

Abstract
Bacterial formyl-CoA:oxalate CoA-transferase (FCOCT) and oxalyl-CoA decarboxylase work in tandem to perform a proton-consuming decarboxylation that has been suggested to have a role in generalized acid resistance. FCOCT is the product of uctB in the acidophilic acetic acid bacterium Acetobacter aceti. As expected for an acid-resistance factor, UctB remains folded at the low pH values encountered in the A. aceti cytoplasm. A comparison of crystal structures of FCOCTs and related proteins revealed few features in UctB that would distinguish it from nonacidophilic proteins and thereby account for its acid stability properties, other than a strikingly featureless electrostatic surface. The apparently neutral surface is a result of a “speckled” charge decoration, in which charged surface residues are surrounded by compensating charges but do not form salt bridges. A quantitative comparison among orthologs identified a pattern of residue substitution in UctB that may be a consequence of selection for protein stability by constant exposure to acetic acid. We suggest that this surface charge pattern, which is a distinctive feature of A. aceti proteins, creates a stabilizing electrostatic network without stiffening the protein or compromising protein–solvent interactions.
Keywords: acid resistance, crystal structure, protein stability, substrate specificity
Introduction
Vinegar production requires acidophilic acetic acid bacteria (AAB) to cope with constant exposure to molar levels of a membrane-permeant weak acid.1 A consequence of habitual cytoplasmic acidification2 is selection for intrinsic acid resistance of Acetobacter aceti proteins, relative to “neutralophile” orthologs.3–5 The primary sequence of acidophile proteins encodes acid stability, just as the primary sequence of thermophile proteins encodes thermal stability. Crystal structures of A. aceti proteins reveal unusual surface electrostatic properties that may have a causal link to acid-resistance properties.3, 4, 6
Acid-resistant and acidophilic microbes deploy overlapping response systems for generalized and specific acid resistance. Formyl-CoA:oxalate CoA-transferase (FCOCT) and oxalyl-CoA decarboxylase (OXC) have an essential role in oxalate assimilation by Oxalobacter formigenes,7 but a less well-defined role in other bacteria.8 Together, FCOCT and OXC mediate the proton-consuming conversion of oxalate to CO2 and formate.9, 10 A role in acid resistance is plausible, given that other decarboxylases function as countermeasures against bacterial acid stress.11 Oxalate catabolic enzymes are induced by acid in other oxalate consumers12, 13 and possibly other bacteria.14, 15
Acidophiles contain specific acid-resistance factors, like membrane pumps or the aarABC variant citric acid cycle.16, 17 The latter process relies on the conversion of acetate to acetyl-CoA by the aarC gene product, succinyl-CoA:acetate CoA-transferase (SCACT).17 The catabolic disposal of membrane-permeant carboxylic acids, each activated by a particular CoA-transferase, may have a broader role in conferring acid resistance phenotypes in the acidophilic AAB.
FCOCT is the gene product of O. formigenesfrc,18Escherichia coliyfdW,19 and, as we confirm here, A. acetiuctB. UctB is overproduced during acetic acid production. Like other A. aceti proteins, UctB has intrinsic acid resistance sufficient for it to remain folded during periods of cytoplasmic acid stress. The surface of the protein has strikingly large regions of near-zero electrostatic potential, in contrast to close structural relatives from nonacidophilic organisms. We suggest that the unusual A. aceti UctB surface enhances protein acid stability to the extent required by a consistently acidic cytoplasm.
Results
Expression of uctB
UctB was first encountered as a contaminant in a native A. aceti alanine racemase preparation.5 Edman sequencing of a prominent 47 kDa band gave GTTSENSKP LDGIKVIDFG GVQ, which matches UctB Gly2–Gln23. A portion of this sequence was used to elicit an UctB-specific antibody.
UctB levels were examined during the A. aceti logarithmic growth phase, which is associated with aerobic ethanol oxidation and the subsequent stationary phase. (Some AAB have a second growth phase that is associated with acetate oxidation.20, 21) The initial ethanol level determines the acetic acid level, and presumably the degree of acid stress, in the first stationary phase. At acetic acid levels above ∼5%, continuous aeration is particularly important for cell viability.22 Western blots with the anti-UctB antibody show two closely spaced bands (47 and 48 kDa), along with a 59 kDa band (Fig. 1). As the ethanol level increases, the amount of the 48 kDa band corresponding to UctB decreases in the log phase but increases in the stationary phase. (A similar pattern is also observed for the 59 kDa band. The 47 kDa band is most abundant in the stationary phase, at all ethanol levels examined.) These observations are consistent with a “housekeeping” role for UctB that escalates during acetic acid stress.
Figure 1.
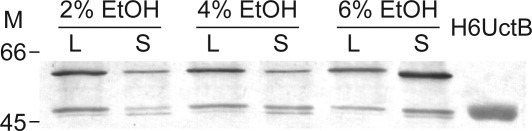
Western blot to detect protein expression in A. aceti strain 1023 cell lysates, using a primary antibody raised against a peptide with the UctB N-terminal sequence. The bands visualized correspond to UctB (two closely spaced bands at 47 and 48 kDa) and an unidentified 59 kDa protein. Lanes marked with an L are from the first logarithmic growth phase, associated with the conversion of ethanol to acetic acid. Lanes marked with an S were harvested during the first stationary phase. The initial ethanol level of the YPDE cultures is indicated above each pair of lanes. A lane containing pure H6UctB (2 kDa larger than native UctB) was used as a standard. Each lane was loaded with 5 μg total protein. M indicates the position of size standards (in kDa).
Isolation and characterization of H6UctB and H6YfdW
Plasmids containing uctB17 were used to overproduce large amounts of soluble, recombinant UctB, or N-terminally hexahistidine-tagged UctB (H6UctB). H6UctB was isolated in good yield (34 mg from a 3 g cell pellet) and high purity by immobilized-metal affinity chromatography. The protein did not aggregate at concentrations up to 70 mg mL−1 (∼0.7 mM dimer). A single peak observed by analytical size-exclusion chromatography was most consistent with a dimeric solution form of H6UctB (83 kDa observed, 99 kDa expected).
As anticipated from sequence comparisons, H6UctB has FCOCT activity. The derived kinetic parameters (Table I) closely resemble those determined for O. formigenes FRC but not E. coli YfdW.19, 23 YfdW displays substrate inhibition by oxalate, unlike FRC. The observation that oxalate inhibits the Trp48 → Gln mutant of FRC indicated that a Gln present at this position in YfdW might account for substrate inhibition.19 However, H6UctB also contains a Gln at the same position (Gln54) but exhibits no inhibition to 50 mM oxalate. We conclude that the identity of the residue at this position is not sufficient to induce substrate inhibition by oxalate in all FCOCTs.
Table I.
