

Abstract
In spite of structural similarities Epithelial- (E-) and Neural- (N-) cadherins are expressed at two types of synapses and differ significantly in dimer disassembly kinetics. Recent studies suggested that the formation of an X-dimer intermediate in E-cadherin is the key requirement for rapid disassembly of the adhesive dimer (Harrison et al., Nat Struct Mol Biol 2010;17:348–357 and Hong et al., J Cell Biol 2011;192:1073–1083). The X-interface in E-cadherin involves three noncovalent interactions, none of which is conserved in N-cadherin. Dimer disassembly is slow at low calcium concentration in N-cadherin, which may be due to the differences in the X-interface residues. To investigate the origin of the slow disassembly kinetics we introduced three point mutations into N-cadherin to provide the opportunity for the formation of X-interface interactions. Spectroscopic studies showed that the triple mutation did not affect the stability or the calcium-binding affinity of the X-enabled N-cadherin mutant. Analytical size exclusion chromatography was used to assay for the effect of the mutation on the rate of dimer disassembly. Contrary to our expectation, the disassembly of dimers of the X-enabled N-cadherin mutant was as slow as seen for wild-type N-cadherin in the apo-state. Thus, the differences in the X-interface residues are not the origin of slow disassembly kinetics of N-cadherin in the apo-state.
Keywords: analytical SEC, strand-crossover dimmer, calcium binding
Introduction
An active search is underway for a unifying molecular code that dictates the stability and dynamics of adherens junctions. This is a compelling physiological question that has its origins in the noncovalent interactions between the cadherin protein components of adherens junctions.1 Cadherins are transmembrane cell–cell adhesion proteins that require calcium for their adhesive function.2–6 The extracellular region of classical cadherins has five modular domains (EC1-5). Three calcium ions bind at the interface between these domains.7,8 The adhesive structure is widely believed to be a strand-crossover between βA-strands from EC1 domains of adhesive partners. They mediate dynamic adhesive processes during embryogenesis, tissue morphogenesis,9,10 cell differentiation,11 and reorganization of adult soft tissues.12 Abnormal cadherin expression, molecular defects in cadherin function and mutations in calcium-binding sites are associated with metastatic cancers.13–15
The two most well studied members of the classical cadherin family are neural- (N-) and epithelial- (E-) cadherin, which show 81% sequence identity or similarity [Fig. 1(A)].16 While both N- and E-cadherin form adhesive dimer through a strand-crossover interface,18 they differ significantly in disassembly kinetics in vitro19–22 and in vivo.23 At high calcium concentrations the disassembly kinetics of both E- and N-cadherin dimers are fast. However, at low calcium concentrations the disassembly kinetics differ significantly; while E-cadherin dissociates rapidly, N-cadherin forms a kinetically trapped dimer.19 This kinetically trapped dimer of N-cadherin may form at excitatory synapses24–27 where there are large fluctuations in extracellular calcium.28
Figure 1.
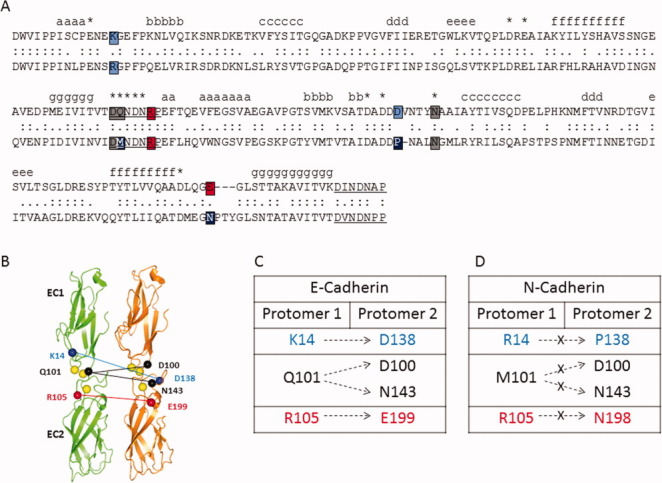
Comparison of X-interface amino acids in E- and N-cadherin. (A) Sequence comparison of E- and N-cadherin. The amino acid sequence of E-cadherin (top) and N-cadherin (bottom) are aligned.16 Identical residues (:) and similar residues (.) are indicated. Amino acids at the X-interface in E-cadherin are color highlighted (K14 to D138-blue, Q101 to D100 and N143- gray, R105 to E199- red). At least one amino acid (black) in each of the four X-interface interactions is not conserved in N-cadherin. (B) Schematic depicting the interactions at the X-interface between two protomers in E-cadherin. Ribbon drawing shows the first two domains of E-cadherin from 1FF5.pdb.17 The four intermolecular ionic interactions between EC1-EC2 of adhesive partners include ionic interactions between K14 and D138 (blue) and between R105 and E199 (red), and hydrogen bonds to N143 and D100 from Q101 (black). Calcium is shown (yellow). (C) X-interface interactions in E-cadherin. (D) Nonconserved X-interface amino acids in N-cadherin.
What are the molecular components that dictate the assembly and disassembly in classical cadherins? Recent studies of E-cadherin proposed that a transient, low affinity dimeric structure termed the X-dimer is the reason for its fast disassembly.29,30 The key noncovalent interactions at the X-interface in E-cadherin are not conserved in N-cadherin [Fig. 1(A)]. Are the observed kinetic differences between the N- and E-cadherin dimers in the apo-state due to differences in the X-interface residues?
