificity
Successful examples of the computational design of protein interaction specificity have been reported. Many of these consisted of redesigning the sequences of two interacting partners to create either obligate heterodimers or orthogonal proteins that could interact with one another but not with the original interacting pair.78,80,101,103–106 In one of the first examples of explicitly designing for specificity, Havranek et al. designed homo and hetero-specific coiled-coil dimers with novel specificity determinants not found in native coiled-coil sequences.80 Bolon et al. redesigned the SspB homodimer into an obligate heterodimer, and demonstrated experimentally the importance of explicit negative design in this example.78 Green et al.,101 Kortemme et al.,103 and Sammond et al.105 also explored the redesign of native protein interfaces to create designed interfaces that were orthogonal. Potapov et al. presented an interesting approach for such interface redesign by considering a protein interface to be made up of independent modules (sets of interconnected residues). A module at the interface between TEM1 β-lactamase and its inhibitor protein BLIP was replaced with another module from an unrelated protein interface. The resulting interface was shown to be orthogonal to the original one and still retained high affinity.106
Other studies have focused on redesigning proteins to selectively bind a desired target in preference to a number of undesired off-targets.33,79,107–110 The protein calmodulin was redesigned to favor binding to one peptide sequence over another. Interestingly, only positive design for binding the target was considered, yet several designs were verified experimentally to have ∼ 150–900-fold increase in specificity.108 It was suggested that explicit negative design might not be necessary in this case because the target and the undesired proteins were significantly different from one another.18 In a contrasting example of a case where explicit consideration of negative design was important, Grigoryan et al. designed specific inhibitor peptides against all 20 human bZIP families, and subsequent experimental testing verified that many of the designed peptides showed the desired specificity.79
Challenges for computational protein design
Significant challenges remain for computational design of protein interaction specificity. A fundamental limitation is that reliably predicting protein–protein interaction specificity is still difficult. Favorable electrostatics at the protein interface appears not to be properly balanced with the energy cost of interfacial charge burial in many cases.111 Insufficient structural sampling can produce artificial steric clashes or fail to identify optimal conformations.89,112 These issues can be rather subtle and difficult to improve upon, and replacing physics-based scoring with statistical potentials does not solve the problem. Various modeling suites adjust the relative weights of physics-based scoring terms, or use different approaches for structural sampling, partly guided and tested by available mutational free energy change data.91,113–115 Predictions made from these models show correlation with experimental data to a certain extent, but the agreement is not impressive.116 Furthermore, it can be problematic to use experimental data sets generated using widely varying protocols.
The imperfections of scoring functions should not prevent attempts at computational protein design. In design, researchers enjoy the advantage of testing only sequences that are predicted to be optimal, allowing a greater tolerance of prediction error. Researchers can also focus on testing designs generated with strategies that they have higher confidence in. For example, Lippow et al. successfully improved the affinities of different antibodies by mainly optimizing energy contributions from electrostatics.117 Sammond et al. presented a series of filters, based on general knowledge of protein–protein interactions, and required that predicted affinity-enhancing mutations pass these before being tested.118 However, as design problems become more challenging, demands on the accuracy of the scoring functions will increase as well.
One potential strategy to address deficiencies in physical or statistical structural modeling is to use information from other sources, such as experimental data.119,120 This was illustrated in a study by Grigoryan et al. that compared the performance of different models for predicting interaction specificity among ∼50 human bZIP coiled coils.76 Two empirical models were tested that each defined a score for a coiled-coil interaction as a sum of weights corresponding to residue–residue interactions at the coiled-coil interface. In the first model, the weights were optimized to reproduce coiled-coil interaction data from the literature, using a machine learning technique called a support vector machine (SVM).121 In the second model, the weights were taken from published experimental double-alanine mutant cycle coupling energies.122 Both models showed better performance than structure-based models for predicting specificity. One caveat for experimentally derived models is that they are unlikely to describe the entire sequence space of interest. For example, coupling energies for many relevant residue-residue interactions in coiled coils have not been measured. Hybrid models that combine physics-based structural models with experimental data have been developed to address this. A hybrid model was constructed for coiled coils that used available coupling energies to score certain residue–residue interactions and a physics-based approach to compute the remaining terms. This model was used in coiled-coil specificity design by Grigoryan et al.,79 generating many designed sequences that were experimentally validated to be specific. It is tempting to generalize such approaches to other interaction specificity design applications. However, this requires a large amount of experimental data for the proteins being studied. Information encoded in evolutionary history can also be used to provide insights into protein–protein interaction specificity,123,124 but such methods often require extensive sequence alignments and at least some knowledge of interaction patterns across different species for the proteins of interest.
Experimental library screening
Like computational protein design, experimental library screening is motivated by the desire to search among a large number of sequences for those with desired properties.125 Below we briefly review key experimental aspects of this approach, including techniques for generating sequence diversity and different screening or selection platforms. We then discuss examples of how library screening can be combined with computational protein design to facilitate the discovery of desired sequences, a promising strategy for engineering specific protein binders.
Generating sequence diversity
The first task in performing a screen for proteins with a desired property is to generate an experimental library. This is typically performed at the DNA level, with diversity translated to protein sequences at a later stage. A simple strategy is to use error-prone PCR or other methods to introduce sequence changes randomly throughout a gene.126,127 However, this approach is not well suited for combination with rational design techniques as discussed below. Genes with sequence variability introduced at predetermined positions can be readily made by PCR-based assembly procedures using partially randomized oligonucleotides containing degenerate codons.128 It is also possible to encode variability at a position using a mixture of oligonucletides129 or trinucleotide synthesis,130 ensuring inclusion of only desired amino acids. Considerations such as the spacing of positions to be varied and how much diversity needs to be introduced at selected positions determine which assembly procedures can be performed cost-effectively. Sequence diversity can also be generated by combining fragments from different native or synthetic genes, mimicking the process of homologous recombination.131 Different strategies give different types of sequence spaces. For example, randomization at selected positions gives a sequence space that is combinatorial with respect to residues encoded at those positions, potentially sampling all possible sequence combinations if a library is large enough. Recombination among gene fragments, on the other hand, results in sequences that are combinatorial with respect to the fragments. This can be advantageous if the individual fragments are thought to be optimal in some way.
Different randomization strategies can be combined. A good example of this in nature is the process leading to diversification of human antibodies.132 The variable region of each class of antibody chain is assembled from different types of gene segments (the V, D, and J segments) in a site-directed recombination event known as V-D-J joining. The presence of variants for each type of gene segment leads to a combinatorial diversity estimated to be greater than 105. Mechanisms such as somatic hypermutation are employed to further increase diversity and generate antibodies with improved affinities for their targets (affinity maturation). This process of generating a preliminary pool of sequence diversity from which selected sequences are further optimized can be mimicked in vitro. For example, randomization can first be introduced in a guided manner by recombining different native or synthetic gene fragments (analogous to V-D-J joining) or by mutating selected positions in a combinatorial manner. Promising sequences can be identified and techniques like error-prone PCR (analogous to somatic hypermutation) can be performed to further optimize protein properties.
Screening and selection platforms
The best library screening or selection platform depends on a number of factors, including the size of the DNA library and the type of protein property or function that is sought. Molecular display technologies133 including phage display,134 bacterial display,135 yeast display,136 mRNA display,137 and ribosome display138 have been widely used to screen for desired protein–protein interactions. Other platforms such as the yeast two-hybrid assay, or various protein complementation assays, can be used to select for interactions in cells.139–142 This provides the advantage of favoring the design of proteins that are well behaved in the complex cellular environment. Cell-free display methods like mRNA and ribosome display are compatible with much larger library sizes (>1014) than those afforded by cell-based display methods (109–1010 for bacterial display and 107–109 for yeast display). However, for bacterial and yeast display, fluorescence activated cell sorting (FACS) can be used to sort cells displaying the desired sequences in solution. This bypasses the need to first immobilize and then elute the desired clones from a surface, which is often required in cell-free display technologies, and also permits real-time observation of changes in the binding characteristics of the library. This monitoring can be advantageous when screening for protein–protein interaction specificity, as conditions for competition or negative selection can be readily tuned by simply varying the concentrations of target and competitors.