Kinetic Constants
| Substrate | Parameter | Valuea |
|---|---|---|
| Oxalateb | kcat | 5.00 ± 0.09 s−1 |
| Km | 1.11 ± 0.09 mM | |
| kcat/Km | (4.5 ± 0.4) × 103 M−1 s−1 | |
| Formyl-CoAc | kcat | 4.5 ± 0.1 s−1 |
| Km | 3.4 ± 0.4 μM | |
| kcat/Km | (1.3 ± 0.2) × 106 M−1 s−1 |
Assuming one active site per 49610 Da subunit (−Met1), 1.0 unit mg−1 corresponds to a turnover number of 0.83 s−1.
Determined at 0.1 mM formyl-CoA.
Determined at 50 mM oxalate.
A set of potential alternative carboxylate substrates (listed in the Experimental section) was screened using formyl-CoA as cosubstrate. Only succinate produced a peak with a retention time different from oxalyl-CoA, but the specific activity (5.9 × 10−3 units mg−1) was 1000-fold lower than that supported by oxalate. YfdW was more active with succinate and succinyl-CoA as alternate substrates.19 Both H6UctB and YfdW are much more stringent than FRC, which actually prefers succinate over oxalate with formyl-CoA as cosubstrate (and succinyl-CoA with formate). Class III CoA-transferases, including FCOCT, simultaneously accommodate both acid/acyl substrates,24, 25 indicating that each substrate can influence specificity for the other substrate.
YfdW with the same N-terminal hexahistidine tag as H6UctB (H6YfdW) was purified in good yield (37 mg from an 8 g cell pellet) and high purity by immobilized-metal affinity chromatography. The specific activity of H6YfdW with formyl-CoA and oxalate was 10 units mg−1, which is comparable to a value reported under similar conditions with a different N-terminally hexahistidine-tagged YfdW fusion protein.19
Electrospray ionization mass spectrometry (ESI-MS) analysis was consistent with the expected molecular masses for both fusion proteins (minus Met1): 49,610 Da observed (49,610.1 Da expected) for H6UctB and 47,860 Da observed (47,860.3 Da expected) for H6YfdW.
Acid stability of H6UctB and H6YfdW
The acid resistance properties of H6UctB were assessed using circular dichroism (CD) to monitor thermal unfolding as a function of pH. Melting temperature (Tm) values (Fig. 2) showed no diminution of protein stability as the pH was lowered to 4.5. Destabilization was noted at pH 3.5, which is slightly below the lowest pH encountered in A. aceti cytoplasm.2 The artificial N-terminal appendage is likely to be a contributing factor, as it contains a His6 sequence that is expected to be positively charged at the lowest pH values examined. Destabilization of a His6-tagged enzyme relative to an untagged form has been previously observed at a similar pH.4 UctB is stable at all pH and temperature (≤35°C) conditions encountered in the A. aceti cytoplasm.
Figure 2.
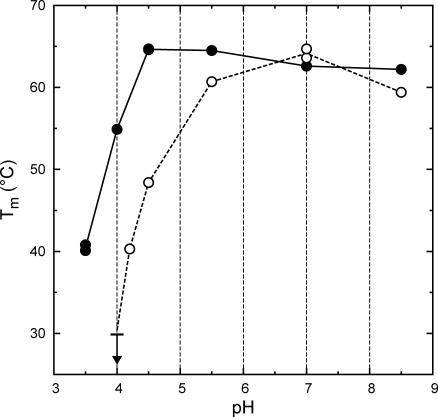
Thermal denaturation of H6UctB (filled circles) and H6YfdW (open circles) monitored by CD as a function of pH. Tm values were determined using changes in molar ellipticity at 222 nm, corresponding to loss of helical structure. The lines connect average Tm values at each pH for H6UctB (solid line) and H6YfdW (dotted lines); H6UctB duplicates at pH 4.5 and 7.0 have completely overlapping symbols. The arrow at pH 4.0 for H6YfdW indicates a maximum for a partially observed melting transition. At pH 3.5, H6YfdW melted completely during equilibration at 20°C. Full CD spectra (recorded at 20°C) and thermal unfolding profiles are given as Figures S4–7 of the Supporting Information.
An identically tagged form of the characterized FCOCT with the highest sequence identity to UctB, H6YfdW, was examined in parallel thermal unfolding experiments. Thermal stabilities were comparable at neutral pH, but H6YfdW is progressively less stable than H6UctB at pH values <5.5 (Fig. 2). Tm values could not be determined for H6YfdW at pH 3.5 (completely unfolded at 20°C) or pH 4.0 (completely unfolded at <30°C, downwards arrow in Fig. 2). The difference in acid stabilities is likely to be due to the ∼30% of the protein sequence that is not identical in UctB and YfdW.
Crystal structure of H6UctB
A crystal structure of H6UctB bound to CoA was solved by molecular replacement (Table II) and refined to a model with good geometry and refinement statistics (Table III). The single Ramachandran outlier, Glu321 in subunit B, has ambiguous side-chain density and is located at a sharp turn between helices α13 and α14. The model extends to the C-terminus (Ala436) in all subunits. The N-terminal appendage and residues 1–6 were not located in any subunit.
Table II.
Summary of X-ray Data Collection Statistics
| H6UctB·CoAa | |
|---|---|
| Space group | P212121 |
| Unit-cell parameters | a = 98.16 Å, b = 121.61 Å, c = 161.38 Å |
| α = β = γ = 90° | |
| Resolution (Å) | 39.64–1.99 (2.10–1.99) |
| Wavelength (Å) | 1.24 |
| No. reflections | 871,173 (137,403) |
| No. unique reflections | 116,953 (17,212) |
| Completeness (%) | 89.1 (90.4) |
| Multiplicity | 7.4 (8.0) |
| Rmergeb | 0.124 (0.397) |
| Rmeasc | 0.134 (0.423) |
| Mean I/σ(I) | 10.3 (5.2) |
| Wilson B factor (Å2) | 21.74 |
| Matthews coefficient (Å3 Da−1) | 2.43 |
Values in parentheses are for the outermost shell.
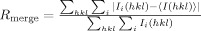 , where Ii(hkl) is the ith measurement of the intensity of reflection hkl and 〈I(hkl)〉 is its mean value.
, where Ii(hkl) is the ith measurement of the intensity of reflection hkl and 〈I(hkl)〉 is its mean value.
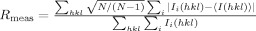 is the multiplicity-weighted Rmerge.26
is the multiplicity-weighted Rmerge.26
Table III.
Summary of Refinement Statistics
| H6UctB·CoAa | |
|---|---|
| Resolution (Å) | 39.64–1.99 (2.01–1.99) |
| No. reflections used | 116,882 (3546) |
| Completeness (%) | 88 (81) |
| Rcrystb (%) | 19.6 (25.9) |
| Rfreec (%) | 24.3 (34.6) |
| Protein atoms | 13,206 |
| CoA atoms | 192 |
| Solvent atoms | 983 |
| R.M.S. deviations from ideality | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.04 |
| Average isotropic B factors (Å2) | |
| Main chain | 21.7 |
| Side chains | 24.7 |
| CoA | 28.4 |
| Solvent | 26.1 |
| Ramachandran plot statistics (%) | |
| Residues in favored regions | 96.96 |
| Residues in allowed regions | 2.98 |
| Outliers | 0.06 |
| MolProbity score [percentile] | 2.01 [73] |
| MolProbity clashscore [percentile] | 12.23 [71] |
Values in parentheses are for the outermost shell.
