

Abstract
The amyloid cascade hypothesis remains a robust model of AD neurodegeneration. However, amyloid deposits contain proteins besides Aβ, such as apolipoprotein E (apoE). Inheritance of the apoE4 allele is the strongest genetic risk factor for late-onset AD. However, there is no consensus on how different apoE isotypes contribute to AD pathogenesis. It has been hypothesized that apoE and apoE4 in particular is an amyloid catalyst or “pathological chaperone”. Alternatively it has been posited that apoE regulates Aβ clearance, with apoE4 been worse at this function compared to apoE3. These views seem fundamentally opposed. The former would indicate that removing apoE will reduce AD pathology, while the latter suggests increasing brain ApoE levels may be beneficial. Here we consider the scientific basis of these different models of apoE function and suggest that these seemingly opposing views can be reconciled. The optimal therapeutic target may be to inhibit the interaction of apoE with Aβ rather than altering apoE levels. Such an approach will not have detrimental effects on the many beneficial roles apoE plays in neurobiology. Furthermore, other Aβ binding proteins, including ACT and apo J can inhibit or promote Aβ oligomerization/polymerization depending on conditions and might be manipulated to effect AD treatment.
1. Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder that is clinically characterized by progressive mental decline and histopathologically defined by highly abundant amyloid deposits and neurofibrillary tangles in the brain parenchyma. The identification of mutations within the amyloid precursor protein (APP) and presenilin (PS) genes that cause autosomal dominantly inherited AD and that result in increased production of amyloid-prone forms of Aβ established beyond doubt that the processing of APP and the production of Aβ peptides are intimately involved in the disease process and led to the proposal and the reinforcement of the Alzheimer Amyloid Cascade Hypothesis [1, 2].
The role of amyloid in neuronal dysfunction has recently been extended by the discovery of small, soluble, oligomers of the Aβ peptide, some forms of which have been termed ADDLs (Aβ-derived diffusible ligands), protofibrils, or Aβ*56 [3–6]. These Aβ oligomers are not only potential intermediates in the formation of amyloid filaments, but they also have been shown to be neurotoxic themselves and to inhibit long-term potentiation (LTP), a cellular model of memory, in hippocampal slices [4, 7, 8]. Thus, the Amyloid Cascade Hypothesis now includes the essential role of Aβ oligomers in the neurodegeneration process.
Despite its strength, the Amyloid Cascade Hypothesis is incomplete without including the essential role of amyloid-associated inflammatory proteins. For example, biochemical and histological studies first showed that, in addition to Aβ, amyloid deposits also contained the inflammation/acute phase protein α1-antichymotrypsin (ACT) [9] and, later, apolipoprotein E (apoE) [10, 11], which were both hypothesized to serve as catalysts or “pathological chaperones” of amyloid formation [9, 11, 12]. These and other results also indicated that Alzheimer's disease and its manifestation in middle-aged Down syndrome may include an inflammatory process, for both ACT and apoE are inflammatory and/or acute phase proteins in other contexts, and both are overexpressed in affected regions of the AD brain (for reviews see [13–15]). Indeed, Alzheimer himself first identified the inflammatory component of Alzheimer's disease when he described reactive astrocytes and microglia in affected brain regions of his first patient [16]. However, until inflammatory proteins such as ACT, IL-1, HLA, and apoE were found to be overexpressed in AD and DS brains, the term “inflammation” was explicitly excluded from the clinical and pathological description of AD because of the lack of edema and lymphocyte infiltration [9–11, 17, 18].
The significance of these biochemical results instigated and was reinforced by parallel genetic discoveries implicating a role for inflammation in AD. In particular, inheritance of the apoE ε4 allele was found to be the strongest known risk factor for AD besides age, with one copy increasing AD risk 3–5-fold and two copies over 10-fold [19–21]. Furthermore, apoE ε4 promotes cognitive decline in middle-aged Down syndrome individuals [22].
Because of apoE's essential genetic, and therefore presumably biochemical, contribution to AD pathology and cognitive decline, it is critical that its role in the AD pathogenic pathway/amyloid cascade be elucidated in order for therapeutics based on apoE to be designed. While recent excellent and encyclopedic literature reviews describe the many potential roles that apoE plays in AD [23–26], this focused review will concentrate on the interaction between Aβ and apoE and other inflammatory proteins, on the effects of such interactions, and on their implications for designing apoE-based AD therapies. The central question we try to answer is whether increasing or decreasing apoE level and/or function will serve best to reduce AD/DS pathology and cognitive decline. Lack of a clear answer may lead to the development of drugs that, rather than serving as an AD therapy, instead potentially exacerbate the disease.
2. Background: ApoE as Amyloid Catalyst
To determine whether inflammation contributes to Alzheimer's disease rather than being merely a correlative pathological feature in the AD brain, we and others tested the hypothesis that ACT and/or apoE serve as amyloid catalysts or pathological chaperones. Numerous in vitro and in vivo studies showed that mature amyloid deposition and the associated cognitive decline is strongly stimulated by apoE and ACT in a dose-dependent and isoform-specific manner, with apoE4 being the strongest promoter of Aβ polymerization and apoE2 being an inhibitor, paralleling the effect of these two isoforms in humans [27–38]. Indeed, without one or the other of these amyloid catalysts expressed in the brain, amyloid deposition is profoundly delayed in APP transgenic mice and does not become filamentous. Such APP+/apoE KO animals also exhibit normal cognition despite levels of Aβ expression equal to the apoE-expressing APP animals. Elegant work by Manelli and colleagues also showed that native lipidated apoE4 from transgene replacement astrocytes increases Aβ neurotoxicity compared to apoE3 or E2, indicating that apoE4 provides a negative gain of function [39]. Finally, Jones and colleagues recently showed that apoE4 also promotes the conversion and enhanced synaptic localization of Aβ as oligomers, the most neurotoxic form of the Alzheimer amyloid peptide [40, 41]. These recent studies extended prior work showing that apoE copurifies with Aβ during biochemical isolation of amyloid from human brains, and that apoE preferentially interacts with Aβ peptides in a β-sheet structure [42–45].