Application to screening for protein–protein interaction specificity
Different groups have used experimental library screening to identify specific protein binders. Using rounds of mutagenesis by error-prone PCR and selection for cell survival, Levin et al. obtained variants of the Im9 protein capable of inhibiting the noncognate ColE7 DNase more strongly than the cognate ColE9.143 Both positive selection and competition selection (positive selection in the presence of undesired off-target competitors) were required to generate mutants showing a >108-fold increase in specificity relative to the wild-type Im9 protein. Structural comparison of Im9 variants obtained during various stages of the selection with the Im7 protein (whose cognate inhibitory target is ColE7) suggested that a selective protein–protein interface was evolved by maintaining promiscuous interactions while gradually transiting to alternative selective configurations stabilized by mutations. In other work, Mason et al. used a cell-survival based protein-complementation assay to select for peptides that bound the leucine-zipper domains of bZIP proteins cFos and cJun.144 They screened a manually designed degenerate-codon library. As in the study by Levin et al., it was necessary to include negative selection, via the inclusion of undesired interaction competitors, to achieve specific binders. Dutta et al. screened for BH3 peptides specific for binding the antiapoptotic protein Bcl-xL in preference to Mcl-1 and vice versa using yeast surface display.145 Explicit negative screening against binding to the undesired partner was combined with screening for binding to the desired target. Interestingly, negative selection was found to be more important for achieving specificity for Bcl-xL than for Mcl-1. Finally, Abe et al. developed a strategy to facilitate competitive screening using phage display.146 Desired target proteins were immobilized on a sensor chip for surface plasmon resonance (SPR) studies. The influence of undesired competitor added in solution was quantified using proteins with known binding affinities for the target and competitor. The concentration of undesired competitor required to reach the desired specificity was calibrated accordingly. Using this approach, the authors isolated mutants of tumor necrosis factor (TNF) that bound selectively to TNF receptor 2 (TNFR2) over TNFR1.
In all examples described above, explicit consideration of interaction specificity was necessary and was addressed in the selection process. Undesired competitors were either included as “decoys” in screening or selection, or were directly selected against. Similar to the computational design of protein–protein interaction specificity, examples of specific binders successfully obtained without explicit consideration of specificity in the screening/selection process have also been reported. For example, Mastumura et al. identified a BH3 peptide that specifically bound the anti-apoptotic protein Bcl-xL over other antiapoptotic proteins using mRNA display with only positive selection.147 On the other hand, many examples have been reported where positive selection leads to nonspecific binders. For example, engineering of numerous PDZ domains for tight binding to target peptides using phage display led to highly nonspecific domain variants in a study by Ernst et al.148
Combining computational protein design and experimental library screening
In experimental screening, desired proteins are identified directly by their exhibition of a particular feature or function. This contrasts with the high risk of computational protein design, where often only a few proteins are made and tested and it is common for these not to possess the desired characteristics. However, relative to computational design, library screening explores a much smaller sequence space, limited to ∼1015 even for the most advanced techniques. This raises concerns about sampling, because mutations picked randomly are rarely beneficial. It is therefore tempting to combine the advantages of these two methods. Instead of computationally designing a few sequences, a protein engineer can computationally design a library. Sequences in the library will not be chosen randomly, but instead will be selected on the basis of computational structural modeling. Although the computational models might not be perfect, they could nevertheless bias the experimental search to a more productive sequence space.
The idea of combining computational protein design and experimental library screening has been explored by several groups.129,149–167 Computationally designing a library presents distinct challenges from designing a fixed number of individual sequences. One is that practical aspects of the experimental strategy should be considered during the computational design phase. As described before, for most experimental library construction protocols, the diversity of library sequences will be combinatorial with respect to residues or gene fragments. The screening platform also places a limit on the number of sequences that can be tested. Another challenge is that the library design objective is no longer obvious; a protein designer must decide whether the library should prioritize inclusion of the predicted best sequences, the largest number of predicted reasonable sequences, or the greatest diversity of sequences. There may be multiple objectives to consider and optimize, and conflicting tradeoffs could exist among these. For example, making a library at lower cost, e.g. by using degenerate codons, imposes restrictions on the types of sequences that can be included. Attempts to include more sequence diversity by screening a larger designed library should also be balanced with the desire to ensure adequate coverage of library sequences predicted to be more favorable. Below we review approaches that have been used to computationally design libraries. Although few studies have focused on designing protein–protein interaction specificity, the general concepts should have broad relevance to a variety of challenging design problems including this one.
Designing a library with selected positions randomized
Several approaches have been suggested for designing protein libraries with selected positions randomized. In the first, a library score is defined and this is optimized. Treynor et al. defined the score as the arithmetic average of the energies of all sequences in the library, calculated using pair-wise decomposable structural models.151 Optimization of this score is analogous to optimizing the energy of a single sequence, with the search being performed in the space of amino acid sets (e.g., amino acids encoded by a degenerate codon) instead of amino acids. Single and pair energies for amino acid sets can be precomputed, allowing the same algorithms to be used. Libraries of green fluorescent protein (GFP) variants were designed this way, and it was observed that these contained a greater fraction of proteins that were functional, as well as a greater diversity of fluorescence emission wavelengths, compared to a library generated using error-prone PCR. In a separate study, Parker et al. also defined library quality to be the average of pair-wise decomposable energies of all library sequences, but proposed an additional objective that represented the novelty of the library sequences when compared with native proteins.160 Optimization was performed using integer programming in the space of degenerate codons and included a constraint on library size. Parker et al. further demonstrated for a few protein systems the tradeoff between predicted quality and novelty. This concept of tradeoffs among different desirable library properties is important.
Recently, we implemented a novel library design strategy that involved library score optimization and also emphasized retention of diversity among library sequences (Chen et al., unpublished). In our scheme, desired residues at each designed position were first predicted by computational structure-based modeling. We then defined the objective to be maximized as the number of unique library sequences with all designed positions occupied by desired residues. Optimal codon selection, under a constraint on library size, was formulated as an integer linear programming problem similar to that described by Parker et al.160 We applied this framework to design a library of Bcl-xL protein variants and screened for those that bound preferentially to a peptide derived from the BH3 motif of Bad over a BH3 peptide from Bim. We obtained Bcl-xL variants showing a large increase in specificity. An important feature of our library design procedure is that we increased diversity not only by including residues that were predicted to be specific for Bad BH3 over Bim BH3, but also by including residues predicted to simply maintain binding to Bad. Analysis of the specific binders identified experimentally suggested that the inclusive approach was important for success, allowing residues important for specificity but missed by the predictions to be included. Interestingly, one of the sequences we identified was globally specific for Bad BH3 over many other BH3 peptides not considered in library design and screening. This raises the intriguing prospect that libraries designed for a simpler task (i.e., maintaining binding to a target while reducing binding to one off-target) can be screened for more demanding objectives (i.e., specificity against multiple off-targets).
In a second approach to computational library design, structure-based computation is first performed to obtain an ensemble of sequences. An amino-acid profile (i.e., the frequency of different amino acids at each designed position) is derived from these sequences, and designed positions in the library are randomized to match the diversity observed in the profile. One caveat is that the library obtained accordingly may not closely resemble the original ensemble of designed sequences. Hayes et al. used this approach to design a library of TEM β-lactamase variants to screen for clones with improved resistance toward the antibiotic cefotaxime.129 Randomization was introduced to the active site, and compatibility with the protein fold (i.e., the crystal structure of TEM β-lactamase) was assessed in designing the sequence ensemble. Variants with a 1280-fold increase in resistance were identified out of a ∼200,000 member library. Guntas et al. also used this approach to design a library of variants of the ubiquitin ligase E6AP to bind to the NEDD8-conjugating enzyme Ubc12, a non-natural partner. They obtained multiple tight binders (Kd < 100 nM) from the screen.155 Degenerate codons were selected at each position by considering their efficiencies in representing the amino-acid diversity profile from design and the library size. One interesting observation was that equally good performance was obtained from a designed library enriched in predicted binders and one enriched simply in predicted well-folded sequences. Both libraries performed better than a random library.
