

Abstract
The armadillo domain is a right-handed super-helix of repeating units composed of three α-helices each. Armadillo repeat proteins (ArmRPs) are frequently involved in protein–protein interactions, and because of their modular recognition of extended peptide regions they can serve as templates for the design of artificial peptide binding scaffolds. On the basis of sequential and structural analyses, different consensus-designed ArmRPs were synthesized and show high thermodynamic stabilities, compared to naturally occurring ArmRPs. We determined the crystal structures of four full-consensus ArmRPs with three or four identical internal repeats and two different designs for the N- and C-caps. The crystal structures were refined at resolutions ranging from 1.80 to 2.50 Å for the above mentioned designs. A redesign of our initial caps was required to obtain well diffracting crystals. However, the structures with the redesigned caps caused domain swapping events between the N-caps. To prevent this domain swap, 9 and 6 point mutations were introduced in the N- and C-caps, respectively. Structural and biophysical analysis showed that this subsequent redesign of the N-cap prevented domain swapping and improved the thermodynamic stability of the proteins. We systematically investigated the best cap combinations. We conclude that designed ArmRPs with optimized caps are intrinsically stable and well-expressed monomeric proteins and that the high-resolution structures provide excellent structural templates for the continuation of the design of sequence-specific modular peptide recognition units based on armadillo repeats.
Keywords: protein structure, armadillo repeat, domain swapping, structure-based design, protein engineering
Introduction
Armadillo repeat proteins (ArmRP) were initially observed in the armadillo locus, the DNA region that codes for a set of segment polarity genes required during Drosophila embryogenesis.1,2 However, it is just a matter of coincidence that the banded structure of the mutant insect larvae, from which the name is derived, and the three-dimensional structure of the corresponding gene product are both emblematized by the armadillo animal. ArmRPs possess modular architectures of repeating structural units. Each armadillo repeat (ArmR) is composed of ∼40 amino acids that fold into a triangular arrangement of three α-helices (helices H1, H2, and H3).3 The stacking of three to over 10 individual ArmRs generates a solenoid-like molecule with an extended hydrophobic core and a concave peptide-binding groove.
Similar to other solenoid proteins, such as ankyrin repeat, leucine-rich repeat, or Sel1-like repeat proteins, ArmRPs are involved in protein/protein interactions. ArmRPs in general recognize an unstructured part of the target protein, which binds in an extended conformation in a peptide-like manner (see below). The modular repeat protein architecture is particularly suitable to generate a large set of different binding interfaces, because the number and the spatial orientation of repeats define the size and the curvature of the target recognition surface. Since the modularity of the protein matches the modularity of the bound peptide, it is of great interest to investigate whether ARM repeat proteins can be used as a general peptide recognition scaffold.
The hydrophobic core, which is indispensable for the thermodynamic stability of a protein, and the target recognition surface are located on opposite sides of secondary structural elements. This topology prevents that the hydrophobic core is affected by the mutation of residues that are required for the recognition of the target molecule. These features explain why in living organisms solenoid proteins are abundant natural signaling modules, which are thus also very attractive for the design of artificial peptide recognition molecules.
The prototypical ArmRPs importin-α and β-catenin are the key molecules for nuclear import and Wnt signaling, respectively.4–6 The recruitment of NLS to importin-α is key to the classical import pathway of cargo molecules into the nucleus. The best characterized NLSs became those which were identified in Simian virus 40 large T-antigen7 and in Xenopus nucleoplasmin.8 Both sequence motifs are characterized by well conserved lysine and arginine residues that are recognized at the concave side of the importin-α super helix. Several crystal structures of ArmRPs in complex with NLSs revealed that the NLS peptide runs antiparallel to the direction of the importin-α main chain and that the NLS peptide crosses helix H3 at an angle of ∼45°. In a first approximation, the complex of the NLS peptide to the ArmRP can be described as an asymmetric antiparallel double helix.
The NLS peptide is recognized by a network of specific hydrogen bonds. The side chains of the NLS lysine residues fit well into surface pockets on the H3 helix of the designed ArmR. These pockets are composed of conserved threonine, tryptophan, and asparagine residues that recognize the lysine side chain by hydrogen bonds and aromatic π-stacking interactions.9 Two classes of NLSs can be distinguished: mono- and bipartite NLSs are characterized by one and two clusters of basic residues, respectively. Only the ArmRs 2–4 and 6–8 of the bipartite NLS binding importin-α contain surface-exposed tryptophan-containing pockets, whereas repeats 5 and 6 separate the concave importin-α surface into two individual binding sites. The detailed structural analysis of many ArmR:peptide complexes revealed a uniform distribution of peptide binding modes, namely each binding site consisting of three ArmRs and recognizing four amino acids of the NLS peptide (reviewed in Ref. 10). The regularity of the peptide recognition mode distinguishes ArmRs from other peptide-binding scaffolds, especially peptide-binding antibodies, and makes them highly attractive for protein engineering.
Previously, artificial binding proteins have been created using different scaffolds by selection from combinatorial libraries.11,12 Although this approach was very successful, it is limited by the need to carry out every selection to a new target as a new experiment. When the target is a folded protein, this will continue to be the case, because the precise structure of the protein:target complex is unpredictable. To overcome these limitations, for peptide targets, Parmeggiani et al. have explored the use of the ArmR scaffold.13 Using a consensus design strategy a set of artificial short ArmRPs with the overall constitution YzIxAz was generated. Here, Y denotes an N-terminal capping repeat that has been derived from yeast importin-α, x denotes the number of internal repeats of type I, and A denotes an artificial C-terminal capping repeat. The subscript z refers to the roman numerals II and III, where II is a second generation capping repeat design based on molecular dynamic simulations (see below) and III is a third generation capping repeat design based on the structure-based design approach presented below. Four different types of internal repeats have been explored. Internal repeats of type-I, -T, and -C were derived from importin-α, β-catenin, and combined importin-α:β-catenin sequence alignments, respectively. The biophysical investigation of consensus design based ArmRPs containing type-I and type-T internal repeats revealed native-like behavior, whereas proteins based on type-C internal repeats showed properties that were similar to a molten globule-like state. To improve the stability of type-C proteins, the hydrophobic core was optimized using a computational modeling approach.13 Three point mutations per repeat were sufficient to overcome the poor folding properties of the type-C internal repeat proteins. The resulting type-M repeat differs from the type-I repeat in six positions and was used to generate YIM4AI, an artificial ArmRP with four internal repeats that showed cooperative unfolding behavior and a well-defined hydrophobic core.
