

Abstract
Villin headpiece (HP67) is a small, autonomously-folding domain that has become a model system for understanding the fundamental tenets governing protein folding. In this communication, we explore the role that Leu61 plays in the structure and stability of the construct. Deletion of Leu61 results in a completely unfolded protein that cannot be expressed in Escherichia coli. Omission of only the aliphatic leucine side chain (HP67 L61G) perturbed neither the backbone conformation nor the orientation of local hydrophobic side chains. As a result, a large, solvent-exposed hydrophobic pocket, a negative replica of the leucine side-chain, was created on the surface. The loss of the hydrophobic interface between leucine 61 and the hydrophobic pocket destabilized the construct by ∼3.3 kcal/mol. Insertion of a single glycine residue immediately before Leu61 (HP67 L61[GL]) was also highly destabilizing and had the effect of altering the backbone conformation (α-helix to π-helix) in order to precisely preserve the wild-type position and conformation of all hydrophobic residues, including Leu61. In addition to demonstrating that the hydrophobic side-chain of Leu61 is critically important for the stability of villin headpiece, our results are consistent with the notion that the precise interactions present within the hydrophobic core, rather than the hydrogen bonds that define the secondary structure, specify a protein's fold.
Keywords: villin headpiece, hydrophobic core, X-ray crystallography, pi-helix, alpha helix, hydrogen bonds, protein folding, protein stability, protein structure, thermodynamics
Introduction
Villin headpiece (HP67) is a small, autonomously-folding domain that has become a model system for understanding the fundamental tenets governing protein folding. The C-terminal hydrophobic core has been of specific interest and the focus of numerous investigations.1–6 Several residues that contribute to the thermostability of villin headpiece have been revealed through extensive point mutagenesis.1–7 These data have established that even small perturbations to the hydrophobic core (e.g. Phe → Leu) destabilize the construct, a result that has been attributed to steric clashes imposed by the nonplanar, branched side-chain of leucine and the void resulting from the smaller volume of leucine relative to phenylalanine.2, 6 Further, with the exception of Phe51, replacing any of the phenylalanines with the more hydrophobic, but larger, perfluorinated derivative was also destabilizing,8 an effect that can be partially offset through the use of incompletely fluorinated derivatives.9 In this report, we investigate the role of Leu61, an extremely well-conserved residue residing in the hydrophobic core of villin headpiece's C-terminal subdomain (Fig. 1).
Figure 1.
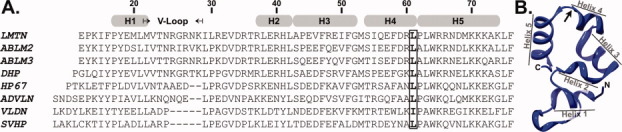
(A) Manual sequence alignment of several villin-type headpiece domains. Labels are as follows: LMTN, limatin; ABLM2, actin binding LIM protein 2; ABLM3, actin-binding LIM protein 3; DHP, dematin headpiece; HP67, chicken villin headpiece; ADVLN, advillin; VLDN, villidin; SVHP, supervillin headpiece. Boxed and bolded letters indicate position 61. Helix designations and numbering schema are based on the crystal structure of HP67 L61G (PDB ID: 3NKJ). (B) Ribbon representation of wild-type villin headpiece identifying each of the five alpha helices. Arrow indicates leucine 61.
Results
Leucine 61 is required for protein expression
We began our investigation of Leu61 by deleting the residue (HP67 Δ61); however, despite multiple attempts with three independently cloned plasmids (the sequence of each was confirmed to code for HP67 Δ61), the protein would not express in Escherichia coli (data not shown). We attribute this result to destabilization of the C-terminal subdomain and, as a result, the entire 66-residue construct because folding of the N-terminal subdomain is intimately dependant on the C-terminal subdomain.10 There are two potential, physical explanations for the presumed destabilization: (1) loss of the leucine side chain from the hydrophobic core and (2) shortening of the protein backbone, which could prevent the proper register of three C-terminal alpha helices. In order to discriminate between these two potential sources of destabilization, we cloned, expressed, and purified HP67 L61G, which restored the 67-residue length of the wild-type construct, but without the hydrophobic leucine side chain.
HP67 L61G expresses but is profoundly thermodynamically destabilized
Unlike the deletion mutant, HP67 L61G expressed well in E. coli, which permitted biophysical characterization of the construct. We found the construct to exhibit significant alpha helicity as demonstrated by circular dichroism spectroscopy [Fig. 2(A)]; however, despite a cooperative unfolding transition, the thermostability of the construct was dramatically decreased relative to wild-type [Tm,L61G is ∼40°C; Fig. 2(B)]. Further, urea denaturation established that the L61G point mutation was profoundly thermodynamically destabilizing [Fig. 2(C); ΔG°H2O,L61G 1.31 kcal/mol; ΔG°H2O,WT 4.62 kcal/mol; Table I]. This result is in line with the work of Pace et al., which demonstrated that the L61A point mutant in HP35 (the C-terminal 35 residues of villin headpiece) was also highly destabilized relative to wild-type.13
Figure 2.