In the third approach, an amino acid diversity profile is derived using a probabilistic framework rather than an ensemble of designed sequences. Voigt et al. applied mean-field theory to capture the structural tolerance of each designed position.149 Using this metric as a guide in selecting positions for randomization, good agreement was observed with prior experimental directed evolution studies of subtilisin E and T4 lysozyme. Saven and coworkers also proposed using a statistical theory for the design of combinatorial libraries.161,162
Designing a library made by combining different gene fragments
One challenge in making a library generated by in vitro recombination among homologous native or synthetic genes is how to select the cross-over points. Voigt et al. developed SCHEMA to help address this.150 Points for cross-over were chosen so as to minimize disruption of important residue-reside interactions observed in the crystal structure. The underlying hypothesis was that hybrid proteins generated this way were more likely to be folded and functional. Correlation between cross-over points predicted from SCHEMA and prior in vitro recombination experiments was observed, and SCHEMA was subsequently applied to a series of different design problems.163,164
One advantage of making a library by combining gene fragments is that favorable combinations of residues within the same fragment can be preserved. This contrasts with designed libraries that are combinatorial in residue substitutions. In an example of recombining synthetic gene fragments obtained from computational protein design, Lippow et al. redesigned a galactose oxidase enzyme to process glucose.157 A set of > 2000 sequences was designed computationally. The 12 designed positions were then grouped into four assembly regions, guided by proximity in sequence. Each region was encoded by a mixture of synthetic oligonucleotides, such that dependencies among different positions in each assembly region were preserved. The library was assembled from these fragments. Using this approach, the authors successfully identified a variant with 400-fold improvement in activity toward glucose from a 10,000-member library.
Improving computational designs by library screening
One final approach for combining computational protein design and experimental library screening is to use library screening to further improve successful or moderately successful designs.165–167 Although computational prediction and design methods can be used to guide library design, most such tools will make assumptions, have biases and introduce errors that prevent discovery of many good sequences. This is especially true for difficult prediction/design problems where the desired function, for example catalysis, is hard to model accurately. In this case, a more random and less guided strategy could prove beneficial in identifying important sequence features missed by the model. This approach was demonstrated by Khersonsky et al. to improve the catalytic activity of an in silico designed Kemp eliminase enzyme.165,168 Error-prone PCR, gene shuffling and site directed randomization were all employed to generate diverse library sequences derived from the initial computationally designed sequence. Mutants with >400-fold improvement in catalytic activity were identified from the screen. Fleishman et al. also applied a similar strategy to optimize the binding affinity of a de novo designed protein targeting the stem region of influenza hemagglutinin, generating binders with low nanomolar affinity.167
One observation from the studies described above is that the metric used for selecting sequences in design can be different from the final, functional objective. Hayes et al.,129 Treynor et al.,151 Guntas et al.155 and we designed library sequences to be compatible with a structural fold but then screened the libraries for function (i.e., improved enzymatic activity, different photophysical properties, protein–protein interaction, and interaction specificity). Although a well-folded sequence is likely necessary but not sufficient for obtaining these types of functions, designing for structure and then screening for additional desired properties could be much easier and may in fact represent a more efficient use of current computational prediction models. This could have general implications for difficult design goals such as protein–protein interaction specificity.
Conclusions
In this review we discussed both computational and experimental approaches for designing protein–protein interaction specificity, especially among proteins with similar sequence and structure. We conclude by suggesting that progress in both fields can be highly synergistic. For example, advances in tri-nucleotide synthesis and gene synthesis will enable the routine screening of large libraries with significantly fewer restrictions on the types of sequences encoded. This should facilitate the testing of a large number of diverse design solutions obtained using very different specificity strategies. Better predictions of specificity strategies can in turn be obtained from improvements in computational sampling and scoring, for example better modeling of protein backbone flexibility. On the other hand, application of machine learning algorithms will in the future enhance our ability to extract information from the ever-growing amount of experimental data. Resulting models can then be used to develop more accurate prediction models for design. We anticipate that such integration of computation and experiment will greatly enhance our design capabilities.
Acknowledgments
The authors thank O. Ashenberg, C. Burge, C. Negron, V. Potapov, A.W. Reinke, R.T. Sauer and B. Tidor for helpful input.
References
- 1.Pawson T, Nash P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000;14:1027–1047. [PubMed] [Google Scholar]
- 2.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dumpelfeld B, Edelmann A, Heurtier MA, Hoffman V, Hoefert C, Klein K, Hudak M, Michon AM, Schelder M, Schirle M, Remor M, Rudi T, Hooper S, Bauer A, Bouwmeester T, Casari G, Drewes G, Neubauer G, Rick JM, Kuster B, Bork P, Russell RB, Superti-Furga G. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- 4.Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Li S, Albala JS, Lim J, Fraughton C, Llamosas E, Cevik S, Bex C, Lamesch P, Sikorski RS, Vandenhaute J, Zoghbi HY, Smolyar A, Bosak S, Sequerra R, Doucette-Stamm L, Cusick ME, Hill DE, Roth FP, Vidal M. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 5.Newman JR, Keating AE. Comprehensive identification of human bZIP interactions with coiled-coil arrays. Science. 2003;300:2097–2101. doi: 10.1126/science.1084648. [DOI] [PubMed] [Google Scholar]
- 6.Stiffler MA, Chen JR, Grantcharova VP, Lei Y, Fuchs D, Allen JE, Zaslavskaia LA, MacBeath G. PDZ domain binding selectivity is optimized across the mouse proteome. Science. 2007;317:364–369. doi: 10.1126/science.1144592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Presser A, Elowitz MB, Kellis M, Kishony R. The evolutionary dynamics of the Saccharomyces cerevisiae protein interaction network after duplication. Proc Natl Acad Sci USA. 2008;105:950–954. doi: 10.1073/pnas.0707293105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee HJ, Zheng JJ. PDZ domains and their binding partners: Structure, specificity, and modification. Cell Commun Signal. 2010;8:8. doi: 10.1186/1478-811X-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tonikian R, Zhang Y, Sazinsky SL, Currell B, Yeh JH, Reva B, Held HA, Appleton BA, Evangelista M, Wu Y, Xin X, Chan AC, Seshagiri S, Lasky LA, Sander C, Boone C, Bader GD, Sidhu SS. A specificity map for the PDZ domain family. PLoS Biol. 2008;6:e239. doi: 10.1371/journal.pbio.0060239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiedemann U, Boisguerin P, Leben R, Leitner D, Krause G, Moelling K, Volkmer-Engert R, Oschkinat H. Quantification of PDZ domain specificity, prediction of ligand affinity and rational design of super-binding peptides. J Mol Biol. 2004;343:703–718. doi: 10.1016/j.jmb.2004.08.064. [DOI] [PubMed] [Google Scholar]