X-dimer is formed between EC1-EC2 of adhesive partners via intermolecular hydrogen bond and ionic interactions.29 Figure 1 shows the aligned sequences of E- and N-cadherin. The residues involved in the X-interface in E-cadherin are highlighted, along with the corresponding residues in N-cadherin. Specifically, X-interface interactions in E-cadherin include intermolecular ionic interactions between the side chains of K14 and D138, and between the side chains of R105 and E199. In addition, the side chain of Q101 participates in intermolecular hydrogen bond interactions with side chains of both N143 and D100. These interactions are illustrated schematically in Figures 1(B,C). Out of the seven amino acids, which participate in the X-interface interactions in E-cadherin, three are not conserved in N-cadherin [Fig. 1(D)]. As such, neither of the ionic interactions nor the hydrogen bonding interactions with Q101 are conserved in N-cadherin.
Recent studies have shown the importance of the X-interface in the disassembly kinetics of E-cadherin. Harrison et al. studied the kinetics of disassembly of two mutants (K14E and K14S) of E-cadherin that targeted one of the four X-interface interactions.29 Through sedimentation velocity, surface plasmon resonance, and crystallographic studies they found that the mutations decreased the rate of association and dissociation of the strand-crossover dimer. From these data they inferred that X-dimer formation in E-cadherin is an intermediate step in the assembly and disassembly of a strand-crossover dimer.29 Later Hong et al. studied two X-interface mutants (K14E and Q101A/N143A) of E-cadherin using photoactivation and coimmunoprecipitation assays. These studies showed that the assembly of E-cadherin is independent of the X-interface, but disassembly occurs via the X-dimer intermediate.30 Hence, E-cadherin assembles and disassembles junctions through different pathways.
Previous studies from our group have noted differences in the kinetics of disassembly for NCAD12 and ECAD12, constructs containing only EC1 and EC2. A schematic of experimentally observable monomeric and dimeric species for E- and N-cadherin is shown in Figure 2. In the apo-state the proteins were monomeric, Mapo. Addition of calcium to Mapo forms Msat leading to formation of the strand-crossover adhesive dimer, Dsat. The kinetics of the equilibrium between Msat and Dsat are fast in the presence of calcium for both proteins. We also observed a dimeric species that is unique to N-cadherin which is formed upon depletion of calcium from Dsat and is kinetically trapped (D*apo; Fig. 2). The D*apo form does not exist for E-cadherin.
Figure 2.
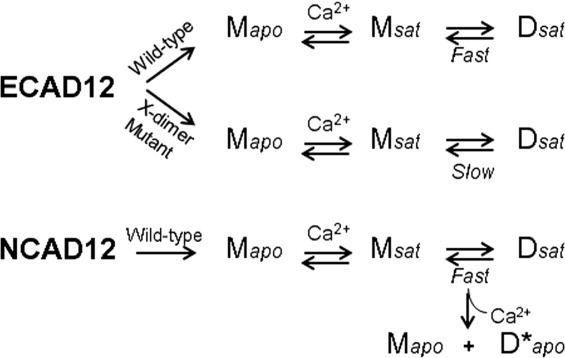
The linkage between different dimeric forms in ECAD12 and NCAD12. Model showing the linked equilibria for wild-type and an X-interface mutant29,30 of ECAD12 and wild-type NCAD12 where Mapo is the calcium-free monomer, Msat is the calcium-saturated monomer, and Dsat is the calcium-saturated dimer, which is the strand-crossover adhesive dimer. D*apo, which is kinetically trapped, is unique to N-cadherin and is formed from decalcification of Dsat.19
E- and N-cadherin have specific physiological niches. They are segregated at synapses in the brain. N-cadherin is considered a “synaptic tag” for actively stimulated excitatory synapses31 that are known to undergo large fluxes in extracellular calcium concentration.28 At an inhibitory synapse where E-cadherin is located, the concentration of calcium is relatively constant. On the basis of their different physiological niches, we might expect that the calcium-dependence of dimer formation or disassembly for E-and N-cadherin would be different. Does the lack of an X-interface influence the formation or disassembly of the kinetically trapped dimer that is unique to N-cadherin, D*apo?
To investigate the link between slow disassembly kinetics in the apo-state and X-interface we created a triple mutant of N-cadherin (M101Q/P138D/N198E) which has all of the components to form the X-interface (X-enabled NCAD12). Sequence alignment of ECAD12 and NCAD12 showed that the D138, Q101, and E199 in E-cadherin are replaced by P138, M101, and N198, respectively, in N-cadherin [Fig. 1(A)]. We expected that the mutant protein would show fast disassembly kinetics in the apo-state like wild-type E-cadherin, since both have an X-interface. Analytical SEC studies were performed on both wild-type and X-enabled NCAD12 to examine the disassembly kinetics. In summary, our studies show that the slow disassembly kinetics in the apo-state in wild-type N-cadherin does not originate from the differences in the X-interface residues.
Results
To study the connection between slow disassembly kinetics and the nonconserved X-interface we created a mutant N-cadherin with residues required for all four X-interface interactions. Spectroscopic studies were performed to monitor the calcium-binding properties and the stability of this construct. We performed analytical SEC experiments to observe the disassembly kinetics of different dimeric forms, Dsat and D*apo, for wild-type and X-enabled NCAD12.
