

Abstract
The objectives of this study are to test the hypothesis that the fatigue and accompanying symptoms of Chronic Myalgic Encephalomyelitis/Fatigue Syndrome are in part due to defects in energy provision at the cellular level, and to understand the pathophysiology of the defects so that effective medical intervention can be implemented. We performed an audit of 138 patients (ages 18-65) diagnosed with ME/CFS and attending a private practice. The patients and 53 normal, healthy controls had the ATP Profile test carried out on neutrophils from a 3-ml venous blood sample. This test yields 6 numerical factors that describe the availability of ATP and the efficiency of oxidative phosphorylation in mitochondria. Other biomedical measurements, including the concentration of cell-free DNA in plasma, were made. The results of the audit are compared with the controls and a previous cohort of 61 patients. We find that all patients tested have measureable mitochondrial dysfunction which correlates with the severity of the illness. The patients divide into two main groups differentiated by how cellular metabolism attempts to compensate for the dysfunction. Comparisons with exercise studies suggest that the dysfunction in neutrophils also occurs in other cells. This is confirmed by the cell-free DNA measurements which indicate levels of tissue damage up to 3.5 times the normal reference range. The major immediate causes of the dysfunction are lack of essential substrates and partial blocking of the translocator protein sites in mitochondria. The ATP Profile is a valuable diagnostic tool for the clinical management of ME/CFS.
Keywords: Chronic fatigue syndrome, myalgic encephalomyelitis, mitochondria, adenosine triphosphate (ATP), oxidative phosphorylation, cellular energetic, glycolysis, cell-free DNA, exercise
Introduction
The devastating illness Myalgic Encephalomyelitis (M.E.) also called Chronic Fatigue Syndrome (CFS) is often misunderstood and considered to be “all in the mind”. E. D. Acheson (later to become Chief Medical Officer of England) and others carefully described the symptom pattern of Myalgic Encephalomyelitis as long ago as 1959 [1], and the World Health Organization recognized M.E. as a neurological illness in 1969. However these facts are ignored by a substantial fraction of the medical community, and a patient can be labeled as having ME/CFS or just M.E. by just being chronically fatigued. The most characteristic and disabling symptom of M.E. is the postexertional malaise following activity or exercise, either physical or mental, often one to three days later. Fortunately, the importance of this key symptom has been recognized in recent years and incorporated into the International Consensus Criteria for Myalgic Encephalomyelitis (ICCME) [2]. In spite of many biomedical studies, there is inadequate understanding of the biochemical processes which lead to this malaise and there is no recognized biomedical test for this illness.
In the first Section, we demonstrate that biochemical tests such as the ATP Profile do exist that can identify those patients who have a cellular biological reason for their symptoms.
In the second Section, we use the results of the various tests which make up the ATP Profile to unravel the basic pathophysiology of the mitochondrial dysfunction and how cellular processes are modified in an attempt to compensate for the dysfunction. We also report on the measurement of DNA fragments in blood plasma (cell-free DNA), produced as a result of cell damage in ME/CFS.
The ATP Profile as a diagnostic tool for ME/CFS
The ATP Profile consists of several biomedical tests on neutrophils extracted from a 3-ml sample of venous blood. The tests, based on published research studies, were developed at Biolab Medical Unit such that they could be used routinely and reproducibly. In 2009, we published (we refer to this paper as MBH) our findings from an audit where we compared deficiencies in the provision of ATP (adenosine triphosphate) in neutrophils with the degree of disability of patients with ME/CFS by multiplying together 5 of the measured factors to produce a Mitochondrial Energy Score (MES) [3]. The patient group spanned a wide range of CFS Ability from 0 to 7 on the Bell 0 to 10 CFS Ability scale [3,4]. The results showed a marked correlation between the MES and the degree of CFS Ability.
Since then we have carried out a revised analysis of that data set. First, we exclude the 10 patients outside the age range 18-65 years of the controls to give Cohort 1 consisting of 61 patients. Secondly, we take into account the fact that one of the 5 factors, TL IN, for some patients is larger than it is for any of the control group. Such behavior is not found for any of the other factors. In addition we present the analysis of a second independent set of 138 patients (Cohort 2). Finally, we demonstrate an alternative method of analysis of the test data which provides a complete separation between the patient and control groups.
