

Abstract
Zinc-mediated inhibition is implicated in global caspase regulation, with relief of zinc-mediated inhibition central to both small-molecule and natively induced caspase activation. As an initiator, caspase-9 regulates the upstream stages of the apoptotic caspase cascade, making it a critical control point. Here we identify two distinct zinc-binding sites on caspase-9. The first site, composed of H237, C239, and C287, includes the active site dyad and is primarily responsible for zinc-mediated inhibition. The second binding site at C272 is distal from the active site. Given the amino-acid conservation in both regions, these sites appear to be present across the caspase family underscoring the importance of zinc-mediated regulation of this class of enzymes.
Keywords: apoptosis, regulation, protease, initiator caspase, exosite binding, active-site inhibition
Introduction
Apoptotic cell death is carried out by caspases, a family of cysteine proteases. Caspases function in a cascade of cleavage events leading to orderly destruction of cells, culminating in cell death. Initiator caspases-8 and -9 activate the executioner caspases-3, -6, and -7, which cleave select targets enabling cell death. Apoptosis is important for both organismal development and homeostasis and is implicated in diseases ranging from ischemic injury to cancer. Understanding the mechanisms of the native regulatory pathways of caspases provides new insights for controlling and harnessing apoptosis.
Caspases are functional as homodimers of monomeric units that comprise an N-terminal prodomain and a catalytic large and small subunit connected by an intersubunit linker. Prodomains observed in the initiator caspases, such as caspase-9, function in recruitment and regulation and are designated as caspase activation and recruitment domains (CARD). Zymogen (uncleaved) caspase-9 as a monomer has very low activity, which is increased upon dimerization.1 In dimeric caspase-9 cleavage at a specific aspartate residue in the intersubuint linker between the large and small subunits is required for caspase-9 to attain increased activity. Full enzymatic activity is achieved only upon interaction with the apoptosome, a multicomponent, heptameric caspase-9 activating platform.2–4
Exposure of the apoptotic machinery to metal ions has been reported to influence apoptosis. Zinc, a metal that is frequently used to stabilize unique protein structures and facilitate protein function (reviewed in Refs. 5 and 6), has also been highlighted as a specific regulator of apoptosis, where even the smallest fluctuations in cellular zinc concentrations can tip a cell toward survival or apoptotic cell death.7 Zinc-based regulation of apoptosis had previously been thought to act upstream of the caspases.8 However, both in vitro and in vivo studies implicate caspases as the targets for zinc-mediated inhibition of apoptosis.9,10
The basis of the necessity for zinc regulation of caspases is not well understood, but may relate to protection of the essential active-site sulfhydryls during oxidative stress (reviewed in Ref. 11). Misregulation of zinc and changes in caspase activity has been linked to a number of diseases. Patients with asthma and chronic bronchitis show a correlation between zinc deficiency and increased levels of apoptosis in airway epithelial cells,12,13 suggesting a protective role for zinc-mediated caspase inhibition. Helicobacter pylori-based infections have also been linked to zinc-mediated regulation of the caspases,14,15 where release of zinc from the bacterium inhibits caspase activity to avoid cell death. PAC-1, a serendipitous small-molecule caspase activator, relieves zinc-mediated caspase-3 inhibition16 further suggesting that regulation of zinc-inhibition of caspases may be therapeutically exploited. In this work, we aim to understand the molecular mechanism of zinc-mediated caspase inhibition.
Results
Influence of metals on caspase-9
Caspase inhibition by zinc has been explored in the executioner caspases -3, -6, and -79; however, little is known about zinc's function on initiator caspases, including caspase-9. Inhibition of the initiator caspases is particularly critical as the initiators control the upstream steps of the proteolytic cascade. Previous studies have suggested that zinc disrupts caspase-9 activation and thus activity17,18; however, a full analysis of zinc's affect on caspase-9 or the mechanism of inhibition has not been investigated. We interrogated the effects of a panel of metal cations on the ability of caspase-9 to cleave its natural substrate, caspase-7. Both full-length caspase-9 [Fig. 1(A)] and caspase-9 lacking its prodomain (caspase activation and recruitment domain, CARD) [Fig. 1(B)] were inhibited by zinc but not by any other cations, including cobalt [Fig. 1(C)]. Full inhibition of both caspase-9 variants by zinc indicates that the CARD domain is not involved in zinc's mechanism of inhibition, but that zinc acts on the catalytic core (large and small subunits) of caspase-9. Other caspases including caspase-3, -6, -7, and -8 have previously been shown to be inhibited in the presence of zinc9; however, other metal cations, including copper also inhibit caspase-3 function.10 In contrast, caspase-9 appears to be inhibited specifically by zinc but not by other metals. Zinc-mediated inhibition was fully reversible by the metal chelator EDTA [Fig. 1(D)] suggesting a regulatory role for zinc rather than a nonspecific mechanism such as promotion of an irreversible protein aggregate.
Figure 1.
