

Abstract
The transmembrane domains of membrane fusogenic proteins are known to contribute to lipid bilayer mixing as indicated by mutational studies and functional reconstitution of peptide mimics. Here, we demonstrate that mutations of a GxxxG motif or of Ile residues, that were previously shown to compromise the fusogenicity of the Vesicular Stomatitis virus G-protein transmembrane helix, reduce its backbone dynamics as determined by deuterium/hydrogen-exchange kinetics. Thus, the backbone dynamics of these helices may be linked to their fusogenicity which is consistent with the known over-representation of Gly and Ile in viral fusogen transmembrane helices. The transmembrane domains of membrane fusogenic proteins are known to contribute to lipid bilayer mixing. Our present results demonstrate that mutations of certain residues, that were previously shown to compromise the fusogenicity of the Vesicular Stomatitis virus G-protein transmembrane helix, reduce its backbone dynamics. Thus, the data suggest a relationship between sequence, backbone dynamics, and fusogenicity of transmembrane segments of viral fusogenic proteins.
Keywords: helix backbone dynamics, hydrogen/deuterium-exchange, membrane fusion, GxxxG, Vesicular Stomatitis virus G-protein, fusion protein, fusogen
Introduction
The fusion of lipid bilayers, which is at the heart of cellular secretion and infection by enveloped viruses, is driven by dedicated fusogenic single-span membrane proteins. While extracellular domains of these proteins mediate apposition of cognate membranes, their transmembrane domains (TMDs) are thought to support actual lipid mixing.1 A recent comprehensive database analysis showed characteristic features of fusion protein TMDs. The TMDs of soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein receptor) (SNARE) proteins are enriched in the β-branched amino acids Ile and Val. High Ile and Val contents have previously been shown to enhance the fusogenicity of synthetic SNARE TMDs2 and of model TMDs that were designed to contain Val and Leu at different ratios.3 Higher membrane fusogenicity correlates with enhanced backbones dynamics, that is with the extent of local and transient helix unfolding,4,5 which is in accord with the known helix-destabilizing role of β-branched residues.6,7
Viral fusogen TMDs exhibit an overabundance of Ile and Gly; Gly frequently occurs at positions four residues apart, thus forming a GxxxG pair.8 Mutating such GxxxG pairs has previously been shown to diminish the fusogenicity of certain viral envelope proteins, including the HIV gp41 protein,9 the Japanese encephalitis virus prM protein,10 and the Vesicular Stomatitis virus (VSV) G-protein.11 In support of the notion that TMDs contribute to fusogenicy, a synthetic version of the VSV G-protein TMD drives liposome fusion and mutating its Gly or Ile residues reduced its fusogenicity.12,13 Similar to Ile, Gly is known to be helix-destabilizing.14–16
Here, we examined a synthetic VSV G-protein TMD for its sequence-specific backbone dynamics. The wild-type (wt) TMD was compared to mutants where either residues G473 and G477 (mutant L2) or I471, I472, I475, I476, and F479 were exchanged for the helix-stabilizing Leu (mutant L5) [Fig. 1(A)].
Figure 1.