The patients attended a private practice specializing in ME/CFS and all satisfied the Centers for Disease Control (CDC) diagnostic criteria for CFS [5]. A review of the clinical notes shows that most, if not all, patients also satisfied the more stringent ICCME [2]. Cohort 1 patients had tests and basic interventions to address sleep problems, food intolerances, and thyroid and adrenal problems. Advice on pacing of activity and stone-age diet was given and simple nutritional supplements were recommended. The patients of Cohort 1 were those who were still ill after these interventions and at this stage the level of disability was assessed jointly by the patient and the clinician using the Bell CFS Ability scale, and a blood sample taken for the ATP Profile test. The nature of the ATP biomedical test was explained and all patients gave written agreement for the results to be used anonymously.
The patients of Cohort 2 were new patients who had not had this basic work-up package. We expected most of these patients to not be as ill as those of Cohort 1. Because the ATP Profile had been found to be so useful, Cohort 2 patients opted to have this test at their first consultation alongside the basic work-up.
In the laboratory neutrophils are separated by centrifugation and several biochemical parameters measured (fuller details are given in Sec. 2 below and in Appendix B of MBH [3]): 1) Whole cell ATP, which we call ATPMg because it is measured by adding excess magnesium (Mg) which is an essential co-factor for the luciferin-luciferase reaction (EC 1.13.12.7) used to measure ATP concentrations. Standard and buffer solutions and also the equipment to measure the luminescence produced by the ATP are commercially available. 2) ATP measured with endogenous Mg only, ATPend. Mg is often complexed with ATP and it is an essential cofactor in the hydrolysis reaction in which energy is released. Intracellular Mg deficiencies are common in ME/CFS and it is important to measure the ratio ATPend/ATPMg which we call ATP Ratio. 3) Ox Phos, a parameter related to the efficiency of the oxidative phosphorylation process which recycles ADP (adenosine diphosphate) back into ATP. In order to measure this, the inhibitor sodium azide is added to a buffer solution containing the neutrophils. After 3 min. an aliquot is taken and the ATP measured. The ratio gives us % ATP inhibited. This quantity, which was ignored in MBH, has proven to be extremely useful. At this same time the remaining cells are washed to remove the inhibitor and 3 min. later the concentration of the recovered ATP is measured and we can calculate the quantity Ox Phos. 4) TL OUT, a measure of the functionality of the translocator protein (TL or ANT for adenine nucleotide translocator or transporter) when its electrogenic antiport faces out to transport ADP into mitochondria for recycling. 5) TL IN, a measure of the TL functionality when the antiport faces into the mitochondrial matrix to transport recycled ATP into the cytosol.
Mitochondrial energy score and Nfn
In MBH we computed a Mitochondrial Energy Score (MES) by multiplying 5 of the measured factors together (ATPMg, ATP Ratio, Ox Phos, TL OUT, and TL IN) and normalizing all patients and controls to unity (or 100%) at the minimum value for the normal controls [3]. This method seems sound because we have found no strong correlations among these 5 factors. We found that the MES correlates well (correlation coefficient r = 0.80) with CFS Ability and only one patient out of 71 (one out of 61, age range 18-65 of the controls) had a value of MES in the normal region. We also found that just counting up the number of factors Nfn which are in the normal region for each subject also correlates with CFS Ability (r = 0.64).
Here we introduce an alternative method. First, we take into account the measurement % ATP inhibited and use it to replace the TL IN factor in the calculation of MES. This solves the problem that TL IN can be higher as well as lower for patients as compared with controls. We label this revised energy score MESinh. Secondly we count up the number of factors Nfn (now including % ATP inhibited) which are in the normal region for each subject. By definition Nfn = 6 for all controls and is in the range 0-6 for patients. We can also ignore the somewhat subjective CFS Ability and plot MESinh vs. Nfn (Figure 1) for both Cohorts 1 and 2. This gives a complete separation between patients and controls because all patients have at least one factor outside the normal region. In terms of a diagnostic test, none of these patients are in the normal region for MESinh and Nfn and none are in the normal region for just Nfn. For Figure 1A we divide patients into the categories ‘moderate’, ‘severe’ and ‘very severe’ which we used in MBH. The categories in Figure 1B will be explained in the next section.
Figure 1.