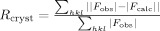 .
.
Rfree is Rcryst with a test set of 5% of the reflections excluded from refinement.
H6UctB has a very similar backbone topology to FRC and YfdW, including a large domain with an N-terminal Rossmann fold, a small αβ domain, and the same interlocking dimer structure (Fig. 3A).27, 28 The large and small domains from different monomers contribute to each active site. The asymmetric unit contains four subunits, corresponding to two dimers. The extensive monomer–monomer interface buries 6300 Å2 of accessible surface area per subunit whereas the dimer–dimer interface buries 780 Å2 per dimer. H6UctB contains a region without canonical secondary structure elements that is not present in related enzymes (residues 163–176, “insert” in Supporting Information, Fig. S8). Together with its pseudo-symmetry mate, this insertion forms part of the monomer–monomer interface between the small domains (bottom of the dimer illustrated in Fig. 3A). In FRC, the same volume is occupied by a different loop (residues 233–243). Neither long loop is present in YfdW, which has a recessed surface in this region. The additional intersubunit contacts may stabilize the dimer of H6UctB (and FRC).
Figure 3.
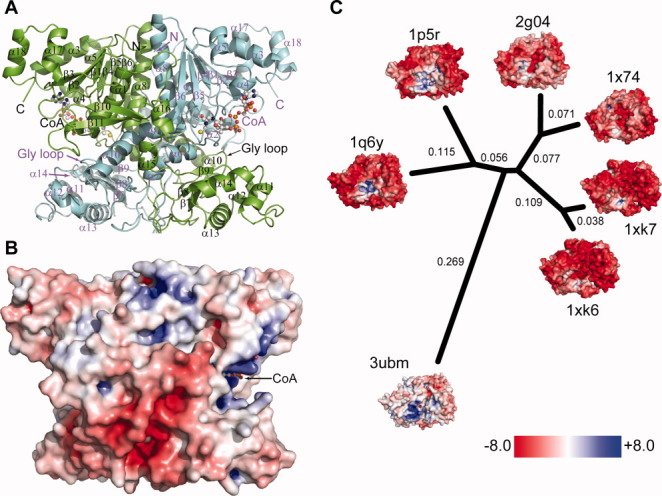
Structure of H6UctB bound to CoA (PDB id 3ubm). A: Elements from both subunits make up the H6UctB active sites, each occupied by CoA (ball-and-stick rendering). Secondary structure elements are labelled; a topology diagram is provided as Figure S8 in the Supporting Information. B: Electrostatic surface potential of H6UctB (subunits A/B) viewed in the same orientation as panel A. Positive and negative regions were scaled as in panel C. Additional orientations are shown in Supporting Information Figure S13. C: Comparison of charge decoration in a set of protein structures with very similar backbone topologies. The indicated phylogenetic tree was generated using Euclidean distances between the 3DZDs. The numbers in black are the computed distance of corresponding branches and the branch lengths are proportional to the distances between the proteins. The PDB identifiers and DALI Z scores are 1q6y, E. coli YfdW, CoA complex (Z = 49.2)30; 1p5r, O. formigenes FRC, CoA complex (Z = 46.6)28; 1xk6 and 1xk7, E. coli CaiB in two crystal forms (Z = 34.2 and 33.8, respectively); 1x74, Mycobacterium tuberculosis α-methyl-CoA racemase (Z = 29.5); and 2g04, M. tuberculosis fatty acid-CoA racemase (Z = 28.7). The thumbnail figures are rotated to show the positively-charged (acyl-)CoA binding pocket.
In the active site region, the CoA thiol(ate) and the Gly-rich loop in subunits B and D have somewhat higher B-factors than surrounding regions, consistent with their dynamic roles in catalysis. The Gly loop (G265GGGQ269) is proposed to shield enzyme-bound anhydride intermediates.25 The Gly loop of H6UctB is even farther from the site of covalent catalysis (Asp188) than in the “open” conformation defined for the FRC aspartyl-CoA thioester adduct (PDB ids 2vjk and 2vjl). H6UctB also lacks the chloride ion(s) present in some FRC active sites.
H6UctB has a larger relative volume and lower packing efficiency than YfdW and FRC that may indicate an increased number of side chain–solvent interactions (Supporting Information, Table SII). This was unexpected as packing efficiency is one mechanism by which proteins are thought to gain stability, especially in thermophiles.29
Electrostatic surfaces of FCOCTs and structural relatives
The surface properties of H6UctB (Fig. 3B) were compared to a set of proteins that have closely related backbone topologies and dimer shapes. Qualitatively, the surface charge decoration of H6UctB is readily distinguished from homologs by having few highly charged regions apart from the basic CoA binding pocket. (CoA was deleted before electrostatic analysis of the three FCOCTs.) The difference in appearance is all the more striking because the three FCOCTs have such similar amino acid compositions: YfdW is 70% identical and FRC is 60% identical in structure-based sequence alignments (Supporting Information, Fig. S9). A comparatively neutral surface appearance also comports with the behavior of native UctB, which passes through anion exchange columns at low ionic strength, like many other A. aceti proteins (data not shown).
Electrostatic surface potentials were described with three-dimensional Zernike descriptors (3DZDs), which is a projection-based encoding method for describing 3D properties such as protein surfaces.31–33 Sael et al.32 showed that 3DZDs can be used to quantitatively evaluate differences in the electrostatic potential of the surface of proteins. Distances between the proteins were then calculated as Euclidian distances between the 3DZD that describes the pattern of electrostatic potentials on each protein surface (Fig. 3C). By this analysis, H6UctB diverges as much from the other FCOCT enzymes as it does from proteins that perform different enzymatic reactions.
We compared H6UctB with the two other FCOCTs, YfdW and FRC, to identify possible reasons for its unusual appearance. Similarities in sequence, amino acid composition, predicted isoelectric points (Table IV), numbers of hydrogen bonds, numbers of salt bridges, and surface areas associated with charged, polar, or neutral regions (Supporting Information, Table SI) rule out several straightforward explanations for the “more neutral” appearance of H6UctB. A similar number of charged surface residues are present in the three FCOCTs, but H6UctB has a higher computed isoelectric point. Many of the charged groups that could account for this difference map to 16 surface residues that are the same (or have the same charge status) in FRC and YfdW, but are different in H6UctB (Supporting Information, Table SII). In this subset of surface residues, a higher proportion of H6UctB side chains extend outward from the protein surface to make polar or charged interactions with solvent and not other protein atoms. These residues are distributed widely on the surface of the protein, primarily in the large domain, and have no obvious role in catalysis or monomer–monomer interactions (Supporting Information Fig. S10). The neutral-appearing regions of the H6UctB surface are the result of well-dispersed charged side chains that are near, but do not directly contact, residues of the opposite charge. This architecture appears to explain why there are few regions of high electrostatic potential on the surface of H6UctB.