To aid the future design of ArmRPs we characterized further M-type proteins with three to six internal repeats. Although biophysical experiments suggested that YIIMxAII proteins are monomeric, the crystal structures of YIIM3AII and YIIM4AII at 2.4 Å and 2.5 Å resolution, respectively, revealed domain-swapping events of the N-terminal capping repeats. To eliminate domain swapping the crystal structures were used to redesign the capping repeats. The subsequent biophysical and structural analysis of YIIIM3AII and YIIIM3AIII confirmed that the redesigned molecules fold into stable monomers that will be extremely helpful for the design of ArmR peptide binding modules in the future.
Results and Discussion
Expression and biophysical characterization of YIIMxAII proteins
To investigate the structures and thermodynamic properties of ArmRPs, four different expression constructs coding for proteins with three to six identical internal type-M repeats between YII- and AII-capping repeats (YIIMxAII, x = 3–6) were assembled following the approach reported previously.13,14 The proteins contain an N-terminal His6-tag (MRGSH6-tag) for efficient expression and purification. Typical expression yields in E. coli XL1-blue were 80–100 mg of pure protein from a 1 L bacterial culture. All YIIMxAII proteins were characterized by size-exclusion chromatography in TBS buffer at pH 7.4 to estimate their oligomerization states. They eluted as single symmetric peaks [Fig. 1(a)] at retention volumes that indicated molecular masses 1.2 ± 0.05 fold higher than the expected monomeric mass values. The increase of the molecular masses is interpreted as an increase of the hydrodynamic radius due to the elongated shape of the molecule, because previously this interpretation was confirmed for the ancestor molecule YIM4AI by multi-angle light scattering (MALS).13
Figure 1.

Biophysical characterization of designed ArmRPs. (a) Size exclusion chromatography of YIIM3-6AII proteins. V0 indicates the void volume of the column and Vtot the total volume. Bovine serum albumin (MW = 66 kDa) and carbonic anhydrase (MW = 29 kDa) were used as molecular weight standards. The corresponding elution volumes are indicated by arrows. (b) ANS fluorescence spectra without buffer subtraction and (c) CD spectra of YIIM3-6AII proteins are shown. (d) GdnHCl-induced and (e) temperature-induced unfolding of designed proteins. The mean residue ellipticity (MRE) at 222 nm was used to follow unfolding of proteins. The protein concentration was 30 μM in (a) and 10 μM in (b)–(e).
Size-exclusion chromatography combined with MALS analysis revealed that oligomerization of YIIM3AII depends on the protein concentration. At a concentration of 5 mg/mL MALS revealed a molecular mass of 21.8 kDa, which agrees with the theoretical molecular mass of 22648 Da. At a concentration of 18 mg/mL, the dominant monomer peak is still present, but it is preceded by a small shoulder, which corresponds to the predicted molecular mass of the of YIIM3AII dimer in the MALS analysis (Supporting Information Fig. S1). Nonetheless, the rather high concentration at which dimers are first visible (∼1 mM) suggests that the equilibrium is on the side of monomers under most experimental conditions.
To test whether the designed ArmRPs fold to a native structure we probed the accessibility of the hydrophobic core for the fluorescent dye 1-anilino-8-naphthalene-sulfonate (ANS) and by circular dichroism (CD) spectroscopy. The binding of ANS to the hydrophobic core of a protein in the molten globule state typically increases the ANS fluorescence by an order of magnitude or more. Figure 1(b) shows the ANS fluorescence after adding YIIMxAII with three to six internal repeats. Since the fluorescence signal of the protein-containing sample is increased by just a factor of 1.5–2, compared to the ANS fluorescence in the absence of any protein, it can be concluded that the hydrophobic cores of all four YIIMxAII proteins are inaccessible for ANS. These results are consistent with the CD spectroscopy measurements, where all four proteins showed pronounced minima a 208 nm and 222 nm. These minima indicate a high content of α-helical secondary structure, as it is expected for ArmRPs [Fig. 1(c)].
The CD signal at 222 nm was used to follow the temperature- and guanidine hydrochloride (GdnHCl)-induced unfolding of YIIMxAII proteins. For all four proteins a clear sigmoidal temperature- and GdnHCl concentration dependency was observed, suggesting a cooperative unfolding behavior of YIIMxAII proteins [Fig. 1(d,e)]. Furthermore, the temperature-induced unfolding was completely reversible as judged by CD spectroscopy (data not shown). The midpoints of the transitions between the folded- and unfolded states increase with the number of internal repeats. For YIIM3AII, YIIM4AII, YIIM5AII, and YIIM6AII the GdnHCl concentrations where 50% of the protein was unfolded were 3.6M, 4.3M, 4.6M, and 4.8M, respectively. An almost linear increase of the transition midpoints with the number of internal repeats has been observed previously for designed ankyrin- and tetratricopeptide-repeat proteins.15,16 Temperature-induced unfolding experiments revealed a similar pattern. All YIIMxAII proteins are rather stable with melting temperatures (Tm) between 76.5°C and 89.0°C for YIIM3AII and YIIM6AII, respectively.
Crystal structures of YIIM3AII and YIIM4AII reveal domain-swapped N-termini
Initial attempts to determine the crystal structures of several different designed ArmRPs with Y- and A-type capping repeats were hampered by the fact that, while large protein crystals were obtained almost immediately under various crystallization conditions, none of them diffracted X-rays to better than 6 Å resolution. Molecular dynamics simulations of ArmRP models suggested five point mutations in the capping repeats [Fig. 2(a)]. Mutations V33R, R36S, ΔR41, and Q38L, F39Q yielded type-YII and -AII capping repeats, respectively (superscripts refer to the positions in the ArmR and not to the residue numbers of the whole protein). These caps thus differ from those in the original publication,13 and this is symbolized by the subscript II for second generation (P. Alfarano, G. V., C. Ewald, F Parmeggiani, R. Pellarin, O. Zerbe, A. P., and A Caflisch, manuscript in preparation).
Figure 2.

Structures of domain-swapped YIIM3AII and YIIM4AII proteins. (a) Sequence alignment of the N-caps (importin-α, YI-, YII-, and YIII-type), internal repeat (M-type), and C-caps (AI-, AII-, and AIII-type). Numbering refers to the position of the amino acid in individual repeats. Mutated residues are highlighted in red. Residues belonging to helices H1, H2, and H3 are indicated by numbers. (b) The 2Fo−Fc (blue, 1σ) and difference electron density maps (red, −4σ; green, +4σ) at the beginning of the refinement process, indicating the domain swapping of the N-caps between neighboring YIIM3AII molecules (cyan and magenta Cα-traces). The loop region, which shows strong negative difference electron density, corresponds to positions 41 and 42 in the YIIM3AII structure. (c) The YIIM3AII dimer is shown in two perpendicular orientations. Subunits are shown as a ribbon that is colored according to B-factor (blue and red indicate low- and high B-factors, respectively) and as a surface representation (YII-, M1-, M2-, M3-, and AII-repeats are shown in magenta, blue, gray, blue, and magenta, respectively). (d) H-bonds at the domain-swapped N-cap. The subunits of the YIIM3AII-dimer are shown with green and salmon carbon atoms. (e) Superposition of YIIM3AII (green) and YIIM4AII (magenta). The N- and C-termini of YIIM4AII are indicated.