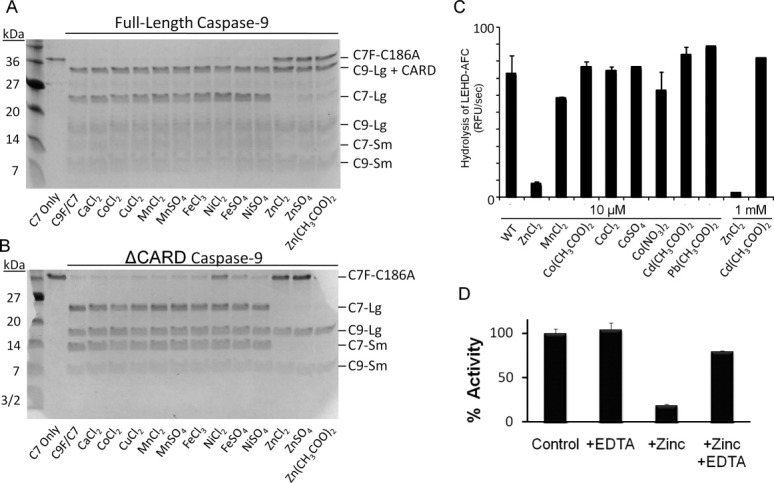
Zinc exclusively inhibits caspase-9 activity. (A,B) Zinc is the predominant metal cation to inhibit full-length caspase-9 (C9 FL) (A) or the N-terminal CARD-domain deleted caspase-9 (C9 ΔN) (B) when monitored by cleavage of a natural caspase-9 substrate, the caspase-7 zymogen (C7 C186A) to the caspase-7 large (C7 Lg) and small (C7 Sm) subunits in an in vitro cleavage assay monitored by gel mobility. (C) Caspase-9 full-length activity was not inhibited by cobalt or cadmium as judged by cleavage of fluorogenic peptide substrate LEHD-AFC. (D) Competition with metal chelator, EDTA, indicates zinc inhibition is reversible, as monitored by the cleavage of fluorogenic peptide substrate LEHD-AFC.
Inhibition of caspase-9 was monitored by titration of zinc in a caspase-9 activity assay. Inhibition of caspase-9 by zinc resulted Michaelis–Menten like curves that fit best to a mixed model of inhibition (Fig. 2). The observed error within the fit of the individual curves was not due to quenching of the fluorophore, AFC, by zinc (data not shown). This suggests zinc inhibition functions allosterically or binds near the active site, thus influencing the binding of substrate. The inhibitory constant, Ki, of 1.5 ± 0.3 μM is similar that reported for zinc-mediated inhibition of caspase-3, −6, −7, and −8 (IC50 = 8.8, 0.3, 1.7, 1.9 μM, respectively).9 Although physiological “free” or unliganded zinc concentrations are reported to be in the femto- to pico-molar range,19,20 the “available” zinc pool is believed to be much higher. As a whole, the eukaryotic cell contains ∼200 μM zinc,20 where small shifts in the glutathione concentration or slight oxidative stress causes release of zinc from the metallothioneins21 or vesicles. Thus inhibition constants for caspases in the low micromolar range are likely to be functionally relevant.
Figure 2.
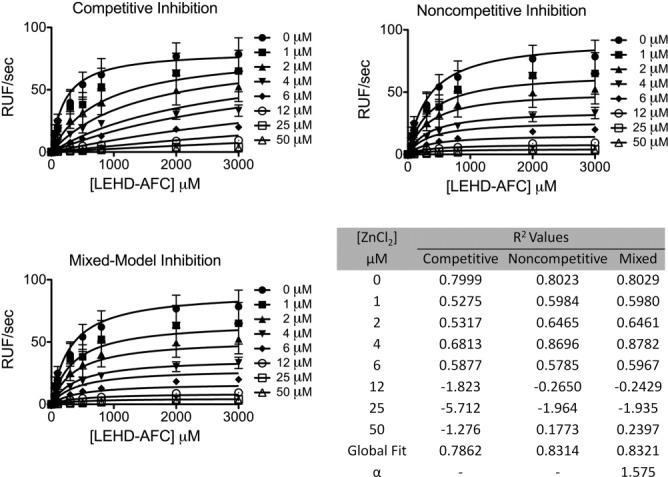
Kinetics of full-length caspase-9 wild type in the presence of 0–50 μM ZnCl2 were fit to competitive, noncompetitive, and mixed models of inhibition. R2 values are presented for individual zinc concentrations and for the overall global fit of the curves fit by each model in which mixed model of inhibition represented the best fit model.
Zinc has been shown to influence oligomerization or structure for a variety of proteins including β-2-microglobulin,22,23 α-synuclein,24 and prion proteins,25 so we likewise investigated the effect of zinc binding on the biophysical properties of caspase-9. Unlike other caspases which are constitutive dimers, caspase-9 is predominantly a monomer in solution, which dimerizes upon binding of substrate.1 Dimerization is coincident with a dramatic increase in the activity of caspase-9.1 The covalent active-site binding substrate mimic VAD-FMK functions as a surrogate for substrate and can drive dimerization of caspase-9. Caspase-9 dimerization can be observed by both native gel analysis [Fig. 3(A)] and size exclusion chromatography [Fig. 3(B)]. The presence of zinc does not influence dimerization [Fig. 3(A,B)], suggesting that inhibition is not related to preventing dimerization, which would also result in inactivation. The slight shift observed in the retention of caspase-9 in the presence of zinc may reflect a small change in the monomer–dimer equilibrium, or may simply reflect a difference in the interaction of zinc-bound caspase-9 monomers with the column. Given the known interactions of caspases with the slightly negative charge on the size exclusion column material, we suspect that zinc is modestly screening some ionic interactions and subtly shifting the retention time. What is abundantly clear is that zinc is not able to full shift caspase-9 to a dimeric state as is the case with VAD-FMK, which locks caspase-9 into the dimeric conformation. Zinc also has no observed effect on the secondary structure of caspase-9 as measured by circular dichroism spectroscopy [Fig. 3(C)], further suggesting that zinc-mediated inhibition does not influence the overall structure of caspase-9 monomers. Thus inhibition of zinc is expected to occur based on binding to a globally folded caspase-9 resulting in subtle changes in the catalytic machinery or substrate-binding groove.
Figure 3.
