

Abstract
BacD is an ATP-dependent dipeptide ligase responsible for the biosynthesis of l-alanyl-l-anticapsin, a precursor of an antibiotic produced by Bacillus spp. In contrast to the well-studied and phylogenetically related d-alanine: d-alanine ligase (Ddl), BacD synthesizes dipeptides using l-amino acids as substrates and has a low substrate specificity in vitro. The enzyme is of great interest because of its potential application in industrial protein engineering for the environmentally friendly biological production of useful peptide compounds, such as physiologically active peptides, artificial sweeteners and antibiotics, but the determinants of its substrate specificity and its catalytic mechanism have not yet been established due to a lack of structural information. In this study, we report the crystal structure of BacD in complex with ADP and an intermediate analog, phosphorylated phosphinate l-alanyl-l-phenylalanine, refined to 2.5-Å resolution. The complex structure reveals that ADP and two magnesium ions bind in a manner similar to that of Ddl. However, the dipeptide orientation is reversed, and, concomitantly, the entrance to the amino acid binding cavity differs in position. Enzymatic characterization of two mutants, Y265F and S185A, demonstrates that these conserved residues are not catalytic residues at least in the reaction where l-phenylalanine is used as a substrate. On the basis of the biochemical and the structural data, we propose a reaction scheme and a catalytic mechanism for BacD.
Keywords: nonribosomal peptide synthetase, BacD, bacilysin, anticapsin, ATP-grasp domain
Introduction
Anticapsin is an antibiotic produced by Bacillus spp. and is secreted in the form of an l-alanyl-l-anticapsin dipeptide known as bacilysin [Fig. 1(A)].1 After being incorporated into target cells, the dipeptide is cleaved by cytoplasmic peptidases. Anticapsin—a nonproteinogenic epoxycyclohexanone-containing amino acid [Fig. 1(A)]—inhibits cell wall biosynthesis by mimicking glutamine and irreversibly binding glucosamine-6-phosphate synthetase.2 The bac and ywf operons encode several genes involved in the biosynthesis of anticapsin using prephenate as a starter molecule, the synthetic pathway of which has not been fully elucidated.3 BacD (EC 6.3.2.28, also termed YwfE) is encoded in the bac cluster and has been identified as an l-amino acid-specific dipeptide synthetase through in silico screening based on amino acid sequence similarity with the ATP-grasp domain (PROSITE accession number: PS 50975), which is characterized by two α/β subdomains to accommodate the ATP molecule between them and is conserved among the members of the ATP-dependent carboxylate-amine/thiol ligase family.4, 5 An in vitro assay has shown that the enzyme utilizes only smaller l-amino acids, such as glycine, alanine, and serine, for the N-terminal residue of the dipeptide and uncharged bulky residues, such as glutamine, phenylalanine, and methionine, for the C-terminal residue.5 Considering this substrate specificity and the result from mutagenesis showing that a deletion of the bacD gene resulted in the accumulation of anticapsin in the cell,6l-alanine and l-anticapsin can be regarded as physiological substrates of BacD.
Figure 1.
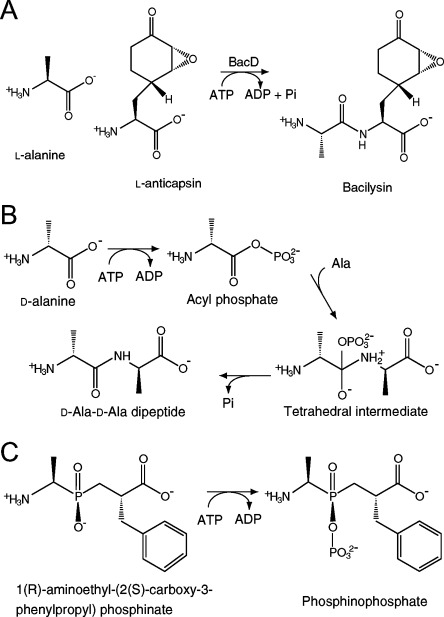
Reactions catalyzed by BacD [(A) and (C)] and Ddl (B). A: The physiological substrates of BacD are considered to be l-alanine and l-anticapsin. B: The previously proposed reaction scheme for Ddls consists of three steps. C: The phosphinate l-alanyl-l-phenylalanine analog (left) binds the active site of BacD, and accepts a phosphate group from ATP to form the phosphorylated phosphinate l-alanyl- l-phenylalanine analog (right, termed “P-analog” in this study).
BacD is phylogenetically related to d-alanine:d-alanine ligase (Ddl) (EC 6.3.2.4), sharing a sequence identity of ∼25% when only the primary structures are considered in generating an alignment (Supporting Information Fig. S1).7 Ddl plays a key role in the biosynthesis of the cell wall in most bacteria by forming the d-alanyl-d-alanine dipeptide that provides the cross-linking site in peptidoglycan. The enzyme can be classified into two groups, DdlA and DdlB, both of which have been found in some bacteria, including Escherichia coli.8 Other groups of enzymes with the same physiological function show different substrate specificities; for example, VanA/B and VanC synthesize, in vivo, the d-alanyl-d-lactate depsipeptide and d-alanyl-d-serine peptide, respectively.9–12 Another class of Ddl-related enzymes from lactic acid bacteria (termed LmDdl2, as an enzyme of Leuconostoc mesenteroides13) synthesizes d-alanyl-d-lactate. Some of these enzymes show low substrate specificities, and the differences in the specificities are closely related to the resistance to glycopeptide antibiotics such as vancomycin and teicoplanin.