Thermal-unfolding studies
Thermal-unfolding studies were performed to assess whether the mutations had an effect on the structure and stability of the protein. The CD signal at 230 nm from thermal-denaturation experiments was plotted as a function of temperature in the apo [Fig. 3(A)] and the calcium-saturated [1 mM; Fig. 3(B)] states. Two transitions were observed for wild-type and X-enabled NCAD12 in both calcium conditions. The first and second transition data from wild-type and X-enabled NCAD12 [Fig. 3(A)] are in agreement with unfolding studies of the isolated domains EC232 and the EC1,33 respectively. Each transition was analyzed separately to provide estimates for the enthalpy and temperature of the unfolding transition.
Figure 3.
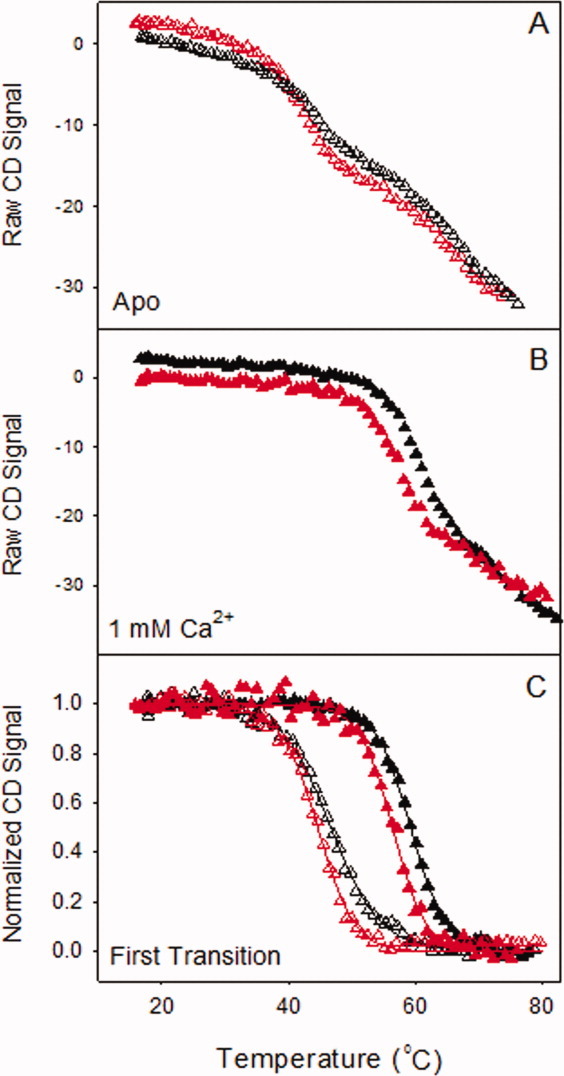
Thermal-unfolding of wild-type and X-enabled NCAD12 at 230 nm. The raw CD signal versus the probe temperature is represented for wild-type (black) and X-enabled NCAD12 (red) in (A) the apo-state (Δ) and (B) the calcium saturated state (▴). (C) Normalized data from the first transition for the apo and calcium-bound states are plotted versus temperature. The solid lines are simulated based on parameters resolved from fits to the Gibbs–Helmholtz equation.
First transition
Unfolding data for the first transition were fit to the Gibbs–Helmholtz equation and resolved parameters are reported in Table I along with calculated values for ΔG° at 25°C. The baseline corrected results from the analysis of the first transition are plotted in Figure 3(C). There was a clear increase in stability upon addition of 1 mM calcium. In the apo- state and the calcium-saturated state, wild-type and X-enabled NCAD12 had similar ΔHm and Tm values (Table I). These results indicate that the triple mutation did not significantly impact the protein structure and calcium-dependent stability of NCAD12.
Table I.
Summary of Parameters Resolved From the First Transition of the Thermal-Unfolding Experiments for Wild-Type and X-Enabled NCAD12 in the Absence and the Presence of Calcium
| Protein | Buffer | ΔHm (kcal mol−1) | Tm (°C) | ΔG° at 25°C (kcal mol−1) |
|---|---|---|---|---|
| Wild-type | Apo | 68 ± 8 | 45 ± 2 | 3.6 ± 0.5 |
| Ca2+ | 79 ± 5 | 59 ± 3 | 6.3 ± 0.7 | |
| X-enabled | Apo | 69 ± 5 | 44 ± 1 | 3.6 ± 0.3 |
| Ca2+ | 82 ± 5 | 57 ± 1 | 6.2 ± 0.9 |
Second transition
Both wild-type and X-enabled NCAD12 showed similar second transitions in the absence and presence of calcium. Because calcium does not impact the stability of the second transition, calcium must dissociate before EC1 domain unfolds. These results suggest that the mutations to create the X-enabled NCAD12 did not affect the EC1 structure and stability. The melting temperatures for the second transition were estimated since they could not be fit to the Gibbs–Helmholtz equation due to insufficient baseline data. Estimated melting temperatures for wild-type and X-enabled NCAD12 (∼70°C) were similar to melting temperature of EC1.33
Calcium-binding studies
Calcium titrations were performed to monitor the impact of the X-enabled NCAD12 mutation on calcium-binding affinity. Representative calcium titrations are shown in Figure 4. The CD signal for both proteins increased as calcium was added. Analysis of binding data for the two proteins yielded similar free energy changes of −6.2 ± 0.2 kcal mol−1. These results indicate that the X-enabled mutation did not affect the calcium binding affinity of the protein.
Figure 4.