Together these results show that inflammatory proteins, particularly apoE, are integral parts of the amyloid cascade, and that without them the cascade would be arrested at the level of the harmless Aβ monomer, and no AD would ensue.
3. Background: ApoE in Aβ/Amyloid Clearance
The view of apoE as an integral and pathological part of the amyloid cascade has been shaken by experiments that suggest that apoE, far from being an amyloid catalyst, serves to clear Aβ from the brain. Under this view, ApoE is protective, with human apoE4 being less protective than apoE3 or E2 (for the most recent discussion, see [46] and commentary at http://www.alzforum.com/).
The first experiments that suggested apoE's role as a neuroprotector examined the pathology and cognition of APP transgenic mice carrying a second transgene expressing one or another human apoE isoform. Contrary to expectations, amyloid deposition in these mice was inhibited by the human apoE transgene, as though human apoE was protective [47]. Ultimately, the mice did develop amyloid, with the apoE4-expressing strain accumulating earlier and more extensive pathology [33, 34, 48, 49]. It was proposed that human apoE might serve to inhibit Aβ clearance from the brain compared to mouse apoE, with apoE4 inhibiting clearance the most. Other experiments showed that indeed, clearance of Aβ species was inhibited by complexing with apoE, especially apoE4 [46, 50].
The possibility that interaction with apoE modulated an Aβ clearance mechanism appeared to be supported by the finding that introduction of anti-Aβ antibodies or other Aβ-binding proteins such as gelsolin, led to a reduced amyloid load in the brain and rapidly improved cognition, with little evidence of Aβ-binding agents invading the brain parenchyma [41–54]. We also introduced apoE itself into the circulation via parabiosis and found that it induced amyloid clearance without entering the brain in AD model mice [38]. Thus the “Peripheral Sink Hypothesis” became a viable alternative or addition to the Amyloid Cascade Hypothesis, with apoE potentially playing an additional role as an Aβ-binding peripheral protein.
Most recently, an approach to therapy has been investigated in AD mice that is based on activating the liver X receptor (LXR), which also exists on other cells including microglia [55–57]. Activation of LXR results in increased expression of many proteins including apoE and its lipidating enzyme, ATP-binding Cassette Transporter A1 (ABCA1). The results indicate that activating LXR with the ligand GW3965 or the FDA-approved antiskin cancer drug bexarotene reduces soluble and insoluble Aβ and improves cognition in APP Tg mice, while knocking out the ABCA1 gene in APP mice showed a tendency to reduced amyloid load. Because apoE expression and lipidation is stimulated by LXR activation, the results were interpreted as proof that increased apoE levels help microglia clear Aβ and amyloid, as indeed some earlier cell culture experiments had suggested. However, it has also been shown that genetic overexpression of ABCA1 reduces amyloid deposition in mice where the apoE levels are unchanged [58]. Hence, because LXR stimulation influences the levels of many proteins, it is problematic to definitively link its in vivo action to the altered level of one particular protein. Furthermore, the increased levels of ABCA1 induced by Bexarotene enhance apoE lipidation, a change that is known to alter apoE/Aβ interactions. Hence, it is important to consider the lipidation state of apoE, which affects its function, in addition to the absolute levels of apoE.
4. Synthesis
When trying to distinguish and weigh the value of two hypotheses, it is instructive to consider their testable predictions. If apoE is an amyloid catalyst, then reducing apoE levels or function in the brain should result in reduced amyloid deposition and reduced cognitive decline. If on the other hand, apoE is involved in Aβ clearance with human apoE4 being a greater inhibitor of clearance (or poorer clearer), then reducing apoE levels or apoE binding to Aβ should increase amyloid deposition and cognitive decline.
All experiments carried out so far in vitro or in transgenic mice indicate that the ability of Aβ to form neurotoxic filaments or oligomers and cause cognitive decline are increased in the presence of apoE, particularly mouse apoE and human apoE4, with apoE2 being protective. In contrast, in the complete absence of apoE, the mutant APP gene and its product Aβ are harmless, generating neither amyloid deposits, synaptic disfunction, or cognitive decline, with one copy of apoE having an intermediate effect, as discussed above. The in vitro experiments in particular indicate that apoE likely acts catalytically to promote Aβ polymerization, as the molar ratio of Aβ to apoE of about 200/1 was appropriate for the formation of neurotoxic products [27–30]. Most recently, earlier work showing that mice expressing only one apoE gene accumulated less amyloid than those with two apoE genes (32) was repeated in two different laboratories using human apoE knock-in mice, and the same result was found, that is, lower doses of apoE3 or apoE4 led to reduced amyloid deposition [59, 60].
The simplest interpretation of the in vitro, cell culture, and transgenic mouse data is that apoE is necessary for Aβ to polymerize into neurotoxic oliogomers/filaments, probably by binding to Aβ and thus altering its structure more toward the β-sheet and more easily allowing successive Aβ peptides to add on to the growing chain. The recent finding that apoE promotes Aβ oligomer formation in vivo reinforces this interpretation [40, 41]. Whether apoE is only needed to initiate the polymerization or also to prepare each peptide for addition to the growing filament is not yet known.
