

Abstract
The aggregation of the protein α-synuclein (AS) is critical to the pathogenesis of Parkinson's disease. Although generally described as an unstructured monomer, recent evidence suggests that the native form of AS may be an α-helical tetramer which resists aggregation. Here, we show that N-terminal acetylation in combination with a mild purification protocol results in an oligomeric form of AS with partial α-helical structure. N-terminal acetylation of AS could have important implications for both the native and pathological structures and functions of AS. Through our demonstration of a recombinant expression system, our results represent an important step toward biochemical and biophysical characterization of this potentially important form of AS.
Keywords: α-synuclein, Parkinson's disease, N-terminal acetylation, amyloid
Introduction
The protein α-synuclein (AS) plays a central role in both sporadic and genetic forms of Parkinson's disease (PD). It is the primary component of Lewy bodies, the neuronal deposits of aggregated protein that are the hallmark of PD.1 Since it was first identified, AS has been described as an unstructured monomeric protein2 and much of our current understanding of AS is based on study in this context. However, two recent publications reported that the native state of AS is an α-helical tetramer.3, 4 Moreover, tetrameric AS was found to be aggregation-resistant. This observation has particular consequences for the role of AS in PD because it suggests that dissociation of the tetramer may be an obligatory step for formation of pathological aggregates.
Motivated by these recent studies, we investigated what conditions are important to stabilize a structured oligomeric form of AS over the disordered monomer. Bartels et al. studied AS purified from several different types of mammalian cells and brain tissue.3 They found the protein to have a molecular mass consistent with the presence of an additional acetyl group,3 and it has been known for several years that AS purified from both normal and pathological brain tissue is N-terminally acetylated.5 Wang et al. purified from E. coli an AS construct that included an additional 10 residue N-terminal fragment4 which may mimic the physical consequences of N-terminal acetylation. Both manuscripts suggested that the mild, non-denaturing conditions used for purification may also preserve the native tetramer structure.
In this work, we purify N-terminally acetylated AS from E. coli. We show that the combination of this post-translational modification and a mild purification protocol results in an oligomeric form of AS with partial α-helical structure that provides support for the previously published tetramer studies as well as insight into the mechanisms of stabilization. Importantly, the bacterially expressed construct we describe is a valuable tool for biochemical and biophysical studies of this potentially physiologically relevant form of AS.
Results and Discussion
To obtain N-terminally acetylated AS from a bacterial expression system, wild-type AS was co-expressed with the components of the fission yeast N-terminal acetylation B complex.6 The resulting purified AS (ASacetyl) was analyzed by electrospray ionization mass spectrometry (Fig. 1) to show that 100% of the protein was acetylated with a molecular weight (MW) consistent with the addition of a single acetyl group (MW 14,502 Da) (Fig. 1).
Figure 1.

Characterization of ASacetyl (gray) by electrospray ionization mass spectrometry, indicating the ∼100% efficiency of the N-terminal acetylation reaction catalyzed by N-terminal acetylation complex B. No unmodified AS is observed in the ASacetyl sample. An ASunmod (black) sample is shown for comparison. Values in parentheses indicate masses calculated from the predominant m/z peak envelope of each sample. Peaks shown correspond to z values of 15, 14, and 13 (from left to right).
To determine whether the purification method had an effect on the final state of the protein, we purified AS following two different protocols. The first method relied on ammonium sulfate cuts to precipitate AS (AMS prep), but does not utilize a boiling step commonly found in purification protocols of recombinant AS. The second method included glycerol and the non-ionic detergent octyl β-d-glucopyranoside (BOG) in all steps of the purification (BOG prep; adapted from Ref.4). We found that both N-terminal acetylation and the BOG purification are necessary for structured, oligomeric AS. By circular dichroism (CD) the spectrum of ASacetyl-BOG is partially α-helical [Fig. 2(A), solid line], with clear negative intensities at 222 and 208 nm, while ASunmod-BOG is a random coil [Fig. 2(B), solid line]. N-terminal acetylation alone does not appear to be sufficient to stabilize a structured form of AS. ASacetyl-AMS [Fig. 2(A), dashed line] results in protein that is random coil by CD with a spectrum very similar to ASunmod-AMS [Fig. 2(B), dashed line]. Incubation of ASacetyl-AMS with BOG buffer after purification was not sufficient to induce structure (data not shown) and ASacetyl-BOG retains its structure even after extensive buffer exchange into CD-compatible phosphate buffer.