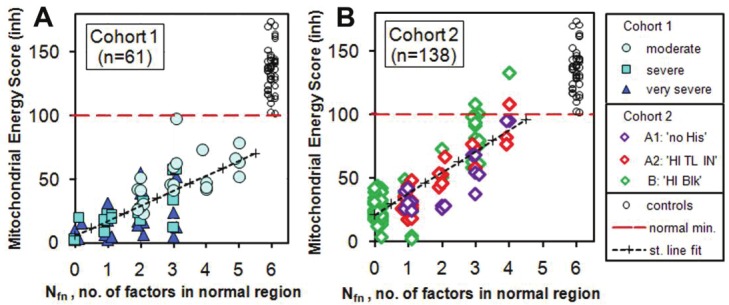
Plots of revised Mitochondrial Energy Score MESinh vs Nfn for controls and the two patient cohorts. For Cohort 1 the patients are divided into the categories ‘moderate’, ‘severe’ and ‘very severe’. For Cohort 2 the patients are divided into Group A (NO BLOCK, with sub-groups A1: 'no HIs' and A2: 'HI TL IN') and Group B (BLOCKED, with label B: 'HI Blk'). Details of the method of computing the revised Mitochondrial Energy Score, and definitions of the Groups are explained in detail in the next section. The linear regression fits to the patient data give correlation coefficients r = 0.72 and r = 0.83 for Cohorts 1 and 2 respectively. The control points extend up as high as 257 with a mean value of 144.
We should still be cautious because of the small sample sizes and because no mildly ill patients were tested. Within these limitations the ATP profile is an inclusive and sensitive test for ME/CFS. However, we cannot claim that it is specific to ME/CFS because there are other neurological illnesses [6,7] and metabolic syndromes [8] associated with mitochondrial dysfunction [9].
Nature of the mitochondrial dysfunction in ME/CFS
Here we discuss the significance and relationships of the quantities that we measure in the ATP Profile test. This test is often supplemented by other tests, for example cell-free DNA, red cell NAD (nicotinamide adenine dinucleotide), Coenzyme Q10 (CoQ10), superoxide dismutase (SOD) function, and in special cases micro-respirometry studies of mitochondria, but we will not discuss these here. The main features that we find from the new Cohort 2 data and a re-analysis of the Cohort 1 data are that there is partial blocking of the standard scenario for ATP production and that the patients divide into two main groups. For one group the poor performance of the standard metabolic scenario is partly offset by increased glycolysis. For the second group we find that there is an alternative process which supplies additional compensatory ATP.
Whole cell ATP, ATP Ratio and ATPend
The ATP Ratio has almost the same value 0.686 ± 0.032 for all the controls, where the error is the standard deviation (SD). We find that about 70% of Cohort 1 and 90% of Cohort 2 patients are deficient in intracellular Mg as compared with the controls. This is physiologically serious because adequate Mg is essential for the enzyme ATPase to function and release the energy stored in ATP.
Also, about 90% of both cohorts are below the minimum value of the controls in ATPend. However, the value of ATPend is not a strong indicator of CFS Ability with a correlation coefficient of only r = 0.086. It is as if a low ATPend is a prerequisite for ME/CFS but does not determine the severity of the illness. A more likely explanation is that the normally vigorous motility of the neutrophils is suppressed such that they are only just alive with less than 2/3 of the useful ATP, ATPend, of the controls. These features are illustrated in Figure 2 where we plot TL IN as a function of ATPend. Note the sharp lower cut-offs in ATPend for both controls and the two patient cohorts.
Figure 2.
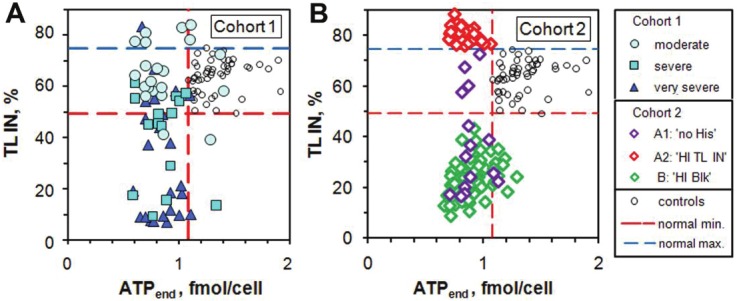
Plots of TL IN vs ATPend for controls and patients in Cohorts 1 and 2. There are few Cohort 1 patients with supernormal values of TL IN but many more in Cohort 2. Cohort 1 has a higher fraction of patients in the normal region of TL IN than has Cohort 2. Note the sharp lower cut-offs of ATPend for the controls and for the 2 patient cohorts, which are much lower in value.
Figure 2 also illustrates the problem of TL IN values which are higher than the normal maximum. Note also that there are no Cohort 2 patients in the normal region of both ATPend and TL IN. The larger fraction of patients in the normal region of TL IN for Cohort 1 is possibly due to the fact that these patients have already had some therapeutic intervention.