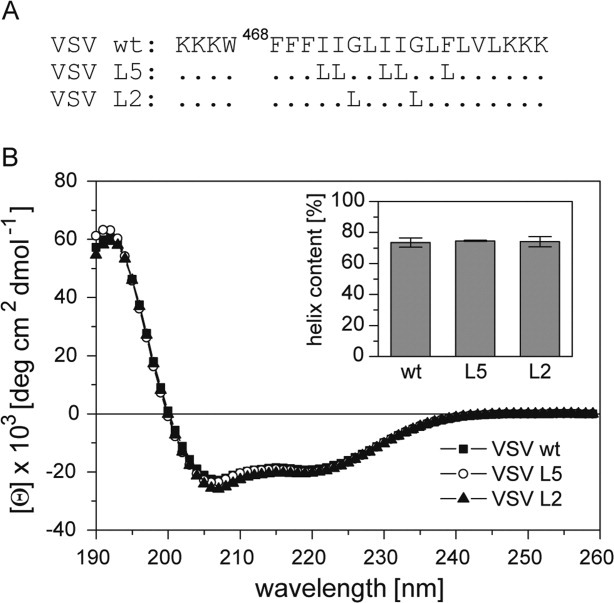
Secondary structure of VSV TMD peptides. A: Primary structures of peptides. Hydrophobic residues of the VSV G-protein (strain San Juan) TMD are flanked by Lys-triplets to enhance solubility and a Trp is added for quantification. Dots correspond to wild-type residues. B: CD spectra and calculated percentages of helix structure (inset); residual secondary structure is accounted for by β-sheet (5–6%), β-turn (∼12%), and random coil (8–9%). All values represent means of three independent measurements + SD.
Results and Discussion
The TMD peptides contain the major part of the predicted G-protein TMD whose hydrophobic residues are flanked by Lys-triplets to enhance solubility and a Trp for quantification [Fig. 1(A)]. All experiments were done in 60% (v/v) 2,2,2-trifluoroethanol (TFE). The advantage of recording DHX kinetics in isotropic solution is that all amide hydrogen atoms are accessible to solvent while a membrane shields part of a transmembrane peptide from exchange.4,5
Circular dichroism (CD) spectroscopy revealed the characteristic line shapes diagnostic of α-helices with minima at 208 and 222 nm. Quantitative evaluation of the spectra indicates α-helix contents ∼70% [Fig. 1(B)]. The backbone dynamics of the helices was investigated by recording deuterium/hydrogen-exchange (DHX) kinetics. Since transient helix unfolding allows for a gradual exchange of amide hydrogen atoms for deuterium atoms or vice versa, exchange rates provide a measure of backbone dynamics.17–19 DHX was determined of exhaustively (>95%) deuterated peptides. In a control experiment, aliquots of the deuterated peptides were diluted under “quench conditions” (pH 2.5, samples on ice) at a final concentration of 5 μM where they are thought to exist in a monomeric state. Under quench conditions, exchange rates are minimal and only deuterium atoms bound to N- and C-termini and to polar amino acid side-chains are expected to back-exchange for hydrogen atoms.20,21 Exchange was monitored by determining the molecular masses of the triply charged peptide ions using electrospray ionization mass spectrometry (ESI-MS). An exemplary mass spectrum of the VSV-wt TMD [Fig. 2(A), t = 0 min] reveals that the isotopic envelope of the triply charged ion shifts from m/z 907.2 to m/z 900.8 under quench conditions to yield a peptide with ∼17 remaining deuterium atoms. A number of 17 deuterium atoms is below the calculated number of potentially hydrogen-bonded amide deuterium atoms in an idealized 22-residue α-helix (=18). Therefore, non-H-bonded deuterium atoms are expected to exchange within seconds at pH 4.5 indicating that DHX kinetics recorded at pH 4.5 exclusively reflect exchange of amide deuterium atoms. As exemplified by the VSV-wt spectra, the envelopes gradually shift with incubation time towards lower m/z values [Fig. 2(A), t = 1 min or 90 min] which suggests uncorrelated exchange of individual deuterium atoms upon local helix unfolding reactions.22,23 In contrast to that, correlated exchange of amides from globally unfolded helices would produce a bimodal distribution of isotopic envelopes corresponding to completely exchanged and unexchanged species, respectively; this is clearly not observed here. Figure 2(B) shows that the velocity of DHX follows the rank order wt > L5 >> L2. For quantitative evaluation, the kinetics were fit with a triple term exponential function which subdivides each peptide's amide deuterium atoms into three classes (A, B, C) that exchange with different mean rate constants (kA, kB, kC). The results show that the three populations contain similar numbers of deuterium atoms for each TMD and that most (=10–11) deuterium atoms are contained in the slow population C compared with the intermediate population B (2.5–3.5 deuterium atoms) and the fast population A (4.5–5.5 deuterium atoms) [Fig. 2(C)]. The TMD-specific differences in the velocity of DHX are reflected by the mean exchange rate constants within populations B and C. Relative to wt, kB values of the mutant peptides L5 and L2 are decreased by 12 and 25%, respectively, while kC values are decreased by even 25 and 80% [Fig. 2(D)]. kA Values are not shown here since the exchange kinetics of population A could not be fit with satisfactory precision which is due to the fact that they are only covered by 2 data points recorded within the first minute and thus inherently imprecise.