Crystals of YIIM3AII that have the symmetry of the space group P1 and diffracted to 2.40 Å resolution were obtained at pH 4.0 (Table I). The positions of four copies of YIIM3AII in the unit cell were determined by molecular replacement using the truncated structure of importin-α as a search molecule. During the refinement process strong negative difference electron density in the supposed loop region of the N-cap and strong positive difference electron density between neighboring molecules suggested a domain swapping event between symmetry-related YIIM3AII molecules [Fig. 2(b)]. Further refinement confirmed the initial assumption, and consequently the final structure is described as a right-handed propeller-shaped homodimer of YIIM3AII subunits with overall dimensions of 60 × 45 × 30 Å [Fig. 2(c)]. The formation of the dimer buries a surface area of 4480 Å2. The dimer interface is formed primarily by the domain-swapped N-cap which covers the hydrophobic core of the first internal repeat M1# from the second subunit (# refers to the symmetry-related YIIM3AII molecule). Minor contacts exist between the loops that are connecting internal repeats M1–M2 and M2–M3 and the same loops from the second subunit.
Table I.
Crystallization, Data Processing, and Refinement Statistics
| YIIM3AII | YIIM4AII | YIIIM3AIII | YIIIM3AII | |
|---|---|---|---|---|
| Crystallization condition | 0.05M succinic acid, pH 4.0, 20% PEG 4000, 0.2M Li2SO4 | 0.2M magnesium chloride, 0.1M HEPES, pH 7.5, 30% PEG 400 | 1.55M sodium malonate, pH 8.03 | 0.1M HEPES, pH 7.5, 1.4M sodium citrate |
| Resolution (Å) | 37–2.4 | 40–2.5 | 117–2.4 | 30–1.8 |
| Space group | P1 | P21 | C2221 | I222 |
| Wavelength (Å) | 1.54 | 1.54 | 1.54 | 0.93 |
| Number of molecules/AU | 4 | 4 | 6 | 1 |
| Unit cell parameters a, b, and c (Å) | a = 56.15 | a = 58.00, | a = 131.92 | a = 42.96 |
| b = 60.60 | b = 113.64, | b = 228.88 | b = 91.58 | |
| c = 61.86 | c = 85.60 | c = 116.53 | c = 92.80 | |
| α, β, and γ (°) | α = 74.8 | α = γ = 90 | α = β = γ = 90 | α = β = γ = 90 |
| β = 89.5 | β = 106.8 | |||
| γ = 75.5 | ||||
| Rmergea (%) | 5.7 (37.7) | 11.0 (40.5) | 10.7 (30.4) | 6.4 (67.9) |
| No. of observations | 74416 (10670) | 120569 (15480) | 347027 (50311) | 194469 (26356) |
| No. of unique reflections | 28399 (4056) | 35110 (4604) | 69088 (9968) | 25635 (3665) |
| (I)/∑ (I) | 8.1 (2.1) | 7.3 (2.3) | 12.6 (5.2) | 15.5 (2.8) |
| Completeness (%) | 94.5 (92.8) | 95.6 (86.3) | 100.0 (100.0) | 99.2 (98.7) |
| Multiplicity | 2.6 (2.6) | 3.4 (3.4) | 5.0 (5.0) | 7.6 (7.2) |
| Refinement | ||||
| Resolution range (Å) | 25–2.4 | 40–2.5 | 117–2.4 | 23–1.8 |
| Rcrystb (%) | 23.7 | 23.6 | 21.3 | 18.4 |
| Rfreeb (%) | 30.1 | 29.8 | 24.6 | 22.3 |
| B factors | ||||
| Wilson B (Å2) | 57.5 | 41.9 | 25.3 | 25.1 |
| Mean B value (Å2) | 72 | 42.1 | 21.3 | 22.5 |
| RMSD from ideal values | ||||
| Bond lengths (Å) | 0.009 | 0.006 | 0.007 | 0.006 |
| Bond angles (°) | 1.203 | 0.999 | 1.036 | 0.951 |
| Total number of atoms | ||||
| Protein | 5876 | 7250 | 9138 | 1554 |
| Water | 70 | 134 | 398 | 156 |
| Glycerol | 30 | – | – | – |
| Mg2+ | – | 4 | – | – |
| Ramachandran Plot | ||||
| Residues in preferred regions | 95.3 | 96.9 | 99.3 | 100 |
| Residues in allowed regions | 4.7 | 3.1 | 0.7 | 0 |
| Residues in generously allowed regions | 0 | 0 | 0 | 0 |
| Outliers | 0 | 0 | 0 | 0 |
Rmerge = ∑hkl∑i||Ii(hkl) – [I(hkl)]||/∑hkl∑iIi(hkl), where Ii(hkl) is the ith observation of reflection hkl and [I(hkl)] is the weighted average intensity for all observations i of reflection hkl. Values in parentheses refer to the highest resolution shell.
Rcryst and Rfree = (∑||Fo| – |Fc||)/(∑|Fo|), where |Fo| is the observed structure-factor amplitude and |FC| is the calculated structure-factor amplitude.
Domain swapping is a well-known mechanism observed during the formation of oligomeric proteins. Although oligomers can be formed by a simple association process of monomeric subunits, an alternative mechanism is to enlarge the interface between subunits by the exchange of secondary structural elements among subunits. The latter process can be observed with several monomeric proteins when brought to very high concentration, and it plays important roles in protein evolution and for the pathogenesis of amyloidogenic proteins.17–19 In YIIM3AII, this process is influenced by residues 26–51. These residues form a continuous α-helix that is spanning the gap between subunits. In other ArmRPs, the corresponding residues form a loop that connects helix H3 from the N-cap to helix H1 from the first internal repeat. However, YIIM3AII residues 40–44 do not adopt the expected loop conformation. Thus, the helix propensity of residues Ser40-Asp41-Gly42-Asn43 appears high enough that the N-caps are swapped between subunits. Residues 41–44 are perfectly suited to extend helix H3 into helix H1 of the next repeat, because in this conformation Asp41-OD1 forms an H-bond with Gln37-NE2 in the preceding turn of helix H3, the small side chain of Gly42 allows a very short distance between subunits, and Asn43-OD1 forms an H-bond with Asn79#-ND2 from the second subunit [Fig. 2(d)]. In the monomeric yeast importin-α (PDB ID: 1bk6) the corresponding loop between helix 3 of the N-cap and helix 1 of the first internal repeat is four amino acids longer and has a completely different sequence, harboring two proline residues that are breaking the α-helix H-bond pattern [Fig. 2(a)].