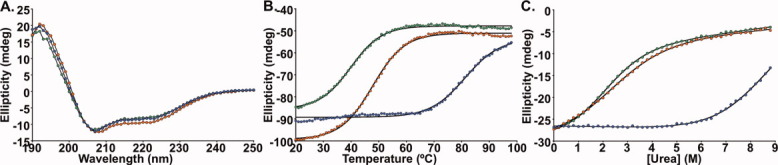
Far-UV circular dichroism spectroscopy. (A) CD wavelength scan at 20°C of wild-type (blue), HP67 L61G (green), HP67 L61[GL] (orange) are very similar to one another and are characteristic of alpha helical proteins. (B and C) Thermal and chemical denaturation was monitored by CD signal at 222 nm (B, thermal; C, urea). Solid lines represents the best fit of thermal denaturation data set to a sigmoidal function (Tm values are 81°C, 40°C, and 48°C and for wild-type, L61G, and L61[GL], respectively) or the urea denaturation data sets to Eq. (1).11, 12 (ΔG°H2O values, which were obtained via Eq. (2), are 4.63, 1.35, 1.16 kcal/mol for wild-type, L61G, and L61[GL], respectively; Table I).
Table I.
Thermodynamic Data for Wild-type HP67, HP67 L61G, and HP67 L61[GL] Determined with Urea Denaturation
| m-Value (kcal/mol·M−1) | [Urea]1/2 (M) | ΔG(H2O) (kcal/mol) | Tm (°C) | |
|---|---|---|---|---|
| Wild-type HP67 | 0.55 | 8.44 | 4.62 | 81 |
| HP67 L61G | 0.67 | 1.95 | 1.31 | 40 |
| HP67 L61[GL] | 0.51 | 2.18 | 1.12 | 48 |
The L61G point mutant does not alter the hydrophobic core
Despite being both thermally and thermodynamically destabilized, the 1H-1H NOESY spectra of HP67 L61G exhibits good peak dispersion and an abundance of cross peaks, which are signatures of well-folded proteins [Supporting Information Fig. 1(A)]. These data prompted us to attempt crystallization of the construct. HP67 L61G crystallized in the P212121 space group and diffracted to 1.6 Å (PDB Accession ID 3NKJ; Table II). In comparing the backbone of the L61G point mutant to wild-type [Fig. 2(A)], it is apparent that the structure of only one region diverges from wild type. This region, termed the variable length loop (V-Loop), is situated well away from the site of mutation in both sequence and space and is known to vary in length among the different villin-type headpiece domains (Fig. 1).14–16 In addition, this region is considerably more dynamic than the remainder of the protein as demonstrated by 15N-relaxation analysis.15, 17, 18 Prior analysis of several different crystal structures of villin headpiece have provided strong evidence that the different conformations in the V-Loop represent distinct substates of a highly dynamic ensemble.5 Despite crystallizing in the P212121 space group, the conformation of the V-Loop region in HP67 L61G is structurally more similar to chain B of wild-type villin headpiece within the P61 crystal lattice than it is to wild-type headpiece crystallized in the P212121 space group (PDB Accession IDs: 2RJX, 2RJY; Supporting Information Fig. 2). This observation is likely the result of crystal contacts as the V-Loop is in close proximity to residue 61 of its −1, 0, 0 lattice mate [Supporting Information Fig. 3(B)]. Thus, we do not believe that the backbone conformation in the V-Loop of L61G can explain the profound destabilization observed of the L61G point mutant.
Table II.