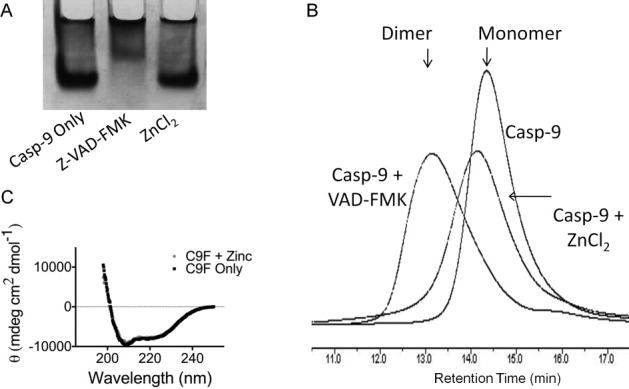
Zinc does not alter the biophysical properties of caspase-9. (A,B) Native gel analysis (A) and size exclusion chromatography (B) both indicate zinc does not alter the oligomeric state of caspase-9 from monomer to dimer like the substrate-mimic z-VAD-FMK. (C) CD spectra of caspase-9 full length in the presence and absence of ZnCl2 indicate that zinc does not alter the caspase-9 secondary structure.
Determining the location of zinc binding
Given that zinc inhibits caspase-9 by interactions with the core domain, we sought to determine the location and stoichiometry of zinc binding. Zinc binding to caspase-9 was assessed by inductively coupled plasma-optical emissions spectroscopy (ICP-OES). Each wild-type caspase-9 monomer was observed to bind two zincs (Table I). Caspase-9 monomers contain 13 cysteine and 8 histidine residues [Fig. 4(A,B)], all potential ligands for zinc. Most of these residues are located in the large subunit, either clustered near the active site or at the bottom of the 210′s helix suggesting the locations of two potential zinc-binding sites, one near the active site and another at an exosite [Fig. 4(C,D)]. The active-site cluster comprises the conserved catalytic residues C287 and H237, in proximity to two cysteines that are unique to caspase-9, C172 and C239. Interestingly, the 210′s helix region contains a conserved cysteine–histidine–cysteine triad of unknown function, comprising residues 272, 224, and 230 respectively. Furthermore, both the active site region and the triad are conserved in the apoptotic class of caspases [Fig. 4(C)].
Table I.
Zinc Binding as Monitored by ICP-OES
| Caspase-9 variant | Zinc:monomer Casp-9 |
|---|---|
| WT | 1.8 ± 0.3 |
| C287A | 0.8 ± 0.1 |
| H237A | 0.8 ± 0.01 |
| C172A | 1.5 ± 0.3 |
| C239S | 1.2 ± 0.3 |
| C287A/C239S | 0.7 ± 0.3 |
| C272A | 0.7 ± 0.3 |
| C272A/C287A | 0.2 ± 0.2 |
S.D. from samples measured independently on three separate days. Although the S.D. for H237A is lower than other variants, the precision of the analysis of all samples is believed to be similar.
Figure 4.
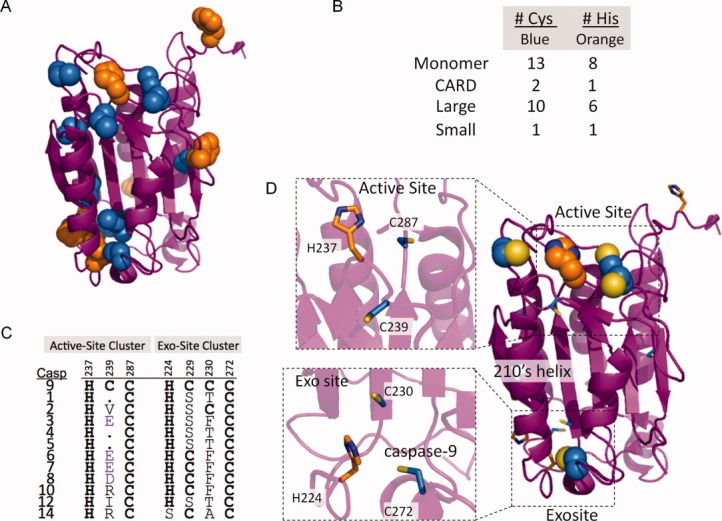
Zinc binding in caspase-9. (A) Caspase-9, monomer, highlights clusters of cys and his metal-binding residues. (B) Cys and his residues are mainly located in the large subunit of caspase-9. (C) Cys-His clusters are conserved throughout the caspase family. (D) Location of the conserved active-site and 210′s helix exosite-ligand clusters on caspase-9 (PDB ID 1JXQ).
We interrogated five positions within these two cys-his clusters in caspase-9 for involvement in zinc binding (Table I). Whereas two zincs bind to one monomer of wild-type caspase-9, caspase-9 variants in which the active-site residues are replaced, C287A or H237A, bind just one zinc. This suggests that the active-site region is one of the zinc binding sites. The two most likely proximal zinc-binding ligands were also substituted. C172A had little effect on zinc binding, but C239S lead to a more substantial loss of zinc binding. The C287A/C239S variant also lost binding of one zinc, suggesting that the active site zinc-binding cluster comprises residues C239, H237, and C287 [Fig. 4(D)]. Although the identity at residue C239 is not conserved across caspases, it is Glu in all the executioner caspases, suggesting that this residue might be a conserved zinc ligand position across the apoptotic caspase family.
Substitution of the residue C272 to alanine in the conserved 210′s-helix triad, also resulted in loss of one zinc (Table I), suggesting that C272 is at the core of the second zinc-binding site. The loss of zinc binding in C272A was not due to unfolding of the protein, as the substitution C272A has no effect on basal activity of the enzyme (Table II). The catalytic efficiency of C272A is nearly the same as wild-type caspase-9, 2.0 ± 0.16 and 2.7 ± 0.25 μM−1 s−1, respectively. Given the proximity, we suggest that the second binding site comprises residues H224, C230, and C272 [Fig. 4(D)]. Together these data suggest that there are two distinct zinc-binding sites in caspase-9. The presence of two independent binding sites was confirmed by caspase-9 C287A/C272A variant in which zinc binding is ablated (Table I).