Figure 2.

CD of ASacetyl (A) and ASunmod (B) purified by AMS (gray dash) and BOG (black solid) protocols. ASacetyl-BOG shows evidence of α-helical secondary structure (∼20%), while the remaining samples are predominantly random coil. While by eye, the ASacetyl sample shows a clear α-helical signal with signature negative intensities at 208 and 222 nm, the relatively low fraction of α-helical structure determined by analysis of the spectrum may be due to the overall low magnitude of the signal. All samples were exchanged into 10 mM phosphate buffer pH 7.4 prior to CD measurements.
ASacetyl-BOG also appears to be oligomeric. Size exclusion chromatography (SEC) at two different concentrations show that 55 μM ASacetyl-BOG elutes with peaks at 14.6 mL (85%) and 19.3 mL (15%) while 1 μM ASacetyl-BOG elutes as a single peak at 19.3 mL, indicating that the larger species present at high protein concentrations disassociates upon dilution. When plotted against a standard curve derived from globular proteins of known MWs, these elution volumes correspond very closely to what is predicted for a globular tetramer (14.6 mL peak) or monomer (19.3 mL peak) protein of the MW of AS [Fig. 3(A)]. Interestingly, both ASacetyl-AMS and ASunmod-AMS elute at around 14.6 mL [Fig. 3(A)]. This is larger than expected for a ∼14.5 kDa monomer globular protein and almost identical to the apparent oligomeric form of ASacetyl-BOG although all of our evidence suggests that both ASacetyl-AMS and ASunmod-AMS are disordered monomers [Fig. 3(A)].
Figure 3.
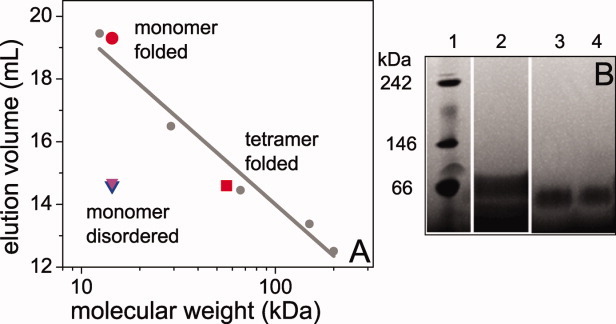
SEC (A) and native gel (B) analysis of AS. For the SEC, a standard curve (gray circles and linear fit) was generated by running cytochrome c (12.4 kDa), carbonic anhydrase (29 kDa), bovine serum albumin (66 kDa), alcohol dehydrogenase (150 kDa), and beta-amylase (200 kDa) on the column under the same conditions as the AS samples. The elution volumes of the ASacetyl-BOG samples were plotted assuming the MW of monomer (1 μM; red circle) or tetramer (55 μM; red square) AS. The elution volumes of ASacetyl-AMS (blue triangle) and ASunmod-BOG (magenta triangle) were plotted against the MW of monomer AS. The native gel shows size standards (1), ASunmod-AMS (2), ASacetyl-AMS (3), and ASacetyl-BOG (4). All samples run close to the 66 kDa reference standard, although they do not run identically. This may reflect the differences in the structures and/or oligomeric states of the different proteins, as well as the charge difference between ASunmod and ASacetyl.