The structural analyses and biochemical studies of Ddl have revealed its reaction scheme. Dipeptide formation is thought to proceed in three steps [Fig. 1(B)]: first, phosphorylation of the N-terminal alanine; second, nucleophilic attack of the amino group in the C-terminal alanine on the carbonyl carbon in the acylphosphate intermediate; and finally, release of the phosphate group from the tetrahedral intermediate.14–16 While the amino group in the second d-alanine is the nucleophile in the second step of d-alanyl-d-alanine synthesis, the hydroxy group in the d-lactate is the nucleophile for d-alanyl-d-lactate synthesis. A key determinant of the preference for d-alanine/d-lactate as the C-terminal residue of the peptide/depsipeptide has been identified to be tyrosine/phenylalanine in a short helix-spanning region referred to as the ω-loop by subsequent mutagenic studies.13, 17, 18
In contrast to Ddl, however, the tertiary structure and the reaction scheme of BacD have not been elucidated thus far, even though an enzymatic characterization has revealed that the enzyme shares some features with Ddl in its reaction properties.5 First, the dipeptide formation is ATP-dependent, where ADP is released as a product. Second, the N-terminal amino acid affinities for the enzyme are higher than those of the C-terminal amino acid. Third, the amide-bond formation activities of the enzymes show an optimum pH higher than 9.5, 17, 19
To investigate structural and functional features of BacD, we have determined the crystal structure of the enzyme in complex with ADP and an intermediate analog, phosphorylated phosphinate l-alanyl-l-phenylalanine. Our structural description and an accompanying enzymatic characterization have implications for substrate specificity and for the reaction mechanism of the dipeptide synthesis catalyzed by BacD.
Results and Discussion
Enzymatic properties of BacD and inhibitory effect of a phosphinate analog
The kinetic parameters of BacD have been determined using l-alanine and l-phenylalanine as substrates (Table I). The pair of these two amino acids showed the highest activity among the combinations of less bulky amino acids (glycine, l-alanine, and l-serine) and uncharged bulky amino acids (l-leucine, l-isolucine, l-methionine, l-phenylalanine, l-tyrosine, l-tryptophane, and l-glutamine), which have been reported to be substrates of BacD.5 Some d-amino acids (d-alanine, d-serine, d-phenylalanine, and d-glutamine) were also tested but showed no activity, consistent with the previous study.5
Table I.
Steady-State Kinetic Constants for Amino Acid Ligases
Interpretation of the results for BacD was much more straightforward than for Ddl, which possesses two amino acid binding sites for a single substrate (i.e., d-alanine), but with different substrate affinities. The plots of activity versus substrate concentration for both l-alanine and l-phenylalanine fit well with Michaelis–Menten kinetics [Supporting Information Fig. S2(A)], confirming that each amino acid is recognized by a unique binding site. In the absence of either amino acid, the enzyme showed no activity, also supporting the simple model for BacD. The Km1 (for l-alanine) and Km2 (for l-phenylalanine) values are significantly higher than those of DdlB from E. coli but are comparable to those of VanA for d-alanine and d-alanine (Table I). VanA is an Enterococcus faecium homolog of Ddl that utilizes d-alanine and d-lactate to produce the d-alanyl-d-lactate depsipeptide in vivo, and the values shown in Table I are for a nonphysiological reaction where only d-alanine was used as the substrate.9 Although the affinities of BacD for l-alanine and l-phenylalanine are significantly lower than those of DdlB for d-alanine, the kcat value is comparable to that of Ddl. The observed high Km values may be related to the use of a nonphysiological substrate in the assay, since the most likely physiological substrates of BacD are l-alanine and l-anticapsin.
In previous structural analyses of Ddls, a synthetic phosphinate d-alanyl-d-alanine analog was used to investigate the binding mode of the substrates to the enzyme.14, 20–22 For the same purpose, we used a phosphinate l-alanyl-l-phenylalanine analog [Fig. 1(C)], and assayed its inhibitory effect on the enzyme. The Dixon plots over a range of the analog compound concentration show a clear sign of competitive inhibition for l-alanine but not for l-phenylalanine [Supporting Information Fig. S2(B,C)]. On the hypothesis that the compound occupies the both amino-acid binding sites, this finding suggests that BacD has a reaction sequence similar to that of Ddls, where the amino acid for the N-terminal residue of the dipeptide (l-alanine) first binds the enzyme to form the acylphosphate intermediate. In this scheme, l-phenylalanine does not compete for the binding to the enzyme with the analog because l-phenylalanine can bind the enzyme only after the binding of l-alanine and the analog competes for the binding to the enzyme with l-alanine. The value of Ki was determined to be 15.4 μM based on a Lineweaver–Burk plot over a range of l-alanine at the inhibitor concentration of 20 μM [Supporting Information Fig. S2(D)]. It should be emphasized that the plot considers only the initial rates of the reactions, and therefore, the possibility of slow onset of inhibition [i.e., phosphorylation of the inhibitor as shown in Fig. 1(C)]23 has not been taken into account.
Overall structure of BacD
The crystal structure of BacD was determined by the multiple-wavelength anomalous dispersion method with a selenomethionine derivative crystal (Table II). One BacD molecule has been found in the asymmetric unit with continuous clear electron density for the polypeptide from Glu2 to the C-terminus (Val472). On a tertiary structural level, BacD shares some structural features with Ddls, as expected from amino acid sequence identities of ∼25%. BacD can be superimposed on DdlB from E. coli (PDB ID: 2DLN)14 with a root-mean-square deviation value of 2.6 Å for 249 Cα atoms, based on secondary structure matching superposition [Fig. 2(A,B)].24 As in Ddls and related enzymes,4, 25 BacD can be divided into three α/β domains: an N-terminal domain (Glu2–Gly126), a central domain (Ala127–Leu229), and a C-terminal domain (Gln230–Gly471). The N-terminal domain includes a parallel β-sheet, whereas the central domain contains an antiparallel β-sheet like all Ddls, whose structures have already been reported. The most prominent difference in the overall structures between BacD and DdlB is the presence of an extra region composed of ∼100 residues in the C-terminal domain of BacD, including an additional smaller antiparallel β-sheet [Fig. 2(B) and Supporting Information Figs. S1 and S3]. Although the BacD-specific region shows no direct interaction with bound substrates, the region unambiguously affects the conformation of two amino-acid recognition loops (Loop 5 and 6 as mentioned below).
Table II.