Table II.
Catalytic Properties of Wild-Type and C272A Caspase-9
| 10−3 × [kcat/Km] (μM−1 s−1) | |
|---|---|
| WT | 2.7 ± 0.25 |
| C272A | 2.0 ± 0.16 |
Km and kcat were measured in triplicate from samples independently prepared on three separate days.
Binding of zinc to the catalytic residues of caspase-9 is expected to block nucleophilic attack of the peptide bond by C287 rendering the enzyme inactive. The potential contribution of the second binding site to inhibition is less intuitive. If the C272 exosite contributes directly to zinc-mediated caspase-9 inhibition, then the caspase-9 C272A variant C272A might be expected to show a competitive mechanism of inhibition. Surprisingly, the C272A variant also shows a mixed model of inhibition (Fig. 5), like wild-type caspase-9 and exhibited a comparable Ki value of 5.0 ± 2.8 μM. The Ki for zinc with the C272A variant is statically indistinguishable from that observed with wild type enzyme (Student's t test). Suggesting that if the exosite is capable of inhibition the Ki for this site is lower than the Ki for the active site.
Figure 5.
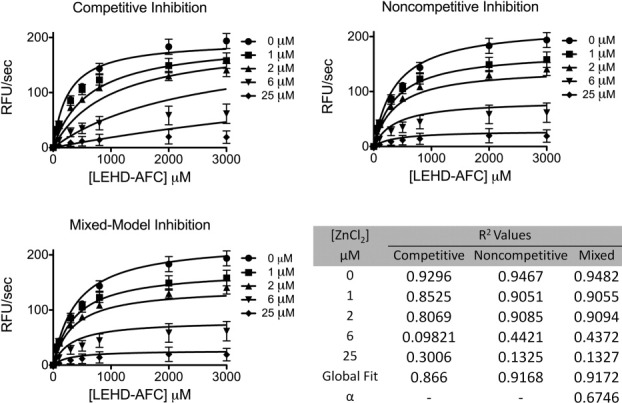
Kinetics of full-length caspase-9 C272A variant in the presence of 0–50 μM ZnCl2 were fit to competitive, noncompetitive, and mixed models of inhibition. R2 values are presented for individual zinc concentrations and for the overall global fit of the curves fit by each model in which mixed model of inhibition represented the best fit model.
To determine whether zinc binding to the exosite is sufficient to inhibit caspase-9, we sought a version of caspase-9 that lacked zinc binding at the active site, but maintained the ability to cleave substrate to assess exosite inhibition. Neither C287A nor H237A, in which the catalytic dyad residues were substituted, could be used as each of these mutations ablates caspase-9 function. On the other hand, C239 is proximal to the active site, but retains proteolytic function in the C239S variant. Moreover, the C239S version of caspase-9 binds only one zinc, presumable at the exosite. C239S can still be inhibited by zinc. The Ki of this is 1.2 ± 0.17 μM (Fig. 6). This suggests either that the exosite can lead to inhibition by zinc independently from the active site or that the active site can bind zinc with one fewer ligands at high zinc concentrations. Thus, it appears that caspase-9 may be inhibited from the exosite in addition to the active site.
Figure 6.
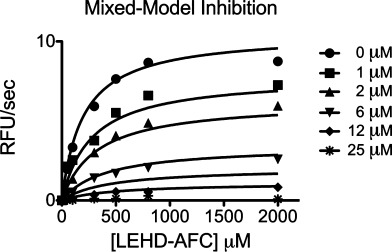
Kinetics of full-length caspase-9 C239S variant in the presence of 0–50 μM ZnCl2 were fit best to a mixed models of inhibition giving a Ki of 1.2 ± 0.17 μM.
Discussion
Although the inhibitory ability of the caspase-9 zinc-binding exosite cannot be unambiguously confirmed, it is informative to note that this 210′s helix region (the 90′s helix in caspase-6) has been implicated in the mechanism of caspase-6 activation.26 A 21° pivot about the bottom of this helix has been observed in caspase-6. The open conformation is adopted when caspase-6 is in an inactive state, whereas the closed conformation is characteristic of the active enzyme. In light of the fact that zinc inhibition is most potent in caspase-6 (IC50 = 0.3 μM),9,27 and that this structural region is sensitive and critical for caspase-6 activity, it is tempting to hypothesize that zinc could control caspase-6 allosterically, via binding to this caspase-conserved exosite triad that we have observed to be present in caspase-9.
Our findings indicate that the reversible binding of zinc to the caspase-9 active site residues H237, C239, and C287 is a dominant mechanism for zinc-mediated caspase-9 inhibition. A plausible model with reasonable metal-to-ligand bond distances between active-site residues and zinc was obtained simply by changing the rotomeric conformations of H237, C239, and C287 (Fig. 7). Like the majority of zinc-binding sites in proteins, the binding site is likely to be four-coordinate, tetrahedral. The fourth ligand could be water, solvent, or potentially a nearby glutamate residue, E290 (Fig. 7). E290 resides on a flexible loop just 3.8 Å from the proposed position of the zinc ion. Optimal E290 ligand-binding distances may be achieved by loop movement or by a water-mediated interaction.
Figure 7.