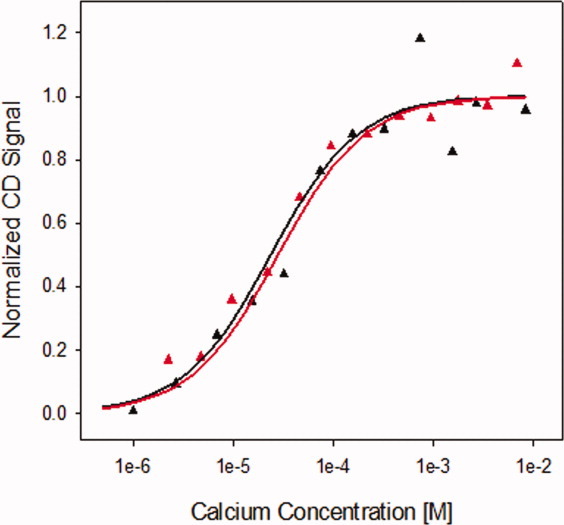
Calcium titrations of wild-type and X-enabled NCAD12. The CD signal changes during the calcium titrations of wild-type (black) and X-enabled NCAD12 (red) monitored by circular dichroism at 229 nm. Protein concentration was 2.5 μM. The lines are simulated based on parameters resolved from fits to Eq. (1).
Analytical SEC studies
Kinetics of monomer–dimer equilibria as a function of calcium concentration
Analytical SEC was used to evaluate the impact of the X-enabled N-cadherin mutant on the disassembly kinetics of D*apo and on the calcium-dependent dimerization. In the apo-mobile phase, both wild-type and X-enabled NCAD12 eluted as two peaks indicating the slow dissociation rate of dimer on the timescale of the chromatographic experiment [30 min; Fig. 5(A)]. It is important to note that although the sample is in high calcium concentration, the assay condition is not. Distinct monomer and dimer peaks in these calcium-added samples result from the apo-mobile phase used in the chromatographic analysis and slow dimer disassembly of apo-protein on the chromatographic time scale. The similarity of the wild-type and X-enabled NCAD12 data indicate that they have similar equilibrium and kinetic properties of dimerization, and that these properties differ significantly from those of ECAD12 under similar conditions [Fig. 5(A) inset]. Thus, the presence of the X-interface residues in X-enabled NCAD12 does not confer fast disassembly kinetics in the apo-condition as seen for ECAD12.
Figure 5.
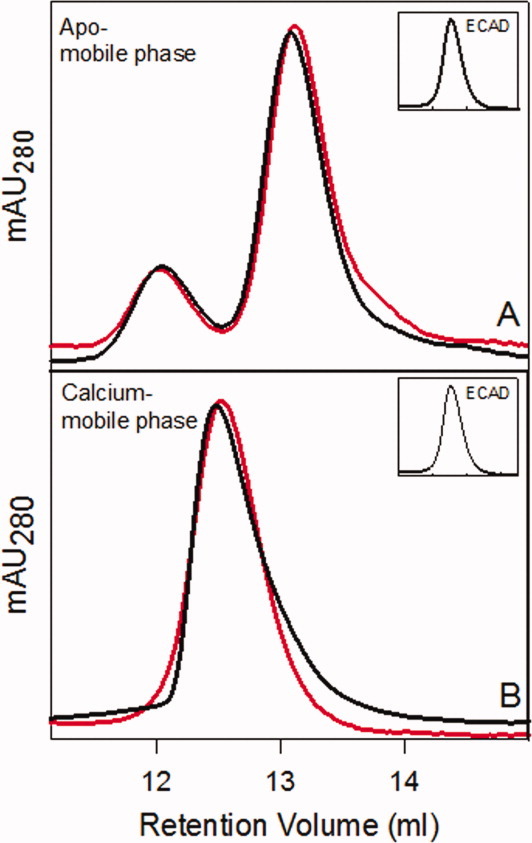
Analytical SEC to determine the kinetics of monomer–dimer equilibria as a function calcium concentration. (A) Calcium-saturated wild-type (black) and X-enabled NCAD12 (red) protein samples (20 μM) were injected on the SEC column and analyzed under apo-buffer conditions. The dimer peak eluted at 12 ± 0.2 mL and the monomer peak eluted at 13.0 ± 0.1 mL, which indicates slow disassembly kinetics. (B) Calcium-saturated wild-type (black) and X-enabled NCAD12 (red) protein samples (20 μM) were injected on the SEC column and analyzed under high calcium-buffer conditions (1 mM). Insets: Calcium-saturated ECAD12 samples analyzed by SEC under same experimental conditions.19
Analysis of calcium-added samples in calcium-mobile phase yielded a single peak [Fig. 5(B)] with an intermediate elution volume. These results showed that monomer and dimer are in rapid equilibrium in presence of calcium. In summary, both wild-type and X-enabled NCAD12 showed slow disassembly in the apo-mobile phase and fast disassembly in the calcium-mobile phase. These results strongly suggest that the X-dimer intermediate is not important for the slow conversion in the absence of calcium. Thus, the differences in the X-interface residues are not the key factor in the slow disassembly kinetics of N-cadherin in the apo-state.