Data Collection, Phasing, and Refinement Statistics
| Dataset | Native | Selenomethionine derivative | |||
|---|---|---|---|---|---|
| Crystal parameters | |||||
| Space group | P6522 | P6522 | |||
| Cell dimensions a, c (Å) | 130.79, 147.74 | 131.58, 148.60 | 131.41, 148.31 | 131.59, 148.61 | 131.72, 148.57 |
| Data collection | |||||
| Wavelength (Å) | 0.9000 | 0.9950 | 0.9793 | 0.9789 | 0.9640 |
| Resolution range (Å)a | 20–2.5 | 20–2.7 | 20–2.7 | 20–2.7 | 20–2.7 |
| Total reflections | 156019 | 395194 | 388809 | 389697 | 390047 |
| Unique reflectionsa | 26252 (1277) | 21282 (1037) | 21353 (1055) | 21258 (1037) | 21387 (1058) |
| Rmergeab | 0.081 (0.487) | 0.070 (0.439) | 0.078 (0.436) | 0.083 (0.417) | 0.091 (0.590) |
| Average I/σ(I)ac | 11.6 (4.4) | 13.6 (6.2) | 13.1 (6.0) | 13.5 (6.3) | 12.1 (5.7) |
| Completenessa | 0.999 (1.000) | 0.999 (1.000) | 0.999 (1.000) | 0.999 (1.000) | 0.999 (1.000) |
| Redundancya | 5.9 (6.0) | 18.6 (19.0) | 18.2 (17.9) | 18.3 (18.8) | 18.2 (17.9) |
| Refinement | MAD phasing | ||||
| Resolution range (Å) | 20–2.5 | No. of selenium sites | 8 | ||
| R-factor / free R-factorde | 0.194/0.230 | Mean figure of merit | |||
| Atoms in an asymmetric unit | acentric | 0.564 | |||
| Protein | 3675 | centric | 0.343 | ||
| Ligand (ADP, P-analog, 2 Mg) | 51 | ||||
| Water | 147 | ||||
| Deviations from ideal geometry | |||||
| Bonds distances (Å) | 0.009 | ||||
| Angles (°) | 1.295 | ||||
| Mean isotropic equivalent B-factors | |||||
| Main-chain (Å2) | 22.3 | ||||
| Side-chain (Å2) | 23.0 | ||||
| Ligand (Å2) | 22.5 | ||||
| Water (Å2) | 22.7 | ||||
| Ramachandran plotf | |||||
| Favored | 0.959 | ||||
| Allowed | 0.041 | ||||
Values in parentheses are for highest resolution shells.
Rmerge = ΣhklΣi(|Ii (hkl)- <I(hkl)>|)/ ΣhklΣiIi(hkl).
Signal-to-noise ratio of intensities.
R = Σ(|Fo − Fc|)/ΣFo.
Five percent of reflections were randomly chosen for calculation of free R value.
Values from Rampage.37
Figure 2.
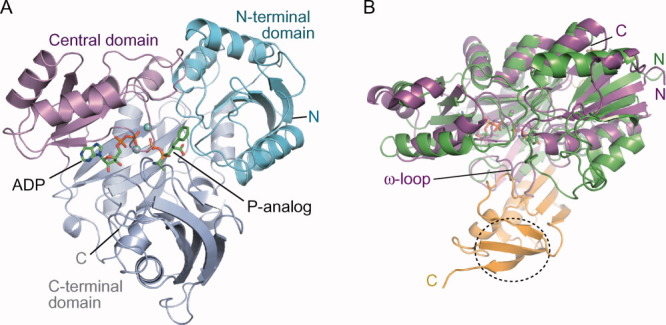
Overall structure of BacD. A: A schematic representation of BacD with a different color for each domain. ADP and the P-analog are drawn with stick models and the two magnesium ions with cyan balls. B: The overlay of DdlB (PDB ID: 2DLN,14 shown with a purple cartoon model) onto BacD (green and yellow) by secondary structure matching superposition is shown. The N- and C-termini of the polypeptides are labeled. The BacD-specific insertion (357-472) at the C-terminal domain are shown with yellow, where the additional smaller antiparallel β-sheet is circled with the dotted line. The ω-loop in DdlB, which includes Tyr216 interacting with d-alanine and Ser150, is labeled.
BacD elutes as a monomer in size-exclusion chromatography, and there is no sign of oligomerization in the crystal, given that the largest contact area with the adjacent molecule shares only 6.5% of the total accessible surface area. This contrasts with Ddl, whose structures have been exclusively reported to be homodimeric, even though the dimerization is unlikely to be of functional/mechanistic relevance. The tertiary structure of the dimerization site identified in Ddls is relatively conserved in BacD. Furthermore, the extra C-terminal region of BacD shows no interference with the dimerization site, implying that the difference in oligomerization state between BacD and Ddls is ascribable to minor amino acid substitutions. A sequence alignment of BacD and Ddls indicates that most of the residues forming hydrophobic interactions in the dimerization interface of DdlB from E. coli are not conserved in BacD (Supporting Information Fig. S1). Specifically, nonhydrophobic properties of Gln130, Glu133, and Asn134 would determine the oligomerization state of BacD to be monomer.