Table IV.
Differential Analysis of Charged Surface Residues
| H6UctB | YfdW | FRC | |
|---|---|---|---|
| 3ubm | 1q6y | 1p5r | |
| Total residues in dimer | 872 | 832 | 856 |
| Surface residues in dimera | 526 | 498 | 503 |
| No. acidic residues (Asp & Glu) | 93 | 94 | 103 |
| No. basic residues (Arg, His & Lys) | 85 | 76 | 90 |
| No. bases–no. acids | −8 | −18 | −13 |
| No. of altered side chain chargesb | 32 | 32 | 32 |
| No. making polar protein contactsc | 8 | 12 | 16 |
| No. of charged residues | 14 | 18 | 18 |
| No. bases–no. acids | +6 | −14 | −10 |
| Computed isoelectric point | |||
| Compositiond | 5.81 | 5.36 | 5.26 |
| Folded proteine | 6.60 | 5.46 | 5.43 |
Residues with relative side chain accessible surface area of ≥5% in chains A and B, computed using NACCESS.
Surface residues that have different charges in 3ubm than in 1q6y/1p5r. A complete residue list is given in Supporting Information Table SII.
Residues with side chains that make polar contacts to other side chains, backbone, or ligand. The other residues appear to make polar contacts only to solvent water.
Computed using a tool at the EXPASY server.
Computed using PROPKA.
Discussion
The idea that electrostatic interactions among protein surface residues are an important determinant of protein stability has attracted broad experimental and theoretical support and has emerged as a design principle of protein engineering.34–37 Extremophilic organisms encounter strong selective pressures to stabilize proteins in an environmentally appropriate manner. Prolonged exposure to membrane-permeant weak acids has compelled acidophilic bacteria to improve the acid stability of cytoplasmic proteins. Crystallographic analysis of H6UctB revealed a distinctive electrostatic surface featuring a network of balanced long-range Coulombic interactions. We suggest that this electrostatic architecture accounts for the increased stability at acidic pH of H6UctB from the acidophile A. aceti relative to H6YfdW from the neutralophile E. coli.
Maintaining a stable network of favorable surface electrostatic interactions over a range of pH values is important in the A. aceti cytoplasm, which has low but variable pH.2 Relative to neutral (apolar or polar) residues, charged side chains interact strongly with water, which favors protein solubility and disfavors protein aggregation. Charged side chain-solvent interactions should be maintained at lower pH values in H6UctB, relative to YfdW or FRC, because the larger number of positive surface charges creates a local electrostatic environment that stabilizes negative surface charges. We speculate that a charged surface may also act as a kinetic barrier against penetration by charged species (e.g., hydronium ions) that might otherwise contribute to acid-mediated protein unfolding.
Thermophiles may counteract thermal protein unfolding with improved hydrophobic packing or an increase in the proportion of hydrogen bonds and salt bridges, which increases the mechanical rigidity of proteins at mesophilic temperatures.38 Decreasing protein flexibility is unsuitable for A. aceti, which grows poorly at temperatures above 35°C.39
A structural comparison of the A. aceti and E. coli orthologs of citrate synthase, PurE, and FCOCT (UctB/YfdW) reveals parallel differences in surface charge decoration.3, 4 All three A. aceti proteins have fractional charged surface areas comparable to E. coli equivalents and higher predicted isoelectric points.3, 4 A. aceti PurE and FCOCT are substantially more stable than E. coli orthologs.3 Among the small set of A. aceti proteins studied, thioredoxin provides the only clear instance of improved catalytic activity at low pH;40, 41 other proteins only show increased stability at low pH.3–5
Conclusion
A. aceti UctB is induced by acid stress and is impervious to all cytoplasmic acid levels encountered by the organism. As a dedicated FCOCT, its most likely role is in the generalized acid-resistance system that couples proton removal to oxalyl-CoA decarboxylation. Prominent surface regions of low electrostatic potential are a consequence of a speckled, granular distribution of oppositely charged residues. This structural motif, found in UctB and other A. aceti proteins, correlates with enhanced acid stability.
Experimental Procedures
Materials and methods
All reagents were obtained from Sigma–Aldrich or Fisher Scientific unless otherwise noted. Oligodeoxynucleotides obtained from IDT (Coralville, IA) were used without further purification. Protein A Sepharose 4 Fast Flow was obtained from Amersham Biosciences. Protein concentrations were measured using a Bradford assay kit (Bio-Rad) with crystalline bovine serum albumin as the standard.42 N-terminal sequencing by Edman degradation of proteins blotted onto polyvinylidene fluoride (PVDF) membranes (Immobilon P) was performed by Midwest Analytical (St. Louis, MO). Mass spectrometric analyses were performed by the Washington University Mass Spectrometry Resource.
Custom antisera were prepared by Sigma–Genosys using rabbits immunized with the peptide GTTSENSKPLD conjugated by a C-terminal Cys to key limpet hemocyanin. The ELISA result on day 63 was 1:300,000. ImmunoPure goat antirabbit IgG alkaline phosphatase antibody (Pierce) was used as a secondary antibody.
Formyl-CoA was synthesized using a slight modification of a published method, in which the final preparative HPLC purification step was omitted.23 HPLC analysis showed peaks for CoA, isoCoA (the 2′-phospho isomer of CoA),43 formyl-CoA, and formyl-isoCoA, typically in a 10:1:23:2 ratio after standing in the quench solution. The isoCoA and formyl-CoA peaks usually overlapped (Supporting Information, Fig. S1). Negative-ion mode ESI-MS showed a large peak at 794.1033 (794.1028 expected for the formyl-CoA monoanion) and a smaller peak at 766.1087 (766.1079 expected for the CoA monoanion).