- 11.Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, et al. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 12.Liu BA, Jablonowski K, Shah EE, Engelmann BW, Jones RB, Nash PD. SH2 domains recognize contextual peptide sequence information to determine selectivity. Mol Cell Proteomics. 2010;9:2391–2404. doi: 10.1074/mcp.M110.001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zarrinpar A, Park SH, Lim WA. Optimization of specificity in a cellular protein interaction network by negative selection. Nature. 2003;426:676–680. doi: 10.1038/nature02178. [DOI] [PubMed] [Google Scholar]
- 14.Tonikian R, Xin X, Toret CP, Gfeller D, Landgraf C, Panni S, Paoluzi S, Castagnoli L, Currell B, Seshagiri S, Yu H, Winsor B, Vidal M, Gerstein MB, Bader GD, Volkmer R, Cesareni G, Drubin DG, Kim PM, Sidhu SS, Boone C. Bayesian modeling of the yeast SH3 domain interactome predicts spatiotemporal dynamics of endocytosis proteins. PLoS Biol. 2009;7:e1000218. doi: 10.1371/journal.pbio.1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 16.Wojtowicz WM, Wu W, Andre I, Qian B, Baker D, Zipursky SL. A vast repertoire of Dscam binding specificities arises from modular interactions of variable Ig domains. Cell. 2007;130:1134–1145. doi: 10.1016/j.cell.2007.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graveley BR, Kaur A, Gunning D, Zipursky SL, Rowen L, Clemens JC. The organization and evolution of the dipteran and hymenopteran Down syndrome cell adhesion molecule (Dscam) genes. RNA. 2004;10:1499–1506. doi: 10.1261/rna.7105504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schreiber G, Keating AE. Protein binding specificity versus promiscuity. Curr Opin Struct Biol. 2011;21:50–61. doi: 10.1016/j.sbi.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaneko T, Sidhu SS, Li SS. Evolving specificity from variability for protein interaction domains. Trends Biochem Sci. 2011;36:183–90. doi: 10.1016/j.tibs.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 20.Kaneko T, Huang H, Zhao B, Li L, Liu H, Voss CK, Wu C, Schiller MR, Li SS. Loops govern SH2 domain specificity by controlling access to binding pockets. Sci Signal. 2010;3:ra34. doi: 10.1126/scisignal.2000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Appleton BA, Zhang Y, Wu P, Yin JP, Hunziker W, Skelton NJ, Sidhu SS, Wiesmann C. Comparative structural analysis of the Erbin PDZ domain and the first PDZ domain of ZO-1. Insights into determinants of PDZ domain specificity. J Biol Chem. 2006;281:22312–22320. doi: 10.1074/jbc.M602901200. [DOI] [PubMed] [Google Scholar]
- 22.Kimber MS, Nachman J, Cunningham AM, Gish GD, Pawson T, Pai EF. Structural basis for specificity switching of the Src SH2 domain. Mol Cell. 2000;5:1043–1049. doi: 10.1016/s1097-2765(00)80269-5. [DOI] [PubMed] [Google Scholar]
- 23.Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J Mol Biol. 1997;273:927–948. doi: 10.1006/jmbi.1997.1354. [DOI] [PubMed] [Google Scholar]
- 24.Meenan NA, Sharma A, Fleishman SJ, Macdonald CJ, Morel B, Boetzel R, Moore GR, Baker D, Kleanthous C. The structural and energetic basis for high selectivity in a high-affinity protein-protein interaction. Proc Natl Acad Sci USA. 2010;107:10080–10085. doi: 10.1073/pnas.0910756107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vinson C, Acharya A, Taparowsky EJ. Deciphering B-ZIP transcription factor interactions in vitro and in vivo. Biochim Biophys Acta. 2006;1759:4–12. doi: 10.1016/j.bbaexp.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 26.Gretes M, Lim DC, de Castro L, Jensen SE, Kang SG, Lee KJ, Strynadka NC. Insights into positive and negative requirements for protein-protein interactions by crystallographic analysis of the beta-lactamase inhibitory proteins BLIP, BLIP-I, and BLP. J Mol Biol. 2009;389:289–305. doi: 10.1016/j.jmb.2009.03.058. [DOI] [PubMed] [Google Scholar]
- 27.Skelton NJ, Koehler MF, Zobel K, Wong WL, Yeh S, Pisabarro MT, Yin JP, Lasky LA, Sidhu SS. Origins of PDZ domain ligand specificity. Structure determination and mutagenesis of the Erbin PDZ domain. J Biol Chem. 2003;278:7645–7654. doi: 10.1074/jbc.M209751200. [DOI] [PubMed] [Google Scholar]
- 28.Songyang Z, Gish G, Mbamalu G, Pawson T, Cantley LC. A single point mutation switches the specificity of group III Src homology (SH) 2 domains to that of group I SH2 domains. J Biol Chem. 1995;270:26029–26032. doi: 10.1074/jbc.270.44.26029. [DOI] [PubMed] [Google Scholar]
- 29.Weng Z, Rickles RJ, Feng S, Richard S, Shaw AS, Schreiber SL, Brugge JS. Structure-function analysis of SH3 domains: SH3 binding specificity altered by single amino acid substitutions. Mol Cell Biol. 1995;15:5627–5634. doi: 10.1128/mcb.15.10.5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li W, Dennis CA, Moore GR, James R, Kleanthous C. Protein-protein interaction specificity of Im9 for the endonuclease toxin colicin E9 defined by homologue-scanning mutagenesis. J Biol Chem. 1997;272:22253–22258. doi: 10.1074/jbc.272.35.22253. [DOI] [PubMed] [Google Scholar]
- 31.Van der Sloot AM, Kiel C, Serrano L, Stricher F. Protein design in biological networks: from manipulating the input to modifying the output. Protein Eng Des Sel. 2009;22:537–542. doi: 10.1093/protein/gzp032. [DOI] [PubMed] [Google Scholar]
- 32.Olive M, Williams SC, Dezan C, Johnson PF, Vinson C. Design of a C/EBP-specific, dominant-negative bZIP protein with both inhibitory and gain-of-function properties. J Biol Chem. 1996;271:2040–2047. doi: 10.1074/jbc.271.4.2040. [DOI] [PubMed] [Google Scholar]
- 33.van der Sloot AM, Tur V, Szegezdi E, Mullally MM, Cool RH, Samali A, Serrano L, Quax WJ. Designed tumor necrosis factor-related apoptosis-inducing ligand variants initiating apoptosis exclusively via the DR5 receptor. Proc Natl Acad Sci USA. 2006;103:8634–8639. doi: 10.1073/pnas.0510187103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee EF, Czabotar PE, van Delft MF, Michalak EM, Boyle MJ, Willis SN, Puthalakath H, Bouillet P, Colman PM, Huang DC, Fairlie WD. A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. J Cell Biol. 2008;180:341–355. doi: 10.1083/jcb.200708096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolon DN, Wah DA, Hersch GL, Baker TA, Sauer RT. Bivalent tethering of SspB to ClpXP is required for efficient substrate delivery: a protein-design study. Mol Cell. 2004;13:443–449. doi: 10.1016/s1097-2765(04)00027-9. [DOI] [PubMed] [Google Scholar]
- 36.Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ, Cathomen T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol. 2007;25:786–793. doi: 10.1038/nbt1317. [DOI] [PubMed] [Google Scholar]