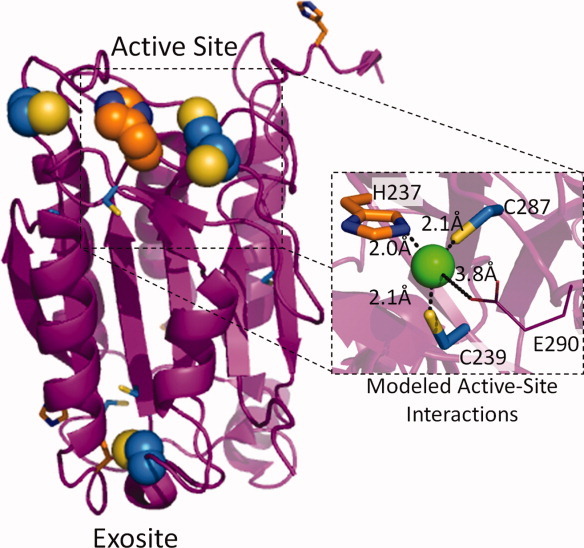
A model of caspase-9 active-site ligand interactions with a modeled zinc ion was obtained by altering the H237, C239, and C287 rotomers.
Taken together, these results suggest a model for how zinc binds to the active site and inhibits caspase-9 and the canonical conformation of caspases generally. The function of the secondary zinc-binding exosite remains an open question; however, the conservation of this site suggests that perhaps zinc does play a physiological role at this site as well. The fact that zinc does not function in a strictly competitive manner with substrate at the active site, may suggest some flexibility in the zinc-binding ligands at the active site. Given that the zinc-mediated inhibition constants for all the caspases are similar and that ligands are conserved in the active site region, suggests that the mechanism of active-site inhibition of the apoptotic caspases is conserved. Thus, utilization of zinc-based inhibition of caspases, like that used by PAC-1,16 remains a promising avenue for controlling caspases and apoptosis.
Materials and Methods
Caspase-9 expression and purification
The caspase-9 full-length gene (human sequence) construct, encoding amino acids 1-416, in pET23b (Addgene plasmid 1182928) was transformed into the BL21 (DE3) T7 Express strain of E. coli (NEB). The cultures were grown in 2xYT media with ampicillin (100 mg L−1, Sigma–Aldrich) at 37°C until they reached an optical density at 600 nm of 1.2. The temperature was reduced to 15°C and cells were induced with 1 mM IPTG (Anatrace) to express soluble 6xHis-tagged full-length protein. Cells were harvested after 3 h to obtain single site processing at Asp315. Cell pellets stored at −20°C were freeze-thawed and lysed in a microfluidizer (Microfluidics) in 50 mM sodium phosphate, pH 8.0, 300 mM NaCl, and 2 mM imidazole. Lysed cells were centrifuged at 17,000 rpm to remove cellular debris. The filtered supernatant was loaded onto a 5-mL HiTrap Ni-affinity column (GE Healthcare). The column was washed with a buffer containing 50 mM sodium phosphate pH 8.0, 300 mM NaCl, 2 mM imidazole until 280-nm absorbance returned to base line. The protein was eluted using a linear imidazole gradient of 2–100 mM over the course of 270 mL. The eluted fractions containing protein of the expected molecular weight and composition were diluted by 10-fold into a buffer composed of 20 mM Tris pH 8.5, 10 mM DTT to reduce the salt concentration. This protein sample was loaded onto a 5-mL Macro-Prep High Q column (Bio-Rad Laboratories). The column was developed with a linear NaCl gradient and eluted in 20 mM Tris pH 8.5, 100 mM NaCl, and 10 mM DTT buffer. The eluted protein was stored at −80°C in the above buffer conditions. The identity of the purified caspase-9 was analyzed by SDS-PAGE and ESI-MS to confirm mass and purity. Caspase-9 variants in the full-length expression construct (C287A, H237A, C172A, C239S, C287A/C239S, C272A, C272A/C287A) were purified by the same method as described here for the wild-type protein.
Caspase-9 ΔCARD was expressed from a two-plasmid expression system. Two separate constructs, one encoding the large subunit, residues 140–305, and the other encoding the small subunit, residues 331–416, each in the pRSET plasmid, were separately transformed into the BL21 (DE3) T7 Express strain of E. coli (NEB). The recombinant large and small subunits were individually expressed as inclusion bodies for subsequent reconstitution. Cultures were grown in 2xYT media with ampicillin (100 mg L−1, Sigma–Aldrich) at 37°C until they reached an optical density at 600 nm of 0.6. Protein expression was induced with 0.2 mM IPTG. Cells were harvested after 3 h at 37°C. Cell pellets stored at −20°C were freeze-thawed and lysed in a microfluidizer (Microfluidics) in 10 mM Tris pH 8.0 and 1 mM EDTA. Inclusion body pellets were washed twice in 100 mM Tris pH 8.0, 1 mM EDTA, 0.5M NaCl, 2% Triton, and 1M urea, twice in 100 mM Tris pH 8.0, 1 mM EDTA and finally resuspended in 6M guanidine hydrochloride. Caspase-9 large and small subunit proteins in guanidine hydrochloride were combined in a ratio of 1:2, large:small subunits, and rapidly diluted dropwise into refolding buffer composed of 100 mM Tris pH 8.0, 10% sucrose, 0.1% CHAPS, 0.15M NaCl, and 10 mM DTT, allowed to stir for 1 h at room temperature and then dialyzed four times against 10 mM Tris pH 8.5, 10 mM DTT, and 0.1 mM EDTA buffer at 4°C. The dialyzed protein was centrifuged for 15 min at 10,000 rpm to remove precipitate and then purified using a HiTrap Q HP ion exchange column (GE Healthcare) with a linear gradient from 0 to 250 mM NaCl in 20 mM Tris buffer pH 8.5, with 10 mM DTT. Protein eluted in 20 mM Tris pH 8.5, 100 mM NaCl, and 10 mM DTT buffer was stored at −80°C. The identity of the purified caspase-9ΔCARD was analyzed by SDS-PAGE and ESI-MS to confirm mass and purity.