Figure 2.
DHX kinetics. A: Exemplary mass spectra of the triply-charged VSV wt ion after different DHX reaction periods. The spectrum at t = 0 min was recorded after exchange under quench conditions (≤2 min on ice at pH 2.5). Low intensity isotopic envelopes at calculated masses of ∼22 Da and ∼38 Da above the dominant envelopes likely originate from Na+- and K+-adducts, respectively. The fully protonated TMD is shown for comparison. B: Exchange kinetics. The data points at t = 0 correspond to the numbers of amide deuterons seen after exchange under quench conditions. The data were fit with a three-term exponential function assuming the presence of 18 deuterium atoms at t = 0 min (continuous lines). Data points (>0 min) are means ± SD, n = 3. C: The sizes of the deuteron populations as taken from the fit. D: Mean exchange rate constants as taken from the fits. Error bars in C and D signify the deviation of the fit functions from the experimental data points.
Our results are consistent with a model where the amide deuterium atoms of the most quickly exchanging population A are located at the invariant Lys-tags at the helix termini, population B corresponds to regions between the termini and the center, and population C represents the center itself. This is suggested by a generally observed decrease of helix backbone dynamics from the termini towards the center.24,25 The strong reduction of the exchange rate constants kB, and in particular kC, upon mutating the GxxxG motif agrees with the known TMD helix-destabilizing effect of Gly.15,16 The finding that kB and kC values of mutant L5 are closer to the respective values of wt than to L2 indicates that the exchange of Ile/Phe→Leu is less helix-stabilizing than the exchange Gly→Leu. DHX was also performed at pH 7.0 where the concentration of the catalytic hydroxyl ions is higher and exchange is therefore much faster. At pH 7.0, kB and kC values of the L2 mutant were also below the respective values for the wild-type TMD while the L5 mutant could not be distinguished from wild-type due to the overall faster exchange reaction (results not shown).
What is the mechanism by which these mutations rigidify the VSV TMD? A previous molecular dynamics study of model TMDs has compared the role of some amino acid types in helix dynamics. In general, the rigidity of the helix backbone is mainly determined by side-chain/side-chain interactions between a residue at position i with its spatial neighbors at i ± 3 and i ± 4 positions.25 Thus, the destabilizing effect of Gly is attributed to its missing side chain and substituting Gly for the large Leu can stabilize the helix backbone. Likewisely, the destabilizing effect of β-branched residues, like Val and Ile, likely originates from weaker side chain packing. The position of the Val γ-methyl group is sterically restricted by the helix backbone thus limiting interactions of the Val side chain with side chains at i ± 3,4 positions. In contrast to that, the γ-branched Leu side chain can rapidly interconvert between different rotameric states such that both of its δ-methyls can explore a larger volume. Consequently, Leu rigidifies the helix backbone by interacting more efficiently with its intrahelical neighbors than Val, and by implication, Ile.25 Although the helix-destabilizing effects of Gly and Ile are likely to be less pronounced in the nonpolar environment of a membrane than in organic solvent,26 the same principles are likely to hold true in a lipid bilayer.