- 37.Czyzyk J, Brogdon JL, Badou A, Henegariu O, Preston Hurlburt P, Flavell R, Bottomly K. Activation of CD4 T cells by Raf-independent effectors of Ras. Proc Natl Acad Sci USA. 2003;100:6003–6008. doi: 10.1073/pnas.1031494100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dreze M, Charloteaux B, Milstein S, Vidalain PO, Yildirim MA, Zhong Q, Svrzikapa N, Romero V, Laloux G, Brasseur R, Vandenhaute J, Boxem M, Cusick ME, Hill DE, Vidal M. 'Edgetic' perturbation of a C. elegans BCL2 ortholog. Nat Methods. 2009;6:843–849. doi: 10.1038/nmeth.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bashor CJ, Helman NC, Yan S, Lim WA. Using engineered scaffold interactions to reshape MAP kinase pathway signaling dynamics. Science. 2008;319:1539–1543. doi: 10.1126/science.1151153. [DOI] [PubMed] [Google Scholar]
- 40.Kiel C, Yus E, Serrano L. Engineering signal transduction pathways. Cell. 2010;140:33–47. doi: 10.1016/j.cell.2009.12.028. [DOI] [PubMed] [Google Scholar]
- 41.Jones S, Thornton JM. Principles of protein-protein interactions. Proc Natl Acad Sci USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cunningham BC, Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 43.Pons J, Rajpal A, Kirsch JF. Energetic analysis of an antigen/antibody interface: alanine scanning mutagenesis and double mutant cycles on the HyHEL-10/lysozyme interaction. Protein Sci. 1999;8:958–968. doi: 10.1110/ps.8.5.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boersma MD, Sadowsky JD, Tomita YA, Gellman SH. Hydrophile scanning as a complement to alanine scanning for exploring and manipulating protein-protein recognition: application to the Bim BH3 domain. Protein Sci. 2008;17:1232–1240. doi: 10.1110/ps.032896.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ponder JW, Richards FM. Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 46.Butterfoss GL, Kuhlman B. Computer-based design of novel protein structures. Annu Rev Biophys Biomol Struct. 2006;35:49–65. doi: 10.1146/annurev.biophys.35.040405.102046. [DOI] [PubMed] [Google Scholar]
- 47.Lippow SM, Tidor B. Progress in computational protein design. Curr Opin Biotechnol. 2007;18:305–311. doi: 10.1016/j.copbio.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karanicolas J, Kuhlman B. Computational design of affinity and specificity at protein-protein interfaces. Curr Opin Struct Biol. 2009;19:458–463. doi: 10.1016/j.sbi.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mandell DJ, Kortemme T. Computer-aided design of functional protein interactions. Nat Chem Biol. 2009;5:797–807. doi: 10.1038/nchembio.251. [DOI] [PubMed] [Google Scholar]
- 50.Boas FE, Harbury PB. Potential energy functions for protein design. Curr Opin Struct Biol. 2007;17:199–204. doi: 10.1016/j.sbi.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 51.Ponder JW, Case DA. Force fields for protein simulations. Adv Protein Chem. 2003;66:27–85. doi: 10.1016/s0065-3233(03)66002-x. [DOI] [PubMed] [Google Scholar]
- 52.Roux B, Simonson T. Implicit solvent models. Biophys Chem. 1999;78:1–20. doi: 10.1016/s0301-4622(98)00226-9. [DOI] [PubMed] [Google Scholar]
- 53.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 54.Bashford D, Case DA. Generalized born models of macromolecular solvation effects. Annu Rev Phys Chem. 2000;51:129–152. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 55.Green DF, Tidor B. Evaluation of electrostatic interactions. Curr Protoc Bioinformatics. 2003 doi: 10.1002/0471250953.bi0803s02. Chapter 8:Unit 8 3. [DOI] [PubMed] [Google Scholar]
- 56.Eisenberg D, McLachlan AD. Solvation energy in protein folding and binding. Nature. 1986;319:199–203. doi: 10.1038/319199a0. [DOI] [PubMed] [Google Scholar]
- 57.Ooi T, Oobatake M, Nemethy G, Scheraga HA. Accessible surface areas as a measure of the thermodynamic parameters of hydration of peptides. Proc Natl Acad Sci USA. 1987;84:3086–3090. doi: 10.1073/pnas.84.10.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 59.Levy RM, Zhang LY, Gallicchio E, Felts AK. On the nonpolar hydration free energy of proteins: surface area and continuum solvent models for the solute-solvent interaction energy. J Am Chem Soc. 2003;125:9523–9530. doi: 10.1021/ja029833a. [DOI] [PubMed] [Google Scholar]
- 60.Chen J, Brooks CL., III Implicit modeling of nonpolar solvation for simulating protein folding and conformational transitions. Phys Chem Chem Phys. 2008;10:471–481. doi: 10.1039/b714141f. [DOI] [PubMed] [Google Scholar]
- 61.Miyazawa S, Jernigan RL. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J Mol Biol. 1996;256:623–644. doi: 10.1006/jmbi.1996.0114. [DOI] [PubMed] [Google Scholar]
- 62.Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002;11:2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carter CW, Jr, LeFebvre BC, Cammer SA, Tropsha A, Edgell MH. Four-body potentials reveal protein-specific correlations to stability changes caused by hydrophobic core mutations. J Mol Biol. 2001;311:625–638. doi: 10.1006/jmbi.2001.4906. [DOI] [PubMed] [Google Scholar]
- 65.Feng Y, Kloczkowski A, Jernigan RL. Four-body contact potentials derived from two protein datasets to discriminate native structures from decoys. Proteins. 2007;68:57–66. doi: 10.1002/prot.21362. [DOI] [PubMed] [Google Scholar]
- 66.DeWitte RS, Shakhnovich EI. Pseudodihedrals: simplified protein backbone representation with knowledge-based energy. Protein Sci. 1994;3:1570–1581. doi: 10.1002/pro.5560030922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lu M, Dousis AD, Ma J. OPUS-PSP: an orientation-dependent statistical all-atom potential derived from side-chain packing. J Mol Biol. 2008;376:288–301. doi: 10.1016/j.jmb.2007.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Summa CM, Levitt M, Degrado WF. An atomic environment potential for use in protein structure prediction. J Mol Biol. 2005;352:986–1001. doi: 10.1016/j.jmb.2005.07.054. [DOI] [PubMed] [Google Scholar]
- 69.Misura KM, Morozov AV, Baker D. Analysis of anisotropic side-chain packing in proteins and application to high-resolution structure prediction. J Mol Biol. 2004;342:651–664. doi: 10.1016/j.jmb.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 70.Morozov AV, Kortemme T, Tsemekhman K, Baker D. Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations. Proc Natl Acad Sci USA. 2004;101:6946–6951. doi: 10.1073/pnas.0307578101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thomas PD, Dill KA. Statistical potentials extracted from protein structures: how accurate are they? J Mol Biol. 1996;257:457–469. doi: 10.1006/jmbi.1996.0175. [DOI] [PubMed] [Google Scholar]
- 72.Skolnick J. In quest of an empirical potential for protein structure prediction. Curr Opin Struct Biol. 2006;16:166–171. doi: 10.1016/j.sbi.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 73.Lu L, Lu H, Skolnick J. MULTIPROSPECTOR: an algorithm for the prediction of protein-protein interactions by multimeric threading. Proteins. 2002;49:350–364. doi: 10.1002/prot.10222. [DOI] [PubMed] [Google Scholar]
- 74.Clark LA, van Vlijmen HW. A knowledge-based forcefield for protein-protein interface design. Proteins. 2008;70:1540–1550. doi: 10.1002/prot.21694. [DOI] [PubMed] [Google Scholar]
- 75.Das R, Baker D. Macromolecular modeling with rosetta. Annu Rev Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 76.Grigoryan G, Keating AE. Structure-based prediction of bZIP partnering specificity. J Mol Biol. 2006;355:1125–1142. doi: 10.1016/j.jmb.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 77.Alvizo O, Mayo SL. Evaluating and optimizing computational protein design force fields using fixed composition-based negative design. Proc Natl Acad Sci USA. 2008;105:12242–12247. doi: 10.1073/pnas.0805858105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bolon DN, Grant RA, Baker TA, Sauer RT. Specificity versus stability in computational protein design. Proc Natl Acad Sci USA. 2005;102:12724–12729. doi: 10.1073/pnas.0506124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458:859–864. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Havranek JJ, Harbury PB. Automated design of specificity in molecular recognition. Nat Struct Biol. 2003;10:45–52. doi: 10.1038/nsb877. [DOI] [PubMed] [Google Scholar]