Crystallization Statistics for HP67 L61G and HP67 L61[GL]
| HP67 L61G | HP67 L61[GL] | |
|---|---|---|
| Wavelength (Å) | 1.54 | 1.54 |
| Resolution range | 13.8–1.60 | 31.70–1.80 |
| Space group | P212121 | P212121 |
| Unit cell dimensions (Å) | 30.0 × 39.5 × 51.4 | 29.8 × 40.3 × 51.6 |
| Cell angles (°) | 90, 90, 90 | 90, 90, 90 |
| Reflections (total/unique) | 357,865/7,855 | 41,253/6,019 |
| I/σ (overall/highest) | 14.0 (7.4) | 19.7 (7.0) |
| Completeness | 92.1 | 94.2 |
| Rsym (overall/ highest) | 0.042 (0.126) | 0.069 (0.166) |
| Test set | 779 (10%) | 597 (9.9%) |
| Rcryst/Rfree | 0.1817/0.2186 | 0.1781/0.1993 |
| Average B-factor | ||
| Protein | 11.87 | 14 |
| Waters | 19.96 | 22.4 |
| Total waters | 132 | 110 |
| Solvent content (%) | 38.8 | 39.5 |
| Vm (Å3/Da) | 2.01 | 2.03 |
| Bond length (Å) | 0.006 | 0.006 |
| Bond angles (°) | 1.101 | 0.795 |
| Accession ID | 3NKJ | 3MYE |
The L61G point mutant creates a solvent exposed hydrophobic pocket
Intriguingly, elimination of the leucine side chain did not alter the conformation of the nearby hydrophobic side-chains [Fig. 3(A); Supporting Information Fig. 4) and thus, when the solvent accessible surface is calculated and compared with wild-type villin headpiece, a large hydrophobic pocket manifests itself in the absence of the leucine 61 side chain (Fig. 4). This pocket is essentially a negative replica of the leucine side chain (Fig. 5). Instead of the protein “relaxing” to hide the hydrophobic surface from the polar solvent, it is apparently more thermodynamically favorable to leave a solvent-exposed hydrophobic pocket on the surface of the protein. The ∼3.3 kcal/mol destabilization can be fully explained by the loss of the hydrophobic interface between leucine 61 and the residues comprising hydrophobic pocket (∼1.1 ± 0.5 kcal/mol per —CH2➖ group).13
Figure 3.
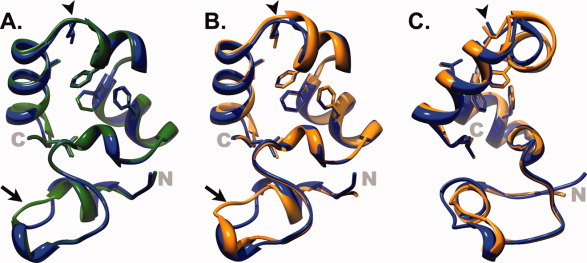
X-Ray crystal structures of HP67 L61G and HP67 L61[GL] overlain on wild-type HP67. (A) The crystal structure of HP67 L61G (green; PDB ID: 3NKJ) aligned to wild-type (blue; PDB ID: 2RJY). (B and C) Overlay of HP67 L61[GL] (orange; PDB ID: 3MYE) with wild-type HP67 (blue PDB ID: 2RJY). (B) and (C) differ in orientation by 45° rotation to better represent the differences in helix 4. The side chains of buried residues Phe47, Phe51, Phe58, Leu61, Leu69, Leu75 in wild-type are shown as sticks; In the HP67 L61[GL] insertion mutant (C) the side chains of Phe47, Phe51, Phe58, and the equivalents of wild-type residues Leu61 (Leu62), Leu69 (Leu70), and Leu75 (Leu76) are shown as sticks. Arrows point to the V-Loop, while arrowheads indicate leucine 61 in wild-type, glycine 61 in L61G, and leucine 62 in L61[GL].
Figure 4.
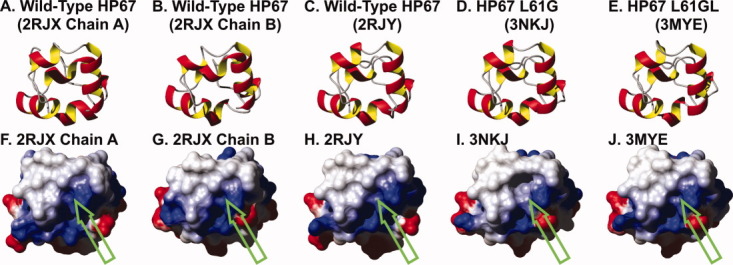
Ribbon diagrams (A–E) and corresponding electrostatic surface potential maps (F–J) for the three crystal structures of wild-type villin headpiece, HP67 L61G, and HP67 L61[GL]. Solvent accessible surfaces in the same orientation as the ribbon diagram are colored by electrostatic potential (computed with MolMol)19 from red (−0.3) through white (0.0) to blue (0.3). Green arrows point to leucine 61 in wild-type HP67, glycine 61 in HP67 L61G, and leucine 62 in HP67 L61[GL].
Figure 5.