Interestingly, dimerization of YIIM3AII was not expected since the protein eluted as a monomer from the size-exclusion chromatography column. However, some dimerization was observed in solution by MALS, albeit at elevated protein concentration (∼1 mM; Supporting Information Fig. S1). Dimerization of YIIM3AII can thus occur at very high concentrations, such as the experimental conditions during protein crystallization. Domain swapping seems to be important to stabilize YIIM3AII in the crystal lattice, which is illustrated by a temperature factor gradient that runs from the N-terminus (<BN-cap> = 57.42 Å2) to the C-terminus (<BC-cap> = 110.28 Å2). The lowest temperature factors are observed in the interface between the N-cap and M1#, indicating that the interaction between these repeats must be very rigid [Fig. 2(c)]. However, this observation is surprising because the interactions seen in the inter-molecular Y:M1# interface are similar to the interactions seen in the intra-molecular M2:M3 interface. These interactions are dominated by van der Waals contacts between hydrophobic side chains. Residues Ala34, Asn37, and Ile38 from the M-repeat form a groove that is filled by the side chain of Leu39 or Ala39 from the N-cap or internal-repeats, respectively. An additional hydrophobic contact is seen between Ala12/Leu16 from the M-repeat and Leu20/Phe35 from the N-cap or Leu20/Leu35 from the preceding internal-repeat. Because of these similarities, the N-cap could also interact with the first internal repeat of the same subunit (M1 instead of M1#)—the desired interaction for a monomeric ArmRP—provided that residues 41–43 would adopt a loop––rather than an α-helix conformation.
The domain swapping could be either an intrinsic feature of the YIIMxAII design or it could be caused by the low pH, the crystalline state or by the instability of YIIM3AII. Therefore, the structure of YIIM3AII was re-determined at a different pH and in a non-isomorphic crystal lattice. Besides in the initial triclinic crystals, YIIM3AII crystallized in the same space group at pH 10.0 and in space group I212121 at pH 9.75, but both crystal forms diffracted merely to 3 Å resolution. Even though the quality of the electron densities were significantly worse than the quality of the electron density of the P1 crystals obtained at pH 4.0, the domain swapping was clearly visible (data not shown), revealing that domain swapping was neither caused by particular crystal lattice forces nor by the acidic pH.
To answer the question if the domain swapping was a consequence of the lower stability of YIIM3AII the structure of the more stable YIIM4AII [Fig. 1(d,e)] was also determined. The YIIM4AII crystals have the symmetry of the space group P21 with four polypeptide chains in the asymmetric unit and diffract to 2.5 Å resolution. In the YIIM4AII crystal structure, the N-caps between symmetry-related molecules are also swapped. A root mean square deviation (RMSD) of 0.61 Å for the 156 Cα atoms coming from the N-cap and three internal repeats confirms that the YIIM3AII and YIIM4AII structures are indeed very similar [Fig. 2(e)]. Furthermore, YIIM4AII reveals a similar temperature factor gradient like YIIM3AII. The lowest temperature factors are observed in the N-caps and the first internal repeats and increase constantly towards the C-termini.
Structure-based optimization of N- and C-caps
The structural analysis of YIIM3AII and YIIM4AII revealed domain-swapped dimeric structures that are unsuitable for the design of peptide-binding modules. To overcome dimerization and to eliminate the increased flexibility of the C-terminus the designs of the N- and C-caps were optimized based on the YIIM3AII and YIIM4AII structures. To eliminate domain swapping we applied the following strategy: in solenoid proteins every cap has two different interfaces: the buried interface, which covers the hydrophobic core of the protein, and the accessible interface, which mediates solvent contacts. The N- and C-caps were redesigned using the conformation of the internal repeat as a scaffold. The sequence of the scaffold was adjusted in such a way that for the buried interface the interactions seen among internal repeats were maintained, whereas hydrophobic residues that would become exposed on the accessible surface were replaced against hydrophilic residues.
Applying this strategy, 9 and 6 mutations were introduced in the N- and C-caps, respectively [Fig. 2(a)]. The newly designed YIII-type N-cap contains the D41G mutation, which decreases the helix propensity of the linker between the N-cap and the first internal repeat. Mutations T17V, Q28L, T32L, F35L, and L39A re-define the hydrophobic contacts in the buried interface. Mutations M25Q and L29Q eliminate surface exposed hydrophobic residues on the accessible interface, and mutation D23P introduces a helix-breaking residue at the C-terminus of helix H2.
For the redesign of the AIII-type C-cap mutations K15A, H22S, and L38I were introduced to improve the fit between the C-cap and the last internal repeat. Furthermore, the mutation L13E should improve the contact with the solvent and the mutations E14P and E23P introduce proline residues at the N-terminus of helix H2 and into the loop between helices H2 and H3, respectively.
Expression and biophysical characterization of ArmRPs with redesigned caps
To investigate the effects of the newly designed YIII- and AIII-type caps three permutations of cap combinations with three internal M-type repeats were expressed in E. coli, purified by IMAC and characterized by size-exclusion chromatography, CD spectroscopy, ANS binding, and unfolding studies. The expression yields of all three modified designs are equally high (∼100 mg purified protein from a 1 L culture) as for the initial YIIM3AII design. All four combinations of caps (YII or YIII with AII or AIII) elute as single peaks at exactly the same retention volumes, indicating the same elongated shape (see above) [Fig. 3(a)]. They all show very moderate increase of ANS fluorescence, indicating well packed proteins, and equal α-helical contents in the CD spectra [Fig. 3(b,c)]. Importantly, neither with the YII/AII- nor with the YIII/AIII-type caps there is evidence for dimer formation in gel filtration experiments at the protein concentrations used (30 μM). The elution profiles of all four cap combinations are virtually superimposable, suggesting that the N-cap may pair in principle intra- or inter-molecularly with the first internal repeat. Yet, it appears that both the YII and YIII cap prefer intra-molecular pairing under the conditions tested in solution, whereas the YII cap, but not the YIII cap, favors inter-molecular pairing at the high molar concentrations within the crystals.
Figure 3.

Biophysical characterization of designed ArmRPs with improved cap designs. (a) Size exclusion chromatography of designed ArmRPs with three internal repeats and permutations of capping repeats. (b) ANS fluorescence spectra without buffer subtraction. (c) CD spectra are shown. (d) GdnHCl-induced and (e) temperature-induced unfolding of designed proteins. The MRE at 222 nm was used to follow unfolding of designed ArmRPs. The protein concentration was 30 μM in (a) and 10 μM in (b)–(e).