Active site structure
Crystallization of BacD has been performed in the presence of ATP, magnesium chloride, and the phosphinate l-alanyl-l-phenylalanine analog. An |Fo| − |Fc| electron density map clearly delineates an ADP molecule and the phosphorylated phosphinate analog (termed “P-analog” hereafter) [Fig. 1(C)] together with two magnesium ions [Fig. 3(A)], demonstrating that the γ phosphate group of ATP has been enzymatically transferred to the phosphinate analog, as observed in Ddls and VanA.14, 21, 22 As in these previously reported structures, ADP is found at the boundary between the central and the C-terminal domains, and the P-analog is situated at the intersection of the three domains [Fig. 2(A)]. The adenine ring lies in a hydrophobic pocket composed of Ile176, Phe228, Leu229, and Phe271 [Fig. 3(B)]. Hydrogen bonds are observed between the amide group of Gln268 and the 2′-hydroxy group in the ribose ring, as well as between the carboxy group in Glu226 and the primary amino group in the adenine ring. The ε-amino groups of two invariant lysines, Lys178 and Lys138, interact electrostatically with the α and β phosphate groups of ADP, respectively. The β phosphate group is also connected through a hydrogen bond with the main-chain amino group of Ser185. The adjacent Ser184 similarly interacts with the phosphate group of the P-analog with its main-chain amino group. Most of the residues involved in the interaction with ADP are localized in the ATP-grasp domain (PROSITE accession number: PS50975) and show high degrees of conservations among Ddls and BacD (Supporting Information Fig. S1). Two magnesium ions, designated Mg2+-1 and Mg2+-2 in Figure 3(B), bridge the β phosphate group of ADP with the phosphate group of the P-analog. Mg2+-1 is further coordinated by the α phosphate group of ADP, the carboxy groups of Glu311 and Glu324, and a water molecule, resulting in a standard octahedral coordination. In contrast, Mg2+-2 shows an asymmetric coordination geometry with the carboxy groups of Glu109 and Glu324, the main-chain carbonyl group of Leu182, and a water molecule. The binding mode of the enzyme with the phosphate groups of ADP, the two magnesium ions, and the phosphate group of the P-analog is quite similar with those of Ddls and VanA as previously reported.14, 21, 22, 26
Figure 3.
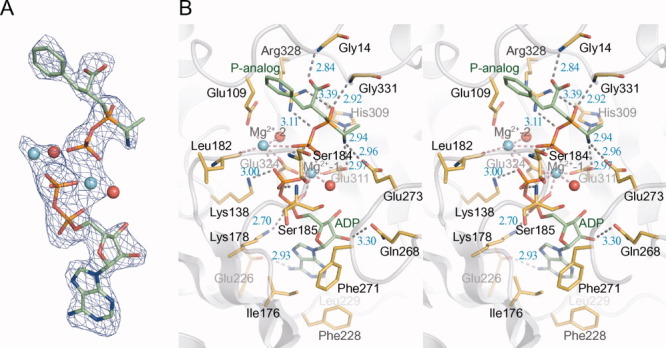
Structure of BacD in complex with ADP, the P-analog, and two magnesium ions. A: The |Fo| − |Fc| electron density map around ADP and the P-analog contoured at 3.0 σ is represented with a blue mesh. Nonprotein atoms were omitted in the calculation of Fc. ADP and the P-analog are drawn with stick models, and magnesium ions and water molecules are shown with cyan and red balls, respectively. B: The stereo view of the substrate-binding site of BacD is drawn with residues involved in the interaction highlighted and labeled. Hydrogen bonds/electrostatic interactions and coordination bonds to magnesium ions are depicted with dashed lines of gray and red, respectively.
In contrast, the binding mode of the dipeptide moiety of the P-analog was found to be significantly different; the direction of the peptide is reversed [Fig. 4(A)]. The amino group of the compound is fixed by three hydrogen bond acceptors: the carboxy groups of Glu273 and Glu311, and the ε-nitrogen of His309. The carboxy group of the P-analog is bound to the main-chain amino group of Gly14 [Fig. 3(B)]. The phosphinate P➖O is bound to the main-chain amino group of Gly331, and the guanidinium group of Arg328 interacts with the P➖O➖P bond in the P-analog. These interactions should contribute to the substrate preference of BacD for l-amino acids, and they would play a critical role in the stabilization of the tetrahedral intermediate state, as proposed in the previous study on DdlB.14
Figure 4.
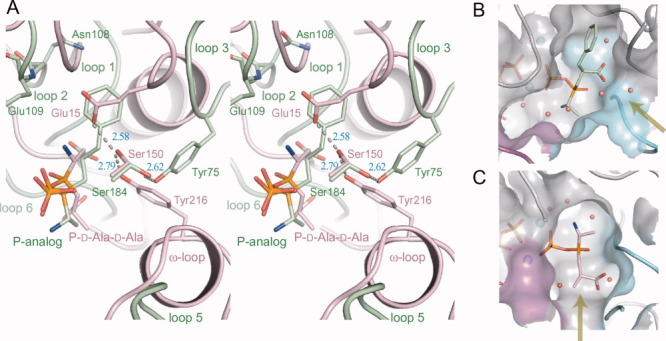
Structural comparison of BacD with DdlB (PDB ID: 2DLN) around the amino acid substrate-binding site. A: The stereo view of the superposition of DdlB (pink) onto BacD (green) is shown. Residues composing the proposed catalytic triad in DdlB and corresponding residues in BacD are highlighted and labeled. Loops 1–6 involved in the formation of the binding cavity in BacD and ω-loop in DdlB are labeled except for Loop 4, which is not shown for clarity. The phosphorylated d-alanyl-d-alanine phosphinate analog in DdlB is abbreviated as “P-D-Ala-D-Ala.” B, C: The amino acid binding cavities of BacD (B) and DdlB (C) are drawn. Water molecules and magnesium ions are shown as balls of red and cyan, respectively. The entrance from the solvent region into each cavity is indicated with yellow arrows. Loop 5 and 6, determinants of the entrance, are colored with magenta and cyan, respectively.
The amino acid binding cavity is formed by six loops: Loop 1 (8–16), Loop 2 (58–84), Loop 3 (105–111), Loop 4 (180–188), Loop 5 (266–274, corresponding to the ω-loop in DdlB27), and Loop 6 (327–333) [Fig. 4(A)]. While all of these loops are also components of the amino acid binding cavity of DdlB with the exception of Loop 2, which lies in an inserted region unique to BacD (Supporting Information Fig. S1), the loop conformations are significantly different between the two enzymes [Fig. 4(A)]. The shape and electrostatic properties of the cavity of BacD appear suitable to enclose the smaller uncharged residues for the N-terminus of the dipeptide and the uncharged bulky residues for the C-terminus [Fig. 4(B)]. The environment surrounding the two amino-acid substrates is almost hydrophobic with the contributions of the side chains in Loop 1–6. The surrounding residues around the methyl group corresponding to the side chain of the N-terminal amino-acid substrate (l-alanine) are Glu273, His276 in and near Loop 5, Trp332 and Met334 in and right next to Loop 6. On the other hand, the side chains of Leu12 and Gly13 in Loop 1, Tyr75 and Trp76 in Loop 2, Asn108, Glu109, and Leu110 in Loop 3, Ala183 and Ser184 in Loop 4 are surrounding the aromatic ring moiety of the P-analog corresponding to the side chain of the C-terminal amino-acid substrate (l-phenylalanine). Among these, the amino groups in the side chain of Asn108 and in the main chain of Glu109 should be hydrogen-bond donors for specific interaction with the polar side chains of anticapsin that possesses additional two oxygen atoms compared with that of phenylalanine [Figs. 1(A) and 4(A)]. It would be possible to change the substrate specificities by modifying the size and properties of the substrate-binding cavities with the replacements of residues mentioned above and/or the insertions of some residues.