What are the functional implications of our results? The mutations studied here had previously been shown to reduce the fusogenicity of the VSV G-protein TMD peptide.12,13 Enhanced backbone dynamics of a fusogen TMD helix might strengthen the binding to lipids which could induce lipid splay where one acyl tail remains aligned with the membrane normal while the other tail can transiently protrude to the surface of the bilayer. If located at a site where membranes contact each other, splayed lipid tails could initiate bilayer mixing as suggested by a number of recent molecular dynamics simulations.27–33 Simulations have also suggested that lipid splay is facilitated by SNARE TMDs34 and a similar function is conceivable of a highly dynamic VSV G-protein TMD. In addition, the GxxxG motif of the G-protein TMD is known to support its homotypic interaction.35 Since the contribution of SNARE TMDs to membrane fusion has also been related to TMD-TMD interaction,30,36 the GxxxG motif of the G-protein TMD could also facilitate fusion by strengthening self-interaction. Recently, homotypic interaction of the HIV gp41 TMD has also been shown to depend on a GxxxG motif.37 Taken together, the known functional relevance of the GxxxG motif in different fusion protein TMDs9–11,38 could relate to the backbone dynamics and/or to self-interaction of the TMD helices at different stages of membrane fusion.
Experimental Procedures
Peptide synthesis
Peptides were synthesized by Fmoc chemistry (PSL, Heidelberg, Germany) and were >90% pure as judged by mass spectrometry. Concentrations were determined via UV spectroscopy using an extinction coefficient at 280 nm of 5500 M−1 cm−1.
Circular dichroism spectroscopy
For CD spectroscopy, peptides were dissolved at 50 μM in 60% (v/v) TFE, 4 mM ammonium acetate, pH 7.4. CD spectra were obtained using a Jasco J-710 CD spectrometer from 190 nm to 260 nm in a 1.0 mm quartz cuvette at 20°C using a response of 1 s, a scan speed of 100 nm min−1 and a sensitivity of 100 mdeg cm−1. Spectra were the signal-averaged accumulation of 10 scans with the baselines (corresponding to solvent) subtracted. Mean molar ellipticities were calculated and secondary structure contents estimated by deconvoluting the spectra using CDNN.39
Recording of DHX kinetics and ESI-MS
Prior to DHX analysis in TFE/buffer solution, peptides were fully deuterated by incubation in 40% (v/v) dTFE, 6 mM ND4Ac, pD 7.4 at 95°C for 20 min prior to lyophilization and re-solubilization in dTFE. To monitor DHX kinetics, the deuterated peptides (100 μM in 60% (v/v) dTFE, 4 mM ND4Ac, pD 4.5, were diluted 1:20 into 60% (v/v) TFE, 4 mM NH4Ac, pH 4.5, and incubated at 20°C. After the indicated time periods, the exchange reactions were quenched by placing samples on ice and adding formic acid to a final concentration of 0.5% (v/v) which results in pH 2.5. In parallel, aliquots of each deuterated sample were diluted directly into 60% (v/v) TFE, 4 mM NH4Ac, pH 2.5 on ice and incubated for ≤ 2 min to determine the number of deuterium atoms exchanging under these “quench conditions”. ESI-MS was done by injecting 50 μL of the samples within about 2 min of reaction quench into the micro electrospray ionization source of a Waters Q-Tof Ultima mass spectrometer. Spectra were acquired in positive-ion mode (capillary voltage 2–3 kV; cone voltage 35 V, source temperature 80°C, desolvation temperature 150°C, desolvation gas 250 L/h) by accumulating ten 1-s scans. Spectra were smoothed with the Savitzky-Golay algorithm (50 channels, 5 iterations) and centered over 80% of the peak areas (100 channels) and peak centers were taken for computation of mass differences. Data analysis was performed using MassLynx 4.0 Software (Waters, Milford, MA).