- 81.Fromer M, Shifman JM. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Comput Biol. 2009;5:e1000627. doi: 10.1371/journal.pcbi.1000627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dunbrack RL., Jr Rotamer libraries in the 21st century. Curr Opin Struct Biol. 2002;12:431–440. doi: 10.1016/s0959-440x(02)00344-5. [DOI] [PubMed] [Google Scholar]
- 83.Baldwin EP, Hajiseyedjavadi O, Baase WA, Matthews BW. The role of backbone flexibility in the accommodation of variants that repack the core of T4 lysozyme. Science. 1993;262:1715–1718. doi: 10.1126/science.8259514. [DOI] [PubMed] [Google Scholar]
- 84.Lim WA, Hodel A, Sauer RT, Richards FM. The crystal structure of a mutant protein with altered but improved hydrophobic core packing. Proc Natl Acad Sci USA. 1994;91:423–427. doi: 10.1073/pnas.91.1.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mandell DJ, Kortemme T. Backbone flexibility in computational protein design. Curr Opin Biotechnol. 2009;20:420–428. doi: 10.1016/j.copbio.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 86.Harbury PB, Plecs JJ, Tidor B, Alber T, Kim PS. High-resolution protein design with backbone freedom. Science. 1998;282:1462–1467. doi: 10.1126/science.282.5393.1462. [DOI] [PubMed] [Google Scholar]
- 87.Desjarlais JR, Handel TM. Side-chain and backbone flexibility in protein core design. J Mol Biol. 1999;290:305–318. doi: 10.1006/jmbi.1999.2866. [DOI] [PubMed] [Google Scholar]
- 88.Fu X, Apgar JR, Keating AE. Modeling backbone flexibility to achieve sequence diversity: the design of novel alpha-helical ligands for Bcl-xL. J Mol Biol. 2007;371:1099–1117. doi: 10.1016/j.jmb.2007.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Smith CA, Kortemme T. Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J Mol Biol. 2008;380:742–756. doi: 10.1016/j.jmb.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Smith CA, Kortemme T. Structure-based prediction of the peptide sequence space recognized by natural and synthetic PDZ domains. J Mol Biol. 2010;402:460–474. doi: 10.1016/j.jmb.2010.07.032. [DOI] [PubMed] [Google Scholar]
- 91.Benedix A, Becker CM, de Groot BL, Caflisch A, Bockmann RA. Predicting free energy changes using structural ensembles. Nat Methods. 2009;6:3–4. doi: 10.1038/nmeth0109-3. [DOI] [PubMed] [Google Scholar]
- 92.Desmet J, De Maeyer M, Hazes B, Lasters I. The dead-end elimination theorem and its use in protein side-chain positioning. Nature. 1992;356:539–542. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- 93.Desmet J, Spriet J, Lasters I. Fast and accurate side-chain topology and energy refinement (FASTER) as a new method for protein structure optimization. Proteins. 2002;48:31–43. doi: 10.1002/prot.10131. [DOI] [PubMed] [Google Scholar]
- 94.Leach AR, Lemon AP. Exploring the conformational space of protein side chains using dead-end elimination and the A* algorithm. Proteins. 1998;33:227–239. doi: 10.1002/(sici)1097-0134(19981101)33:2<227::aid-prot7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 95.Kingsford CL, Chazelle B, Singh M. Solving and analyzing side-chain positioning problems using linear and integer programming. Bioinformatics. 2005;21:1028–1036. doi: 10.1093/bioinformatics/bti144. [DOI] [PubMed] [Google Scholar]
- 96.Lee C. Predicting protein mutant energetics by self-consistent ensemble optimization. J Mol Biol. 1994;236:918–39. doi: 10.1006/jmbi.1994.1198. [DOI] [PubMed] [Google Scholar]
- 97.Voigt CA, Gordon DB, Mayo SL. Trading accuracy for speed: a quantitative comparison of search algorithms in protein sequence design. J Mol Biol. 2000;299:789–803. doi: 10.1006/jmbi.2000.3758. [DOI] [PubMed] [Google Scholar]
- 98.Georgiev I, Keedy D, Richardson JS, Richardson DC, Donald BR. Algorithm for backrub motions in protein design. Bioinformatics. 2008;24:i196–204. doi: 10.1093/bioinformatics/btn169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Georgiev I, Lilien RH, Donald BR. The minimized dead-end elimination criterion and its application to protein redesign in a hybrid scoring and search algorithm for computing partition functions over molecular ensembles. J Comput Chem. 2008;29:1527–1542. doi: 10.1002/jcc.20909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Barth P, Alber T, Harbury PB. Accurate, conformation-dependent predictions of solvent effects on protein ionization constants. Proc Natl Acad Sci USA. 2007;104:4898–4903. doi: 10.1073/pnas.0700188104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Green DF, Dennis AT, Fam PS, Tidor B, Jasanoff A. Rational design of new binding specificity by simultaneous mutagenesis of calmodulin and a target peptide. Biochemistry. 2006;45:12547–12559. doi: 10.1021/bi060857u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Grigoryan G, Zhou F, Lustig SR, Ceder G, Morgan D, Keating AE. Ultra-fast evaluation of protein energies directly from sequence. PLoS Comput Biol. 2006;2:e63. doi: 10.1371/journal.pcbi.0020063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kortemme T, Joachimiak LA, Bullock AN, Schuler AD, Stoddard BL, Baker D. Computational redesign of protein-protein interaction specificity. Nat Struct Mol Biol. 2004;11:371–379. doi: 10.1038/nsmb749. [DOI] [PubMed] [Google Scholar]
- 104.Ali MH, Taylor CM, Grigoryan G, Allen KN, Imperiali B, Keating AE. Design of a heterospecific, tetrameric, 21-residue miniprotein with mixed alpha/beta structure. Structure. 2005;13:225–234. doi: 10.1016/j.str.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 105.Sammond DW, Eletr ZM, Purbeck C, Kuhlman B. Computational design of second-site suppressor mutations at protein-protein interfaces. Proteins. 2010;78:1055–1065. doi: 10.1002/prot.22631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Potapov V, Reichmann D, Abramovich R, Filchtinski D, Zohar N, Ben Halevy D, Edelman M, Sobolev V, Schreiber G. Computational redesign of a protein-protein interface for high affinity and binding specificity using modular architecture and naturally occurring template fragments. J Mol Biol. 2008;384:109–119. doi: 10.1016/j.jmb.2008.08.078. [DOI] [PubMed] [Google Scholar]
- 107.Barth P, Schoeffler A, Alber T. Targeting metastable coiled-coil domains by computational design. J Am Chem Soc. 2008;130:12038–12044. doi: 10.1021/ja802447e. [DOI] [PubMed] [Google Scholar]
- 108.Yosef E, Politi R, Choi MH, Shifman JM. Computational design of calmodulin mutants with up to 900-fold increase in binding specificity. J Mol Biol. 2009;385:1470–1480. doi: 10.1016/j.jmb.2008.09.053. [DOI] [PubMed] [Google Scholar]
- 109.Reinke AW, Grigoryan G, Keating AE. Identification of bZIP interaction partners of viral proteins HBZ, MEQ, BZLF1, and K-bZIP using coiled-coil arrays. Biochemistry. 2010;49:1985–1997. doi: 10.1021/bi902065k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen TS, Reinke AW, Keating AE. Design of peptide inhibitors that bind the bZIP domain of Epstein-Barr virus protein BZLF1. J Mol Biol. 2011;408:304–320. doi: 10.1016/j.jmb.2011.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sharabi O, Dekel A, Shifman JM. Triathlon for energy functions: who is the winner for design of protein-protein interactions? Proteins. 2011;79:1487–1498. doi: 10.1002/prot.22977. [DOI] [PubMed] [Google Scholar]