For consistency with the other published studies of tetramer AS, a native gel was run which showed that ASacetyl-BOG migrates with an apparent MW ≍ 60 kDa [Fig. 3(B)], as was also seen in the studies of tetramer AS.3, 4 However, we also found that disordered monomers ASunmod-AMS and ASacetyl-AMS migrate at the same apparent MW as structured oligomer ASacetyl-BOG [Fig. 3(B)]. Both SEC and native gels separate proteins based on molecular size and shape, and our findings illustrate a key physical feature of disordered proteins—that they are significantly more extended than globular proteins of the same MW. This has been shown to be true for disordered monomer AS by several different experimental methods,7, 8 including native gel electrophoresis.9 Moreover, it emphasizes that caution must be used in interpreting molecular size from methods that are dependent upon the shape of the molecules measured.
Finally, sedimentation equilibrium analytical ultracentrifugation (SE-AUC), which is independent of molecular shape, yielded results also consistent with the conclusion that ASacetyl-BOG is an oligomer with biophysical properties that distinguish it from ASacetyl-AMS. ASacetyl-AMS fit reasonably well to a one component analysis consistent with monomer protein [Fig. 4(A)] while ASacetyl-BOG required a two component fit [Fig. 4(B)] reflecting the presence of monomer as well as a larger, oligomeric species. Although we use caution in directly comparing the size of the oligomer observed by SE-AUC and the other methods, these results qualitatively support our findings by SEC and native gel electrophoresis.
Figure 4.

SE-AUC of ASacetyl-AMS (A) and ASacetyl-BOG (B). In each case 85 μM protein was analyzed at three different rotor speeds. MWs are calculated from one component (A) or two component (B) fits (solid lines in both panels) to the data, yielding MW = 14.3 kDa (rmsd = 0.013) for ASacetyl-AMS and MW = 14.3 and 93 kDa (rmsd = 0.007) for ASacetyl-BOG. For clarity, every 10 data points are plotted, and residuals for each fit are shown in the inset.
Many eukaryotic proteins are N-terminally acetylated.10 While the functional implications of this modification are not fully understood, a number of roles have been observed including enhancing protein–protein11–13 and protein–membrane interactions14, 15 both of which are relevant to AS. Acetylation removes the N-terminal charge, making this region of the protein more hydrophobic. Increased hydrophobicity would favor protein–protein interactions of AS involving hydrophobic packing, either in folding to a native tetramer or in aggregation found in PD. Furthermore, N-terminal acetylation has been shown to be a general mechanism for stabilizing α-helical structure in proteins and peptides.16 Both the putative tetramer4 and membrane-bound17 forms of AS involve α-helix formation by the N-terminus of the protein, which may be facilitated by stabilization of nascent N-terminal α-helical structure. Lastly, we also consider the possibility that as a result of the mild purification protocol either lipids or detergent molecules remain associated with ASacetyl-BOG throughout purification. The binding of individual detergent molecules is capable of generating α-helical structure in AS.18 This, in combination with the properties conferred by N-terminal acetylation, may also favor oligomerization of AS. Ongoing work in our lab is aimed at clarifying this issue.
Here, we have demonstrated that N-terminal acetylation of AS and purification in the presence of glycerol and BOG results in an oligomeric, partially α-helical form of the protein. Our results provide insight into why the established bacterial expression and purification methods have resulted in the well-studied disordered monomer form of AS rather than a structured oligomer. Moreover, our construct provides a convenient platform that is amenable to mutagenesis techniques for studying this oligomeric form of AS not only to better characterize it, but also to determine whether it has relevance to the native and pathological roles of AS.
Materials and Methods
Protein expression and purification
ASunmod was expressed from a T7-7 plasmid in E. coli BL21 cells and purified following two different protocols. The first, the AMS prep, we have described previously.19 The second, the BOG prep, was adapted from Ref.4. Briefly, the cell pellet was resuspended in BOG buffer A (100 mM HEPES, 20 mM NaCl, 10% glycerol, 0.1% BOG pH 7.4) and one half Complete protease inhibitor tablet (Roche). The suspension was sonicated (microtip at 10 cycles of 10 s on/10 s off, power output 5) to lyse cells. After a clearance spin, the lysate was filtered and loaded onto an anion exchange column (GE HiTrap Q FF). AS was eluted using a gradient to 100% BOG buffer B (100 mM HEPES, 1 M NaCl, 10% glycerol, 0.1% BOG pH 7.4). Fractions containing AS were pooled and concentrated using Amicon 3K MWCO concentrators and loaded onto a Superdex75 (HiLoad 16/60 prep grade) column for further purification. Fractions containing AS were analyzed by SDS-PAGE for purity, pooled, and concentrated prior to freezing at −80°C for storage.