Evaluation of exchange kinetics and curve fitting
The molecular masses of the triply charged ions were determined as described.4 To evaluate DHX kinetics, the data points corresponding to the corrected masses were approximated with the function D(t) = A·e−kA·t + B·e−kB·t + C·e−kC·t where A, B, and C represent the numbers of amide deuterium atoms (D) that exchange with the time constants kA, kB, and kC, respectively, and t is time. Since we are interested in the exchange kinetics of potentially hydrogen-bonded amide deuterium atoms, their theoretical number (18 D = 21 N-D – 3 nonbonded N-D of an idealized α-helix of 22 residues) was taken as the initial data point (t = 0) for curve fitting with Origin™ 7.5 software (OriginLab, Northhampton, MA).
Glossary
Abbreviations
- CD
circular dichroism
- (d)TFE
(mono-deuterated) 2,2,2-trifluoroethanol
- DHX
deuterium/hydrogen-exchange
- ESI-MS
electrospray ionization mass spectrometry
- MRE
mean residue ellipticity
- P/L
peptide/lipid
- TMD
transmembrane domain
- VSV
vesicular stomatitis virus
References
- 1.Langosch D, Hofmann MW, Ungermann C. The role of transmembrane domains in membrane fusion. Cell Mol Life Sci. 2007;64:850–864. doi: 10.1007/s00018-007-6439-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Langosch D, Crane JM, Brosig B, Hellwig A, Tamm LK, Reed J. Peptide mimics of SNARE transmembrane segments drive membrane fusion depending on their conformational plasticity. J Mol Biol. 2001;311:709–721. doi: 10.1006/jmbi.2001.4889. [DOI] [PubMed] [Google Scholar]
- 3.Hofmann MW, Weise K, Ollesch J, Agrawal A, Stalz H, Stelzer W, Hulsbergen F, deGroot H, Gerwert K, Reed J, Langosch D. De novo design of conformationally flexible transmembrane peptides driving membrane fusion. Proc Natl Acad Sci USA. 2004;101:14776–14781. doi: 10.1073/pnas.0405175101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stelzer W, Poschner BC, Stalz H, Heck AJ, Langosch D. Sequence-specific conformational flexibility of SNARE transmembrane helices probed by hydrogen/deuterium exchange. Biophys J. 2008;95:1326–1330. doi: 10.1529/biophysj.108.132928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poschner BC, Quint S, Hofmann M, Langosch D. Sequence-specific conformational dynamics of model transmembrane domains determines their membrane fusogenic function. J Mol Biol. 2009;386:733–741. doi: 10.1016/j.jmb.2008.12.077. [DOI] [PubMed] [Google Scholar]
- 6.Creamer TP, Rose GD. Side-chain entropy opposes alpha-helix formation but rationalizes experimentally determined helix-forming propensities. Proc Natl Acad Sci USA. 1992;89:5937–5941. doi: 10.1073/pnas.89.13.5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blaber M, Zhang X, Matthews BW. Structural basis for amino acid alpha helix propensity. Science. 1993;260:1637–1640. doi: 10.1126/science.8503008. [DOI] [PubMed] [Google Scholar]
- 8.Neumann S, Langosch D. Conserved conformational dynamics of membrane fusion protein transmembrane domains and flanking regions indicated by sequence statistics. Proteins. 2011;79:2418–2427. doi: 10.1002/prot.23063. [DOI] [PubMed] [Google Scholar]
- 9.Miyauchi K, Curran R, Matthews E, Komano J, Hoshino T, Engelman DM, Matsuda Z. Mutations of conserved glycine residues within the membrane-spanning domain of human immunodeficiency virus type 1 gp41 can inhibit membrane fusion and incorporation of Env onto Virions. Jpn J Infect Dis. 2006;59:77–84. [PubMed] [Google Scholar]