Inhibition study
Following the basic ATP measurements of ATPMg and ATPend we use sodium azide to inhibit ATP production prior to a two-stage re-measurement of ATP. The azide ion inhibits cytochrome c oxidase (Complex IV, EC 1.9.3.1) and F0F1-ATPase (Complex V, EC 3.6.3.14) of the mitochondrial respiratory electron transfer chain (ETC) thereby blocking the ETC and oxidative phosphorylation. The limited store of ATP in the cells is rapidly depleted - the ATP the cells need for their own functioning, not the ATP produced in response to external demands. In a separate experiment, using a Strathkelvin Mitocell™, we have measured oxygen uptake by mitochondria isolated from leucocytes and suspended in a phosphate solution to which normal substrates and ADP are added, and verified that introduction of azide completely stops the production of ATP within 3 minutes. The azide is a very useful inhibitor because it can be washed away before apoptosis is triggered. This allows us to explore the recovery phase and derive two clinically useful factors for each patient: a measure of the degree to which the active sites of the Krebs cycle and ETC are already blocked (before the addition of the azide) and the efficiency of ADP to ATP re-conversion (when the azide is removed), the parameter Ox Phos. In practice, we see for the controls a rapid (in less than 3 min) fall in measured ATP, usually to just a few percent (7.5 ± 3.4 %) of the starting value, where the quoted error is the standard deviation (SD). The results are shown in Figure 3A. In the standard metabolic scenario the process of glycolysis is the precursor to the Krebs cycle and oxidative phosphorylation via the ETC, and takes place in the cytosol and is not inhibited by the azide. Glycolysis of a molecule of glucose produces a net 2 molecules of ATP. The Krebs cycle plus the ETC normally produce another 30 molecules of ATP. Thus with the ETC inhibited we expect 1/16 or 6.3 % of the ATP production to remain. This is consistent with the average of 7.5 % which we measure for the controls (and 9.2 ± 3.4 % for Cohort 1 and 8.2 ± 2.7 % for Cohort 2), but also the rate of glycolysis may increase in order to make up some of the shortfall while the ETC is inhibited.
Figure 3.
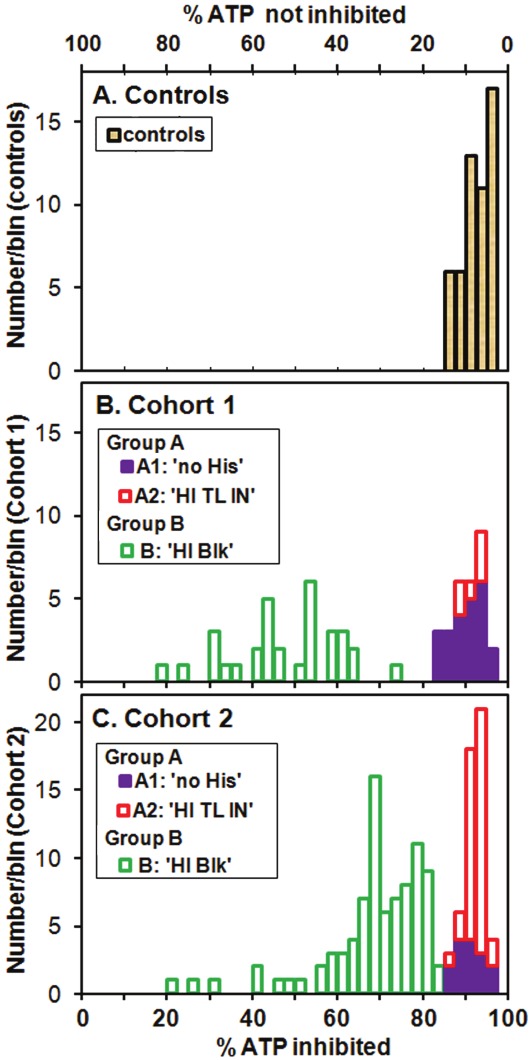
Histograms of percentage of whole cell ATPMg inhibited 3 min after azide inhibitor is added, for A. the control group, B. patient Cohort 1 and C. patient Cohort 2. For all the controls and for the patients with more than about 84 % of the ATP inhibited, the remaining up to 16 % ATP production with the inhibitor present is consistent with that expected from glycolysis. For values lower than about 84 % inhibited another compensatory pathway for ATP generation must exist.
We should remark on our ability to accurately measure such small concentrations of ATP. On a routine basis quadruplicate measurements are made and the reproducibility is about 2 % on the scale of 0 to 100 % of Figure 3. This is roughly consistent with the SDs we find.
After removal of the inhibitor we see (within 3 min) in the control group the total ATP levels recover in a systematic way to between 60 % and 90% of their original values (see Figure 3B of MBH) [3]. We will discuss the recovery of the patient cohorts later.