These data suggest that the proteins with all four combinations of caps fold into stable α-helical conformations and native molecules. However, differences were observed in the GdnHCl- and temperature-induced unfolding experiments. Although the sigmoidal shapes of the curves confirm the co-operativities of the unfolding processes, differences exist in the transition midpoints. For the GdnHCl-induced unfolding, the transition midpoints for YIIM3AIII, YIIIM3AIII, YIIM3AII, and YIIIM3AII were 3.2M, 3.4M, 3.6M, and 3.8M, respectively [Fig. 3(d)]. Thus, the redesign of the N-cap improved the GdnHCl-stability by 0.2M (for YIIM3AII → YIIIM3AII), but simultaneously the redesign of the C-cap decreased the stability by 0.4M (for YIIM3AII → YIIM3AIII and YIIIM3AII → YIIIM3AIII). The same trend was observed in the temperature-induced unfolding experiments. YIIIM3AII is the most stable design with a melting temperature of 81°C, which is 4.5°C higher than the melting temperature of the parent molecule YIIM3AII [Fig. 3(e)]. The AII- to AIII-type replacement of the C-cap decreased the melting temperature by 5.5°C, which is consistent with the GdnHCl-induced unfolding experiments. The temperature-induced unfolding was completely reversible for all four designs (data not shown).
Redesign of the N-cap eliminates domain swapping
To answer the question whether the replacement of the YII-type with the YIII-type N-cap has eliminated the domain swapping the crystal structures of YIIIM3AII and YIIIM3AIII have been determined at 1.8 Å and 2.4 Å resolution, respectively. None of them showed domain-swapped N-caps, revealing that the redesign was successful [Fig. 4(a,b)]. The temperature factor gradients with rigid N-caps and flexible C-caps, as they were observed in the domain-swapped YIIM3AII and YIIM4AII structures, were also eliminated by the redesign. The structures of YIIIM3AII and YIIIM3AIII possess low temperature factors for the internal repeats (<Binternal> = 25.97 Å2 for YIIIM3AII and <Binternal> = 14.75 Å2 for YIIIM3AIII) and elevated temperature factors for the N- (<BN-cap> = 42.01 Å2 for YIIIM3AII and <BN-cap> = 34.69 Å2 for YIIIM3AIII) and C-caps (<BC-cap> = 30.40 Å2 for YIIIM3AII and <BC-cap> = 23.47 Å2 for YIIIM3AIII). Similar distributions of temperature factors are commonly observed in other solenoid proteins.20–22 Because the sequences of the structures differ by only six positions in the C-caps both structures are very similar. The YIIIM3-parts can be superimposed with a RMSD of 0.56 Å (Cα atoms of residues 14–169). The major differences between both structures are observed for the loops between helices H2 and H3 of the internal repeats. Figure 4(c) shows that in YIIIM3AII these loops are shifted towards the N-terminus compared to YIIIM3AIII, whereas the same loop of the C-cap is shifted in the opposite direction. These differences can be explained by the presence of residues with bulkier side chains, such as Lys183 and His190 in the M3:AII interface compared to Ala183 and Ser190 in the redesigned M3:AIII interface.
Figure 4.

(a) Structure of YIIIM3AII. The YIII-type, three M-type, and AII-type repeats are shown in green, light blue, and orange, respectively. The side chains of tryptophan residues that are potentially able to bind target peptides are shown. (b) Superposition of N-cap helices H2 and H3 and the first internal repeat helix H1 of YIIM4AII (magenta), YIIIM3AII (green), and importin-α (blue, PDB ID: 1bk6). The artificial N-terminal His6-tag from YIIM4AII is shown in gray. (c) Superposition of YIIIM3AIII (gray tube) onto YIIIM3AII (tube colored according to temperature factor). (d) Sketch to illustrate the effect of a non-glycine residue in the loop between N-cap helix H3 and helix H1 from the first internal repeat. The main chain of YIIIM3AII is shown with green carbon atoms and the modeled Cβ-atom in gray. The distance between the carbonyl oxygen from position 38 and the Cβ-atom at position 41 is indicated by a gray dashed line. Hydrogen bonds are shown as yellow dashed lines. Spheres are drawn at 1.2 × rvdW to account for the Cβ hydrogen atoms.
Why does the redesign of the N-cap eliminate the domain swapping? The analysis of the interface between the N-cap (residues 13–40) and M1 (residues 43–84) in YIIIM3AII or M1# in YIIM3AII revealed buried surface areas and surface complementarity indices (SC) of 660 Å2 and 0.695 in YIIIM3AII and 760 Å2 and 0.750 in YIIM3AII, respectively. Thus, the domain swapping event buries a larger area and provides a better fit between surfaces than the intra-molecular interaction in the non-domain-swapped YIIIM3AII monomer. On the other hand, the interface of YIIIM3AII contains four H-bonds, compared to two H-bonds in the domain-swapped YIIM3AII interface. In addition, the nature of the short linker between the N-cap and M1 is probably the most important feature for domain swapping. In YIIIM3AII this linker is formed by Gly41 and Gly42, which adopt ϕ/ψ–angles of −76°/−157° and −77°/−175°, respectively. Since both glycine residues adopt main chain torsion angles that are close to the β-sheet region of the Ramachandran diagram, non-glycine residues, such as Asp41 from YIIM3AII, could theoretically adopt very similar conformations. However, at position 41 any side chain bigger than a hydrogen atom would clash with the main chain oxygen of the residue at position 38. Because Ile38 participates in helix H3 from the N-cap there is little flexibility to escape such a clash [Fig. 4(d)]. Therefore, Gly41 seems indispensable for an extremely short linker that still allows an intra-molecular interaction between the N-cap and the first internal repeat.
The peptide binding site
The final goal of this protein engineering endeavor is the design of a stable ArmR module with identical internal repeats (except for residues directly contacting the bound peptide), which is capable of recognizing peptide epitopes in an extended conformation. Indeed, the internal repeats of YIIIM3AII are most similar to the minor NLS-binding site of importin-α (residues 289–414 of PDB ID 1bk6 match with a RMSD of 0.71 Å). Most residues, which are crucial for NLS binding, such as the conserved tryptophan and asparagine residues, are also present in YIIIM3AII but, due to the absence of a peptide ligand, they show multiple conformations in YIIIM3AII [Fig. 5(a)]. The conformations of these tryptophan and asparagine residues are all very similar in the YIIM3AII, YIIM4AII, YIIIM3AII, and YIIIM3AIII structures, because they are not directly affected by domain swapping or mutations of the C-cap. However, one important residue from the NLS binding site is absent in the M-type repeat. In importin-α, Thr334 (or its equivalent Thr166 in the major NLS binding site) forms a short H-bond with the amino group of Lys128 from the NLS peptide. In M-type internal repeats the corresponding residue is Ile88, because the consensus design favored isoleucines over threonine residues at position 4 of the ArmRs [Fig. 5(a)].