Even though the key predictions of the polymerization hypothesis, that is apoE serving as an Aβ filament catalyst, have been borne out, the compelling experiments demonstrating that human apoE inhibits filament formation in a mouse background require explanation. Furthermore, data from LaDu and colleagues and by others have shown that lipidated apoE, presumably the prevalent form in vivo, binds Aβ with an affinity of E2 > E3 > E4 [61–64]. Finally, the elegant and thorough experiments of Castellano and colleagues show very convincingly that expression of a human apoE4 transgene (in the absence of mouse apoE) leads to a longer half-life, (i.e., slower clearance) of Aβ in the brain interstitial fluid compared to E2 or E3 [46].
The apoE-Aβ binding studies might be interpreted as support for apoE functioning in Aβ clearance because apoE2, for example, would bind Aβ tightly and could thereby promote its removal from the interstitial fluid via LRP receptors [50, 61–64]. However, an important feature of any catalyst is that it must bind its substrate only tightly enough to convert it to the transition state structure and then release it as the reaction is completed [65, 66]. If a mutation leads to an overly tight substrate binding, then no further reaction can occur. Thus apoE2 could indeed bind Aβ most tightly, and thereby not only prevent apoE4 from binding and promoting Aβ oligo/polymerization, but also prevent the spontaneous polymerization of the peptide.
The ability of different apoE isoforms to bind Aβ with different strengths can also explain why human apoE isoforms slow amyloid deposition in the presence of the endogenous mouse apoE, for they may bind Aβ more tightly or differently than mouse apoE and slow the catalytic conversion of Aβ into oligomers/polymers in the mouse background.
The data showing that human apoE inhibits Aβ clearance can also be interpreted as reflecting apoE's role in catalyzing Aβ oligo/polymerization. Pathologic macromolecular structures are often resistant to various clearance mechanisms designed for monomeric species, whether by intracellular proteasome degradation or cross-membrane/BBB transfer, thus allowing their accumulation. Only when oligo/polymeric structures are anticipated and physiological clearance mechanisms are in place to handle them, as for antibody-antigen complexes, will clearance be facilitated by conversion to larger structures. Because apoE clearly has the ability to catalyze the conversion of Aβ into oligomeric and polymeric structures, it is reasonable to assume that those structures will be more difficult to clear, and that such difficulty will be detected as clearance inhibition in the brain, for instance, by apoE4, in pulse chase type experiments, while the higher apoE levels in blood may aid the clearance of Aβ from the circulations (Figure 1).
Figure 1.
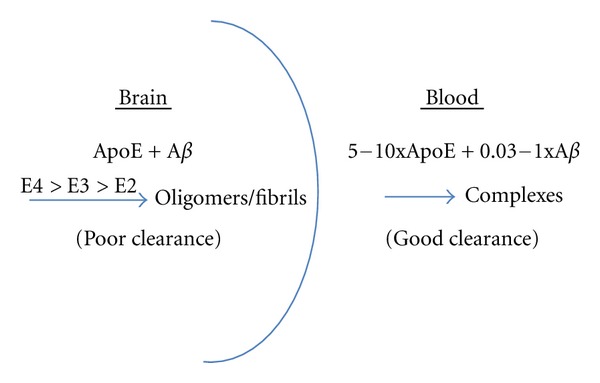
ApoE promotes Aβ Fibril formation in brain and Aβ clearance from the blood. Data from both in vitro and in vivo experiments indicate that apoE, especially apoE4 promotes the polymerization of Aβ into oligomers and polymers that accumulate in the brain and are difficult to clear. In contrast, the concentration of apoE is higher in the blood, while those of Aβ species are equivalent to or lower than in the brain, promoting the formation and clearance of equimolar apoE-Aβ complexes.
Finally, the ability of GW3965 and Bexarotene to reduce soluble and insoluble Aβ in the brain of Tg APP mice and improve cognition is most easily understood as resulting from a general activation of the phagocytic activity of microglia. Previous work showed that activation of microglia by acute intracerebral treatment of APP mice with LPS or with Granulocyte-macrophage stimulating factor can similarly reduce amyloid load and improve cognition [67–69] but that long-term peripheral treatment with LPS exacerbated amyloid deposition in an apoE-dependent manner [70]. Stimulation of microglial activity via induction of Toll-like receptor 9 (TLR9) has also been shown to greatly reduce amyloid load and improve cognition [71]. Clearly the interaction of neuroinflammation, microglia, and amyloid load is complex, and the fact that bexarotene “cures” AD in mice is more likely to be despite, rather than because it stimulates expression of apoE.
5. Aβ Binding Proteins and AD Therapy
A good test of any hypothesis about the pathogenesis of a disease is whether it successfully predicts how the pathogenesis can be inhibited or reversed. For example, small fragments of Aβ corresponding to the amino acid sequences to which ACT (Aβ1-12) and apoE (Aβ12-28) bind can serve as decoy peptides that prevent the binding of apoE to Aβ and its catalysis of Aβ into neurotoxic species [30]. This early in vitro work has recently been repeated and confirmed in other laboratories [72, 73]. The decoy principal was extended in vivo by preparing a version of Aβ12-28 that has a better plasma 1/2 life and is nonfibrillogenic/nontoxic. It was shown that this peptide could be peripherally introduced into a transgenic APP mouse, where it effectively entered the brain and prevented/reversed oligomer formation, amyloid deposition, and cognitive decline [74–76]. Similarly, amyloid plaques in APP mice contain mouse ACT and injecting Aβ1-11 into one side of the APP mouse brain to block ACT's binding site with endogenous Aβ rapidly reduced amyloid load compared to the other, vehicle-injected side of the brain. Furthermore the inflammatory cytokine IL-1 that is overexpressed in AD brain [18] induces astrocyte expression of ACT [77], and blocking IL-1 expression in APP transgenic mice by Ibuprofen treatment, thereby reducing mouse ACT expression, lowers amyloid formation and restores cognition [78]. Evidently, blocking ACT or apoE expression or function, both in vitro or in vivo, successfully prevents Aβ pathology and neurotoxicity.