Another critical difference between the two enzymes has been observed in the position of the entrance to the cavity [Fig. 4(B,C)]. The solvent region is connected to the C-terminal side of the dipeptide in both enzymes. This finding further supports the idea that the N-terminal amino acid first binds the enzyme to form an acylphosphate intermediate in BacD as well as Ddls. Furthermore, the entrance in BacD is much wider than that in DdlB [Fig. 4(B,C)], which should be correlated with the size of the second amino acid since BacD utilizes bulkier amino acids for the C-terminal amino acid.
Proposed catalytic mechanism of BacD
The function of the catalytic triad [Glu15, Ser150, and Tyr216 in Fig. 4(A)] as a proton acceptor for the amino group of the second d-alanine has been proposed in the previous structural study of DdlB from E. coli.14 However, a subsequent mutagenic study by the same group has revealed that the kcat values of three variants—Glu15Gln, Ser150Ala, and Tyr216Phe—are not significantly changed but that the substitutions increase the Km1 and Km2 values by one to three orders of magnitude.17, 27 The substitution of the proposed proton acceptor (Tyr216Phe) increases the Km2 value by only eightfold at pH 7.5, suggesting that the residue does not function as the catalytic base for the deprotonation of the second d-alanine. Based on the result, it has been proposed that the free-base state (➖NH2) of d-alanine selectively binds to the enzyme while the zwitterion state of d-alanine (➖NH3+) is excluded.27, 28 However, the calculation of the pKa value for the ligand in its binding site has implied that the zwitterion state of d-alanine is more likely than the free-base state.29 Although the function of Tyr216 in the dipeptide synthesis is still not clear and other candidate as the catalytic base has not been identified, the Tyr216Phe variant of DdlB gained a small but a measurable depsipeptide-forming activity with using d-lactate as the second molecule; at a higher pH (>∼7.5), the formation of d-alanyl-d-lactate predominates whereas d-alanyl-d-lactate is the main product at a lower pH (<∼7.5) in the presence of d-alanine and d-lactate.17 This pH-dependent enzymatic property resembles VanA from Enterococcus faecium, whose structure has also been reported in complex with the phosphorylated intermediate analog (PDB ID: 1E4E).17, 21 VanA lacks the ω-loop found in DdlB, and Tyr216 in DdlB is spatially replaced with His244, which is not hydrogen-bonded with the conserved Ser177 and Glu16. LmDdl2 from Leuconostoc mesenteroides also shows both the d-alanyl-d-alanine and d-alanyl-d-lactate forming activity and has the ω-loop that includes Phe261 corresponding to Tyr216 in DdlB (PDB ID: 1EHI).13 The Phe261Tyr variant has been reported to retain the depsipeptide-forming activity but lost the dipeptide-forming activity.
While a glutamic acid residue corresponding to Glu15 of DdlB has not been found in the BacD structure, two other residues, Tyr75 and Ser184, occur in a similar position to those of Tyr216 and Ser150 of DdlB with forming a hydrogen bond to each other [Fig. 4(A)]. No other residue that could function as the catalytic base is found around the carbon atom of the P-analog in the place of amino nitrogen of l-phenylalanine, although a terminal oxygen atom of the phosphate group of the P-analog and a water molecule occur within the distance of 3.5 Å. To investigate the function of the two conserved residues in BacD, we have prepared two variants, Tyr75Phe and Ser184Ala, and have determined their steady-state kinetic constants (Table III). As with DdlB, there was no significant difference in kcat values between the wild type and the two variants, but, in contrast to DdlB, only twofold increases in Km1 for Ser184Ala and in Km2 for Tyr75Phe were found. Therefore, it is likely that Tyr75 and Ser184 are not directly involved in the deprotonation of the amino group in l-phenylalanine, but that the property of each residue affects the substrate recognition. We have also investigated the effect of pH on the enzymatic activities and have found that only the Km2 values increased upon decreasing the pH. These results suggest that deprotonation of the amino group in the second amino acid substrate is a critical step but is not actively catalyzed by the enzyme, in contrast to the originally proposed reaction mechanism of DdlB.14, 27 An alternative candidate for the catalyst of the deprotonation is a terminal oxygen atom in the transferred phosphate group from ATP, but the oxygen atom should be required for the deprotonation of the dipeptide product.18, 28 The proposed reaction mechanism of BacD is summarized in Figure 5. As in the case of DdlB-related enzymes, the N-terminal amino acid (l-alanine) first binds to the ATP-bound state of the enzyme. The previous structural study of Ddl from Thermus thermophilus HB8 in various substrate bound states (PDB IDs: 2YZG, 2YZN, 2ZDG, 2ZDH, and 2ZDQ) has suggested that the binding of the first d-alanine should be preceded by the binding of ATP because the loops forming the active site cannot take the conformation suitable for the amino-acid binding without the ATP binding.26 The bound l-alanine is oriented for the phosphate transfer reaction with the aid of Glu273, His301, Glu311, and Gly331, and the carboxy oxygen atom nucleophilically attacks the phosphorous atom in the γ phosphate group [Figs. 4(A) and 5]. After the transfer of the phosphate group, the amino nitrogen of the second amino acid in the free-base state attacks the carbonyl carbon atom of the acylphosphate intermediate. The geometrical configuration of the second amino acid is confined by the main-chain amino groups of Gly14 and Trp332, where the latter indirectly interacts with the carboxy group of the amino-acid substrate through a water molecule (not shown in Fig. 5). Furthermore, the dipole moment of the α-helix (334–342) should also attract the carboxy group of the substrate. The α-helix is conserved among Ddl-related enzymes, and the carboxy group of the tetrahedral intermediate corresponding to the C-terminal alanine in DdlB-related enzymes also points toward the N-terminal side of the α-helix. Namely, the orientation of the charged group in the tetrahedral intermediate is conserved between BacD and DdlB even though the direction of the dipeptide is reversed. The observed binding manners should be critical in the chiral discrimination of the amino-acid substrates. Finally, the phosphate group is released with depriving proton from the amide group of the dipeptide product.