Figure 5.
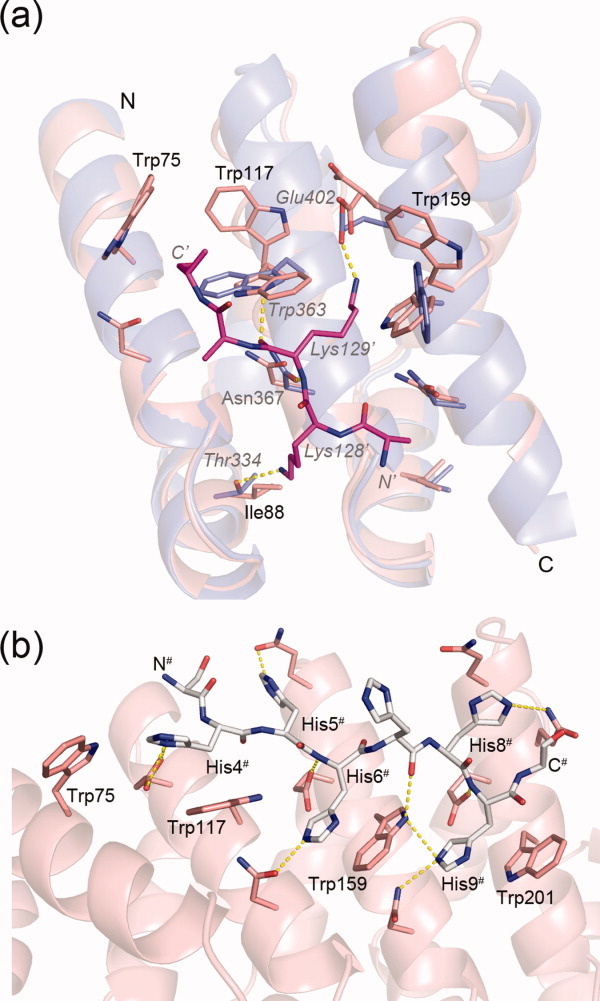
(a) Superposition of YIIIM3AII on the importin-α:NLS-peptide complex (PDB ID: 1bk6).9 Shown are residues 68–168 from YIIIM3AII in salmon, residues 313–413 from importin-α in blue, and residues 127–131 from the NLS-peptide with carbon atoms colored in magenta. Residue numbers referring to YIIIM3AII and importin-α are given in black and gray letters in italics, respectively. The prime indicates residues from the NLS peptide. (b) His6-tag of the YIIM4AII molecule (chain A with white carbon atoms) and the peptide binding site (chain B with salmon carbon atoms). Hydrogen bonds are shown as yellow dotted lines. N- and C- termini of the His6-tag are labeled.
Even though designed ArmRPs do not bind the NLS peptide appreciably, the structures of YIIM4AIII and YIIIM3AIII provide models for peptide recognition by designed ArmRPs. In both structures the N-terminal His6-tags are involved in crystal contacts and interact with the conserved tryptophan residues from the peptide binding sites of symmetry-related molecules (Supporting Information Table S1). In contrast to the NLS peptide, which runs antiparallel to the direction of importin-α, the main chains of the His6-tags run parallel to YIIM4AII and YIIIM3AIII and occupy similar positions in the peptide binding sites. The His6-tags form specific H-bonds and aromatic stacking interactions with the conserved tryptophan residues. In the YIIM4AII crystal, the side chains of His4#, His6#, and His9# form π-stacking interactions with the side chains of Trp117, Trp159, and Trp201 [Fig. 5(b)]. Furthermore, the side chains of Glu114, Glu156, and Glu198 form polar H-bonds with His4#, His6#, and His9#, respectively. Thus, a designed molecule with a repetitive architecture, such as YIIM4AII, is structurally well suited to bind repetitive peptides like hexahistidine peptides.
Super-helical parameters of designed ArmRPs
The spatial distribution of binding pockets for the peptide side chains and hydrogen bonds to the main chain is crucial for the affinity and selectivity of ArmRPs. The stacking interactions of individual ArmRs define the super-helical parameters of the solenoid and thereby the distribution of binding pockets for the targeted peptides. Thus, the peptide binding properties of designed ArmRPs are influenced by their super-helical parameters. A solenoid protein with a modular architecture can be described by the curvature, twist, and lateral bending angles that define the relative spatial orientations of adjacent repeats (Supporting Information Fig. S2).23 The curvature is defined as a rotation around an axis that lies in the repeat plane and runs almost parallel to helix H3, the twist is defined as a rotation around an axis that points perpendicular to the repeat plane, and the lateral bending is defined as a rotation around an axis that lies in the repeat plane and points perpendicular to helix H3.
We compared the super-helical parameters of designed ArmRPs to the repeats of importin-α that are involved in NLS binding, and the data are summarized in Supporting Information Table S2. The average curvature, twist, and lateral bending angles for importin-α within the minor NLS binding site are 19.9°, −24.8°, and −13.3°, respectively. They are independent of the bound peptide, as deduced from a comparison of the structures in the free and complexed state. For all four designed ArmRPs, the curvature and twist values are almost equal with values around 16.8° and −24.1°, respectively. The redesign of the N-cap affected primarily the lateral bending, especially around the first internal repeat, which still influences the average. With −9.22° (YIIIM3AII) and −7.52° (YIIIM3AIII) the lateral bending angles for the ArmRPs with redesigned N-caps are significantly smaller than for domain-swapped ArmRPs (−10.26° and −10.60° for YIIM4AII and YIIM3AII, respectively).
Thus, whereas the twist angles are similar between importin-α and designed ArmRPs, the curvature and lateral bending angles of importin-α are significantly larger in the minor NLS binding region than in designed ArmRPs, giving the ArmRPs in their current version a very slightly more stretched-out shape. This analysis based on several experimental structures will be very important for the future fine-tuning of the super-helical parameters by protein engineering, to make the structures match the unit length of peptides as closely as possible.
Materials and Methods
General molecular biology methods
Unless stated otherwise, experiments were performed according to Sambrook and Russell.24 Vent Polymerase (New England Biolabs) was used for all DNA amplifications. Enzymes and buffers were from New England Biolabs. The cloning and production strain was E. coli XL1-blue (Stratagene). The cloning and protein expression vector was pPANK (GenBank accession number AY327140).14 From this, the vector pPANK-YM-MA was constructed by cloning the capping repeats and two M-type internal repeats joined by a short DNA linker. pPANK-YM-MA contains the BsaI and BpiI restriction sites between the consensus M-type repeats for receiving further repeat modules and also encodes a MRGSH6-tag at the N-terminus of the construct.