Prediction of metal ligands and construction of ligand substitution variants
Potential zinc-binding ligands were predicted via the computational server which predicts metal ion-binding sites, HotPatch version 4.0, by a computational program specific for zinc binding sites, PREDZINC version 1.1, and by visual inspection of zinc ligand clusters and distances based on the caspase-9ΔCARD crystal structure (PDB ID: 1JXQ) via the PyMOL Molecular Graphics System, Version 1.3 (Schrödinger, LLC). Sites predicted as potential zinc ligands were C172, C239, C272, H237, and C287. Individual active-site knockout variants, H237A and C287A, and cysteine knockout variants, C172A, C239S, and C272A, in the caspase-9 full-length gene were made by a single round of QuikChange site-directed mutagenesis (Stratagene). Double knockout variants, C239S/C287A and C272A/C287A, were made by an additional round of QuikChange site directed mutagenesis in the background of the caspase-9 full-length C287A variant. Formation of the proper variant was confirmed by sequence analysis (Genewiz).
Caspase-7 C186A expression and purification
The active site knockout variant, C186A, was made by QuikChange mutagenesis (Stratagene) of the human caspase-7 gene in pET23b (gift of Guy Salvesen). The construct was transformed into BL21 (DE3) T7 Express strain of E. coli and protein expression was induced with 1 mM IPTG at 18°C for 18 h. The protein was purified as described previously for caspase-7. The eluted protein was stored at −80°C in the buffer in which they eluted. The identity of purified caspase-7 C186A was assessed by SDS-PAGE and ESI-MS to confirm mass and purity.
Activity assays
For measurements of caspase activity, proteolytic cleavage gel-based caspase-9 activity assays were performed for testing the ability of a panel of metal cations to inhibit caspase-9 activity against a natural substrate. Cleavage of the full-length procaspase-7 variant, C186A, which is catalytically inactive and incapable of self-cleavage, was used to report caspase-9 activity. The 1 μM caspase-9 full-length in a minimal assay buffer containing 100 mM MES pH 6.5 and 10 mM DTT was incubated in the presence of 1 mM metal cation for 1 h at room temperature. The 1 μM full-length procaspase-7 C186A was added to the reaction mix and incubated in a 37°C water bath for 1 h. Samples were analyzed by 16% SDS-PAGE to confirm the exact lengths of the cleavage products.
To test the reversibility of zinc binding, 700 nM freshly purified caspase-9 full-length was incubated in the presence and absence of five equivalents excess ZnCl2 for 1 h at room temperature. Half of each protein sample was treated with 25-fold excess EDTA, just prior to fluorescence activity assay measurements. Samples were assayed for activity over the course of 10 min in a caspase-9 activity assay buffer containing 100 mM MES pH 6.5, 10% PEG 8000, and 10 mM DTT. A 300 μM fluorogenic substrate, LEHD-AFC (N-acetyl-Leu-Glu-His-Asp-7-amino-4-fluorocoumarin) (Enzo Lifesciences) Ex395/Em505, was added to initiate the reaction. Assays were performed in duplicate at 37°C in 100 μL volumes in 96-well microplate format using a Molecular Devices Spectramax M5 spectrophotometer.
The inhibitory constant for zinc, Ki, was determined by incubating 700 nM caspase-9 full-length protein, diluted in 100 mM MES pH 6.5, 10% PEG 8000, and 5 mM DTT, in the presence of 0–50 μM ZnCl2 for 1.5 h at room temperature. Samples from each ZnCl2 concentration were subjected to a substrate titration, performed in the range of 0–300 μM fluorogenic substrate, LEHD-AFC (Ex365/Em495), which was added to initiate the reaction. Assays were performed in duplicates at 37°C in 100 μL volumes in 96-well microplate format using a Molecular Devices Spectramax M5 spectro-photometer. Initial velocities versus substrate concentration were globally fit to competitive, noncompetitive and mixed models of inhibition using GraphPad Prism (Graphpad Software) to determine inhibitory constant, Ki. Determination for the type of inhibition was based on four criteria. R2 values for the individual curve fits, criterion for the α-value reported by the mixed model of inhibition, overall visual inspection of the curve fits with the data and knowledge of the system were all taken into consideration. The R2 values and visual inspection of the curve fits with the data ruled out competitive inhibition, whereas, these criteria were less clear between the noncompetitive and mixed models of inhibition. However, a slight improvement in the R2 values for the individual curve fits for the mixed model of inhibition was observed, suggesting that this is the best fit for the data. The α-value obtained from the mixed model of inhibition fits also reports on the inhibition mechanism. If the α-value is equal to 1, the mixed model of inhibition is identical to the noncompetitive model, whereas, if this value is very large, the model becomes identical to the competitive model of inhibition. The reported α-value for the fits of both full-length caspase-9 wild-type and variant C272A match neither of these criteria, therefore the mechanism of inhibition appears to follow a mixed model. Furthermore, based on our enzymatic system and the determined location of metal binding, a mixed model of inhibition appears to be the most likely.
Oligomeric-state determination
Caspase-9 wild-type, full-length protein samples in 20 mM Tris pH 8.5, 110 mM NaCl, and 5 mM DTT were incubated alone (monomer), with covalent inhibitor z-VAD-FMK (carbobenzoxy-Val-Ala-Asp-fluoromethylkeytone) (dimer) or with five equivalents excess of ZnCl2 for 2 h at room temperature. The oligomeric state of the caspase-9 samples was determined via gel filtration and native gel analysis. About 100 μL of 0.75 mg mL−1 protein sample was loaded onto a Superdex 200 10/300 GL (GE Healthcare) gel-filtration column. Apo and z-VAD-FMK incubated protein samples were eluted with 20 mM Tris pH 8.0, 100 mM NaCl, and 2 mM DTT, while ZnCl2-incubated protein was eluted in the same buffer and one containing 50 μM ZnCl2 to ensure zinc was not be lost during the gel filtration process. Eluted peaks were analyzed by SDS-PAGE to ensure protein identity. Four different molecular weight standards from the gel-filtration calibration kit LMW (GE Healthcare) were run in the same conditions and a standard plot was generated to determine whether the peaks were caspase-9 monomer or dimer. For native gel analysis, samples were mixed with glycerol loading dye and fractionated on a 10% Tris/Glycine pH 8.3 polyacrylamide gel. The oligomeric state of the ZnCl2-treated caspase-9 was identified by comparison to the Apo (monomer) and z-VAD-FMK (dimer) caspase-9 full-length samples.