Characterization of the kinetically trapped dimer species
Analytical SEC was used to determine whether X-enabled NCAD12 forms kinetically trapped dimer, D*apo, which is formed by stripping calcium from the protein sample. When the calcium-depleted protein sample (20 μM) was diluted to 5 μM in low calcium buffer, the ratio of monomer to dimer was unchanged (Fig. 6). This ratio was constant for 3 days after dilution at 4°C, indicating that the D*apo is formed and it is kinetically trapped since it is not in equilibrium with monomer. X-enabled NCAD12 showed 45% dimer at 20 μM protein concentration, which is consistence with Kd of 25 μM.19,34 Similar results were observed for the kinetically trapped dimeric form of wild-type.19 Taken together the results of the analytical SEC studies indicate that the presence of X-interface residues has no role in disassembly kinetics and has no impact on either D*apo of N-cadherin. Hence, our data confirm that the differences in the X-interface residues are not a factor in the disassembly kinetics of the N-cadherin in the apo-state.
Figure 6.
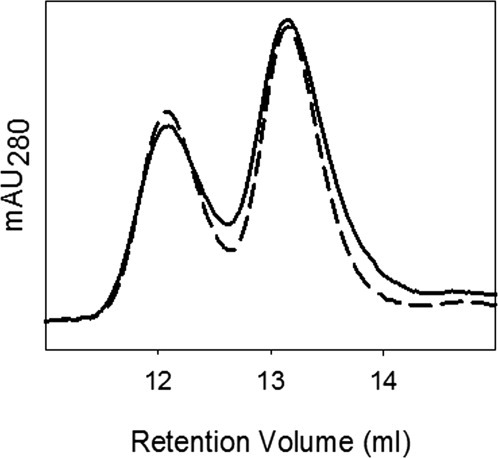
Analytical SEC analysis of kinetically trapped dimer in X-enabled NCAD12 stocks. Kinetically trapped dimer was formed by depleting calcium by addition of EGTA (5 mM). The calcium-depleted concentrated X-enabled NCAD12 stock (20 μM) (solid, 45% dimer) and a 1–4 dilution (5 μM) (dashed, 45% dimer) were injected on the SEC column in apo-buffer conditions.
Discussion
There has been intense interest recently over a dimeric structure that has been proposed to be a necessary intermediate on the pathway for the conversion of the strand-crossover dimer in E-cadherin to monomer. There is great enthusiasm over the possibility that formation of the X dimer is the governing factor for controlling the disassembly kinetics for members of the classical cadherin family. Recent studies of E-cadherin have shown that its fast disassembly kinetics originates from X-interface interactions.29,30 In our previous studies on the first two domains of E- and N-cadherin, we showed that they form dimeric species which differ in their disassembly kinetics.19 Because the noncovalent interactions in the X-interface are not strictly conserved in N-cadherin, our current studies investigate the link between the nonconserved X-interface residues and the slow disassembly kinetics in N-cadherin.
On the basis of a sequence comparison of E- and N-cadherin, we found that all four noncovalent interactions that comprise the X-interface are compromised in N-cadherin. Out of the seven amino acids, which participate in the X-interface interactions in E-cadherin, three are not conserved in N-cadherin (Fig. 1D). Here, we introduced three point mutations into N-cadherin to provide the component residues for X-interface interactions and expected this X-enabled mutant to show fast dimer disassembly behavior in the apo-state like E-cadherin. We find that the X-enabled NCAD12 has similar disassembly kinetics as wild-type NCAD12. Thus, the differences in the X-interface residues are not responsible for the slow disassembly kinetics of apo N-cadherin.
Spectroscopic and analytical SEC studies showed that the triple mutation did not affect the integrity of the protein. The X-enabled NCAD12 had very similar calcium binding properties and thermal denaturation profiles as wild-type NCAD12. Analytical SEC studies were performed in the apo- and calcium added mobile phases to characterize and compare the disassembly kinetics of X-enabled NCAD12 to wild-type. X-enabled NCAD12 showed slow disassembly behavior in the absence of calcium in contrast to the fast disassembly behavior of E-cadherin [Fig. 5(A)]. In the presence of calcium both wild-type and X-enabled NCAD12 disassembled rapidly like E-cadherin [Fig. 5(B)] even though the wild-type N-cadherin has differences in the X-interface residues. In spite of the presence of the X-interface residues, X-enabled NCAD12 behaved like wild-type N-cadherin in absence and presence of calcium. These observations suggest that calcium-binding influences the dynamics of adhesive interactions in a different way in N-and E-cadherin; the disassembly of N-cadherin dimers depends on the binding of calcium rather than the X-interface interactions, whereas disassembly of E-cadherin dimer depends on the formation of the X-interface rather than calcium binding.29,30
D*apo is a dimeric form that is unique to N-cadherin.19 We expected that the X-enabled NCAD12 would not make this kinetically trapped dimeric form. Surprisingly, it did make D*apo (Fig. 6). This indicates that the introduction of the X-interface residues does not affect either the formation or the disassembly of the D*apo form in N-cadherin.
Kinetically trapped dimer (D*apo; Fig. 2) of N-cadherin, which is formed upon depletion of calcium from Dsat, disassembles upon addition of calcium. Thus, Dsat is the necessary intermediate in the formation and disassembly of D*apo. Our data suggest that the D*apo will form during normal neural activity, where there is a significant decrease in the extracellular calcium levels.28 The disassembly of D*apo occurs during the resting nerve activity, where the extracellular calcium levels return to normal. Taken together our data suggest that the disassembly of D*apo requires the binding of calcium to lower the activation energy barrier35 just as X-dimer formation lowers the activation energy barrier for dimer disassembly for E-cadherin. Hence, the kinetic advantage provided by the X-dimer in E-cadherin is equivalent to that provided by calcium binding in N-cadherin.