Apolipoprotein J also binds Aβ and can be shown to aid its passage across the blood brain barrier [79–83]. Interestingly, knocking out either apoJ or apoE reduces amyloid deposition in APP transgenic animals, yet knocking out both leads to robust amyloid deposition at an even earlier age than arises in nonmanipulated APP animals [84]. This result may reflect the ability of mouse ACT to promote amyloid formation, but that in the presence of the stronger binding apoE and apoJ proteins mouse ACT is prevented from exhibiting its catalytic activity.
6. Potential Efficacy and Dangers of Aβ-Binding Antibodies as AD Therapy
The role of apoE and ACT in the Alzheimer pathogenic pathway has potentially general implications. One of the most studied classes of Aβ binding proteins are specific anti-Aβ antibodies, which form the basis of both passive and active immunization therapies for Alzheimer's disease (for review see [85]). The finding that apoE and ACT can catalyze Aβ oligo/polymerization begs the question of whether Aβ antibodies might also promote or inhibit Aβ polymerization. Indeed we found that two Aβ antibodies, 6E10 which is directed to the same the N-terminal sequence bound by ACT, and 13 M, which binds to the C-terminus, function very differently in the in vitro Aβ polymerization assay. 6E10 inhibits ACT-catalyzed polymerization of Aβ while 13 M inhibits ACT catalysis much less and even promotes some polymerization itself. Interestingly, the N-terminus of Aβ is also the target of many attempts at AD immunotherapy with the aim of inducing microglial phagocytosis of neurotoxic Aβ species. Yet removing the microglial-binding Fc portion of 3D6 antibodies to Aβ1-5 to generate Fab'2 fragments does not reduce the antibody's ability to remove diffuse amyloid in APP mice [86]. Evidently, only its Aβ-binding feature is required to allow the antibody to remove amyloid. A possible explanation for this result is that the antibody functions by blocking Aβ interaction with mouse ACT. The consequent suppression of ACT-catalyzed oligo/polymerization could thus tilt the dynamic process of plaque development toward depolymerization.
These results illustrate the fact that Aβ-binding proteins can have multiple effects on polymerization and that their full range of activities must be considered when using them as potential targets or tools for therapeutic intervention.
7. Potential Toxic Mechanism of ApoE-Induced Aβ Oligomers
Although Aβ oligomers have been shown to be highly neurotoxic in vitro and in vivo, and their formation is promoted by apoE4, the mechanism of their toxicity is still being elucidated. The data reviewed above coupled to other recent findings suggest a novel mechanism for Aβ toxicity that encompasses the essential role of apoE. Specifically, Aβ oligomers bind to and inhibit certain microtubule motors that are essential for the function and stability of the mitotic spindle—Eg5/kinesin5, Kif4A, and MCAK [87]. Similar motors, including kinesin 5, are also present in mature neurons [88, 89]. We have found recently that inhibition of MT motor function by Aβ or by the specific kinesin 5 inhibitor Monastrol prevents the efficient transport of receptors such as the LDLR, the NMDA neurotransmitter receptor, and the p75 neurotrophin receptor to the cell surface, resulting in reduced function ([90]; in preparation). Similarly, apoE, particularly apoE4, has been shown to reduce the cell surface levels and function of NMDA, AMPA, and apoEr2 receptors in neurons [91]. This latter finding can now be understood as potentially reflecting the ability of apoE4 to promote the conversion of endogenous neuronal Aβ into oligomers, which then inhibit MT-based transport of key cellular components such as receptors to their functional location.
8. Conclusion
In sum, it appears that the preponderance of the data can be most consistently interpreted as showing that the brain inflammatory protein apoE plays a catalytic role in the AD/DS amyloid cascade and consequent cognitive decline, with binding and clearance differences between the apoE isoforms reflecting their differing abilities to bind to Aβ and catalyze its conversion into neurotoxic macromolecular species (Figure 2). This conclusion, and the in vivo demonstration that blocking apoE-Aβ interaction prevents AD in a mouse model, suggests that this decoy approach should be translatable into human patients and serve as an effective new approach to AD therapy.
Figure 2.
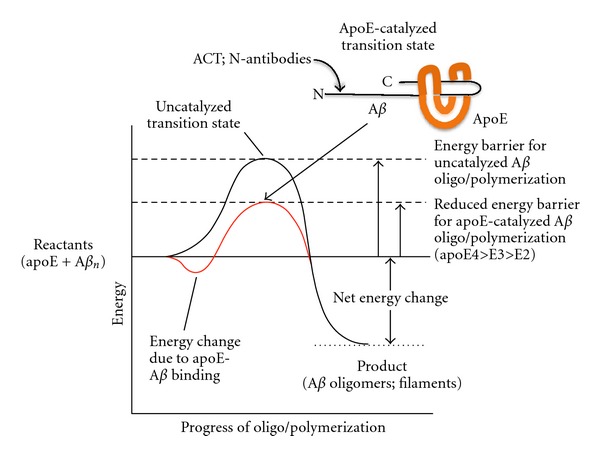
Conceptual energy diagram of ApoE-catalyzed Aβ oligo/polymerization. Although Aβ can polymerize spontaneously, the reaction is greatly promoted by apoE in vitro and in vivo. This catalysis can be understood in terms of the energy diagram shown. The first energy change, a reduction, occurs as apoE binds to amino acids 12–28 of Aβ, with different apoE isoforms binding with different affinities. Then apoE apparently alters the structure of its bound Aβ to a higher-energy β-sheet conformation (the transition state), which allows additional Aβ molecules to add and form a larger oligomer or fibril. These products have lower energy than either the transition state or the initial reactants (apoE and Aβ), thus driving the reaction to completion. Because the energy of the apoE-Aβ transition state is lower than either the transition state of monomeric Aβ in a β-sheet conformation, the oligo/polymerization reaction is effectively catalyzed by apoE. ApoE4 evidently forms the lowest energy transition state and thus strongly catalyzes the reaction, apoE3 catalyzes the reaction less well, and apoE2 likely forms such a high energy transition state that it effectively inhibits the spontaneous Aβ polymerization reaction. Antichymotrypsin (ACT), which binds to Aβ amino acids 1–12, also catalyzes Aβ polymerization, while Aβ antibodies can either promote Aβ fibrillization themselves or interfere with ACT or apoE-catalyzed polymerization. Molecules, including antibodies, that prevent apoE or ACT binding to Aβ are being developed as AD therapies that leave the normal physiological functions of Aβ and apoE or ACT intact, while blocking their pathological interaction.