- 112.Ramachandran S, Kota P, Ding F, Dokholyan NV. Automated minimization of steric clashes in protein structures. Proteins. 2011;79:261–270. doi: 10.1002/prot.22879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 114.Pokala N, Handel TM. Energy functions for protein design: adjustment with protein-protein complex affinities, models for the unfolded state, and negative design of solubility and specificity. J Mol Biol. 2005;347:203–227. doi: 10.1016/j.jmb.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 115.Yin S, Ding F, Dokholyan NV. Modeling backbone flexibility improves protein stability estimation. Structure. 2007;15:1567–1576. doi: 10.1016/j.str.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 116.Potapov V, Cohen M, Schreiber G. Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein Eng Des Sel. 2009;22:553–560. doi: 10.1093/protein/gzp030. [DOI] [PubMed] [Google Scholar]
- 117.Lippow SM, Wittrup KD, Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol. 2007;25:1171–1176. doi: 10.1038/nbt1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sammond DW, Eletr ZM, Purbeck C, Kimple RJ, Siderovski DP, Kuhlman B. Structure-based protocol for identifying mutations that enhance protein-protein binding affinities. J Mol Biol. 2007;371:1392–1404. doi: 10.1016/j.jmb.2007.05.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chen JR, Chang BH, Allen JE, Stiffler MA, MacBeath G. Predicting PDZ domain-peptide interactions from primary sequences. Nat Biotechnol. 2008;26:1041–1045. doi: 10.1038/nbt.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gfeller D, Butty F, Wierzbicka M, Verschueren E, Vanhee P, Huang H, Ernst A, Dar N, Stagljar I, Serrano L, Sidhu SS, Bader GD, Kim PM. The multiple-specificity landscape of modular peptide recognition domains. Mol Syst Biol. 2011;7:484. doi: 10.1038/msb.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fong JH, Keating AE, Singh M. Predicting specificity in bZIP coiled-coil protein interactions. Genome Biol. 2004;5:R11. doi: 10.1186/gb-2004-5-2-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Acharya A, Rishi V, Vinson C. Stability of 100 homo and heterotypic coiled-coil a-a' pairs for ten amino acids (A, L, I, V, N, K, S, T, E, and R) Biochemistry. 2006;45:11324–11332. doi: 10.1021/bi060822u. [DOI] [PubMed] [Google Scholar]
- 123.Skerker JM, Perchuk BS, Siryaporn A, Lubin EA, Ashenberg O, Goulian M, Laub MT. Rewiring the specificity of two-component signal transduction systems. Cell. 2008;133:1043–1054. doi: 10.1016/j.cell.2008.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ashenberg O, Rozen-Gagnon K, Laub MT, Keating AE. Determinants of homodimerization specificity in histidine kinases. J Mol Biol. 2011;413:222–235. doi: 10.1016/j.jmb.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jackel C, Kast P, Hilvert D. Protein design by directed evolution. Annu Rev Biophys. 2008;37:153–173. doi: 10.1146/annurev.biophys.37.032807.125832. [DOI] [PubMed] [Google Scholar]
- 126.Cirino PC, Mayer KM, Umeno D. Generating mutant libraries using error-prone PCR. Methods Mol Biol. 2003;231:3–9. doi: 10.1385/1-59259-395-X:3. [DOI] [PubMed] [Google Scholar]
- 127.Shivange AV, Marienhagen J, Mundhada H, Schenk A, Schwaneberg U. Advances in generating functional diversity for directed protein evolution. Curr Opin Chem Biol. 2009;13:19–25. doi: 10.1016/j.cbpa.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 128.Mena MA, Daugherty PS. Automated design of degenerate codon libraries. Protein Eng Des Sel. 2005;18:559–561. doi: 10.1093/protein/gzi061. [DOI] [PubMed] [Google Scholar]
- 129.Hayes RJ, Bentzien J, Ary ML, Hwang MY, Jacinto JM, Vielmetter J, Kundu A, Dahiyat BI. Combining computational and experimental screening for rapid optimization of protein properties. Proc Natl Acad Sci USA. 2002;99:15926–15931. doi: 10.1073/pnas.212627499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kayushin AL, Korosteleva MD, Miroshnikov AI, Kosch W, Zubov D, Piel N. A convenient approach to the synthesis of trinucleotide phosphoramidites—synthons for the generation of oligonucleotide/peptide libraries. Nucleic Acids Res. 1996;24:3748–3755. doi: 10.1093/nar/24.19.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhao H, Arnold FH. Optimization of DNA shuffling for high fidelity recombination. Nucleic Acids Res. 1997;25:1307–1308. doi: 10.1093/nar/25.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Frieder D, Larijani M, Tang E, Parsa JY, Basit W, Martin A. Antibody diversification: mutational mechanisms and oncogenesis. Immunol Res. 2006;35:75–88. doi: 10.1385/IR:35:1:75. [DOI] [PubMed] [Google Scholar]
- 133.Levin AM, Weiss GA. Optimizing the affinity and specificity of proteins with molecular display. Mol Biosyst. 2006;2:49–57. doi: 10.1039/b511782h. [DOI] [PubMed] [Google Scholar]
- 134.Sidhu SS, Koide S. Phage display for engineering and analyzing protein interaction interfaces. Curr Opin Struct Biol. 2007;17:481–487. doi: 10.1016/j.sbi.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 135.Georgiou G, Poetschke HL, Stathopoulos C, Francisco JA. Practical applications of engineering gram-negative bacterial cell surfaces. Trends Biotechnol. 1993;11:6–10. doi: 10.1016/0167-7799(93)90068-K. [DOI] [PubMed] [Google Scholar]
- 136.Feldhaus MJ, Siegel RW, Opresko LK, Coleman JR, Feldhaus JM, Yeung YA, Cochran JR, Heinzelman P, Colby D, Swers J, Graff C, Wiley HS, Wittrup KD. Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat Biotechnol. 2003;21:163–170. doi: 10.1038/nbt785. [DOI] [PubMed] [Google Scholar]
- 137.Roberts RW, Szostak JW. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci USA. 1997;94:12297–12302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hanes J, Pluckthun A. In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci USA. 1997;94:4937–4942. doi: 10.1073/pnas.94.10.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Norman TC, Smith DL, Sorger PK, Drees BL, O'Rourke SM, Hughes TR, Roberts CJ, Friend SH, Fields S, Murray AW. Genetic selection of peptide inhibitors of biological pathways. Science. 1999;285:591–595. doi: 10.1126/science.285.5427.591. [DOI] [PubMed] [Google Scholar]
- 140.Pelletier JN, Arndt KM, Pluckthun A, Michnick SW. An in vivo library-versus-library selection of optimized protein-protein interactions. Nat Biotechnol. 1999;17:683–690. doi: 10.1038/10897. [DOI] [PubMed] [Google Scholar]
- 141.Magliery TJ, Wilson CG, Pan W, Mishler D, Ghosh I, Hamilton AD, Regan L. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: scope and mechanism. J Am Chem Soc. 2005;127:146–157. doi: 10.1021/ja046699g. [DOI] [PubMed] [Google Scholar]
- 142.Mason JM, Schmitz MA, Muller KM, Arndt KM. Semirational design of Jun-Fos coiled coils with increased affinity: universal implications for leucine zipper prediction and design. Proc Natl Acad Sci USA. 2006;103:8989–8994. doi: 10.1073/pnas.0509880103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Levin KB, Dym O, Albeck S, Magdassi S, Keeble AH, Kleanthous C, Tawfik DS. Following evolutionary paths to protein-protein interactions with high affinity and selectivity. Nat Struct Mol Biol. 2009;16:1049–1055. doi: 10.1038/nsmb.1670. [DOI] [PubMed] [Google Scholar]
- 144.Mason JM, Muller KM, Arndt KM. Positive aspects of negative design: simultaneous selection of specificity and interaction stability. Biochemistry. 2007;46:4804–4814. doi: 10.1021/bi602506p. [DOI] [PubMed] [Google Scholar]