What surprised us most when we examined the Cohort 2 data (Figure 2B) is the large number of patients, 37 out of 139 (27%), with supernormal values of TL IN which for Cohort 1 (Figure 2A) is only 7 of 61 (11%). We also started to look more carefully at how much of the ATP production is inhibited when the azide is present. We find that for many patients the inhibitor has a much smaller effect than for the controls. In fact 62 % of the patients of Cohort 2 (and 52 % of Cohort 1) have inhibitor effects outside the range of the normal controls (Figure 3B and 3C).
In Figure 3 we use two horizontal scales: % ATP not inhibited (the quantity we measure) at the top, and its difference from 100 %, % ATP inhibited at the bottom. It is important to note that the value of % ATP inhibited is also the percentage of ATP generated by oxidative phosphorylation in the ETC and this can be as low as about 20 % for both cohorts. We are forced to conclude that, for patients with low values of % ATP inhibited, the ETC is already partially inhibited (or BLOCKED, and hence the introduction of the category label ‘B: 'HI Blk'). This could be due to blockages within the Krebs Cycle or ETC, or of the TL protein. The TL protein is investigated in the third part of the ATP Profile. It is important to understand that, in spite of such blockages, the cells still produce ATP and we must conclude that there is another source or pathway for the production of ATP which attempts to compensate for the dysfunction resulting from the blockages. We need to understand the sources of blockage and the compensatory pathways.
Figure 3 also shows that the patients of both cohorts divide into two main groups: Group A (NO BLOCK), with sub-groups A1: 'no HIs' and A2: 'HI TL IN', and Group B (BLOCKED), with label B: 'HI Blk'. We now use these instead of the categories 'moderate', 'severe' and 'very severe' used in Cohort 1 and which have become redundant. However, the new groups roughly correlate in a similar way with the CFS Ability.
Patients in Group A have values of % ATP inhibited in the normal region of the control group, i.e. they have no pre-blocking of the ADP-ATP conversion process. Their only compensatory pathway for ATP generation when the inhibitor is present appears to be glycolysis. Any increased demand for ATP will increase the rate of glycolysis. Patients in Group B: 'HI Blk' have depressed ATP production with below normal values of % ATP inhibited. Group B patients appear to have a different compensatory pathway which can produce 60 % or more of their ATP.
Group A patients in sub-group A2: 'HI TL IN' have supernormal values of TL IN while sub-group A1: 'no HIs' includes all remaining patients because, with the exception of one patient in Cohort 1, none of the patients satisfy both the 'HI Blk' and 'HI TL IN' definitions. The two cohorts have similar fractions in Group B: 'HI Blk' but different proportions in A1 and A2. The main features are summarized in Table 1.
Table 1.
Numbers of patients and controls divided into Group A (NO BLOCK, with sub-groups A1: 'no HIs' and A2: 'HI TL IN'), and Group B (BLOCKED), with label B: 'HI Blk').
| Groups | Patients | Controls | |
|---|---|---|---|
|
| |||
| Cohort 1 | Cohort 2 | ||
| Percent females | 79 % | 70 % | 76 % |
| Mean age (years) ± SD | 45.1 ± 11.8 | 41.1 ± 12.1 | 35.9 ± 13.4 |
| Numbers in Group A (NO BLOCK) | |||
| A1: 'no HIs' | 23 | 15 | 53 |
| A2: 'HI TL IN' | 6 | 37 | 0 |
| Total for Group A | 29 (48%) | 52 (38%) | 53 (100%) |
| Numbers in Group B (BLOCKED) | |||
| B: 'HI Blk' | 31 | 86 | 0 |
| ‘HI TL IN’ and ‘HI Blk’* | 1 | 0 | 0 |
| Total for Group B | 32 (52%) | 86 (62%) | 0 (0%) |
| Total number of subjects | 61 | 138 | 53 |
Merged into ‘Hi Blk’ category which then has n=32
Other features of Groups A and B, and their correlations with Ox Phos, % ATP inhibited, and the amount of the original ATP which is recovered when the inhibitor is removed, will be discussed later. Next, because of its close association with these new categories, we consider the role of the translocator protein TL.
ADP - ATP translocator (TL) study
In many studies of mitochondrial function, mainly concerning the individual complexes I-V of the ETC, the role of the ADP-ATP translocator protein TL is largely ignored [10-12]. This is unfortunate because TL plays an essential physiological role. In order for ADP to be recycled it must pass from the cytosol through the mitochondrial impermeable inner membrane into the matrix, and the ATP produced must pass from the matrix into the cytosol where its energy can be used, and both functions are provided by TL [13]. If TL is not working properly, oxidative phosphorylation will be inhibited, pyruvate dehydrogenase becomes inhibited and also the Krebs cycle [14].