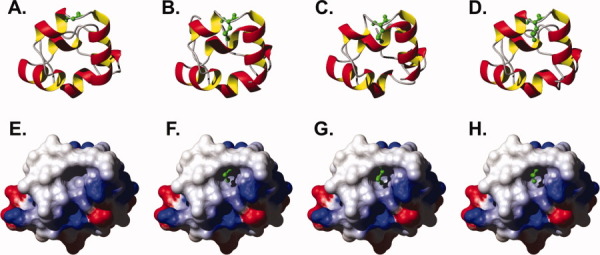
(A–D) Ribbon diagram with residue 61 represented as ball and stick for (A) HP67 L61G, 3NKJ; (B) wild-type HP67, 2RJX chain A; (C) wild-type HP67, 2RJX chain B; (D) wild-type HP67, 2RJY. (E–H) Solvent-accessible surface maps of HP67 L61G colored by electrostatic potential (computed with MolMol)19 from red (−0.3) through white (0.0), to blue (0.3). The leucine 61 side chain of wild-type villin headpiece (F, 2RJX chain A; G 2RJX chain B; H wild-type HP67, 2RJY), appropriately aligned (all atom via Topofit algorithm)20, 21 to HP67 L61G, are drawn to demonstrate that the leucine side chain fills the vacant solvent exposed hydrophobic pocket of HP67 L61G.
The L61[GL] point mutation is profoundly thermodynamically destabilizing
In addition to traditional point and deletion mutants of residue 61, described above, we also employed a different strategy, insertion mutagenesis. Insertion mutagenesis does not technically eliminate any potential favorable interactions that exist within parent construct. Instead, if strategically positioned, it requires the protein to adapt by altering certain aspects of its structure in order to preserve other essential interactions. We chose to insert a single glycine residue because it is not large enough to permit the formation of a loop; however, it does represent a significant proportional increase to total sequence length of the minute villin headpiece domain. A single glycine residue was inserted immediately before residue 61 (HP67 L61[GL]) for two reasons. First, this position resides within the alpha helix that Leu61 both terminates and presumably stabilizes by way of acting as a hydrophobic C-terminal “staple.” An additional residue within this secondary structural motif would have the effect of imposing a 100° rotation (3.6 residues per alpha helical turn) between the residues proximal and distal to the site of insertion. Second, as demonstrated above, leucine 61 is important to the stability of the villin headpiece and therefore the inserted glycine would reside between two, essential hydrophobic residues (Phe58 and Leu61). In this example, maintenance of alpha helicity and the precise position of the hydrophobic side chains are mutually exclusive, and thus the L61[GL] insertion mutant will force the construct to choose between these favorable interactions.
Circular dichroism spectroscopy was initially used to assess the structure and stability of the L61[GL] insertion mutant. Our data revealed that L61[GL] exhibits significant helicity [Fig. 2(A)] and undergoes a cooperative thermal unfolding transition with a midpoint ∼30°C below that of wild-type villin headpiece [Tm,L61[GL] of ∼48°C; Fig. 2(B)]. The cooperative unfolding transition is significant in that it suggests the presence of a well-structured hydrophobic core that is exposed to water upon unfolding. Urea denaturation demonstrated a dramatic thermodynamic destabilization, similar in magnitude to that of the L61G point mutant [ΔG°H2O,L61[GL] 1.12 kcal/mol; ΔG°H2O,WT 4.62 kcal/mol; Fig. 2(C); Table I].
Conversion of an alpha helix to a pi helix to preserve the hydrophobic core
Similar to L61G, the 1H-1H NOESY spectra of L61[GL] is characteristic of a well-folded protein [Supporting Information Fig. 1(B)], which prompted us to attempt crystallization of the construct. HP67 L61[GL] was crystallized in the P212121 space group and diffracted to 1.8 Å (PDB Accession ID 3MYE; Table II). When the structure of L61[GL] is overlain on wild-type villin headpiece crystallized under similar conditions and in the same space group (PDB Accession ID 2RJY), it is apparent that, although the overall structure of the L61[GL] insertion mutant is very similar to wild-type, there are two regions that differ between the two proteins: (1) the V-Loop region and (2) helix 4, the site of glycine insertion [Fig. 3(B,C)].
Analogous to the structure of HP67 L61G presented above, the conformation of the V-Loop in L61[GL] is more similar to wild-type villin headpiece crystallized in the P61 space group than it is to wild-type in the P212121 space group. The same reasoning previously applied to L61G is also relevant here; specifically, the conformation of the V-Loop region in L61[GL] is a consequence of crystallizing a different substate of a conformationally flexible region. Again, the V-Loop of HP67 L61[GL] is in close proximity to Leu61 and helix 4 of the −1, 0, 0 crystal contact [Supporting Information Fig. 3(C)].