- 145.Dutta S, Gulla S, Chen TS, Fire E, Grant RA, Keating AE. Determinants of BH3 binding specificity for Mcl-1 versus Bcl-xL. J Mol Biol. 2010;398:747–762. doi: 10.1016/j.jmb.2010.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Abe Y, Yoshikawa T, Inoue M, Nomura T, Furuya T, Yamashita T, Nagano K, Nabeshi H, Yoshioka Y, Mukai Y, Nakagawa S, Kamada H, Tsutsumi Y, Tsunoda S. Fine tuning of receptor-selectivity for tumor necrosis factor-alpha using a phage display system with one-step competitive panning. Biomaterials. 2011;32:5498–5504. doi: 10.1016/j.biomaterials.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 147.Matsumura N, Tsuji T, Sumida T, Kokubo M, Onimaru M, Doi N, Takashima H, Miyamoto-Sato E, Yanagawa H. mRNA display selection of a high-affinity, Bcl-X(L)-specific binding peptide. FASEB J. 2010;24:2201–2210. doi: 10.1096/fj.09-143008. [DOI] [PubMed] [Google Scholar]
- 148.Ernst A, Gfeller D, Kan Z, Seshagiri S, Kim PM, Bader GD, Sidhu SS. Coevolution of PDZ domain-ligand interactions analyzed by high-throughput phage display and deep sequencing. Mol Biosyst. 2010;6:1782–1790. doi: 10.1039/c0mb00061b. [DOI] [PubMed] [Google Scholar]
- 149.Voigt CA, Mayo SL, Arnold FH, Wang ZG. Computational method to reduce the search space for directed protein evolution. Proc Natl Acad Sci USA. 2001;98:3778–3783. doi: 10.1073/pnas.051614498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Voigt CA, Martinez C, Wang ZG, Mayo SL, Arnold FH. Protein building blocks preserved by recombination. Nat Struct Biol. 2002;9:553–558. doi: 10.1038/nsb805. [DOI] [PubMed] [Google Scholar]
- 151.Treynor TP, Vizcarra CL, Nedelcu D, Mayo SL. Computationally designed libraries of fluorescent proteins evaluated by preservation and diversity of function. Proc Natl Acad Sci USA. 2007;104:48–53. doi: 10.1073/pnas.0609647103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Allen BD, Nisthal A, Mayo SL. Experimental library screening demonstrates the successful application of computational protein design to large structural ensembles. Proc Natl Acad Sci USA. 2010;107:19838–19843. doi: 10.1073/pnas.1012985107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chica RA, Moore MM, Allen BD, Mayo SL. Generation of longer emission wavelength red fluorescent proteins using computationally designed libraries. Proc Natl Acad Sci USA. 2010;107:20257–20262. doi: 10.1073/pnas.1013910107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Barderas R, Desmet J, Timmerman P, Meloen R, Casal JI. Affinity maturation of antibodies assisted by in silico modeling. Proc Natl Acad Sci USA. 2008;105:9029–9034. doi: 10.1073/pnas.0801221105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Guntas G, Purbeck C, Kuhlman B. Engineering a protein-protein interface using a computationally designed library. Proc Natl Acad Sci USA. 2010;107:19296–19301. 156. doi: 10.1073/pnas.1006528107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Grove TZ, Hands M, Regan L. Creating novel proteins by combining design and selection. Protein Eng Des Sel. 2010;23:449–455. doi: 10.1093/protein/gzq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lippow SM, Moon TS, Basu S, Yoon SH, Li X, Chapman BA, Robison K, Lipovsek D, Prather KL. Engineering enzyme specificity using computational design of a defined-sequence library. Chem Biol. 2010;17:1306–1315. doi: 10.1016/j.chembiol.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 158.Saraf MC, Moore GL, Goodey NM, Cao VY, Benkovic SJ, Maranas CD. IPRO: an iterative computational protein library redesign and optimization procedure. Biophys J. 2006;90:4167–4180. doi: 10.1529/biophysj.105.079277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Pantazes RJ, Saraf MC, Maranas CD. Optimal protein library design using recombination or point mutations based on sequence-based scoring functions. Protein Eng Des Sel. 2007;20:361–373. doi: 10.1093/protein/gzm030. [DOI] [PubMed] [Google Scholar]
- 160.Parker AS, Griswold KE, Bailey-Kellogg C. Optimization of combinatorial mutagenesis. J Comput Biol. 2011;18:1743–1756. doi: 10.1089/cmb.2011.0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wang W, Saven JG. Designing gene libraries from protein profiles for combinatorial protein experiments. Nucleic Acids Res. 2002;30:e120. doi: 10.1093/nar/gnf119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kono H, Saven JG. Statistical theory for protein combinatorial libraries. Packing interactions, backbone flexibility, and the sequence variability of a main-chain structure. J Mol Biol. 2001;306:607–628. doi: 10.1006/jmbi.2000.4422. [DOI] [PubMed] [Google Scholar]
- 163.Otey CR, Landwehr M, Endelman JB, Hiraga K, Bloom JD, Arnold FH. Structure-guided recombination creates an artificial family of cytochromes P450. PLoS Biol. 2006;4:e112. doi: 10.1371/journal.pbio.0040112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Heinzelman P, Snow CD, Smith MA, Yu X, Kannan A, Boulware K, Villalobos A, Govindarajan S, Minshull J, Arnold FH. SCHEMA recombination of a fungal cellulase uncovers a single mutation that contributes markedly to stability. J Biol Chem. 2009;284:26229–26233. doi: 10.1074/jbc.C109.034058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Khersonsky O, Rothlisberger D, Wollacott AM, Murphy P, Dym O, Albeck S, Kiss G, Houk KN, Baker D, Tawfik DS. Optimization of the in-silico-designed kemp eliminase KE70 by computational design and directed evolution. J Mol Biol. 2011;407:391–412. doi: 10.1016/j.jmb.2011.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Karanicolas J, Corn JE, Chen I, Joachimiak LA, Dym O, Peck SH, Albeck S, Unger T, Hu W, Liu G, Delbecq S, Montelione GT, Spiegel CP, Liu DR, Baker D. A de novo protein binding pair by computational design and directed evolution. Mol Cell. 2010;42:250–260. doi: 10.1016/j.molcel.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, Wilson IA, Baker D. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science. 2011;332:816–821. doi: 10.1126/science.1202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rothlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, Albeck S, Houk KN, Tawfik DS, Baker D. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 169.Musacchio A, Saraste M, Wilmanns M. High-resolution crystal structures of tyrosine kinase SH3 domains complexed with proline-rich peptides. Nat Struct Biol. 1994;1:546–551. doi: 10.1038/nsb0894-546. [DOI] [PubMed] [Google Scholar]
- 170.Poy F, Yaffe MB, Sayos J, Saxena K, Morra M, Sumegi J, Cantley LC, Terhorst C, Eck MJ. Crystal structures of the XLP protein SAP reveal a class of SH2 domains with extended, phosphotyrosine-independent sequence recognition. Mol Cell. 1999;4:555–561. doi: 10.1016/s1097-2765(00)80206-3. [DOI] [PubMed] [Google Scholar]
- 171.Birrane G, Chung J, Ladias JA. Novel mode of ligand recognition by the Erbin PDZ domain. J Biol Chem. 2003;278:1399–13402. doi: 10.1074/jbc.C200571200. [DOI] [PubMed] [Google Scholar]
- 172.Glover JN, Harrison SC. Crystal structure of the heterodimeric bZIP transcription factor c-Fos-c-Jun bound to DNA. Nature. 1995;373:257–261. doi: 10.1038/373257a0. [DOI] [PubMed] [Google Scholar]
- 173.Fire E, Gulla SV, Grant RA, Keating AE. Mcl-1-Bim complexes accommodate surprising point mutations via minor structural changes. Protein Sci. 2010;19:507–519. doi: 10.1002/pro.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Meijers R, Puettmann-Holgado R, Skiniotis G, Liu JH, Walz T, Wang JH, Schmucker D. Structural basis of Dscam isoform specificity. Nature. 2007;449:487–491. doi: 10.1038/nature06147. [DOI] [PubMed] [Google Scholar]