Other Aβ-binding proteins may be similarly manipulated by a decoy approach to reduce oligomerization and polymerization of Aβ into neurotoxic species. However, the finding that different antibodies to Aβ can both inhibit ACT-catalyzed Aβ polymerization and promote polymerization of Aβ itself, argues that immunotherapy must be approached with care to avoid the use or induction of antibodies that can catalyze further oligo/polymerization of Aβ, instead of inducing its phagocytosis and removal. Furthermore, human and mouse intracerebral environments may differ in important ways with respect to the pattern and activities of Aβ-binding proteins and may also respond differently to intervention or inflammation. Such differences may explain why so many treatments that were successful in reducing amyloid-dependent cognitive decline in transgenic mice have failed to translate into human AD patients.
Finally, the ability of Aβ oligomers to inhibit key microtubule motors and prevent the transport of neurotrophin, neurotransmitter, and other receptors to the cell surface may underlie their neuronal toxicity. It is apparently the ApoE-, especially E4-dependent formation of such Aβ oligomers, that constitutes the key catalytic step in the AD pathogenic pathway.
Acknowledgments
Funding provided by the Byrd Alzheimer's Institute, the Eric Pfeiffer Chair for Research on Alzheimer's Disease, and the NIH (AG025711, AG037942, NS073502 and AG020245).
References
- 1.Hardy J. Alzheimer’s disease: the amyloid cascade hypothesis—an update and reappraisal. Journal of Alzheimer’s Disease. 2006;9(3):151–153. doi: 10.3233/jad-2006-9s317. [DOI] [PubMed] [Google Scholar]
- 2.Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nature Reviews Drug Discovery. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 3.Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid β-protein fibrillogenesis: detection of a protofibrillar intermediate. Journal of Biological Chemistry. 1997;272(35):22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 4.Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(11):6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Podlisny MB, Walsh DM, Amarante P, et al. Oligomerization of endogenous and synthetic amyloid β-protein at nanomolar levels in cell culture and stabilization of monomer by Congo red. Biochemistry. 1998;37(11):3602–3611. doi: 10.1021/bi972029u. [DOI] [PubMed] [Google Scholar]
- 6.Lesné S, Ming TK, Kotilinek L, et al. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440(7082):352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 7.Wang HW, Pasternak JF, Kuo H, et al. Soluble oligomers of β amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Research. 2002;924(2):133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- 8.Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo . Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 9.Abraham CR, Selkoe DJ, Potter H. Immunochemical identification of the serine protease inhibitor α1-antichymotrypsin in the brain amyloid deposits of Alzheimer’s disease. Cell. 1988;52(4):487–501. doi: 10.1016/0092-8674(88)90462-x. [DOI] [PubMed] [Google Scholar]
- 10.Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Research. 1991;541(1):163–166. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- 11.Wisniewski T, Frangione B. Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neuroscience Letters. 1992;135(2):235–238. doi: 10.1016/0304-3940(92)90444-c. [DOI] [PubMed] [Google Scholar]
- 12.Wisniewski T, Lalowski M, Golabek A, Vogel T, Frangione B. Is Alzheimer’s disease an apolipoprotein E amyloidosis? The Lancet. 1995;345(8955):956–958. doi: 10.1016/s0140-6736(95)90701-7. [DOI] [PubMed] [Google Scholar]
- 13.Nilsson L, Rogers J, Potter H. The essential role of inflammation and induced gene expression in the pathogenic pathway of Alzheimer’s disease. Frontiers in Bioscience. 1998;3:d436–d446. doi: 10.2741/a290. [DOI] [PubMed] [Google Scholar]
- 14.Potter H, Wefes IM, Nilsson LNG. The inflammation-induced pathological chaperones ACT and apo-E are necessary catalysts of Alzheimer amyloid formation. Neurobiology of Aging. 2001;22(6):923–930. doi: 10.1016/s0197-4580(01)00308-6. [DOI] [PubMed] [Google Scholar]
- 15.Rozemuller AJM, van Gool WA, Eikelenboom P. The neuroinflammatory response in plaques and amyloid angiopathy in Alzheimer’s disease: therapeutic implications. Current Drug Targets. 2005;4(3):223–233. doi: 10.2174/1568007054038229. [DOI] [PubMed] [Google Scholar]
- 16.Alzheimer A. Uber eine eigenartige Erkrankung der Hirnride. Allgemeine Zeitschrift fur Psychiatrie und Psychisch-gerichtliche Medizin. 1907;64:146–148. [Google Scholar]
- 17.Rogers J, Luber-Narod J, Styren SD, Civin WH. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer’s disease. Neurobiology of Aging. 1988;9(4):339–349. doi: 10.1016/s0197-4580(88)80079-4. [DOI] [PubMed] [Google Scholar]
- 18.Griffin WST, Stanley LC, Ling C, et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(19):7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strittmatter WJ, Saunders AM, Schmechel D, et al. Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(5):1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 21.Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:4725–4727. doi: 10.1073/pnas.92.11.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubinsztein DC, Hon J, Stevens F, et al. Apo E genotypes and risk of dementia in Down syndrome. American Journal of Medical Genetics. 1999;88:344–347. doi: 10.1002/(sici)1096-8628(19990820)88:4<344::aid-ajmg10>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 23.Mahley RW, Huang Y, Weisgraber KH. Detrimental effects of apolipoprotein E4: potential therapeutic targets in Alzheimer’s disease. Current Alzheimer Research. 2007;4(5):537–540. doi: 10.2174/156720507783018334. [DOI] [PubMed] [Google Scholar]