There is ample evidence for X-dimer type conformations in the literature for E-cadherin.17,36 However, to our knowledge, no such structure exists for N-cadherin. This means that the structural components of an X-interface in N-cadherin, if it exists, can only be discerned from sequence comparison to E-cadherin. It is interesting to note that there is evidence of D*apo in the structural literature. The original crystal structure of N-cadherin was of a single domain construct that formed a strand crossover structure between protomers.37 The authors noted that this construct was purified as a homogeneous solution of dimer as assessed by size exclusion chromatography. Thus, one might consider this a structure similar to D*apo. D*apo is likely a different conformation than Dsat. Hence, the D*apo form of N-cadherin may preclude the orientational changes required to exploit the X-interface, when it is present.
Although the concept of an X-interface is compelling, the specifics of its constitution are an open question. A survey of the classical cadherin family7 shows that the four sets of interactions noted for the X-interface in E-cadherin are not conserved in the other members of the family. Further, the X-interface is also not conserved between E-cadherins from mammals and amphibians. While it is possible that there is no consensus sequence for the X-interface, it is also possible that alternative mechanisms will emerge in the search for a unifying molecular code that dictates the stability and dynamics of adherens junctions.
Materials and Methods
Site-directed mutagenesis
The construction of the first two ectodomains (residues 1–221), designated as NCAD12 (EC1, linker 1, EC2, and linker 2) was described previously.19 The amino acid sequences of mouse NCAD12 and ECAD12 were aligned using LALIGN16 to find the amino acids in N-cadherin which correspond to the X-dimer interface in E-cadherin. Three mutations were introduced into the NCAD12 sequence by site-directed mutagenesis by using the Quickchange kit (Stratagene) with the following sense primers, M101Q Sense: 5′ GACATTGTCATCAATGTTATTGACCAGAATGATAACAGACCTGAG 3′, P138D Sense: 5′ GCGGA TGATGACAATGCCCTGAATGGAATGCTGCGG 3′, N198E Sense: 5′ GCCACAGACATGGAAGGCGAGCCCACTTATGGCCTTTCAAACAC 3′. Amplification of the two-domain constructs, digestion of template DNA, ligation into pET30 Xa/LIC, and subsequent transformation into E. coli BL21 (DE3) were performed according to standard protocols, utilizing KOD HiFi DNA polymerase (Stratagene) and Xa/LIC cloning kit (Novagen). The mutations were confirmed by plasmid sequencing. This M101Q/P138D/N198E triple mutant NCAD12 protein has an X-interface and is called X-enabled NCAD12 in subsequent discussions.
Overexpression and purification
Protocols for protein overexpression and purification were described previously.19 Digested NCAD12 (wild-type), and X-enabled NCAD12 proteins were further purified using size exclusion chromatography (SEC) with a Sephacyl S-100 (Amersham) 1.2 cm × 0.5 m (∼100 mL) column in 140 mM NaCl and 10 mM HEPES, pH 7.4. Purity of proteins was assessed by SDS-PAGE in 17% polyacrylamide gels. Extinction coefficients were determined experimentally38 at 280 nm to be 17,700 ± 500 M−1 cm−1 for wild-type and 17,200 ± 300 M−1 cm−1 for X-enabled NCAD12.
Thermal-unfolding studies
Thermal unfolding of wild-type and X-enabled NCAD12 was monitored with AVIV 202SF CD spectrometer. Experimental conditions were described previously.19 To monitor the effect of calcium binding on protein stability, studies were performed at two calcium concentrations, apo (50 μM EGTA) in 140 mM NaCl, 10 mM HEPES, pH 7.4 and the saturated state with 1 mM added calcium. The thermal-unfolding transitions were fit to the Gibbs–Helmholtz equation as described previously.39 The ΔCp for apo wild-type NCAD12 was determined previously19 from the Kirchoff plot of the observed enthalpy of denaturation versus transition temperature and found to be 1 kcal (mol K−1)−1.
Calcium binding studies
Calcium titrations were monitored by the change in the circular dichroism (CD) signal using an AVIV 202SF circular dichroism spectrometer at 229 nm. The protein solution (2.5 μM) in 10 mM HEPES and 140 mM NaCl, pH 7.4 was placed in a 1-cm path cuvette at 25°C while stirring and titrated with calcium by addition of small volumes of different calcium chloride stocks (1, 10, 100, and 700 mM). The total calcium concentration was presumed to be equivalent to the free calcium concentration. The spectral signal of the sample before calcium was added was given a free calcium concentration of 1 μM.19 The calcium titrations data were fitted to a model of equal and independent binding shown in Eq. (1), where Y is the fractional saturation of sites and Ka is the calcium association constant.
| 1 |
Analytical size exclusion chromatography (SEC) studies
Analytical SEC was used to assess the overall differences in the kinetics of dimer disassembly of wild-type and X-enabled NCAD12 in the apo (140 mM NaCl, 10 mM HEPES, pH 7.4) and calcium (140 mM NaCl, 10 mM HEPES, 1 mM Calcium, pH 7.4) mobile phase. The protein concentration was varied (5–20 μM) to control the level of dimer in the samples. These experiments were performed using an ÄKTA Purifier HPLC (GE LifeSciences) with a Superose-12 10/300 GL Column (GE LifeSciences) with detection at 280 nm and 0.5 mL min−1 flow rate. The column volume is ∼25 mL and the total time to elute monomer was 30 min. Calibration of the SEC column was described previously.19 We monitored the level of monomer and dimer as the height of the peaks detected at 280 nm. Each experiment was repeated at least three times. This method was validated using sedimentation velocity and used successfully to illustrate the calcium-dependent monomer-dimer equlibria in E- and N-cadherins.19
Kinetics of monomer–dimer equilibria as a function of calcium concentration
To monitor the kinetics of the monomer–dimer equilibrium as a function of calcium concentration, protein samples (20 μM) in 1 mM calcium were injected on the SEC column in both the apo- and calcium-mobile phases (∼0 μM and 1 mM). If the X-dimer interface is the key factor in the kinetics of dimer disassembly, we should observe a single peak in the SEC runs under both conditions in X-enabled NCAD12.