Analysis of UctB levels in A. aceti
A. aceti strain 1023 was obtained from Dr. Koichi Kondo (Mizkan Group, Aichi, Japan).39, 44A. aceti was propagated at 30°C on yeast extract–peptone–dextrose (YPD) medium supplemented with 2.5% (v/v) ethanol (YPDE). After 2 d, a single colony was used to inoculate a 50 mL YPDE culture. After overnight growth with agitation at 35°C, the starter culture was used to inoculate YPD supplemented with 2%, 4%, or 6% ethanol (200 mL) in 1 L Erlenmeyer flasks. These cultures were agitated at 220 rpm for 12–14 h at 35°C. Aliquots (10 mL) were withdrawn during the first mid-logarithmic (OD600 = 0.35–0.4) and steady-state (OD600 = 0.7–0.8) growth phases. Cells were harvested by centrifugation at 3000g for 20 min at 4°C, resuspended in 1 mL 50 mM Tris•HCl, pH 8.0, and 100 mM KCl, and disrupted by sonication. Soluble cell lysates (5 μg protein) were blotted onto PVDF and cross-reacting proteins were visualized using 5-bromo-4-chloro-3-indolyl phosphate and nitroblue tetrazolium as described.45
Cloning and purification of H6UctB
Genomic DNA was isolated from an A. aceti cell pellet (1 g; Promega Wizard Genomic DNA purification kit). PCRs were performed using A. aceti genomic DNA template; oligodeoxynucleotides 1022 (5′-CTG GTA AGC TTA GTC TCT CAA GAT GCA GTC) and 1168 (5′-AGG TAG ACA TAT GGG CAC GAC), which introduce HindIII and NdeI sites (bold text), respectively; and Taq DNA polymerase in MBP HotStart Micro 100 PCR tubes. A 1.4 kb PCR product was cloned into the NdeI and HindIII sites of pET23a (Novagen) to create UctB expression vector pJK286 or pET28a (Novagen) to create H6UctB expression vector pJK287. The DNA sequence of pJK286 corresponded to the deposited sequence (GenBank™ accession number DQ668372). However, the DNA sequence of pJK287 contained two silent mutations: Pro138 (CCA → CCC) and Val376 (GTT → GTC). H6UctB contains an additional 20 amino acids (MGSSHHHHHHSSGLVPRGSH) appended to the N-terminus.
A starter culture of E. coli BL21(DE3) transformed with pJK287 was grown 18 h at 37°C in LB medium supplemented with 70 mg L−1 kanamycin. Production cultures (1 L of the same medium) in 2.8 L Fernbach flasks were inoculated with 20 mL of starter culture and grown at 37°C to mid-log phase (OD600 = 0.6). Protein overproduction was induced by the addition of isopropyl β-d-1-thiogalactopyranoside to 0.4 mM. After an additional 4–5 h growth period, cells (typically 7 g L−1 culture) were harvested by centrifugation at 6000g and either used immediately or stored at −80°C. All subsequent steps were performed at 4°C.
Cells were resuspended in TK buffer (20 mM Tris·HCl, pH 8.0, and 100 mM KCl) at 5 mL per g and disrupted by three cycles of sonication. After removing cell debris by centrifugation (27,000g for 30 min), streptomycin sulfate was added to a final concentration of 1% from a 10% (w/v) stock solution and the solution was stirred another 10 min. After removing solids by centrifugation, the supernatant was applied to an iminodiacetate–Ni2+ Sepharose column (2.5 × 5.0 cm), which was washed with eight column volumes of TK buffer containing 20 mM imidazole. The column was then developed in a linear gradient of imidazole (20–500 mM, 120 × 120 mL) in TK buffer. Fractions containing pure H6UctB were pooled (ca. 60 mL) and concentrated by ultrafiltration over a YM10 membrane. The concentration of imidazole was reduced to <10 mM by several cycles of dilution–reconcentration in TK buffer. Small single-use aliquots were stored at −80°C.
Cloning and purification of H6YfdW
Genomic DNA was isolated from an E. coli BL21(DE3) cell pellet (0.02 g; Qiagen Genomic-tip 20/G). PCR was performed using the genomic DNA template; oligodeoxynucleotides 2229 (5′-GGT ATT CAT ATG TCA ACT CCA CTT CAA GG) and 2230 (5′-CCC CGT TGA ATT CAG ATG GCG TGG TTT TGC TTC ATT GC), which introduce NdeI and EcoRI sites (bold text), respectively; and Vent DNA polymerase. A 1.3 kb PCR product was cloned into the NdeI and EcoRI sites of pET28a (Novagen) to create H6YfdW expression vector pJK597, which encodes the same 20 residue N-terminal sequence present in H6UctB fused to the N-terminus of YfdW. The DNA sequence in the coding region matches GenBank™ accession number YP_003035548. However, during cloning 0.66 kb from the vector 3′ untranslated region (a region that includes the T7 terminator, F1 origin, and M13 origin) was deleted by an unknown mechanism.
E. coli BL21(DE3) cells transformed with pJK597 were grown and processed as described for E. coli BL21(DE3)/pJK287. The poststreptomycin supernatant was applied to an nitrilotriacetate–Ni2+ agarose column (1.0 × 3.0 cm), which was washed with 10 column volumes of TK buffer containing 20 mM imidazole. The column was then developed in a linear gradient of imidazole (20–500 mM, 30 × 30 mL) in TK buffer. Fractions containing pure H6YfdW were pooled (ca. 20 mL) and concentrated by ultrafiltration over a YM10 membrane. The concentration of imidazole was reduced to <10 mM by several cycles of dilution–reconcentration in TK buffer. Single-use aliquots were stored at −80°C.
Kinetic characterization of H6UctB and H6YfdW
Methods for acyl-CoA preparation and analysis were designed to minimize spontaneous formyl-CoA hydrolysis (half-life of about 2 h in quenched reaction mixtures; E.A. Mullins, unpublished observations). FCOCT activity was monitored using a discontinuous HPLC assay to quantitate the conversion of formyl-CoA to oxalyl-CoA. Reaction mixtures (25°C, final volume 0.5 mL) contained 50 mM potassium phosphate, pH 6.7, 0.5–100 μM formyl-CoA, and 0.15–50 mM sodium oxalate, with 20–100 ng H6UctB used to initiate the reaction. After 5 min, an aliquot of the reaction mixture (oxalate saturation, 0.1 mL; formyl-CoA saturation, 0.225 mL) was quenched by the addition of trichloroacetic acid to 5% (w/v; oxalate saturation, 0.4 mL of a 6.25% solution; formyl-CoA saturation, 25 μL of a 50% solution), vortexed briefly, and centrifuged at 16,100g for 3 min. The soluble portion was analyzed by HPLC. No-enzyme controls were processed in the same manner. The unusual formyl-CoA quenching conditions were developed to accommodate a low Km value and to keep substrate conversion under 25%.
H6YfdW was assayed using the above procedure at 100 μM formyl-CoA and 50 mM sodium oxalate with 100 ng H6YfdW.
CoA and acyl-CoA thioesters were resolved on a Waters Breeze HPLC system equipped with a Symmetry C18 column (4.6 × 75 mm, 3.5 μm) in a column heater. Samples (0.1–0.2 mL) were injected using an autosampler (Waters 717plus). Elution was performed at 30°C and a flow rate of 0.8 mL min−1 with detection at 260 nm. A mixture of 200 mM sodium phosphate and 150 mM sodium acetate, pH 4.6, was used in a water/methanol solvent system. The gradient program was 0–5 min, 0% methanol (isocratic); 5–15 min, 0 to 15% methanol (linear gradient); 15–17 min, 15% methanol (isocratic); 17–22 min, 15–0% methanol (linear gradient); 22–27 min, 0% methanol (isocratic). Acyl-CoAs and CoA were quantitated by integrated peak areas referenced to an acetyl-CoA standard (ε260 = 16.4 mM−1 cm−1).46
Alternate acid substrates were screened in a reaction mixture containing 50 mM carboxylate (acetate, glycolate, glyoxylate, l-lactate, malonate, oxamate, propionate, pyruvate, or succinate), 0.1 mM formyl-CoA, 50 mM potassium phosphate, pH 6.7, and 13.9 μg H6UctB. Aliquots were removed for HPLC analysis before the addition of enzyme and 5 and 30 min after the addition of enzyme. HPLC analysis was performed as described above except the initial and final isocratic phases of reactions containing propionate, l-lactate, or pyruvate used 4.5% methanol. A new peak was noted in glyoxyate reaction mixtures, either with or without enzyme addition; this peak was not identified.