Secondary structure analysis by circular dichroism
Caspase-9 full-length protein was buffer exchanged via dialysis against 100 mM sodium phosphate pH 7.0, 110 mM NaCl, and 2 mM DTT, and diluted to 7 μM. The sample was split in half and incubated in the presence or absence of five equivalents of ZnCl2 for 1 h at room temperature. The circular dichroism spectra of caspase-9 full-length in the presence and absence of zinc was monitored from 250 to 190 nm, measured on a J-720 circular dichroism spectrometer (Jasco) with a peltier controller.
Zinc binding analysis by ICP-OES
Purified caspase-9 full-length, wild-type, and variants (15 μM) in 20 mM Tris pH 8.5, 110 mM NaCl, and 5 mM DTT were incubated with five equivalents excess of ZnCl2 for 1 h at room temperature. Unbound zinc was removed by incubating samples of 1 mL volume in the presence of ∼125 mg of Chelex® 100 for 1 h, mixing every 20 min. A control sample of 1 mL of 20 mM Tris pH 8.5, 110 mM NaCl, and 5 mM DTT buffer only was treated in the exact same manner to judge the completeness of unbound zinc chelation. Wild-type caspase-9 full-length samples either treated with zinc or untreated were also used as controls for metal content pre and postzinc incubation. Zinc content of the sample supernatant was quantified using a PerkinElmer Optima 4300 DV inductively coupled plasma-optical emission spectrometer (ICP-OES) equipped with a 40 MHz free-running generator and a segmented-array charge-coupled device (SCD) detector and a sample introduction system consisted of a concentric nebulizer with a cyclonic spray chamber. The concentration of zinc in each sample was determined at 206.2 nm and converted from ppm to μM to obtain the zinc to monomer of caspase-9 full-length ratio. Post ICP-OES analysis, remaining samples were tested via absorbance at 280 nm determine protein concentration. A 16% SDS-PAGE gel visualized with the Pierce Silver Stain Kit (Thermo Scientific) was used to determine the percentage of low concentration contaminants so the total caspase-9 concentration could be adjusted accordingly. This adjustment did not alter the overall trend observed.
Model of zinc binding to caspase-9
A proposed model for zinc binding at the active site of caspase-9 ΔCARD was developed in PyMOL Molecular Graphics System, Version 1.3 (Schrödinger, LLC). The zinc ionic radius was contoured to the reported values for a tetrahedral geometry of ligand binding. The rotomeric conformations of the ligands which coordinate the zinc ion, H237, C239, and C287, as well as a putative ligand, E290, were sampled to obtain the best metal to ligand geometry and allowed bond distances for ligand character.
Acknowledgments
The authors thank Kelly Ryan and Khadine Higgins for assistance with ICP and Julian Tyson for use of his ICP instrument. They gratefully acknowledge Michael Maroney and Michael Knapp for advice and helpful discussions about zinc.
Glossary
Abbreviations
- AFC
amino fluoro coumarin
- CARD
caspase activation and recruitment domain
- CD
circular dichroism spectroscopy
- FMK
fluoromethyl ketone
- LEHD
canonical caspase-9 recognition site leucine-glutamate-histidine-aspartate
- VAD
broad spectrum caspase recognition site valine-alanine-aspartate
References
- 1.Renatus M, Stennicke HR, Scott FL, Liddington RC, Salvesen GS. Dimer formation drives the activation of the cell death protease caspase 9. Proc Natl Acad Sci USA. 2001;98:14250–14255. doi: 10.1073/pnas.231465798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pop C, Timmer J, Sperandio S, Salvesen GS. The apoptosome activates caspase-9 by dimerization. Mol Cell. 2006;22:269–275. doi: 10.1016/j.molcel.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Rodriguez J, Lazebnik Y. Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev. 1999;13:3179–3184. doi: 10.1101/gad.13.24.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 5.Coleman JE. Zinc proteins: enzymes, storage proteins, transcription factors, and replication proteins. Annu Rev Biochem. 1992;61:897–946. doi: 10.1146/annurev.bi.61.070192.004341. [DOI] [PubMed] [Google Scholar]
- 6.Vallee BL, Auld DS. Zinc: biological functions and coordination motifs. Acc Chem Res. 1993;26:543–551. [Google Scholar]
- 7.Zalewski PD, Forbes IJ, Betts WH. Correlation of apoptosis with change in intracellular labile Zn(II) using zinquin [(2-methyl-8-p-toluenesulphonamido-6-quinolyloxy)acetic acid], a new specific fluorescent probe for Zn(II) Biochem J. 1993;296:403–408. doi: 10.1042/bj2960403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolf CM, Morana SJ, Eastman A. Zinc inhibits apoptosis upstream of ICE/CED-3 proteases rather than at the level of an endonuclease. Cell Death Differen. 1997;4:125–129. doi: 10.1038/sj.cdd.4400218. [DOI] [PubMed] [Google Scholar]
- 9.Stennicke HR, Salvesen GS. Biochemical characteristics of caspases-3, −6, −7, and −8. J Biol Chem. 1997;272:25719–25723. doi: 10.1074/jbc.272.41.25719. [DOI] [PubMed] [Google Scholar]