- 10.Lin YJ, Peng JG, Wu SC. Characterization of the GXXXG motif in the first transmembrane segment of Japanese encephalitis virus precursor membrane (prM) protein. J Biomed Sci. 2010;17:39. doi: 10.1186/1423-0127-17-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cleverley DZ, Lenard J. The transmembrane domain in viral fusion: essential role for a conserved glycine residue in vesicular stomatitis virus G protein. Proc Natl Acad Sci USA. 1998;95:3425–3430. doi: 10.1073/pnas.95.7.3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langosch D, Brosig B, Pipkorn R. Peptide mimics of the Vesicular Stomatitis virus G-protein transmembrane segment drive membrane fusion in vitro. J Biol Chem. 2001;276:32016–32021. doi: 10.1074/jbc.M102579200. [DOI] [PubMed] [Google Scholar]
- 13.Dennison SM, Greenfield N, Lenard J, Lentz BR. VSV transmembrane domain (TMD) peptide promotes PEG-mediated fusion of liposomes in a conformationally sensitive fashion. Biochemistry. 2002;41:14925–14934. doi: 10.1021/bi0203233. [DOI] [PubMed] [Google Scholar]
- 14.O'Neil KT, DeGrado WF. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science. 1990;250:646–651. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 15.Li SC, Deber CM. A measure of helical propensity for amino acids in membrane environments. Nature Struct Biol. 1994;1:368–373. doi: 10.1038/nsb0694-368. [DOI] [PubMed] [Google Scholar]
- 16.Bocharov EV, Mayzel ML, Volynsky PE, Goncharuk MV, Ermolyuk YS, Schulga AA, Artemenko EO, Efremov RG, Arseniev AS. Spatial structure and pH-dependent conformational diversity of dimeric transmembrane domain of the receptor tyrosine kinase EphA1. J Biol Chem. 2008;283:29385–29395. doi: 10.1074/jbc.M803089200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bahar I, Wallqvist A, Covell DG, Jernigan RL. Correlation between native-state hydrogen exchange and cooperative residue fluctuations from a simple model. Biochemistry. 1998;37:1067–1075. doi: 10.1021/bi9720641. [DOI] [PubMed] [Google Scholar]
- 18.Englander SW. Protein folding intermediates and pathways studied by hydrogen exchange. Annu Rev Biophys Biomol Struct. 2000;29:213–238. doi: 10.1146/annurev.biophys.29.1.213. [DOI] [PubMed] [Google Scholar]
- 19.Kaltashov IA, Eyles SJ. Studies of biomolecular conformations and conformational dynamics by mass spectrometry. Mass Spec Rev. 2002;21:37–71. doi: 10.1002/mas.10017. [DOI] [PubMed] [Google Scholar]
- 20.Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins-Struct Funct Gene. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dempsey CE. Hydrogen exchange in peptides and proteins using NMR spectroscopy. Progr Nucl Magn Res Spectr. 2001;39:135–170. [Google Scholar]
- 22.Ferraro DM, Lazo ND, Robertson AD. EX1 hydrogen exchange and protein folding. Biochemistry. 2004;43:587–594. doi: 10.1021/bi035943y. [DOI] [PubMed] [Google Scholar]
- 23.Xiao H, Hoerner JK, Eyles SJ, Dobo A, Voigtman E, Mel'Cuk AI, Kaltashov IA. Mapping protein energy landscapes with amide hydrogen exchange and mass spectrometry. I. A generalized model for a two-state protein and comparison with experiment. Protein Sci. 2005;14:543–557. doi: 10.1110/ps.041001705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fierz B, Reiner A, Kiefhaber T. Local conformational dynamics in alpha-helices measured by fast triplet transfer. Proc Natl Acad Sci USA. 2009;106:1057–1062. doi: 10.1073/pnas.0808581106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quint S, Widmaier S, Minde D, Langosch D, Scharnagl C. Residue-specific side-chain packing determines backbone dynamics of transmembrane model helices. Biophys J. 2010;99:2541–2549. doi: 10.1016/j.bpj.2010.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu L-P, Deber CM. Uncoupling hydrophobicity and helicity in transmembrane segments. J Biol Chem. 1998;273:23645–23648. doi: 10.1074/jbc.273.37.23645. [DOI] [PubMed] [Google Scholar]