Characterization of the kinetically trapped dimer species
To observe the kinetically trapped dimer (D*apo) in X-enabled NCAD12 60 μL of 20 μM of calcium-depleted protein sample and 240 μL of 1 to 4 dilution of 20 μM was injected on the SEC column in the apo-mobile phase. Thus, an equal amount of protein was injected regardless of the concentration, and hence the UV absorbance should be same at 280 nm. If X-enabled NCAD12 forms D*apo, there will be no change in the monomer to dimer ratio in this experiment.
Glossary
Abbreviations
- Apo
calcium-depleted
- EC1
extracellular domain 1
- EC2
extracellular domain 2
- ECAD12
epithelial-cadherin domains 1 and 2 (residues 1 to 219)
- EGTA
ethylene glycol tetraacetic acid
- HEPES
N-(2-hydroxyethyl) piperazine-N′-2-ethanesulfonic acid
- NCAD12
neural-cadherin domains 1 and 2 (residues 1 to 221)
- SDS-PAGE
sodium dodecylsulfate polyacrylamide gel electrophoresis
- SEC
size exclusion chromatography
References
- 1.Arikkath J, Reichardt LF. Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends Neurosci. 2008;31:487–494. doi: 10.1016/j.tins.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alattia JR, Kurokawa H, Ikura M. Structural view of cadherin-mediated cell-cell adhesion. Cell Mol Life Sci. 1999;55:359–367. doi: 10.1007/s000180050297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ringwald M, Schuh R, Vestweber D, Eistetter H, Lottspeich F, Engel J, Dolz R, Jahnig F, Epplen J, Mayer S, Muller C, Kemler R. The structure of cell adhesion molecule uvomorulin. Insights into the molecular mechanism of Ca2+-dependent cell adhesion. EMBO J. 1987;6:3647–3653. doi: 10.1002/j.1460-2075.1987.tb02697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451–1455. doi: 10.1126/science.2006419. [DOI] [PubMed] [Google Scholar]
- 5.Vleminckx K, Kemler R. Cadherins and tissue formation: integrating adhesion and signaling. Bioessays. 1999;21:211–220. doi: 10.1002/(SICI)1521-1878(199903)21:3<211::AID-BIES5>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 6.Ozawa M, Engel J, Kemler R. Single amino acid substitutions in one Ca2+ binding site of uvomorulin abolish the adhesive function. Cell. 1990;63:1033–1038. doi: 10.1016/0092-8674(90)90506-a. [DOI] [PubMed] [Google Scholar]
- 7.Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science. 2002;296:1308–1313. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- 8.Koch AW, Pokutta S, Lustig A, Engel J. Calcium binding and homoassociation of E-cadherin domains. Biochemistry. 1997;36:7697–7705. doi: 10.1021/bi9705624. [DOI] [PubMed] [Google Scholar]
- 9.Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- 10.Takeichi M. Morphogenetic roles of classic cadherins. Curr Opin Cell Biol. 1995;7:619–627. doi: 10.1016/0955-0674(95)80102-2. [DOI] [PubMed] [Google Scholar]
- 11.Rowlands TM, Symonds JM, Farookhi R, Blaschuk OW. Cadherins: crucial regulators of structure and function in reproductive tissues. Rev Reprod. 2000;5:53–61. doi: 10.1530/ror.0.0050053. [DOI] [PubMed] [Google Scholar]
- 12.Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 13.Takeichi M. Cadherins in cancer: implications for invasion and metastasis. Curr Opin Cell Biol. 1993;5:806–811. doi: 10.1016/0955-0674(93)90029-p. [DOI] [PubMed] [Google Scholar]
- 14.Handschuh G, Candidus S, Luber B, Reich U, Schott C, Oswald S, Becke H, Hutzler P, Birchmeier W, Hofler H, Becker KF. Tumour-associated E-cadherin mutations alter cellular morphology, decrease cellular adhesion and increase cellular motility. Oncogene. 1999;18:4301–4312. doi: 10.1038/sj.onc.1202790. [DOI] [PubMed] [Google Scholar]
- 15.Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene. 2008;27:6920–6929. doi: 10.1038/onc.2008.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang X, Miller W. A time-efficient, linear-space local similarity algorithm. Adv Appl Math. 1991;12:337–357. [Google Scholar]
- 17.Pertz O, Bozic D, Koch AW, Fauser C, Brancaccio A, Engel J. A new crystal structure, Ca2+ dependence and mutational analysis reveal molecular details of E-cadherin homoassociation. EMBO J. 1999;18:1738–1747. doi: 10.1093/emboj/18.7.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harrison OJ, Jin X, Hong S, Bahna F, Ahlsen G, Brasch J, Wu Y, Vendome J, Felsovalyi K, Hampton CM, Troyanovsky RB, Ben-Shaul A, Frank J, Troyanovsky SM, Shapiro L, Honig B. The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure. 2011;19:244–256. doi: 10.1016/j.str.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vunnam N, Flint J, Balbo A, Schuck P, Pedigo S. Dimeric states of neural- and epithelial-cadherins are distinguished by the rate of disassembly. Biochemistry. 