- 10.Perry DK, Smyth MJ, Stennicke HR, Salvesen GS, Duriez P, Poirier GG, Hannun YA. Zinc is a potent inhibitor of the apoptotic protease, caspase-3. A novel target for zinc in the inhibition of apoptosis. J Biol Chem. 1997;272:18530–18533. doi: 10.1074/jbc.272.30.18530. [DOI] [PubMed] [Google Scholar]
- 11.Truong-Tran AQ, Carter J, Ruffin RE, Zalewski PD. The role of zinc in caspase activation and apoptotic cell death. Biometals. 2001;14:315–330. doi: 10.1023/a:1012993017026. [DOI] [PubMed] [Google Scholar]
- 12.Carter JE, Truong-Tran AQ, Grosser D, Ho L, Ruffin RE, Zalewski PD. Involvement of redox events in caspase activation in zinc-depleted airway epithelial cells. Biochem Biophys Res Commun. 2002;297:1062–1070. doi: 10.1016/s0006-291x(02)02292-1. [DOI] [PubMed] [Google Scholar]
- 13.Truong-Tran AQ, Grosser D, Ruffin RE, Murgia C, Zalewski PD. Apoptosis in the normal and inflamed airway epithelium: role of zinc in epithelial protection and procaspase-3 regulation. Biochem Pharmacol. 2003;66:1459–1468. doi: 10.1016/s0006-2952(03)00498-2. [DOI] [PubMed] [Google Scholar]
- 14.Kohler JE, Dubach JM, Naik HB, Tai K, Blass AL, Soybel DI. Monochloramine-induced toxicity and dysregulation of intracellular Zn2+ in parietal cells of rabbit gastric glands. Am J Physiol Gastro Liver Physiol. 2010;299:G170–G178. doi: 10.1152/ajpgi.00355.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohler JE, Mathew J, Tai K, Blass AL, Kelly E, Soybel DI. Monochloramine impairs caspase-3 through thiol oxidation and Zn2+ release. J Surg Res. 2009;153:121–127. doi: 10.1016/j.jss.2008.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peterson QP, Goode DR, West DC, Ramsey KN, Lee JJ, Hergenrother PJ. PAC-1 activates procaspase-3 in vitro through relief of zinc-mediated inhibition. J Mol Biol. 2009;388:144–158. doi: 10.1016/j.jmb.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chimienti F, Seve M, Richard S, Mathieu J, Favier A. Role of cellular zinc in programmed cell death: temporal relationship between zinc depletion, activation of caspases, and cleavage of Sp family transcription factors. Biochem Pharmacol. 2001;62:51–62. doi: 10.1016/s0006-2952(01)00624-4. [DOI] [PubMed] [Google Scholar]
- 18.Schrantz N, Auffredou MT, Bourgeade MF, Besnault L, Leca G, Vazquez A. Zinc-mediated regulation of caspases activity: dose-dependent inhibition or activation of caspase-3 in the human Burkitt lymphoma B cells (Ramos) Cell Death Differen. 2001;8:152–161. doi: 10.1038/sj.cdd.4400772. [DOI] [PubMed] [Google Scholar]
- 19.Bozym RA, Thompson RB, Stoddard AK, Fierke CA. Measuring picomolar intracellular exchangeable zinc in PC-12 cells using a ratiometric fluorescence biosensor. ACS Chem Biol. 2006;1:103–111. doi: 10.1021/cb500043a. [DOI] [PubMed] [Google Scholar]
- 20.Krezel A, Maret W. Zinc-buffering capacity of a eukaryotic cell at physiological pZn. J Biol Inorgan Chem. 2006;11:1049–1062. doi: 10.1007/s00775-006-0150-5. [DOI] [PubMed] [Google Scholar]
- 21.Krezel A, Hao Q, Maret W. The zinc/thiolate redox biochemistry of metallothionein and the control of zinc ion fluctuations in cell signaling. Arch Biochem Biophys. 2007;463:188–200. doi: 10.1016/j.abb.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 22.Eakin CM, Knight JD, Morgan CJ, Gelfand MA, Miranker AD. Formation of a copper specific binding site in non-native states of beta-2-microglobulin. Biochemistry. 2002;41:10646–10656. doi: 10.1021/bi025944a. [DOI] [PubMed] [Google Scholar]
- 23.Morgan CJ, Gelfand M, Atreya C, Miranker AD. Kidney dialysis-associated amyloidosis: a molecular role for copper in fiber formation. J Mol Biol. 2001;309:339–345. doi: 10.1006/jmbi.2001.4661. [DOI] [PubMed] [Google Scholar]
- 24.Goers J, Uversky VN, Fink AL. Polycation-induced oligomerization and accelerated fibrillation of human alpha-synuclein in vitro. Protein Sci. 2003;2:702–707. doi: 10.1110/ps.0230903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Millhauser GL. Copper binding in the prion protein. Acc Chem Res. 2004;37:79–85. doi: 10.1021/ar0301678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vaidya S, Velazquez-Delgado EM, Abbruzzese G, Hardy JA. Substrate-induced conformational changes occur in all cleaved forms of caspase-6. J Mol Biol. 2011;406:75–91. doi: 10.1016/j.jmb.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takahashi A, Alnemri ES, Lazebnik YA, Fernandes-Alnemri T, Litwack G, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC. Cleavage of lamin A by Mch2 alpha but not CPP32: multiple interleukin 1 beta-converting enzyme-related proteases with distinct substrate recognition properties are active in apoptosis. Proc Natl Acad Sci USA. 1996;93:8395–8400. doi: 10.1073/pnas.93.16.8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stennicke HR, Deveraux QL, Humke EW, Reed JC, Dixit VM, Salvesen GS. Caspase-9 can be activated without proteolytic processing. J Biol Chem. 1999;274:8359–8362. doi: 10.1074/jbc.274.13.8359. [DOI] [PubMed] [Google Scholar]