Table III.
Steady-State Kinetic Constants for Wild-Type and Variants
| Enzymea | pH | Km1 (mM)b | Km2 (mM)c | kcat (min−1) |
|---|---|---|---|---|
| Wild-type | 7.7 | 2.0 ± 0.17 | 18 ± 0.78 | 580 ± 8.9 |
| Wild-type | 7.1 | 2.6 ± 0.32 | 44 ± 1.6 | 460 ± 7.6 |
| Tyr75Phe | 7.7 | 1.7 ± 0.10 | 45 ± 0.48 | 580 ± 2.8 |
| Tyr75Phe | 7.1 | 1.5 ± 0.078 | 92 ± 10.0 | 430 ± 27 |
| Ser184Ala | 7.7 | 4.6 ± 0.63 | 16 ± 1.4 | 650 ± 19 |
| Ser184Ala | 7.1 | 4.9 ± 0.78 | 38 ± 2.7 | 470 ± 14 |
Hexahistidine has not been cleaved off.
For l-alanine.
For l-phenylalanine.
Figure 5.
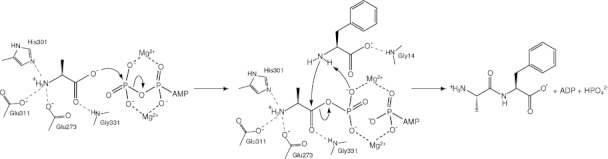
Schematic drawing of the reaction mechanism for the dipeptide synthesis catalyzed by BacD. The residues involved in the interaction with the dipeptide moiety of the P-analog are shown. The first step is the formation of the acylphosphate intermediate where the carboxy oxygen atom in l-alanine is nucleophile. The second step includes the formation of the tetrahedral intermediate and the release of the phosphate group, where the carbonyl carbon is electrophile and the phosphate group deprives the amido nitrogen atom of a proton.
Based on the proposed mechanism, BacD utilizes only the free-base state of l-phenylalanine as the substrate. The extraordinary low affinity for l-phenylalanine might be related to the low concentration of the free-base state of amino acids at a physiological condition. On the other hand, given the physiological function of BacD, the enzyme most likely utilizes l-alanine and l-anticapsin as amino acid substrates in vivo, and therefore, the low affinity for l-phenylalanine found in this study may be caused by our use of a nonphysiological substrate. Moreover, there remains a possibility that a different reaction mechanism is applied to the synthesis of l-alanyl-l-anticapsin, where Tyr75 might function as the base catalyst.
Further study will help elucidate the enzymatic properties of BacD under physiological conditions, and our structural data will form the basis for protein engineering of BacD toward the enzymatic synthesis of dipeptides bearing l-amino acids.
Materials and Methods
Cloning, expression, and purification
The gene encoding BacD was amplified by PCR from pColdI-ywfE, a derivative of the previously reported pQE60ywfE,5 by using the primers listed in Supporting Information Table SI with the Pfu Ultra II DNA polymerase (Stratagene), followed by insertion into the expression vector pPROExHtb (Invitrogen). BacD was produced in E. coli BL21(DE3) cells (Novagen) transformed with the plasmid. After culture in LB medium containing 100 μg mL−1 ampicillin to an OD600 of 0.6, expression was induced by the addition of 200 μM isopropyl-β-d-thiogalactopyranoside, followed by further incubation for 16 h at 24°C. The protein was purified with a Ni-nitrilotriacetic acid resin (Qiagen), HiTrapQ anion exchange column and with a HiLoad Superdex 200 16/60 gel filtration column (GE Healthcare) equilibrated with 10 mM HEPES-NaOH (pH 7.4) and 50 mM NaCl. The N-terminal hexahistidine was cleaved by tobacco etch virus protease prior to anion exchange chromatography. The protein was concentrated to 60 mg mL−1 using a Vivaspin concentrator with a 10 kDa cutoff (Sartorius) and was stored at −80°C. Selenomethionine-substituted BacD was produced as described previously,30 and was purified using the same procedure as for the native enzyme.
Crystallization and structure determination of BacD
Crystals of both native and selenomethionine-substituted BacD were obtained under the same conditions. Crystallization was performed at 293 K by the sitting-drop vapor-diffusion method by using a CrystalQuick 96-well plate (Greiner). The drop was prepared by mixing 1 μL of the protein solution, containing 20 mg mL−1 BacD, 10 mM HEPES-NaOH (pH 7.4), 50 mM NaCl, 4 mM ATP, 8 mM MgCl2, and 4 mM phosphinate l-alanyl-l-phenylalanine analog [left compound in Fig. 1(C)], with 1 μL of reservoir solution containing 100 mM Bis-Tris propane, 60 mM sodium citrate, and 18% (w/v) PEG3, 350. Crystals grown to a maximum size of 0.1 × 0.1 × 0.2 mm3 were soaked in a cryoprotectant buffer composed of 100 mM Bis-Tris propane, 60 mM sodium citrate, 25% (w/v) PEG3,350, and 20% glycerol, prior to flash cooling with liquid nitrogen. Single-wavelength native data and multiple-wavelength selenium derivative data were collected at SPring-8 beamline BL44XU, with the crystals maintained at 90 K using a gaseous nitrogen stream. All data were processed and scaled with HKL2000.31 Eight selenium sites were found, of which the positions, occupancies, and B-factors were refined and the initial phases calculated with autoSHARP.32 After density modification using SOLOMON33 and automatic model building by Buccaneer,34 an initial model covering ∼80% of the BacD molecule was obtained. Subsequent iterative manual model building/corrections and refinement were performed with Coot24 and Refmac5,35 respectively. Statistics for data collection, phasing, and refinement are summarized in Table II. Secondary structural elements were assigned with DSSP,36 and main-chain dihedral angles were checked with RAMPAGE.37 The coordinates and the structure factors have been deposited in the Protein Data Bank, under the accession code of 3VMM. Graphical representations of the model were prepared with PyMOL (DeLano Scientific).