Unlike the dynamic V-Loop, the C-terminal subdomain has been shown by 15N relaxation analysis to be comparatively rigid.15, 17, 18 As a result, the structural perturbation in the C-terminal subdomain of L61[GL] is significant. Relative to the wild-type structure, helix 4 of L61[GL] is unwound (Figs. 3 and 6). A physical explanation for the altered backbone conformation in L61[GL] is apparent when the side chains of the residues comprising the hydrophobic core are depicted on the ribbon backbone (Fig. 6). The position of each hydrophobic side chain in HP67 L61[GL] precisely overlays with the respective residue in wild-type HP67. In order to achieve this end in the context of a sequence containing an additional glycine residue, the backbone conformation of residues Phe58 through Leu62 (Leu61 in wild-type HP67) is underwound by one residue, effectively transforming the C-terminal half of helix 4 from an alpha helix (i → i + 4 hydrogen bonds) into a pi-helix (i → i + 5 hydrogen bonds). This result is consistent with the notion that the hydrophobic interactions within the C-terminal subdomain of villin headpiece are rigid, centrally-directed anchors and are more specific to the ground-state, native structure of villin-type headpiece domains than the electrostatic hydrogen bonds that define its secondary structure.
Figure 6.
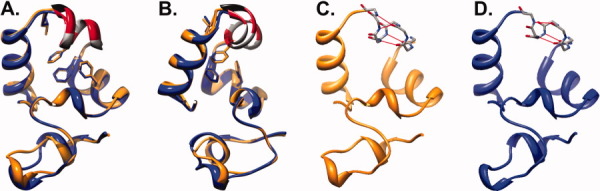
Effect of glycine insertion on the backbone conformation and hydrogen bonding network. (A) Ribbon diagram of wild-type (blue) and the L61[GL] mutant (orange) of villin headpiece with hydrophobic side chains drawn as sticks. (B) Is the same as (A) except rotated 45° degrees to emphasize the conformational change in helix 4 induced by the inserted glycine. The ribbon of helix 4 is colored in alternating grey and red colors to emphasize the position of each residue (56, 58, 60, grey; 57, 59, and 61, red; inserted glycine, black). (C and D) Ribbon diagram of the L61[GL] and wild-type villin headpiece domain with the ribbon of helix 4 replaced with a backbone ball and stick representation. Backbone hydrogen bonds present within helix 4 were computed with chimera22 and displayed as red lines.
Discussion
Numerous studies have described the physical and thermodynamic consequences of altering the hydrophobic core of both the C-terminal subdomain1–6, 13 and more recently the N-terminal subdomain7 of villin headpiece. In the studies of the C-terminal subdomain, it has been demonstrated that even relatively small perturbations within the hydrophobic core are of significant thermodynamic consequence. In the current work, we focused our attention on Leu61, an extremely well-conserved residue present within the hydrophobic core of villin's C-terminal subdomain.
In this communication, we discovered that deleting leucine 61 resulted in a construct that could not be expressed in E. coli most likely because the shorter sequence disallowed the wild-type orientation of the helices and assembly of the hydrophobic core. Omission of the aliphatic leucine side chain (HP67 L61G) perturbed neither the conformation of the backbone nor the orientation of local hydrophobic side chains. As a result, a large, solvent-exposed hydrophobic pocket, a negative replica of the leucine side-chain, was created on the surface. Finally, when we inserted a glycine residue immediately before leucine 61, we found that villin headpiece responded by altering the conformation of its backbone in order to preserve the precise position of all hydrophobic side chains including that of the leucine.
Both the L61G and L61[GL] mutants are profoundly thermodynamically destabilized relative to wild-type villin headpiece (ΔG°H2O,L61G 1.31 kcal/mol; ΔG°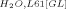 1.12 kcal/mol; ΔG°
1.12 kcal/mol; ΔG°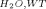 4.62 kcal/mol). The ∼3.3 kcal/mol destabilization of the L61G point mutation can be explained by the loss of the hydrophobic interface between leucine 61 and the residues comprising hydrophobic pocket (∼1.1 ± 0.5 kcal/mol per ➖CH2➖ group).13 The loss of ∼3.5 kcal/mol between the L61[GL] and the wild-type construct is likely due to the conformational strain imposed by transforming the C-terminal end of helix 4 from an α-helix into π-helix. It is interesting that 3.5 kcal/mol destabilization is very close to the 3.3 kcal/mol destabilization attributable to the loss of the hydrophobic interface. This near equivalence explains why the formation of a π-helix is not a universal occurrence and that, at least in lysozyme, maintenance of alpha helicity appears to be more common.23, 24 The fact that the π-helical conformation is favored in villin headpiece despite being destabilized to a greater degree than the energetic cost of exposing the interface between leucine 61 and the hydrophobic pocket to water (3.5 vs. 3.3 kcal/mol) suggests that extending the alpha helix would displace helix 5 and disrupt the overall topology of villin headpiece (similar to the deletion mutant, HP67 Δ61).