ASacetyl was produced by co-transfecting the E. coli with both AS and N-terminal acetylation B complex plasmids, using ampicillin and chloramphenicol to select for colonies containing both plasmids. Growth and purification were the same as for ASunmod.
Electrospray ionization mass spectrometry
Samples consisting of 10–20 μM protein were exchanged into 50% acetonitrile spiked with 0.2% trifluoroacetic acid using C-18 Ziptips (Millipore), and analyzed using a Waters Platform LCT electrospray ionization mass spectrometer. Capillary, sample cone, and extraction cone voltages were set to 1500 kV, 50 V, and 10 V, respectively. Data were analyzed in MassLynx 3.5.
Circular dichroism
BOG is incompatible with far-UV CD, so for uniformity all protein samples were extensively buffer exchanged into 10 mM phosphate buffer pH 7.4 prior to CD measurements. An Applied Photophysics Chirascan instrument was used with a 0.1 cm pathlength quartz cuvette to collect scans from 190 to 250 nm of 45 or 65 μM protein. Each spectrum is an average of five scans with background of phosphate buffer alone subtracted. Secondary structure analysis of the spectra was performed using CDNN 2.1 software (developed by Gerald Böhm) supplied by Applied Photophysics.
Size exclusion chromatography
For MW determination by SEC we used a Superdex200 10/30 column equilibrated in 20 mM Tris, 100 mM NaCl, 1 mM EDTA pH 8.0, and run at 0.5 mL/min. To allow for comparison between the various purification conditions, all protein samples were first concentrated in BOG buffer A and then diluted into the column equilibration buffer prior to loading onto the column. Proteins were injected in 100 μL aliquots and eluted from the column at a flow rate of 0.5 mL/min. Globular protein chromatography standards were used for column calibration.
Gel electrophoresis
Native gel electrophoresis was carried out using Native PAGE Novex 4–16% Bis-Tris gels (Invitrogen). The gels were run at 150V at 4°C according to the manufacturer's instructions and using the anode and cathode buffers provided by the manufacturer. Native MARK (Invitrogen) MW markers were used. Proteins were visualized with Coomassie stain, present in the cathode buffer during running and destained from the gel until bands were readily visible.
Sedimentation equilibrium analytical ultracentrifugation
SE-AUC was performed using a Beckman Coulter Proteome XL-1 instrument with an An-60 Ti rotor. Samples of 111 μM protein in 50 mM ammonium acetate buffer, 1mM EDTA, pH 8.5 were run at 20°C at 11,600 rpm, 20,600 rpm, and 32,000 rpm until they reached equilibrium. The presence of EDTA was found to prevent degradation of the protein. Sedphat was used to globally fit the data from all three speeds for each protein sample. The Species Analysis method was used to calculate a MW for each sample.
Acknowledgments
The authors thank Dr. Daniel Mulvihill for the N-acetylation B complex construct and Dr. Abhinav Nath for the mass spectrometry measurements.