Cloning of designed ArmRPs
Oligonucleotides were purchased from Microsynth AG (Balgach, Switzerland). A complete list of all oligonucleotides is given in Supporting Information Table S3. An approach that was similar to Binz et al.14 and Parmeggiani et al.13 was adopted for gene assembly. All single repeat modules were assembled from oligonucleotides by assembly PCR. As an example, for the AIII-type of the C-cap, pairs of partially overlapping oligonucleotides (1–2, 3–4, and 5–6) were annealed and the double strand was completed by PCR. Then, 2 μL from these PCR reaction mixtures were used as templates for a second PCR reaction in the presence of oligonucleotides 1 and 6. All the oligonucleotides were used at final concentrations of 1 μM. The annealing temperature was 50°C for the first and second reaction. Thirty PCR cycles were performed with an extension time of 30 s. The same procedure was applied for the internal and other capping repeats. Four oligonucleotides were used for the N-terminal capping repeats. BamHI and KpnI restriction sites were used for direct insertion of modules into plasmid pQE30. The single modules were PCR amplified from the vectors, using external primers pQE_f_1 and pQE_r_1 (Qiagen, Switzerland). Neighboring modules were digested with restriction enzymes BpiI and BsaI and directly ligated together. The genes coding for the whole proteins were assembled by stepwise ligation of the internal and capping modules. BamHI and KpnI restriction sites were used for insertion of whole genes into the vector pPANK. Proper assembly of constructs was validated by DNA sequencing.
Protein purification
YIIM3-6AII, YIIM3AIII, YIIIM3AII, and YIIIM3AIII were expressed in E. coli, and purified as described previously.13 Protein size and purity were assessed by 15% SDS-PAGE, stained with Coomassie PhastGel Blue R-350 (GE Healthcare, Switzerland). The expected protein masses were confirmed by SDS-PAGE (Supporting Information Fig. S3) and mass spectroscopy. Elution fractions from IMAC were passed over a desalting column (PD-10, GE Healthcare). Proteins used for crystallization trials were further purified by size exclusion chromatography on a Superdex 200 Hi-load 16/60 column using an ÄKTA prime chromatography system (GE Healthcare, Switzerland). Proteins in 10 mM Tris-HCl, 100 mM NaCl, pH 7.4 were used for crystallization trials. The proteins were finally concentrated to 14 mg/mL using Amicon Ultra centrifugation filters (Millipore, Switzerland).
Circular dichroism spectroscopy
All CD measurements were performed on a Jasco J-810 spectropolarimeter (Jasco, Japan) using a 0.5 mm or 1 mm circular thermo cuvette. CD spectra were recorded from 190 to 250 nm with a data pitch of 1 nm, a scan speed of 20 nm/min, a response time of 4 s and a band width of 1 nm. Each spectrum was recorded three times and averaged. Measurements were performed at room temperature unless stated differently. The CD signal was corrected by buffer subtraction and converted to mean residue ellipticity (MRE). Heat denaturation curves were obtained by measuring the CD signal at 222 nm with temperatures increasing from 20°C to 95°C (data pitch, 1 nm; heating rate, 1°C/min; response time, 10 s; bandwidth, 1 nm). GdnHCl-induced denaturation measurements were performed after overnight incubation at 20°C with increasing concentrations of GdnHCl (99.5% purity, Fluka) in phosphate buffered saline (pH 7.4).
ANS fluorescence spectroscopy
The fluorophore 1-anilino-naphthalene-8-sulfonate (ANS) binds to exposed hydrophobic patches or pockets in proteins. Upon binding the fluorescence of ANS increases significantly. In this study, ANS fluorescence was used to probe the packing of the designed hydrophobic cores. The measurements were performed at 20°C by adding ANS (final concentration 100 μM) to 10 μM of purified protein in 20 mM Tris-HCl, 50 mM NaCl, pH 8.0. The fluorescence signal was recorded using a PTI QM-2000-7 fluorimeter (Photon Technology International). The emission spectrum from 400 to 650 nm (1 nm/s) was recorded with an excitation wavelength of 350 nm. For each sample, three spectra were recorded and averaged.
Crystallization, X-ray data collection, and refinement
Preliminary crystallization conditions were identified using sparse-matrix screens from Hampton Research (California) and Molecular Dimensions (Suffolk, UK) in 96-well Corning plates (Corning Incorporated, New York) at 4°C and 20°C. Sitting-drop vapor-diffusion experiments were pipetted using a Phoenix crystallization robot (Art Robbins Instruments). Protein solutions were mixed with reservoir solutions at 1:1, 1:2, or 2:1 ratios (200 nL final volume) and the mixtures were equilibrated against 50 μL of reservoir solution. Crystallization conditions, data collection and refinement statistics are summarized in Table I. After adding 20% glycerol to the reservoir solution crystals were flash-cooled in liquid nitrogen. This procedure was used for all crystals except for YIIIM3AII crystals, where no cryo-protection was required.
Data were collected using either a MAR-345dtb image plate detector (MAR Research, Hamburg, Germany) mounted on a rotating anode X-ray generator equipped with a Helios optical system (Microstar Generator, Bruker AXS, Germany) or a MAR-CCD detector system on beam line X06DA at the Swiss Light Source (Paul Scherrer Institute, Villigen, Switzerland). Data were processed using programs MOSFLM25 and SCALA.26
The structures were solved by molecular replacement using program PHASER.27 Models for molecular replacement were prepared as follows. For YIIM3AII a homology model created from the crystal structure of importin-α (PDB ID: 1bk6, Chain A)9 was used. The YIIM3AIII and YIIIM3AIII structures were solved using the truncated structure of YIIM3AII (residues 42–195). The YIIM4AII structure was solved using a full-length poly-alanine model of YIIM3AII. Refinement was done using programs REFMAC528 and COOT29 with 5% of data that were set aside to calculate Rfree. The refinement of the YIIM3AII and YIIM4AII structures converged at relatively high Rcryst values and also the gap between Rcryst and Rfree is higher than expected. This increased gap can be explained by the extremely high B-factors of the C-cap, which cause electron densities of poor qualities and finally a poor fit between the final structures of the C-caps and the experimental diffraction data. Water molecules were added to well-defined difference electron density peaks at H-bond distance from the protein (between 2.2 Å and 3.6 Å from oxygen or nitrogen atoms). The final structures were validated using program PROCHECK.30 Figures were prepared using program PYMOL.31 N-caps were analyzed by eliminating residues at positions 41 and 42 and calculating the surface complementarities using program SC.32 Super-helical parameters were calculated using the program CUTLAT.23
Acknowledgments
X-ray diffraction experiments were performed on the X06DA beamline at the Swiss Light Source (Paul Scherrer Institut, Villigen, Switzerland) and the authors thank the beam line staff for skillful technical advice.