Enzymatic assay
The enzymatic activity of the wild type and BacD point mutants was assayed spectrophotometrically by using an ATP/NADH coupled system with pyruvate kinase (PK) and lactate dehydrogenase (LDH).38 The assay solution contained 10 mM magnesium acetate, 10 mM potassium acetate, 5 mM ATP, 2.5 mM phosphoenolpyruvate, 0.8 mM NADH, 34.1 U mL−1 PK, and 49.5 U mL−1 LDH (Sigma). The pH was measured to be 7.7 and 7.1 for reaction solutions containing 50 mM Tris-HCl (pH 8.5) and 100 mM HEPES-NaOH (pH 7.4), respectively. All assays were performed at 310 K with a 96-well plate reader (BioRad), by monitoring the decrease in absorbance at 340 nm. The values of Km1 for l-alanine and of Km2 for l-phenylalanine were determined in the presence of 100 mMl-phenylalanine and of 25 mMl-alanine, respectively, over a range of the concentration of the counterpart amino acid. The value of kcat was determined from the latter plot because a saturating concentration of l-phenylalanine is unattainable for some conditions, because of its low solubility. All measurements were performed at least three times.
Site-directed mutagenesis
Point mutations were introduced to prepare two variants (Tyr75Phe and Ser185Ala) by PCR using the Pfu Ultra II DNA polymerase (Stratagene) with the primers listed in Table SI. The mutations were verified by DNA sequencing. In the enzymatic assay for comparing the wild-type and the two variants, proteins purified by Ni-affinity chromatography and by dialysis with 10 mM HEPES-NaOH (pH 7.4) and 50 mM NaCl were used. The purified proteins were tagged with an N-terminal hexahistidine, whose effect on steady-state kinetic parameters has been found to be negligible.
Acknowledgments
The authors acknowledge the assistance of the staff at the SPring-8 beamline BL44XU (Proposal No.: 2011B6623). The MX225-HE (Rayonix) CCD detector at BL44XU was financially supported by Academia Sinica and by the National Synchrotron Radiation Research Center (Taiwan, ROC). The authors are also grateful to Prof. Jun Hiratake at Kyoto University for the synthesis of the phosphinate analog.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Foster JW, Woodruff HB. Bacillin, a new antibiotic substance from a soil isolate of Bacillus subtilis. J Bacteriol. 1946;51:363–369. doi: 10.1128/JB.51.3.363-369.1946. [DOI] [PubMed] [Google Scholar]
- 2.Kenig M, Vandamme E, Abraham EP. The mode of action of bacilysin and anticapsin and biochemical properties of bacilysin-resistant mutants. J Gen Microbiol. 1976;94:46–54. doi: 10.1099/00221287-94-1-46. [DOI] [PubMed] [Google Scholar]
- 3.Mahlstedt SA, Walsh CT. Investigation of anticapsin biosynthesis reveals a four-enzyme pathway to tetrahydrotyrosine in Bacillus subtilis. Biochemistry. 2010;49:912–923. doi: 10.1021/bi9021186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fan C, Moews PC, Shi Y, Walsh CT, Knox JR. A common fold for peptide synthetases cleaving ATP to ADP: glutathione synthetase and d-alanine:d-alanine ligase of Escherichia coli. Proc Natl Acad Sci USA. 1995;92:1172–1176. doi: 10.1073/pnas.92.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tabata K, Ikeda H, Hashimoto S. ywfE in Bacillus subtilis codes for a novel enzyme, l-amino acid ligase. J Bacteriol. 2005;187:5195–5202. doi: 10.1128/JB.187.15.5195-5202.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinborn G, Hajirezaei MR, Hofemeister J. bac genes for recombinant bacilysin and anticapsin production in Bacillus host strains. Arch Microbiol. 2005;183:71–79. doi: 10.1007/s00203-004-0743-8. [DOI] [PubMed] [Google Scholar]
- 7.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zawadzke LE, Bugg TD, Walsh CT. Existence of two d-alanine:d-alanine ligases in Escherichia coli: cloning and sequencing of the ddlA gene and purification and characterization of the DdlA and DdlB enzymes. Biochemistry. 1991;30:1673–1682. doi: 10.1021/bi00220a033. [DOI] [PubMed] [Google Scholar]
- 9.Bugg TD, Wright GD, Dutka-Malen S, Arthur M, Courvalin P, Walsh CT. Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry. 1991;30:10408–10415. doi: 10.1021/bi00107a007. [DOI] [PubMed] [Google Scholar]
- 10.Billot-Klein D, Gutmann L, Sable S, Guittet E, van Heijenoort J. Modification of peptidoglycan precursors is a common feature of the low-level vancomycin-resistant VANB-type Enterococcus D366 and of the naturally glycopeptide-resistant species Lactobacillus caseiPediococcus pentosaceusLeuconostoc mesenteroides, and Enterococcus gallinarum. J Bacteriol. 1994;176:2398–2405. doi: 10.1128/jb.176.8.2398-2405.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reynolds PE, Snaith HA, Maguire AJ, Dutka-Malen S, Courvalin P. Analysis of peptidoglycan precursors in vancomycin-resistant Enterococcus gallinarum BM4174. Biochem J. 1994;301:5–8. doi: 10.1042/bj3010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bugg TD, Dutka-Malen S, Arthur M, Courvalin P, Walsh CT. Identification of vancomycin resistance protein VanA as a d-alanine:d-alanine ligase of altered substrate specificity. Biochemistry. 1991;30:2017–2021. doi: 10.1021/bi00222a002. [DOI] [PubMed] [Google Scholar]