Glossary
Abbreviations
- AS
α-synuclein
- BOG
octyl β-d-glucopyranoside
- CD
circular dichroism
- MW
molecular weight
- PD
Parkinson's disease
- SE-AUC
sedimentation equilibrium analytical ultracentrifugation
- SEC
size exclusion chromatography
References
- 1.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- 3.Bartels T, Choi JG, Selkoe DJ. Alpha-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LTT, Liao JL, Auclair JR, Johnson D, Landeru A, Simorellis AK, Ju SL, Cookson MR, Asturias FJ, Agar JN, Webb BN, Kang CH, Ringe D, Petsko GA, Pochapsky TC, Hoang QQ. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci USA. 2011;108:17797–17802. doi: 10.1073/pnas.1113260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang JP, Kling K, Lee M, Diep L, Keim PS, Shen XF, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281:29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- 6.Johnson M, Coulton AT, Geeves MA, Mulvihill DP. Targeted amino-terminal acetylation of recombinant proteins in E. coli. PLoS One. 2010;5:e15801. doi: 10.1371/journal.pone.0015801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho MK, Nodet G, Kim HY, Jensen MR, Bernado P, Fernandez CO, Becker S, Blackledge M, Zweckstetter M. Structural characterization of alpha-synuclein in an aggregation prone state. Prot Sci. 2009;18:1840–1846. doi: 10.1002/pro.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sevcsik E, Trexler AJ, Dunn JM, Rhoades E. Allostery in a disordered protein: Oxidative modifications to α-synuclein act distally to regulate membrane binding. J Am Chem Soc. 2011;133:7152–7158. doi: 10.1021/ja2009554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fauvet B, Mbefo MK, Fares MB, Desobry C, Michael S, Ardah MT, Tsika E, Coune P, Prudent M, Lion N, Eliezer D, Moore DJ, Schneider B, Aebischer P, El-Agnaf OM, Masliah E, Lashuel HA. Alpha-synuclein in the central nervous system and from erythrocytes, mammalian cells and E. coli exists predominantly as a disordered monomer. J Biol Chem. 2012 doi: 10.1074/jbc.M111.318949. jbc.M111.318949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arnesen T, Van Damme P, Polevoda B, Helsens K, Evjenth R, Colaert N, Varhaug JE, Vandekerckhove J, Lillehaug JR, Sherman F, Gevaert K. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc Natl Acad Sci USA. 2009;106:8157–8162. doi: 10.1073/pnas.0901931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott DC, Monda JK, Bennett EJ, Harper JW, Schulman BA. N-Terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science. 2011;334:674–678. doi: 10.1126/science.1209307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urbancikova M, Hitchcock-DeGregori SE. Requirement of amino-terminal modification for striated-muscle alpha-tropomyosin function. J Biol Chem. 1994;269:24310–24315. [PubMed] [Google Scholar]
- 13.Caesar R, Blomberg A. The stress-induced Tfs1p requires NatB-mediated acetylation to inhibit carboxypeptidase Y and to regulate the protein kinase A pathway. J Biol Chem. 2004;279:38532–38543. doi: 10.1074/jbc.M402939200. [DOI] [PubMed] [Google Scholar]
- 14.Behnia R, Panic B, Whyte JR, Munro S. Targeting of the Arf-like GTPase Arl3p to the golgi requires N-terminal acetylation and the membrane protein Sys1p. Nat Cell Biol. 2004;6:405–413. doi: 10.1038/ncb1120. [DOI] [PubMed] [Google Scholar]
- 15.Murthi A, Hopper AK. Genome-wide screen for inner nuclear membrane protein targeting in Saccharomyces cerevisiae: roles for N-acetylation and an integral membrane protein. Genetics. 2004;170:1553–1560. doi: 10.1534/genetics.105.043620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chakrabartty A, Doig AJ, Baldwin RL. Helix capping propensities in peptides parallel those in proteins. Proc Natl Acad Sci USA. 1993;90:11332–11336. doi: 10.1073/pnas.90.23.11332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eliezer D, Kutluay E, Bussell R, Jr, Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 18.Ferreon AC, Gambin Y, Lemke EA, Deniz AA. Interplay of alpha-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc Natl Acad Sci USA. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trexler AJ, Rhoades E. Alpha-synuclein binds large unilamellar vesicles as an extended helix. Biochemistry. 2009;48:2304–2306. doi: 10.1021/bi900114z. [DOI] [PMC free article] [PubMed] [Google Scholar]