- 24.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63(3):287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. The Lancet Neurology. 2011;10(3):241–252. doi: 10.1016/S1474-4422(10)70325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hauser PS, Narayanaswami V, Ryan RO. Apolipoprotein E: from lipid transport to neurobiology. Progress in Lipid Research. 2011;50(1):62–74. doi: 10.1016/j.plipres.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma J, Yee A, Brewer HB, Das S, Potter H. Amyloid-associated proteins α1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature. 1994;372(6501):92–94. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- 28.Wisniewski T, Castano EM, Golabek A, Vogel T, Frangione B. Acceleration of Alzheimer’s fibril formation by apolipoprotein E in vitro. American Journal of Pathology. 1994;145(5):1030–1035. [PMC free article] [PubMed] [Google Scholar]
- 29.Sanan DA, Weisgraber KH, Russell SJ, et al. Apolipoprotein E associates with β amyloid peptide of Alzheimer’s disease to form novel monofibrils. Isoform ApoE4 associates more efficiently than ApoE3. Journal of Clinical Investigation. 1994;94(2):860–869. doi: 10.1172/JCI117407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma J, Brewer HB, Jr., Potter H. Alzheimer Aβ neurotoxicity: promotion by antichymotrypsin, ApoE4; inhibition by Aβ-related peptides. Neurobiology of Aging. 1996;17(5):773–780. doi: 10.1016/0197-4580(96)00112-1. [DOI] [PubMed] [Google Scholar]
- 31.Bales KR, Verina T, Dodel RC, et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nature genetics. 1997;17(3):263–264. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- 32.Bales KR, Verina T, Cummins DJ, et al. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(26):15233–15238. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holtzman DM, Fagan AM, Mackey B, et al. Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer's disease model. Annals of Neurology. 2000;47(6):739–747. [PubMed] [Google Scholar]
- 34.Holtzman DM, Bales KR, Tenkova T, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(6):2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mucke L, Yu GQ, McConlogue L, Rockenstein EM, Abraham CR, Masliah E. Astroglial expression of human α1-antichymotrypsin enhances Alzheimer-like pathology in amyloid protein precursor transgenic mice. American Journal of Pathology. 2000;157(6):2003–2010. doi: 10.1016/s0002-9440(10)64839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nilsson LNG, Bales KR, DiCarlo G, et al. α-1-Antichymotrypsin promotes β-sheet amyloid plaque deposition in a transgenic mouse model of Alzheimer’s disease. Journal of Neuroscience. 2001;21(5):1444–1451. doi: 10.1523/JNEUROSCI.21-05-01444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Potter H. Beyond beta protein—the essential role of inflammation in Alzheimer Amyloid formation. In: Bondy SC, Cambell A, editors. Inflammatory Events in Neurodegeneration. Prominent Press; 2001. [Google Scholar]
- 38.Nilsson LNG, Gografe S, Costa DA, Hughes T, Dressler D, Potter H. Use of fused circulations to investigate the role of apolipoprotein E as amyloid catalyst and peripheral sink in Alzheimer’s disease. doi: 10.3727/194982412X13462021398010. Technology and Innovation. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manelli AM, Bulfinch LC, Sullivan PM, LaDu MJ. Aβ42 neurotoxicity in primary co-cultures: effect of apoE isoform and Aβ conformation. Neurobiology of Aging. 2007;28(8):1139–1147. doi: 10.1016/j.neurobiolaging.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones PB, Adams KW, Rozkalne A, et al. Apolipoprotein E: isoform specific differences in tertiary structure and interaction with amyloid-β in human Alzheimer brain. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0014586. Article ID e14586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koffie RM, Hashimoto T, Tai H-C, et al. Apolipoprotein E4 effects in Alzheimer disease are mediated by synaptotoxic oligomeric amyloid-beta. Brain. 2012;135:2155–2168. doi: 10.1093/brain/aws127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wisniewski T, Lalowski M, Golabek A, Vogel T, Frangione B. Is Alzheimer’s disease an apolipoprotein E amyloidosis? The Lancet. 1995;345(8955):956–958. doi: 10.1016/s0140-6736(95)90701-7. [DOI] [PubMed] [Google Scholar]
- 43.Naslund J, Thyberg J, Tjernberg LO, et al. Characterization of stable complexes involving apolipoprotein E and the amyloid β peptide in Alzheimer’s disease brain. Neuron. 1995;15(1):219–228. doi: 10.1016/0896-6273(95)90079-9. [DOI] [PubMed] [Google Scholar]
- 44.Golabek AA, Soto C, Vogel T, Wisniewski T. The Interaction between apolipoprotein E and Alzheimer’s amyloid β-peptide is dependent on β-peptide conformation. Journal of Biological Chemistry. 1996;271(18):10602–10606. doi: 10.1074/jbc.271.18.10602. [DOI] [PubMed] [Google Scholar]
- 45.Wahrle SE, Holtzman DM. Apolipoprotein E, amyloid beta-peptide and Alzheimer's disease. In: Dawbarn D, Allen SJ, editors. Neurobiology of Alzheimer's Disease. Oxford University Press; 2007. pp. 161–172. [Google Scholar]