In order to study the functionality of the two translocation processes we separate mitochondria from neutrophils by a standard technique and make 3 aliquots of the separated mitochondria. The first is used to measure ATP within the mitochondria (mtATP with units of pmol/106 cells) by the same method used to measure the whole-cell ATP. The second aliquot is provided with excess ADP and the third deprived of ADP (see Appendix B of MBH for details [3]). The analysis conditions are designed to maximize the production of mtATP from cytosolic ADP (parameter TL OUT), and the provision of ATP to the external artificial cytosol (parameter TL IN). In this way we explore the two transfer efficiencies of the ADP-ATP TL sites in the mitochondrial inner membrane.
As a first step to understanding the high values of TL IN and the nature of the partial blocking of TL sites we plot TL IN and TL OUT vs. % ATP inhibited for controls and Cohort 2 patients (Figure 4).
Figure 4.

Translocator protein factors compared with % ATP inhibited for controls and Cohort 2 patients. A. TL IN, B. TL OUT, C. Correlation between TL OUT and TL IN.
In the TL IN plot (Figure 4A) there is a striking separation between the Group A ‘HI TL IN’ and Group B ‘HI Blk’ patients. All of the ‘HI Blk’ patients have subnormal values of TL IN indicating that there is reduced transfer of ATP from the mitochondria to the cytosol. Also, there is a marked correlation between TL IN and the degree of blocking (decreased blocking corresponds to increased % ATP inhibited). Although we cannot exclude the possibility of partial blocking of the Krebs cycle or the ETC, these two features strongly suggest that their origin is blocking of the TL protein on the matrix side. For the A1: 'no HIs' sub-group, 11 (of 16) patients have low TL IN and 4 have normal values. The super-normal values of TL IN for the ‘HI TL IN’ patients are most likely due to below normal ATP levels in the mitochondria due to shortage of substrate, either the well-known substrates of the Krebs cycle or those of the ETC, principally ADP and inorganic phosphate, Pi, but also the essential co-factors CoQ10, reduced Niacinamide (NADH) and Mg. Lack of the main substrate ADP could be due to low values of TL OUT, and in fact 33 of 37 (89 %) of the ‘HI TL IN’ patients have below normal minimum values of TL OUT (Figure 4B). This plot also shows that 78 % of the Group B ‘HI Blk’ patients have subnormal values of TL OUT indicating reduced transfer of ADP into the mitochondria because of blocking of TL sites on the cytosol side. This is also the case for 13 of the 15 (87%) A1: 'no HIs' patients.
Also, the mtATP concentration (not shown here) correlates well with TL OUT for all 3 categories and 78 % of all the patients of Cohort 2 have mtATP below the normal minimum of 295 pmol/106 cells, and all below the normal average of 490 pmol/106 cells.
Clearly, the partial blocking of the TL sites is a very important aspect of the mitochondrial dysfunction and this has not been considered in any other study. Although we cannot exclude the possibility of blocking or dysfunction of the Krebs cycle or ETC, the fact that for Cohort 2 there are no patients (out of n=138) in the normal region of both TL IN and TL OUT (Figure 4C) suggests that it is mainly the TL sites that are blocked. One of us (JMH) is making further studies of TL function and has found chemical blocking in those cases with subnormal values. Possible sources of blocking agents are byproducts of viral or bacterial pathogens, cellular debris due to oxidative damage, and some environmental chemicals. Results from molecular level fluorescence microscopy, and the identification of the blocking agents by Micro Raman Spectroscopy and Fourier Transform Infrared Spectroscopy, will be the subject of a further paper.
ADP to ATP reconversion by oxidative phosphorylation
We have already discussed the first part of the measurement of Ox Phos, the parameter related to the efficiency of the oxidative phosphorylation of ADP to ATP in the sub-section Inhibition study. At the same time that the ATP concentration with inhibitor present is measured, the azide inhibitor in the remaining sample is washed away and 3 min. later an aliquot (again in quadruplicate) is taken and the amount of ATP recovered is measured. The recovery phase has been studied in detail by one of us (JMH) and this procedure has been optimized for use on many samples on a routine basis. In general terms, if the fraction of ATP recovered after 3 min. is high, the oxidative phosphorylation process is working well, and if the recovery is low it is not. In the calculation of Ox Phos we subtract out the part that is not inhibited when the azide is added and take it as the baseline value. This gives the initial concentration of ATP produced by the ETC, ADPETC, and this should not be influenced by any blocking. The same baseline is subtracted from the total amount recovered to give ATPrec, the amount recovered of the ATP due to the ETC. We then calculate Ox Phos = (ATPrec / ADPETC) x 100 %. Figure 5 shows a plot of Ox Phos vs ADPETC and also plots vs TL OUT and vs TL IN.