- 27.Kinnunen PKJ, Holopainen JM. Mechanisms of initiation of membrane fusion: role of lipids. Biosci Rep. 2000;20:465–482. doi: 10.1023/a:1010402819509. [DOI] [PubMed] [Google Scholar]
- 28.Smeijers AF, Markvoort AJ, Pieterse K, Hilbers PAJ. A detailed look at vesicle fusion. J Phys Chem B. 2006;110:13212–13219. doi: 10.1021/jp060824o. [DOI] [PubMed] [Google Scholar]
- 29.Grafmuller A, Shillcock J, Lipowsky R. Pathway of membrane fusion with two tension-dependent energy barriers. Physical Review Letters. 2007;98:218101-218101–218101-218104. doi: 10.1103/PhysRevLett.98.218101. [DOI] [PubMed] [Google Scholar]
- 30.Tong J, Borbat PP, Freed JH, Shin YK. A scissors mechanism for stimulation of SNARE-mediated lipid mixing by cholesterol. Proc Natl Acad Sci USA. 2009;106:5141–5146. doi: 10.1073/pnas.0813138106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kasson PM, Lindahl E, Pande VS. Atomic-resolution simulations predict a transition state for vesicle fusion defined by contact of a few lipid tails. PLoS Comput Biol. 2010;6:e1000829. doi: 10.1371/journal.pcbi.1000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mirjanian D, Dickey AN, Hoh JH, Woolf TB, Stevens MJ. Splaying of aliphatic tails plays a central role in barrier crossing during liposome fusion. J Phys Chem B. 2010;114:11061–11068. doi: 10.1021/jp1055182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smirnova YG, Marrink SJ, Lipowsky R, Knecht V. Solvent-exposed tails as prestalk transition states for membrane fusion at low hydration. J Am Chem Soc. 2010;132:6710–6718. doi: 10.1021/ja910050x. [DOI] [PubMed] [Google Scholar]
- 34.Risselada HJ, Kutzner C, Grubmuller H. Caught in the act: Visualization of SNARE-mediated fusion events in molecular detail. Chembiochem. 2011;12:1049–1055. doi: 10.1002/cbic.201100020. [DOI] [PubMed] [Google Scholar]
- 35.Unterreitmeier S, Fuchs A, Schäffler T, Heym RG, Frishman D, Langosch D. Phenylalanine promotes interaction of transmembrane domains via GxxxG Motifs. J Mol Biol. 2007;374:705–718. doi: 10.1016/j.jmb.2007.09.056. [DOI] [PubMed] [Google Scholar]
- 36.Hofmann MW, Peplowska K, Rohde J, Poschner B, Ungermann C, Langosch D. Self-interaction of a SNARE transmembrane domain promotes the hemifusion-to-fusion transition in lipid mixing. J Mol Biol. 2006;364:1048–1060. doi: 10.1016/j.jmb.2006.09.077. [DOI] [PubMed] [Google Scholar]
- 37.Reuven EM, Dadon Y, Viard M, Manukovsky N, Blumenthal R, Shai Y. HIV-1 gp41 Transmembrane domain interacts with the fusion peptide: implication in lipid mixing and inhibition of virus-cell fusion. Biochemistry. 2012;51:2867–2878. doi: 10.1021/bi201721r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fink A, Sal-Man N, Gerber D, Shai Y. Transmembrane domains interactions within the membrane milieu: principles, advances and challenges. Biochimica et Biophysica Acta. 2012;1818:974–983. doi: 10.1016/j.bbamem.2011.11.029. [DOI] [PubMed] [Google Scholar]
- 39.Bohm G, Muhr R, Jaenicke R. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 1992;3:191–195. doi: 10.1093/protein/5.3.191. [DOI] [PubMed] [Google Scholar]