4.62 kcal/mol). The ∼3.3 kcal/mol destabilization of the L61G point mutation can be explained by the loss of the hydrophobic interface between leucine 61 and the residues comprising hydrophobic pocket (∼1.1 ± 0.5 kcal/mol per ➖CH2➖ group).13 The loss of ∼3.5 kcal/mol between the L61[GL] and the wild-type construct is likely due to the conformational strain imposed by transforming the C-terminal end of helix 4 from an α-helix into π-helix. It is interesting that 3.5 kcal/mol destabilization is very close to the 3.3 kcal/mol destabilization attributable to the loss of the hydrophobic interface. This near equivalence explains why the formation of a π-helix is not a universal occurrence and that, at least in lysozyme, maintenance of alpha helicity appears to be more common.23, 24 The fact that the π-helical conformation is favored in villin headpiece despite being destabilized to a greater degree than the energetic cost of exposing the interface between leucine 61 and the hydrophobic pocket to water (3.5 vs. 3.3 kcal/mol) suggests that extending the alpha helix would displace helix 5 and disrupt the overall topology of villin headpiece (similar to the deletion mutant, HP67 Δ61).
The L61G point mutant has a significantly larger m-value (the change in ΔG as a function of denaturant concentration) than either the wild-type or the L61[GL] point mutation. As the m-value is roughly proportional to the change in surface area between the native structure and the denatured state ensemble, our data suggest that the L61G point mutant unfolds to a greater extent than either wild-type or the L61[GL] insertion mutant. This result is unlikely to be due to the greater conformational flexibility afforded by glycine relative to leucine in HP67 L61G because the L61[GL] point mutant has an additional residue (glycine) in a similar position within the primary sequence. Instead, our results are more consistent with interactions between leucine 61 and other hydrophobic residues that are present in the denatured state ensembles of both wild-type and L61[GL] but absent in L61G. A glimpse of these presumed interactions might have previously been described in a small fragment of the C-terminal alpha helical subdomain.25
The term “hydrophobic effect” is typically used to describe the sum of van der Waal's interactions that shape the hydrophobic core and stabilize the overall fold of the protein. The results of this study strongly support the notion that unlike the hydrophobic interactions present in an oil droplet, those at the heart of villin headpiece as in other proteins are highly structurally specific and would be better described as “hydrophobic bonds”26 as the term “bond” generally implies a highly specific arrangement of atoms.
Materials and Methods
Sample preparation
HP67 Δ61, HP67 L61G, and HP67 L61[GL] constructs were generated from wild-type villin headpiece using QuikChange® Site-Directed mutagenesis (Stratagene). Before expression, the plasmid was verified by sequencing at SeqWright. The expression and purification protocols have been described elsewhere.16 Briefly, wild-type, HP67 L61G and HP67 L61[GL] point mutants were expressed in and purified from E. coli strain BL21(DE3). After expression the cells were lysed via sonication and the centrifugally cleared soluble supernatant was run through size exclusion chromatography column (Sephadex G-50). The fractions that contain the desired protein were then further purified to homogeneity with C18 reversed-phase chromatography. Purified protein was stored as lyophilized powder. Protein concentrations were determined by UV absorbance.
Circular dichroism spectroscopy
CD wavelength scans and thermal denaturation experiments were acquired from a solution containing 20 μM protein, 10 mM phosphate (pH 7.0), and 1 mM dithiothreitol on an Aviv 62DS spectrometer. Far-ultraviolet wavelength scans at 20°C were recorded from 250 to 190 nm at 1 nm increments. A 1 nm bandwidth and 15 s averaging time were used for wavelength scans in a 1 mm cell. Thermal denaturation experiments recorded the CD signal at 222 nm as the temperature was increased from 20 to 98°C in 1 or 2°C increments. A 1 min equilibration time, 3 nm bandwidth, and 20 s averaging time for the sample in a 1 cm cell were employed for all thermal unfolding experiments.
Urea denaturation experiments were acquired from samples of 5 μM protein, 10 mM phosphate, without dithiothreitol. CD signal at 222 nm was recorded at 20°C using a bandwidth of 3 nm and 20 second sample averaging time as the concentration of urea was increased from 0 to 9M in 0.2M increments using an autotitrator. The slope of the unfolded baseline from HP67 L61G was used to fit the wild-type data and, conversely, the slope of the folded baseline from wild-type HP67 was used to fit the urea unfolding data of the mutants to Eq. (1), where [D] is the concentration of urea, y is the CD signal, yf and yu are intercepts for the folded and unfolded baselines, af and au are the slopes of the folded and unfolded baseline, [D]1/2 is the denaturant concentration halfway through the transition, and m is the m-value.11, 12 The ΔG was computed with Eq. (2) using the m-value and [D]1/2 obtained from refining Eq. (1).11, 12
was computed with Eq. (2) using the m-value and [D]1/2 obtained from refining Eq. (1).11, 12
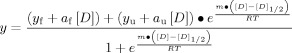 |
1 |
| 2 |
Multidimensional nuclear magnetic resonance spectroscopy
The 1H-1H NOESY spectrum (200 ms mixing time) of HP67 L61G and HP67 L61[GL] were acquired on a Bruker DMX500 at 20°C from a 1 mM sample of the respective protein in 10 mM phosphate, pH 7.0.