Figure 5.

Plots of Ox Phos vs ADPETC and also vs TL OUT and TL IN for controls and Cohort 2 patients.
Figure 5A shows that for all three groups there are strong correlations between Ox Phos and ATPETC. The ‘HI TL IN’ and ‘no HIs’ patients have a slightly steeper recovery than the ‘HI Blk’ patients. The control group has a different behaviour, with those subjects with initial ATP values close to the sharp minimum value having higher recoveries than those with higher initial values of ATP. At present we do not fully understand these features, but for the patients with subnormal values of ATPETC we do expect a positive correlation between Ox Phos and ATPETC. The plot vs TL OUT suggests partial blocking on the cytosol side and the TL IN plot shows that Ox Phos increases as TL IN gets closer to the normal region both from below B: 'HI Blk' and most of the A1: 'no HIs') and above (A2: 'HI TL IN').
Comparison with some other mitochondrial studies
We should comment on two recent studies one of which measured the activities of two of the electron transfer complexes in peripheral blood mononuclear cells (PBMCs) and concluded that CFS patients have normal oxidative phosphorylation capacity [15]. We have already published a Comment on this paper (http://www.translational-medicine.com/content/8/1/93/comments).
The second study also measured the activities of electron chain complexes but in cells from skeletal muscle biopsies, not only of healthy controls and patients with ME/CFS, but also of two groups of patients with known mitochondrial DNA mutations [16]. The authors concluded that mitochondrial content was decreased in patients with ME/CFS compared to controls whereas the ETC activities were not. The method of investigating energy turnover by measuring the activities of electron transfer complexes can give misleading results and much more reliable methods, including the ones that we use, are available [11]. Moreover, both studies completely ignore the possibility of dysfunction of the TL protein and we have found this to be very important in patients with ME/CFS.
ATP compensatory pathways for Group A and Group B patients
We have identified increased glycolysis as the compensatory ATP generation pathway for Group A (A1 and A2) patients. This is what is traditionally identified for both patients and healthy controls in exercise studies. However there are limits to how much additional glycolysis can take place and for how long. Glycolysis without the ETC results in lactate production, acidosis, and cellular damage. Athletes usually recover quickly because via training they have adjusted the proportions of type I and type II muscle fibers according to their discipline, and they normally use increased glycolysis for only short time intervals. If patients with ME/CFS have to use increased glycolysis on a regular basis they will suffer from its results. The conversion of lactate back to pyruvate (via the Cori cycle) requires 6 molecules of ATP which must be made available and for patients this extends the time-scale of the lactic acid burn.
The new question is – what is the compensatory ATP generating pathway used by the Group B: 'HI Blk' patients? There may be a number of possibilities. The most likely one is the adenylate kinase (ADK, EC 2.7.4.7) reaction in which two molecules of ADP combine to make one of ATP and one of AMP (adenosine monophosphate) [11,17-19]. This reaction has a very small free energy change, needs no oxygen or glucose, and can provide ATP close to where it is needed. The problem is that for every molecule of ATP generated, one molecule of the ADP pool is changed to AMP and then, in skeletal muscle, to IMP (inosine monophosphate, via AMP deaminase, EC 3.5.4.6) which is not recycled but mainly lost in urine [11,19]. The replacement of the lost adenine nucleotides can take several days. This may explain one clinical feature of ME/CFS, namely delayed fatigue. Recycling by Group A patients of the lactate generated by glycolysis into glucose and glycogen takes place on a shorter time scale, but Group A patients will suffer from increased acidosis due to build-up of H+ ions from ATP hydrolysis which are not recycled by the ETC, and will also exhibit excess lactate in the blood.
Our experimental results are all obtained from neutrophils. Neutrophils are similar to skeletal muscle cells and most other cells (but not cardiac muscle cells) in that the proton gradient across the mitochondrial inner membrane is about 50 % electrical and 50 % chemical. However, at this stage we cannot claim that the mitochondria in other cell types behave similarly, even though mitochondria are systemic. However, some of the features that we observe are very similar to some of the effects seen in exercise studies of patients with ME/CFS.
Comparison with exercise studies
There have been a number of studies of postexertional malaise following exercise [20] and two small studies have also found that patients divide into 2 groups. The first study [21] used magnetic resonance spectroscopy (MRS) to measure ATP and phosphocreatine (PCr) in sub-anaerobic threshold exercise tests (SATET) and found one group with excess acidosis and low ATP synthesis rate during recovery. The authors termed this group (SATET +ve) and concluded that oxidative phosphorylation was dysfunctional and there was a compensatory high rate of glycolysis. The second group (SATET –ve) did not have excess acidosis, but had a higher concentration of ADP during recovery. The authors concluded that there was another compensatory mechanism. However, the ADP concentration was not measured but was calculated by assuming that the creatine kinase reaction (EC. 2.7.3.2) is always in equilibrium and this may not be valid when the mitochondria are dysfunctional [22].