For the determination of FCOCT kinetic parameters, HPLC injections were performed 7 min after quenching the reaction mixture to minimize the nonenzymatic hydrolysis of formyl-CoA in the quenching solution. Enzyme activity determinations were based on the peak area of oxalyl-CoA, which has a long half-life (29 h; E.A. Mullins, unpublished observations) relative to the time spent in quenching buffer. Enzyme kinetic parameters were obtained by nonlinear least-squares fitting to the Michaelis–Menten equation using gnuplot 4.4. No attempt was made to account for competitive inhibition by CoA. A unit is defined as the amount of enzyme that forms 1 μmol of oxalyl-CoA product per minute under the specified conditions.
Biophysical characterization of H6UctB and H6YfdW
The solution form of H6UctB in 50 mM Tris•HCl, pH 8.0, and 100 mM KCl was determined using a Superdex 200 column (1.6 × 60 cm) that was developed at 1 mL min−1 and calibrated with Sigma MW-GF-1000 standards as previously described.5
Thermal unfolding associated with changes in the molar ellipicity at 222 nm was detected with a Chirascan spectropolarimeter (Applied Photophysics). A 10-mm pathlength quartz cuvette containing 50 mM potassium phosphate (pH values ranging from 3.5 to 8.5), 100 mM KCl, and either 26 μg mL−1 H6UctB or 24 μg mL−1 H6YfdW (each 0.5 μM subunits) was equilibrated for 30 min at 20°C before recording a spectrum (average of three scans). The sample was then heated from 20 to 90°C at a continuous rate of 0.5°C min−1. Solution temperature was detected using a probe inserted through the cuvette stopper. Melting profiles were fit to a double-sigmoid equation implemented within the Pro-Data Viewer software (Applied Photophysics) and the steepest part of each curve was taken to be Tm. Melting profiles for H6UctB obtained at pH 3.5 and H6YfdW obtained at pH 4.0 and 4.2 were truncated at 47°C and 60°C, respectively, before fitting. Thermal unfolding was apparently irreversible for both proteins at all pH values tested. The postexperiment cuvette cleaning procedure included a 12 h soak in concentrated HNO3. The cuvette was also silanized (AquaSil, Thermo Scientific) before each H6YfdW experiment to inhibit protein adhesion to the cuvette surface.
H6UctB crystallization, data collection, and processing
H6UctB was crystallized using the hanging-drop method with 1 μL protein (20 mg mL−1 in 0.3× TK buffer) mixed with an equal volume of well solution [24% (w/v) PEG 4000 and 100 mM bicine, pH 8.8]. Plate-like crystals that formed over 4 weeks were soaked in well solution supplemented with 10 mM CoA for 5 h, briefly dipped in well solution containing 20% (v/v) ethylene glycol, and then flash-cooled in liquid nitrogen.47
Single-wavelength X-ray diffraction data were collected at the Advanced Light Source beamline 4.2.2 in two passes using different crystal orientations. Initial attempts to merge the two data sets using d*TREK48 failed. The indexed and integrated reflection file and processing header file from each data set were converted to CCP4 format49 using dtrek2scala and the merged mtz file was scaled with Scala.50 Data-collection statistics are shown in Table II.
Structure determination and refinement
The starting model for molecular replacement was a structure of E. coli YfdW (YfdW fused to a C-terminal His6 appendage that was not found in electron density maps; PDB id 1q6y).30 SEAMAN was used to remove regions with the greatest sequence differences, and to trim the remaining side chains to minimal conserved shapes.51 Molecular replacement was performed using MOLREP52 in spacegroup P212121 specifying that four monomers should be located in each asymmetric unit. Initial model refinement was performed with CNS53 using successive rounds of rigid body refinement, simulated-annealing refinement, grouped temperature-factor refinement, and model building with strict noncrystallographic symmetry (NCS) restraints. Torsion angle simulated-annealing refinement and individual temperature-factor refinement, alternating with model building in O, were performed in the absence of NCS restraints. Waters were added using CNS or ARP/wARP.54 CoA was added to each monomer and additional refinement was performed using Refmac55 with increased geometric restraint weighting. Final model adjustments and water additions were performed with COOT56 using PHENIX57 for refinement.
Computational analyses
Proteins for surface comparisons were selected among high-scoring DALI hits.58 The surface and the electrostatic potential of protein structures stripped of ligands were computed with APBS software (http://www.poissonboltzmann.org/apbs/) version 1.0.059 using a protein dielectric of 2.0, a solvent dielectric of 78.4, and a temperature of 298.15 Kelvin. Structural and surface32 renderings were created using PyMOL.60 Phylogenetic trees based on 3DZD values were generated with Phylip 3.66.61 Structure-based multiple sequence alignments assembled with PDBeFold/SSM were illustrated with ESPript.62, 63 Secondary structure assignments were made with the assistance of DSSP.64 Surface areas were computed using PyMOL. All subsequent analyses used the dimeric form of each protein as the input model, except where indicated. Relative sidechain surface accessible areas were computed using NACCESS version 2.1.1.65 Hydrogen-bonding interactions were analyzed using HBPLUS version 3.1566 or the PDBSUM server.67 Volumes and packing efficiencies were analyzed using VADAR.68 Protein isoelectric points were computed with PROPKA version 3.1.69 In some cases, missing sidechain atoms were inserted before PROPKA analysis using PDB2PQR version 1.7.1.70
Acknowledgments
The authors thank Kelly Sullivan and Stas Zakharov for help with gel filtration and CD experiments, Mark Hermodson and Barb Golden for comments on the manuscript, and Koichi Kondo for A. aceti strain 1023.