2011;50:2951–2961. doi: 10.1021/bi2001246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baumgartner W, Golenhofen N, Grundhofer N, Wiegand J, Drenckhahn D. Ca2+ dependency of N-cadherin function probed by laser tweezer and atomic force microscopy. J Neurosci. 2003;23:11008–11014. doi: 10.1523/JNEUROSCI.23-35-11008.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perret E, Benoliel AM, Nassoy P, Pierres A, Delmas V, Thiery JP, Bongrand P, Feracci H. Fast dissociation kinetics between individual E-cadherin fragments revealed by flow chamber analysis. EMBO J. 2002;21:2537–2546. doi: 10.1093/emboj/21.11.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perret E, Leung A, Feracci H, Evans E. Trans-bonded pairs of E-cadherin exhibit a remarkable hierarchy of mechanical strengths. Proc Natl Acad Sci USA. 2004;101:16472–16477. doi: 10.1073/pnas.0402085101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mysore SP, Tai CY, Schuman EM. N-cadherin, spine dynamics, and synaptic function. Front Neurosci. 2008;2:168–175. doi: 10.3389/neuro.01.035.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bekirov IH, Needleman LA, Zhang W, Benson DL. Identification and localization of multiple classic cadherins in developing rat limbic system. Neuroscience. 2002;115:213–227. doi: 10.1016/s0306-4522(02)00375-5. [DOI] [PubMed] [Google Scholar]
- 25.Redies C. Cadherins in the central nervous system. Prog Neurobiol. 2000;61:611–648. doi: 10.1016/s0301-0082(99)00070-2. [DOI] [PubMed] [Google Scholar]
- 26.Hirano S, Suzuki ST, Redies C. The cadherin superfamily in neural development: diversity, function and interaction with other molecules. Front Biosci. 2003;8:d306–d355. doi: 10.2741/972. [DOI] [PubMed] [Google Scholar]
- 27.Benson DL, Tanaka H. N-cadherin redistribution during synaptogenesis in hippocampal neurons. J Neurosci. 1998;18:6892–6904. doi: 10.1523/JNEUROSCI.18-17-06892.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rusakov DA, Fine A. Extracellular Ca2+ depletion contributes to fast activity-dependent modulation of synaptic transmission in the brain. Neuron. 2003;37:287–297. doi: 10.1016/s0896-6273(03)00025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harrison OJ, Bahna F, Katsamba PS, Jin X, Brasch J, Vendome J, Ahlsen G, Carroll KJ, Price SR, Honig B, Shapiro L. Two-step adhesive binding by classical cadherins. Nat Struct Mol Biol. 2010;17:348–357. doi: 10.1038/nsmb.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong S, Troyanovsky RB, Troyanovsky SM. Cadherin exits the junction by switching its adhesive bond. J Cell Biol. 2011;192:1073–1083. doi: 10.1083/jcb.201006113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mendez P, De Roo M, Poglia L, Klauser P, Muller D. N-cadherin mediates plasticity-induced long-term spine stabilization. J Cell Biol. 2010;189:589–600. doi: 10.1083/jcb.201003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vunnam N, McCool JK, Williamson M, Pedigo S. Stability studies of extracellular domain two of neural-cadherin. Biochim Biophys Acta. 2011;14:1841–1845. doi: 10.1016/j.bbapap.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Vunnam N, Pedigo S. Prolines in betaA-sheet of neural cadherin act as a switch to control the dynamics of the equilibrium between monomer and dimer. Biochemistry. 2011;50:6959–6965. doi: 10.1021/bi2007788. [DOI] [PubMed] [Google Scholar]
- 34.Katsamba P, Carroll K, Ahlsen G, Bahna F, Vendome J, Posy S, Rajebhosale M, Price S, Jessell TM, Ben-Shaul A, Shapiro L, Honig BH. Linking molecular affinity and cellular specificity in cadherin-mediated adhesion. Proc Natl Acad Sci USA. 2009;106:11594–11599. doi: 10.1073/pnas.0905349106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen CP, Posy S, Ben-Shaul A, Shapiro L, Honig BH. Specificity of cell–cell adhesion by classical cadherins: critical role for low-affinity dimerization through beta-strand swapping. Proc Natl Acad Sci USA. 2005;102:8531–8536. doi: 10.1073/pnas.0503319102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagar B, Overduin M, Ikura M, Rini JM. Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature. 1996;380:360–364. doi: 10.1038/380360a0. [DOI] [PubMed] [Google Scholar]
- 37.Shapiro L, Fannon AM, Kwong PD, Thompson A, Lehmann MS, Grubel G, Legrand JF, Als-Nielsen J, Colman DR, Hendrickson WA. Structural basis of cell–cell adhesion by cadherins [see comments] Nature. 1995;374:327–337. doi: 10.1038/374327a0. [DOI] [PubMed] [Google Scholar]
- 38.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar extinction coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prasad A, Housley NA, Pedigo S. Thermodynamic stability of domain 2 of epithelial cadherin. Biochemistry. 2004;43:8055–8066. doi: 10.1021/bi049693c. [DOI] [PubMed] [Google Scholar]