- 46.Castellano JM, Kim J, Stewart FR, et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Science Translational Medicine. 2011;3(89) doi: 10.1126/scitranslmed.3002156. Article ID 89ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holtzman DM, Bales KR, Wu S, et al. Expression of human apolipoprotein E reduces amyloid-β deposition in a mouse model of Alzheimer’s disease. Journal of Clinical Investigation. 1999;103(6):R15–R21. doi: 10.1172/JCI6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM. Human and murine apoE markedly alters Aβ metabolism before and after plaque formation in a mouse model of Alzheimer’s Disease. Neurobiology of Disease. 2002;9(3):305–318. doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- 49.Dodart JC, Marr RA, Koistinaho M, et al. Gene delivery of human apolipoprotein E alters brain Aβ burden in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(4):1211–1216. doi: 10.1073/pnas.0409072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deane R, Sagare A, Hamm K, et al. apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. Journal of Clinical Investigation. 2008;118(12):4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(15):8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dodart JC, Bales KR, Gannon KS, et al. Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer’s disease model. Nature Neuroscience. 2002;5(5):452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- 53.DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-β efflux: a measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science. 2002;295(5563):2264–2267. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- 54.Matsuoka Y, Saito M, LaFrancois J, et al. Novel therapeutic approach for the treatment of Alzheimer’s disease by peripheral administration of agents with an affinity to β-amyloid. Journal of Neuroscience. 2003;23(1):29–33. doi: 10.1523/JNEUROSCI.23-01-00029.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang Q, Lee CYD, Mandrekar S, et al. ApoE promotes the proteolytic degradation of Aβ . Neuron. 2008;58(5):681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Donkin JJ, Stukas S, Hirsch-Reinshagen V, et al. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. Journal of Biological Chemistry. 2010;285(44):34144–34154. doi: 10.1074/jbc.M110.108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cramer PE, Cirrito JR, Wesson DW, et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335(6075):1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wahrle SE, Jiang H, Parsadanian M, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. Journal of Clinical Investigation. 2008;118(2):671–682. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim J, Jiang H, Park S, et al. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. The Journal of Neuroscience. 2011;31:18007–18012. doi: 10.1523/JNEUROSCI.3773-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bien-Ly N, Gillespie AK, Walker D, Yuon SY, Huang Y. Reducing human apolipoprotein E levels attenuates age dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. The Journal of Neuroscience. 2012;32(14):4803–4811. doi: 10.1523/JNEUROSCI.0033-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE. Isoform-specific binding of apolipoprotein E to β-amyloid. Journal of Biological Chemistry. 1994;269(38):23403–23406. [PubMed] [Google Scholar]
- 62.Aleshkov S, Abraham CR, Zannis VI. Interaction of nascent apoe2, apoe3, and apoe4 isoforms expressed in mammalian cells with amyloid peptide β (1–40). Relevance to Alzheimer’s disease. Biochemistry. 1997;36(34):10571–10580. doi: 10.1021/bi9626362. [DOI] [PubMed] [Google Scholar]
- 63.Tokuda T, Calero M, Matsubara E, et al. Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer’s amyloid β peptides. Biochemical Journal. 2000;348(2):359–365. [PMC free article] [PubMed] [Google Scholar]
- 64.Deane R, Wu Z, Sagare A, et al. LRP/amyloid β-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron. 2004;43(3):333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 65.Dressler D, Potter H. Discovering Enzymes. New York, NY, USA: Scientific American Library, W. H. Freeman; 1991. [Google Scholar]
- 66.Fersht A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York, NY, USA: W. H. Freeman; 1999. [Google Scholar]
- 67.Herber DL, Roth LM, Wilson D, et al. Time-dependent reduction in Aβ levels after intracranial LPS administration in APP transgenic mice. Experimental Neurology. 2004;190(1):245–253. doi: 10.1016/j.expneurol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 68.Herber DL, Mercer M, Roth LM, et al. Microglial activation is required for Aβ clearance after intracranial injection of lipopolysaccharide in APP transgenic mice. Journal of Neuroimmune Pharmacology. 2007;2(2):222–231. doi: 10.1007/s11481-007-9069-z. [DOI] [PubMed] [Google Scholar]
- 69.Boyd TD, Bennett SP, Mori T, et al. GM-CSF upregulated in rheumatoid arthritis reverses cognitive impairment and amyloidosis in Alzheimer mice. Journal of Alzheimer’s Disease. 2010;21(2):507–518. doi: 10.3233/JAD-2010-091471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qiao X, Cummins DJ, Paul SM. Neuroinflammation-induced acceleration of amyloid deposition in the APPV717F transgenic mouse. European Journal of Neuroscience. 2001;14(3):474–482. doi: 10.1046/j.0953-816x.2001.01666.x. [DOI] [PubMed] [Google Scholar]