Glossary
Abbreviations
- AAB
acetic acid bacteria
- CD
circular dichroism
- CoA
coenzyme A
- FCOCT
formyl-CoA:oxalate CoA-transferase
- FRC
O. formigenes FCOCT
- H6UctB
UctB with an N-terminal hexahistidine tag
- H6YfdW
YfdW with an N-terminal hexahistidine tag
- SCACT
succinyl-CoA:acetate CoA-transferase
- UctB
A. aceti FCOCT
- YfdW
E. coli FCOCT.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Nakano S, Fukaya M. Analysis of proteins responsive to acetic acid in Acetobacter: Molecular mechanisms conferring acetic acid resistance in acetic acid bacteria. Int J Food Microbiol. 2008;125:54–59. doi: 10.1016/j.ijfoodmicro.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 2.Menzel U, Gottschalk G. The internal pH of Acetobacterium wieringae and Acetobacter aceti during growth and production of acetic acid. Arch Microbiol. 1985;143:47–51. [Google Scholar]
- 3.Constantine CZ, Starks CM, Mill CP, Ransome AE, Karpowicz SJ, Francois JA, Goodman RA, Kappock TJ. Biochemical and structural studies of N5-carboxyaminoimidazole ribonucleotide mutase (PurE) from the acidophilic bacterium Acetobacter aceti. Biochemistry. 2006;45:8193–8208. doi: 10.1021/bi060465n. [DOI] [PubMed] [Google Scholar]
- 4.Francois JA, Starks CM, Sivanuntakorn S, Jiang H, Ransome AE, Nam J-W, Constantine CZ, Kappock TJ. Structure of a NADH-insensitive hexameric citrate synthase that resists acid inactivation. Biochemistry. 2006;45:13487–13499. doi: 10.1021/bi061083k. [DOI] [PubMed] [Google Scholar]
- 5.Francois JA, Kappock TJ. Alanine racemase from the acidophile Acetobacter aceti. Protein Expr Purif. 2007;51:39–48. doi: 10.1016/j.pep.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 6.Settembre EC, Chittuluru JR, Mill CP, Kappock TJ, Ealick SE. Acidophilic adaptations in the structure of Acetobacter aceti N5-carboxyaminoimidazole ribonucleotide mutase (PurE) Acta Crystallogr D Biol Crystallogr. 2004;60:1753–1760. doi: 10.1107/S090744490401858X. [DOI] [PubMed] [Google Scholar]
- 7.Allison MJ, Dawson KA, Mayberry WR, Foss JG. Oxalobacter formigenes gen. nov., sp. nov.: oxalate-degrading anaerobes that inhabit the gastrointestinal tract. Arch Microbiol. 1985;141:1–7. doi: 10.1007/BF00446731. [DOI] [PubMed] [Google Scholar]
- 8.Svedružić D, Jónsson S, Toyota CG, Reinhardt LA, Ricagno S, Lindqvist Y, Richards NGJ. The enzymes of oxalate metabolism: unexpected structures and mechanisms. Arch Biochem Biophys. 2005;433:176–192. doi: 10.1016/j.abb.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 9.Baetz AL, Allison MJ. Purification and characterization of oxalyl-coenzyme A decarboxylase from Oxalobacter formigenes. J Bacteriol. 1989;171:2605–2608. doi: 10.1128/jb.171.5.2605-2608.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baetz AL, Allison MJ. Purification and characterization of formyl-coenzyme A transferase from Oxalobacter formigenes. J Bacteriol. 1990;172:3537–3540. doi: 10.1128/jb.172.7.3537-3540.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foster JW. Escherichia coli acid resistance: tales of an amateur acidophile. Nat Rev Microbiol. 2004;2:898–907. doi: 10.1038/nrmicro1021. [DOI] [PubMed] [Google Scholar]
- 12.Azcarate-Peril MA, Bruno-Bárcena JM, Hassan HM, Klaenhammer TR. Transcriptional and functional analysis of oxalyl-coenzyme A (CoA) decarboxylase and formyl-CoA transferase genes from Lactobacillus acidophilus. Appl Environ Microbiol. 2006;72:1891–1899. doi: 10.1128/AEM.72.3.1891-1899.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turroni S, Bendazzoli C, Dipalo SCF, Candela M, Vitali B, Gotti R, Brigidi P. Oxalate-degrading activity in Bifidobacterium animalis subsp. lactis: impact of acidic conditions on the transcriptional levels of the oxalyl coenzyme A (CoA) decarboxylase and formyl-CoA transferase genes. Appl Environ Microbiol. 2010;76:5609–5620. doi: 10.1128/AEM.00844-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masuda N, Church GM. Escherichia coli gene expression responsive to levels of the response regulator EvgA. J Bacteriol. 2002;184:6225–6234. doi: 10.1128/JB.184.22.6225-6234.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masuda N, Church GM. Regulatory network of acid resistance genes in Escherichia coli. Mol Microbiol. 2003;48:699–712. doi: 10.1046/j.1365-2958.2003.03477.x. [DOI] [PubMed] [Google Scholar]
- 16.Fukaya M, Takemura H, Okumura H, Kawamura Y, Horinouchi S, Beppu T. Cloning of genes responsible for acetic acid resistance in Acetobacter aceti. J Bacteriol. 1990;172:2096–2104. doi: 10.1128/jb.172.4.2096-2104.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mullins EA, Francois JA, Kappock TJ. A specialized citric acid cycle requiring succinyl-coenzyme A (CoA):acetate CoA-transferase (AarC) confers acetic acid resistance on the acidophile Acetobacter aceti. J Bacteriol. 2008;190:4933–4940. doi: 10.1128/JB.00405-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sidhu H, Ogden SD, Lung H-Y, Luttge BG, Baetz AL, Peck AB. DNA sequencing and expression of the formyl coenzyme A transferase gene, frc, from Oxalobacter formigenes. J Bacteriol. 1997;179:3378–3381. doi: 10.1128/jb.179.10.3378-3381.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toyota CG, Berthold CL, Gruez A, Jónsson S, Lindqvist Y, Cambillau C, Richards NGJ. Differential substrate specificity and kinetic behavior of Escherichia coli YfdW and Oxalobacter formigenes formyl coenzyme A transferase. J Bacteriol. 2008;190:2556–2564. doi: 10.1128/JB.01823-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saeki A, Taniguchi M, Matsushita K, Toyama H, Theeragool G, Lotong N, Adachi O. Microbiological aspects of acetate oxidation by acetic acid bacteria, unfavorable phenomena in vinegar fermentation. Biosci Biotechnol Biochem. 1997;61:317–323. [Google Scholar]
- 21.Saeki A, Matsushita K, Takeno S, Taniguchi M, Toyama H, Theeragool G, Lotong N, Adachi O. Enzymes responsible for acetate oxidation by acetic acid bacteria. Biosci Biotechnol Biochem. 1999;63:2102–2109. doi: 10.1271/bbb.63.2102. [DOI] [PubMed] [Google Scholar]
- 22.Muraoka H, Watabe Y, Ogasawara N. Effect of oxygen deficiency on acid production and morphology of bacterial cells in submerged acetic fermentation by Acetobacter aceti. J Ferment Technol. 1982;60:171–180. [Google Scholar]
- 23.Jonsson S, Ricagno S, Lindqvist Y, Richards NGJ. Kinetic and mechanistic characterization of the formyl-CoA transferase from Oxalobacter formigenes. J Biol Chem. 2004;279:36003–36012. doi: 10.1074/jbc.M404873200. [DOI] [PubMed] [Google Scholar]
- 24.Heider J. A new family of CoA-transferases. FEBS Lett. 2001;509:345–349. doi: 10.1016/s0014-5793(01)03178-7. [DOI] [PubMed] [Google Scholar]
- 25.Berthold CL, Toyota CG, Richards NGJ, Lindqvist Y. Reinvestigation of the catalytic mechanism of formyl-CoA transferase, a class III CoA-transferase. J Biol Chem. 2008;283:6519–6529. doi: 10.1074/jbc.M709353200. [DOI] [PubMed] [Google Scholar]