Glossary
Abbreviations
- ArmRPs
Armadillo repeat proteins
- AU
asymmetric unit
- NLS
nuclear localization sequence
- TBS
tris buffered saline
Supplementary material
Additional Supporting Information may be found in the online version of the article.
References
- 1.Perrimon N, Mahowald AP. Multiple functions of segment polarity genes in Drosophila. Dev Biol. 1987;119:587–600. doi: 10.1016/0012-1606(87)90061-3. [DOI] [PubMed] [Google Scholar]
- 2.Wieschaus E, Riggleman R. Autonomous requirements for the segment polarity gene armadillo during Drosophila embryogenesis. Cell. 1987;49:177–184. doi: 10.1016/0092-8674(87)90558-7. [DOI] [PubMed] [Google Scholar]
- 3.Peifer M, Berg S, Reynolds AB. A repeating amino acid motif shared by proteins with diverse cellular roles. Cell. 1994;76:789–791. doi: 10.1016/0092-8674(94)90353-0. [DOI] [PubMed] [Google Scholar]
- 4.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mason DA, Stage DE, Goldfarb DS. Evolution of the metazoan-specific importin alpha gene family. J Mol Evol. 2009;68:351–365. doi: 10.1007/s00239-009-9215-8. [DOI] [PubMed] [Google Scholar]
- 6.Moroianu J, Blobel G, Radu A. Nuclear protein import: Ran-GTP dissociates the karyopherin alpha-beta heterodimer by displacing alpha from an overlapping binding site on beta. Proc Natl Acad Sci USA. 1996;93:7059–7062. doi: 10.1073/pnas.93.14.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalderon D, Richardson WD, Markham AF, Smith AE. Sequence requirements for nuclear location of simian virus 40 large-T antigen. Nature. 1984;311:33–38. doi: 10.1038/311033a0. [DOI] [PubMed] [Google Scholar]
- 8.Dingwall C, Robbins J, Dilworth SM, Roberts B, Richardson WD. The nucleoplasmin nuclear location sequence is larger and more complex than that of SV-40 large T antigen. J Cell Biol. 1988;107:841–849. doi: 10.1083/jcb.107.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conti E, Uy M, Leighton L, Blobel G, Kuriyan J. Crystallographic analysis of the recognition of a nuclear localization signal by the nuclear import factor karyopherin alpha. Cell. 1998;94:193–204. doi: 10.1016/s0092-8674(00)81419-1. [DOI] [PubMed] [Google Scholar]
- 10.Marfori M, Mynott A, Ellis JJ, Mehdi AM, Saunders NF, Curmi PM, Forwood JK, Boden M, Kobe B. Molecular basis for specificity of nuclear import and prediction of nuclear localization. Biochim Biophys Acta. 2011;1813:1562–1577. doi: 10.1016/j.bbamcr.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 11.Binz HK, Amstutz P, Plückthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol. 2005;23:1257–1268. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- 12.Boersma YL, Plückthun A. DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr Opin Biotechnol. 2011;22:849–857. doi: 10.1016/j.copbio.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 13.Parmeggiani F, Pellarin R, Larsen AP, Varadamsetty G, Stumpp MT, Zerbe O, Caflisch A, Plückthun A. Designed armadillo repeat proteins as general peptide-binding scaffolds: consensus design and computational optimization of the hydrophobic core. J Mol Biol. 2008;376:1282–1304. doi: 10.1016/j.jmb.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 14.Binz HK, Stumpp MT, Forrer P, Amstutz P, Plückthun A. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J Mol Biol. 2003;332:489–503. doi: 10.1016/s0022-2836(03)00896-9. [DOI] [PubMed] [Google Scholar]
- 15.Kajander T, Cortajarena AL, Main ER, Mochrie SG, Regan L. A new folding paradigm for repeat proteins. J Am Chem Soc. 2005;127:10188–10190. doi: 10.1021/ja0524494. [DOI] [PubMed] [Google Scholar]
- 16.Wetzel SK, Settanni G, Kenig M, Binz HK, Plückthun A. Folding and unfolding mechanism of highly stable full-consensus ankyrin repeat proteins. J Mol Biol. 2008;376:241–257. doi: 10.1016/j.jmb.2007.11.046. [DOI] [PubMed] [Google Scholar]
- 17.Ostermeier M, Benkovic SJ. Evolution of protein function by domain swapping. Adv Protein Chem. 2000;55:29–77. doi: 10.1016/s0065-3233(01)55002-0. [DOI] [PubMed] [Google Scholar]
- 18.Bennett MJ, Eisenberg D. The evolving role of 3D domain swapping in proteins. Structure. 2004;12:1339–1341. doi: 10.1016/j.str.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 19.Bennett MJ, Sawaya MR, Eisenberg D. Deposition diseases and 3D domain swapping. Structure. 2006;14:811–824. doi: 10.1016/j.str.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 20.Lüthy L, Grütter MG, Mittl PR. The crystal structure of Helicobacter cysteine-rich protein C at 2.0 Å resolution: similar peptide-binding sites in TPR and SEL1-like repeat proteins. J Mol Biol. 2004;340:829–841. doi: 10.1016/j.jmb.2004.04.055. [DOI] [PubMed] [Google Scholar]
- 21.Merz T, Wetzel SK, Firbank S, Plückthun A, Grütter MG, Mittl PR. Stabilizing ionic interactions in a full-consensus ankyrin repeat protein. J Mol Biol. 2008;376:232–240. doi: 10.1016/j.jmb.2007.11.047. [DOI] [PubMed] [Google Scholar]
- 22.Kramer MA, Wetzel SK, Plückthun A, Mittl PR, Grütter MG. Structural determinants for improved stability of designed ankyrin repeat proteins with a redesigned C-capping module. J Mol Biol. 2010;404:381–391. doi: 10.1016/j.jmb.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 23.Forwood JK, Lange A, Zachariae U, Marfori M, Preast C, Grubmuller H, Stewart M, Corbett AH, Kobe B. Quantitative structural analysis of importin-beta flexibility: paradigm for solenoid protein structures. Structure. 2010;18:1171–1183. doi: 10.1016/j.str.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 24.Sambrook J, Russell DW. Molecular cloning: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 25.Leslie AGW. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography. 1992.
- 26.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 27.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D Biol Crystallogr. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- 29.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 31.DeLano WL. 2002. PyMOL. http://www.pymol.org.
- 32.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.