X-ray crystallography
L61[GL] was initially crystallized by vapor diffusion method (hanging drop at room temperature) by mixing an equal proportion of 5 mM L61[GL] at pH 7.0 with the mother liquor (0.2M sodium acetate, 0.1M sodium cacodylate, pH 6.5, 30% PEG 8000). After three rounds of seeding crushed crystals into sitting drops containing equal proportions of 2 mM L61[GL] pH 7.0 with the mother liquor (0.2M sodium acetate, 0.1M sodium cacodylate, pH 6.5, 30% PEG 10,000) diffraction quality crystals were obtained.
L61G was crystallized by vapor diffusion method (hanging drop) by mixing an equal proportion of 2 mM L61G pH 7.0 with the mother liquor (0.2M sodium acetate, 0.1M sodium cacodylate, pH 6.5, 30% PEG 10,000). Crystals formed spontaneously after several weeks incubation at 4°C.
Crystals were looped out of crystallization drops and frozen immediately. Crystals were maintained at 93K using an X-stream cryogenic system. X-rays were generated by a Rigaku RU-H3RHB X-ray generator using a rotating copper anode operated at 50 kV and 100 mA and collected with an MSC R-Axis IV++ detector. The data were then processed using d*TREK (Rigaku). The crystals were determined to be orthorhombic space group P212121 with unit cell dimensions of 30.0 × 39.5 × 51.4 Å for L61G and 29.8 × 40.2 × 51.6 Å for L61[GL]. Molecular replacement was carried out with Phenix.AutoMR27 using the structure of wild-type HP67 structure. In solving the structure of L61[GL], residues Asn60, Leu61, and Pro62 from the wild-type structure were omitted during molecular replacement to prevent bias. This region of the resulting solution was manually rebuilt using Coot28 and then refined using Phenix.Refine.27
Glossary
Abbreviations
- CD
circular Dichroism
- HP35
C-terminal 35 residues of villin (G. gallus)
- HP67
C-terminal 67 residues of villin headpiece (G. gallus)
- NMR
nuclear magnetic resonance
- PDB
Protein Data Bank
- RMS
root mean square
- V-Loop
variable length loop.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Doering DS, Matsudaira P. Cysteine scanning mutagenesis at 40 of 76 positions in villin headpiece maps the F-actin binding site and structural features of the domain. Biochemistry. 1996;35:12677–12685. doi: 10.1021/bi9615699. [DOI] [PubMed] [Google Scholar]
- 2.Frank BS, Vardar D, Buckley DA, McKnight CJ. The role of aromatic residues in the hydrophobic core of the villin headpiece subdomain. Protein Sci. 2002;11:680–687. doi: 10.1110/ps.22202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meng J, Vardar D, Wang Y, Guo H-C, Head JF, McKnight CJ. High-resolution crystal structures of villin headpiece and mutants with reduced F-actin binding activity. Biochemistry. 2005;44:11963–11973. doi: 10.1021/bi050850x. [DOI] [PubMed] [Google Scholar]
- 4.Vermeulen W, Van Troys M, Bourry D, Dewitte D, Rossenu S, Goethals M, Borremans FA, Vanderkerckhove J, Martins JC, Ampe C. Identification of the PXW sequence as a structural gatekeeper of the headpiece C-terminal subdomain fold. J Mol Biol. 2006;359:1277–1292. doi: 10.1016/j.jmb.2006.04.042. [DOI] [PubMed] [Google Scholar]
- 5.Meng J, McKnight CJ. Heterogeneity and dynamics in villin headpiece crystal structures. Acta Cryst D. 2009;65:470–476. doi: 10.1107/S0907444909008646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao S, Bi Y, Shan B, Raleigh DP. Analysis of core packing in a cooperatively folded miniature protein: the ultrafast folding villin headpiece helical subdomain. Biochemistry. 2009;48:4607–4616. doi: 10.1021/bi8021763. [DOI] [PubMed] [Google Scholar]
- 7.Brown JW, Farelli JD, McKnight CJ. On unsatisfied hydrogen bonds in the N-terminal subdomain of villin headpiece. J Mol Biol. 2011;413:543–547. doi: 10.1016/j.jmb.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woll MG, Hadley EB, Mecozzi S, Gellman SH. Stabilizing and destabilizing effects of phenylalanine → F5-phenylalanine mutations on the folding of a small protein. J Am Chem Soc. 2006;128:15932–1593. doi: 10.1021/ja0634573. [DOI] [PubMed] [Google Scholar]