- 13.Park IS, Walsh CT. d-Alanyl-d-lactate and d-alanyl-d-alanine synthesis by d-alanyl-d-alanine ligase from vancomycin-resistant Leuconostoc mesenteroides. Effects of a phenylalanine 261 to tyrosine mutation. J Biol Chem. 1997;272:9210–9214. doi: 10.1074/jbc.272.14.9210. [DOI] [PubMed] [Google Scholar]
- 14.Fan C, Moews PC, Walsh CT, Knox JR. Vancomycin resistance: structure of d-alanine:d-alanine ligase at 2.3 A resolution. Science. 1994;266:439–443. doi: 10.1126/science.7939684. [DOI] [PubMed] [Google Scholar]
- 15.Mullins LS, Zawadzke LE, Walsh CT, Raushel FM. Kinetic evidence for the formation of d-alanyl phosphate in the mechanism of d-alanyl-d-alanine ligase. J Biol Chem. 1990;265:8993–8998. [PubMed] [Google Scholar]
- 16.Neuhaus FC. The enzymatic synthesis of d-alanyl-d-alanine. II. Kinetic studies on d-alanyl-d-alanine synthetase. J Biol Chem. 1962;237:3128–3135. [PubMed] [Google Scholar]
- 17.Park IS, Lin CH, Walsh CT. Gain of d-alanyl-d-lactate or d-lactyl-d-alanine synthetase activities in three active-site mutants of the Escherichia colid-alanyl-d-alanine ligase B. Biochemistry. 1996;35:10464–10471. doi: 10.1021/bi9603128. [DOI] [PubMed] [Google Scholar]
- 18.Lessard IA, Healy VL, Park IS, Walsh CT. Determinants for differential effects on d-Ala-d-lactate vs d-Ala-d-Ala formation by the VanA ligase from vancomycin-resistant enterococci. Biochemistry. 1999;38:14006–14022. doi: 10.1021/bi991384c. [DOI] [PubMed] [Google Scholar]
- 19.Wu D, Zhang L, Kong Y, Du J, Chen S, Chen J, Ding J, Jiang H, Shen X. Enzymatic characterization and crystal structure analysis of the d-alanine-d-alanine ligase from Helicobacter pylori. Proteins. 2008;72:1148–1160. doi: 10.1002/prot.22009. [DOI] [PubMed] [Google Scholar]
- 20.Fan C, Park IS, Walsh CT, Knox JR. d-alanine:d-alanine ligase: phosphonate and phosphinate intermediates with wild type and the Y216F mutant. Biochemistry. 1997;36:2531–2538. doi: 10.1021/bi962431t. [DOI] [PubMed] [Google Scholar]
- 21.Roper DI, Huyton T, Vagin A, Dodson G. The molecular basis of vancomycin resistance in clinically relevant Enterococci: crystal structure of d-alanyl-d-lactate ligase (VanA) Proc Natl Acad Sci USA. 2000;97:8921–8925. doi: 10.1073/pnas.150116497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuzin AP, Sun T, Jorczak-Baillass J, Healy VL, Walsh CT, Knox JR. Enzymes of vancomycin resistance: the structure of d-alanine-d-lactate ligase of naturally resistant Leuconostoc mesenteroides. Structure. 2000;8:463–470. doi: 10.1016/s0969-2126(00)00129-5. [DOI] [PubMed] [Google Scholar]
- 23.Duncan K, Walsh CT. ATP-dependent inactivation and slow binding inhibition of Salmonella typhimurium d-alanine:d-alanine ligase (ADP) by (aminoalkyl)phosphinate and aminophosphonate analogues of d-alanine. Biochemistry. 1988;27:3709–3714. doi: 10.1021/bi00410a028. [DOI] [PubMed] [Google Scholar]
- 24.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 25.Artymiuk PJ, Poirrette AR, Rice DW, Willett P. Biotin carboxylase comes into the fold. Nat Struct Biol. 1996;3:128–132. doi: 10.1038/nsb0296-128. [DOI] [PubMed] [Google Scholar]
- 26.Kitamura Y, Ebihara A, Agari Y, Shinkai A, Hirotsu K, Kuramitsu S. Structure of d-alanine-d-alanine ligase from Thermus thermophilus HB8: cumulative conformational change and enzyme-ligand interactions. Acta Cryst. 2009;D65:1098–1106. doi: 10.1107/S0907444909029710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi Y, Walsh CT. Active site mapping of Escherichia colid-Ala-d-Ala ligase by structure-based mutagenesis. Biochemistry. 1995;34:2768–2776. doi: 10.1021/bi00009a005. [DOI] [PubMed] [Google Scholar]
- 28.Ellsworth BA, Tom NJ, Bartlett PA. Synthesis and evaluation of inhibitors of bacterial d-alanine:d-alanine ligases. Chem Biol. 1996;3:37–44. doi: 10.1016/s1074-5521(96)90082-4. [DOI] [PubMed] [Google Scholar]
- 29.Carlson HA, Briggs JM, McCammon JA. Calculation of the pKa values for the ligands and side chains of Escherichia colid-alanine:d-alanine ligase. J Med Chem. 1999;42:109–117. doi: 10.1021/jm980351c. [DOI] [PubMed] [Google Scholar]
- 30.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 31.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 32.Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods Mol Biol. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- 33.Abrahams JP, Leslie AG. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Cryst. 1996;D52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 34.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Cryst. 2006;D62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 36.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 37.Lovell SC, Davis IW, Arendall WB, III, de Bakker, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 38.Neuhaus FC. The enzymatic synthesis of d-alanyl-d-alanine. I. Purification and properties of d-alanyl-d-alanine synthetase. J Biol Chem. 1962;237:778–786. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.