- 26.Diederichs K, Karplus PA. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat Struct Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]
- 27.Gruez A, Roig-Zamboni V, Valencia C, Campanacci V, Cambillau C. The crystal structure of the Escherichia coli YfdW gene product reveals a new fold of two interlaced rings identifying a wide family of CoA transferases. J Biol Chem. 2003;278:34582–34586. doi: 10.1074/jbc.C300282200. [DOI] [PubMed] [Google Scholar]
- 28.Ricagno S, Jonsson S, Richards N, Lindqvist Y. Formyl-CoA transferase encloses the CoA binding site at the interface of an interlocked dimer. EMBO J. 2003;22:3210–3219. doi: 10.1093/emboj/cdg333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levitt M, Gerstein M, Huang E, Subbiah S, Tsai J. Protein folding: The endgame. Annu Rev Biochem. 1997;66:549–579. doi: 10.1146/annurev.biochem.66.1.549. [DOI] [PubMed] [Google Scholar]
- 30.Gogos A, Gorman J, Shapiro L. Structure of Escherichia coli YfdW, a type III CoA transferase. Acta Crystallogr D Biol Crystallogr. 2004;60:507–511. doi: 10.1107/S0907444904000034. [DOI] [PubMed] [Google Scholar]
- 31.Sael L, Kihara D. Improved protein surface comparison and application to low-resolution protein structure data. BMC Bioinformatics. 2010;11:S2. doi: 10.1186/1471-2105-11-S11-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sael L, La D, Li B, Rustamov R, Kihara D. Rapid comparison of properties on protein surface. Proteins. 2008;73:1–10. doi: 10.1002/prot.22141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sael L, Li B, La D, Fang Y, Ramani K, Rustamov R, Kihara D. Fast protein tertiary structure retrieval based on global surface shape similarity. Proteins. 2008;72:1259–1273. doi: 10.1002/prot.22030. [DOI] [PubMed] [Google Scholar]
- 34.Grimsley GR, Shaw KL, Fee LR, Alston RW, Huyghues-Despointes BMP, Thurlkill RL, Scholtz JM, Pace CN. Increasing protein stability by altering long-range coulombic interactions. Protein Sci. 1999;8:1843–1849. doi: 10.1110/ps.8.9.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torrez M, Schultehenrich M, Livesay DR. Conferring thermostability to mesophilic proteins through optimized electrostatic surfaces. Biophys J. 2003;85:2845–2853. doi: 10.1016/S0006-3495(03)74707-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strickler SS, Gribenko AV, Gribenko AV, Keiffer TR, Tomlinson J, Reihle T, Loladze VV, Makhatadze GI. Protein stability and surface electrostatics: a charged relationship. Biochemistry. 2006;45:2761–2766. doi: 10.1021/bi0600143. [DOI] [PubMed] [Google Scholar]
- 37.Gribenko AV, Patel MM, Liu J, McCallum SA, Wang C, Makhatadze GI. Rational stabilization of enzymes by computational redesign of surface charge-charge interactions. Proc Natl Acad Sci U S A. 2009;106:2601–2606. doi: 10.1073/pnas.0808220106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vieille C, Zeikus GJ. Hyperthermophilic enzymes: Sources, uses, and molecular mechanisms for thermostability. Microbiol Mol Biol Rev. 2001;65:1–43. doi: 10.1128/MMBR.65.1.1-43.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohmori S, Masai H, Arima K, Beppu T. Isolation and identification of acetic acid bacteria for submerged acetic acid fermentation at high temperature. Agric Biol Chem. 1980;44:2901–2906. [Google Scholar]
- 40.Starks CM, Francois JA, MacArthur KM, Heard BZ, Kappock TJ. Atomic-resolution crystal structure of thioredoxin from the acidophilic bacterium Acetobacter aceti. Protein Sci. 2007;16:92–98. doi: 10.1110/ps.062519707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez-Jimenez R, Inglés-Prieto A, Zhao Z-M, Sanchez-Romero I, Alegre-Cebollada J, Kosuri P, Garcia-Manyes S, Kappock TJ, Tanokura M, Holmgren A, Sanchez-Ruiz JM, Gaucher EA, Fernandez JM. Single-molecule paleoenzymology probes the chemistry of resurrected enzymes. Nat Struct Mol Biol. 2011;18:592–596. doi: 10.1038/nsmb.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 43.Burns KL, Gelbaum LT, Sullards MC, Bostwick DE, May SW. Iso-coenzyme A. J Biol Chem. 2005;280:16550–16558. doi: 10.1074/jbc.M411898200. [DOI] [PubMed] [Google Scholar]
- 44.Ohmori S, Uozumi T, Beppu T. Loss of acetic acid resistance and ethanol oxidizing ability in an Acetobacter strain. Agric Biol Chem. 1982;46:381–389. [Google Scholar]
- 45.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Short protocols in molecular biology: a compendium of methods from Current protocols in molecular biology. 4th ed. New York: Wiley; 1999. [Google Scholar]
- 46.Dawson RMC. Data for biochemical research. Oxford: Clarendon Press; 1986. [Google Scholar]
- 47.Teng T-Y. Mounting of crystals for macromolecular crystallography in a free-standing thin film. J Appl Cryst. 1990;23:387–391. [Google Scholar]
- 48.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 49.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D: Biol Crystallogr. 1994;50:760–770. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 50.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 51.Kleywegt GJ, Jones TA. Making the most of your search model. CCP4/ESF-EACBM Newsletter on Protein Crystallography. 1996;32:32–36. [Google Scholar]
- 52.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 53.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR System: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 54.Perrakis A, Sixma TK, Wilson KS, Lamzin VS. wARP: improvement and extension of crystallographic phases by weighted averaging of multiple-refined dummy atomic models. Acta Crystallogr D Biol Crystallogr. 1997;53:448–455. doi: 10.1107/S0907444997005696. [DOI] [PubMed] [Google Scholar]
- 55.Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 56.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeLano WL. The PyMOL molecular graphics system. Palo Alto, CA: DeLano Scientific; 2002. [Google Scholar]
- 61.Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 62.Gouet P, Courcelle E, Stuart DI, Métoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 63.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 64.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 65.Hubbard SJ, Thornton J. NACCESS computer program. London: Department of Biochemistry and Molecular Biology, University College London; 1993. [Google Scholar]
- 66.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J Mol Biol. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 67.Laskowski RA. PDBsum new things. Nucleic Acids Res. 2009;37:D355–D359. doi: 10.1093/nar/gkn860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Willard L, Ranjan A, Zhang H, Monzavi H, Boyko RF, Sykes BD, Wishart DS. VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 2003;31:3316–3319. doi: 10.1093/nar/gkg565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 70.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.