- 9.Zheng H, Comeforo K, Gao J. Expanding the fluorous arsenal: tetrafluorinated phenylalanines for protein design. J Am Chem Soc. 2009;131:18–19. doi: 10.1021/ja8062309. [DOI] [PubMed] [Google Scholar]
- 10.Packer LE, Song B, Raleigh DP, McKnight CJ. Competition between intradomain and interdomain interactions: a buried salt bridge is essential for villin headpiece folding and actin binding. Biochemistry. 2011;50:3706–3712. doi: 10.1021/bi1020343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santoro MM, Bolen DW. A test of linear extrapolation of unfolding free energy changes over an extended denaturation concentration range. Biochemistry. 1992;31:4901–4907. doi: 10.1021/bi00135a022. [DOI] [PubMed] [Google Scholar]
- 12.Grimsley GR, Huyghues-Despointes BMP, Pace CN, Scholtz JM. Measuring the conformational stability of a protein in purifying proteins for proteomics: a laboratory manual. Cold Spring Harbor Laboratory Press; 2004. Chapter 23; pp. 535–566. [Google Scholar]
- 13.Pace CN, Fu H, Fryar KL, Landua J, Trevino SR, Shirley BA, Hendricks MM, Iimura S, Gajiwala K, Scholtz JM, Grimsley GR. Contributions of hydrophobic interactions to protein stability. J Mol Biol. 2011;408:514–528. doi: 10.1016/j.jmb.2011.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vardar D, Chishti AH, Frank BS, Luna EJ, Noegel AA, Oh SW, Schleicher M, McKnight CJ. Villin-type headpiece domains show a wide range of F-actin-binding affinities. Cell Motil Cytoskeleton. 2002;52:9–21. doi: 10.1002/cm.10027. [DOI] [PubMed] [Google Scholar]
- 15.Frank BS, Vardar D, Chishti AH, McKnight CJ. The NMR structure of dematin headpiece reveals a dynamic loop that is conformationally altered upon phosphorylation at a distal site. J Biol Chem. 2004;279:7909–7916. doi: 10.1074/jbc.M310524200. [DOI] [PubMed] [Google Scholar]
- 16.Brown JW, Vardar-Ulu D, McKnight CJ. How to arm a supervillin: designing f-actin binding activity into supervillin headpiece. J Mol Biol. 2009;393:608–618. doi: 10.1016/j.jmb.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang ZG, McKnight CJ. A phosphorylation-induced conformation change in dematin headpiece. Structure. 2006;14:379–387. doi: 10.1016/j.str.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Vugmeyster L, McKnight CJ. Phosphorylation-induced changes in backbone dynamics of the dematin headpiece C-terminal domain. J Biomol NMR. 2009;43:39–50. doi: 10.1007/s10858-008-9289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 20.Abyzov A, Errami M, Leslin CM, Ilyin VA. Friend, an integrated analytical front-end application for bioinformatics. Bioinformatics. 2005;21:3677–3678. doi: 10.1093/bioinformatics/bti602. [DOI] [PubMed] [Google Scholar]
- 21.Ilyin VA, Abyzov A, Leslin CM. Structural alignment of proteins by a novel TOPOFIT method, as a superimposition of common volumes at a topomax point. Protein Sci. 2004;13:1865–1874. doi: 10.1110/ps.04672604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 23.Heinz DW, Baase WA, Dahlquist FW, Matthews BW. How amino-acid insertions are allowed in an α-helix of T4 lysozyme. Nature. 1993;361:561–564. doi: 10.1038/361561a0. [DOI] [PubMed] [Google Scholar]
- 24.Baase WA, Liu L, Tronrud DE, Matthews BW. Lessons from the lysozyme of phage T4. Protein Sci. 2010;19:631–641. doi: 10.1002/pro.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meng W, Shan B, Tang Y, Raleigh DP. Native like structure in the unfolded state of the villin headpiece helical subdomain, an ultrafast folding protein. Protein Sci. 2009;18:1692–1701. doi: 10.1002/pro.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fersht A. Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. New York: W.H. Freeman and Company; 2003. [Google Scholar]
- 27.Adams PD, Afonine PV, Gunkoczi G, Chen VB, Davis IW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.