- 71.Scholtzova H, Kascsak RJ, Bates KA, et al. Induction of toll-like receptor 9 signaling as a method for ameliorating alzheimer’s disease-related pathology. Journal of Neuroscience. 2009;29(6):1846–1854. doi: 10.1523/JNEUROSCI.5715-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hao J, Zhang W, Zhang P, et al. Aβ20-29 peptide blocking apoE/Aβ interaction reduces full-length Aβ42/40 fibril formation and cytotoxicity in vitro. Neuropeptides. 2010;44(4):305–313. doi: 10.1016/j.npep.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 73.Liu Q, Wu WH, Fang CL, et al. Mapping ApoE/Abeta binding regions to guide inhibitor discovery. Molecular BioSystems. 2011;7:1693–1700. doi: 10.1039/c1mb05019b. [DOI] [PubMed] [Google Scholar]
- 74.Sadowski M, Pankiewicz J, Scholtzova H, et al. A synthetic peptide blocking the apolipoprotein E/β-amyloid binding mitigates β-amyloid toxicity and fibril formation in vitro and reduces β-amyloid plaques in transgenic mice. American Journal of Pathology. 2004;165(3):937–948. doi: 10.1016/s0002-9440(10)63355-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sadowski MJ, Pankiewicz J, Scholtzova H, et al. Blocking the apolipoprotein E/amyloid-β interaction as a potential therapeutic approach for Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(49):18787–18792. doi: 10.1073/pnas.0604011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang J, Ji Y, Mehta P, Bates KA, Sun Y, Wisniewski T. Blocking the apolipoprotein E/Amyloid-β interaction reduces fibrillar vascular amyloid deposition and cerebral microhemorrhages in TgSwDI mice. Journal of Alzheimer’s Disease. 2011;24(2):269–285. doi: 10.3233/JAD-2011-101401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Das S, Potter H. Expression of the Alzheimer amyloid-promoting factor antichymotrypsin is induced in human astrocytes by IL-1. Neuron. 1995;14(2):447–456. doi: 10.1016/0896-6273(95)90300-3. [DOI] [PubMed] [Google Scholar]
- 78.Morihara T, Teter B, Yang F, et al. Ibuprofen suppresses interleukin-Iβ induction of pro-amyloidogenic α1-antichymotrypsin to ameliorate β-amyloid (aβ) pathology in Alzheimer’s models. Neuropsychopharmacology. 2005;30(6):1111–1120. doi: 10.1038/sj.npp.1300668. [DOI] [PubMed] [Google Scholar]
- 79.Ghiso J, Matsubara E, Koudinov A, et al. The cerebrospinal-fluid soluble form of Alzheimer’s amyloid beta is complexed to SP-40,40 (apolipoprotein J), an inhibitor of the complement membrane-attack complex. Biochemical Journal. 1993;293(1):27–30. doi: 10.1042/bj2930027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Koldamova RP, Lefterov IM, Lefterova MI, Lazo JS. Apolipoprotein A-I directly interacts with amyloid precursor protein and inhibits Aβ aggregation and toxicity. Biochemistry. 2001;40(12):3553–3560. doi: 10.1021/bi002186k. [DOI] [PubMed] [Google Scholar]
- 81.Paula-Lima AC, Tricerri MA, Brito-Moreira J, et al. Human apolipoprotein A-I binds amyloid-β and prevents Aβ-induced neurotoxicity. International Journal of Biochemistry and Cell Biology. 2009;41(6):1361–1370. doi: 10.1016/j.biocel.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 82.Bell RD, Sagare AP, Friedman AE, et al. Transport pathways for clearance of human Alzheimer’s amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. Journal of Cerebral Blood Flow and Metabolism. 2007;27(5):909–918. doi: 10.1038/sj.jcbfm.9600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ito S, Ohtsuki S, Kamiie J, Nezu Y, Terasaki T. Cerebral clearance of human amyloid-β peptide (1-40) across the blood-brain barrier is reduced by self-aggregation and formation of low-density lipoprotein receptor-related protein-1 ligand complexes. Journal of Neurochemistry. 2007;103(6):2482–2490. doi: 10.1111/j.1471-4159.2007.04938.x. [DOI] [PubMed] [Google Scholar]
- 84.DeMattos RB, O’dell MA, Parsadanian M, et al. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(16):10843–10848. doi: 10.1073/pnas.162228299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-β immunotherapy? Nature Reviews Neurology. 2010;6(2):108–119. doi: 10.1038/nrneurol.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bacskai BJ, Kajdasz ST, McLellan ME, et al. Non-Fc-mediated mechanisms are involved in clearance of amyloid-βin vivo by immunotherapy. Journal of Neuroscience. 2002;22(18):7873–7878. doi: 10.1523/JNEUROSCI.22-18-07873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Borysov SI, Granic A, Padmanabhan J, Walczak CE, Potter H. Alzheimer Aβ disrupts the mitotic spindle and directly inhibits mitotic microtubule motors. Cell Cycle. 2011;10(9):1397–1410. doi: 10.4161/cc.10.9.15478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takemura R, Nakata T, Okada Y, Yamazaki H, Zhang Z, Hirokawa N. mRNA expression of KIF1A, KIF1B, KIF2, KIF3A, KIF3B, KIF4, KIF5, and cytoplasmic dynein during axonal regeneration. Journal of Neuroscience. 1996;16(1):31–35. doi: 10.1523/JNEUROSCI.16-01-00031.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Baas PW. The role of motor proteins in establishing the microtubule arrays of axons and dendrites. Journal of Chemical Neuroanatomy. 1998;14(3-4):175–180. doi: 10.1016/s0891-0618(98)00012-x. [DOI] [PubMed] [Google Scholar]
- 90.Abisambra JF, Fiorelli T, Padmanabhan J, Neame P, Wefes I, Potter H. LDLR expression and localization are altered in mouse and human cell culture models of Alzheimer’s disease. PLoS ONE. 2010;5(1) doi: 10.1371/journal.pone.0008556. Article ID e8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen Y, Durakoglugil MS, Xian X, Herz J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(26):12011–12016. doi: 10.1073/pnas.0914984107. [DOI] [PMC free article] [PubMed] [Google Scholar]