A more recent study [23] also found one group with excess acidosis and normal PCr depletion and a second group with low PCr depletion (compared to controls) and no excess acidosis. It is difficult to compare this with our results because the PCr-Cr shuttle is not very relevant for neutrophils because ATP moves from the distributed mitochondria along actin filaments to sites that require energy for chemotaxis and motility. In skeletal muscles PCr acts as a quick access energy store for fight-or-flight situations and the shuttle provides rapid transfer of ADP and ATP between mitochondria and myofibrils. However if the ADK reaction becomes important in the exercise study, the PCr-Cr shuttle is not needed because new ATP is generated from excess ADP at the same myofibrils.
If what we see in neutrophil mitochondria also applies to muscle cells we would predict the following: Group A patients will have large PCr depletion, excess lactate production and high acidosis (depressed pH). Group B patients will have low PCr depletion (the shuttle is not needed for the ADK reaction), no excess lactate production and less acidosis for the same work load. For repeat exercise tests patients of Group B will have great difficulty in reaching the same maximal voluntary contraction not because of the behavioral reason of exercise avoidance, but because the lost substrate has not been replaced. These predictions are close to what is observed in this recent exercise study [23]. We therefore have some evidence that the effects that we see occur not just in neutrophils. Future exercise studies should combine MRS measurements with ATP Profile tests before and after the exercise, and should also make use of the new technique of using near-infrared spectroscopy to measure oxygen uptake in individual muscles and near-surface organs in real-time during the exercise [24].
Cell-free DNA shows that cells other than neutrophils are affected
Most of our patients have some other tests in addition to those of the ATP profile. We pick out just one of these, namely ‘Cell-free DNA’ because it strongly suggests that whatever is causing the mitochondrial dysfunction in neutrophils is also associated with the degeneration of other cells. This test uses blood plasma and is independent of our studies of neutrophils. Very low levels of cell free DNA are present in healthy people (< 95 μg/l) and increased levels are associated with serious illnesses such as malignancy, stroke, auto-immune diseases and severe infections [25]. During the normal cell-death process of apoptosis the cell components are largely recycled. However, when cells are damaged and die (necrosis), they release their contents into the blood stream, and Cell-free DNA measures the extent of this damage. This test was developed at Biolab using a published method [26] and is now carried out by one us (JMH) at Acumen. Most (132 of 138) patients of Cohort 2 had this test done together with their initial ATP Profile test. The results demonstrate that some patients have abnormally high levels of up to 3½ times those of normal subjects, and that there is a strong correlation with the degree of illness as indicated by the associated low levels of ATPend and Ox Phos and also of MESinh (Figure 6). These high levels of damaged and necrotic cells put ME/CFS firmly in the realm of major organic pathology.
Figure 6.
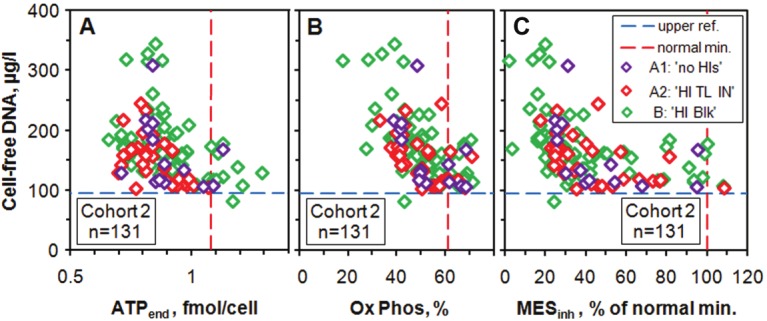
Plots of Cell-free DNA vs ATPend and vs Ox Phos and vs MESinh for 131 patients of Cohort 2. Almost all patients are above the reference range of up to 95 μg/l and there is a strong anti-correlation as ATPend and Ox Phos decrease to the lowest values measured, and these anti-correlations are reflected in the MESinh plot. For Cell-free DNA we use the SI unit μg/l although the traditional unit of μg/dl (which gives numbers lower by a factor of 10) is still widely used. Note that the scale for ATPend starts at 0.5 fmol/cell rather than at zero; this is because ATPend has a rather sharp minimum value of 0.66 fmol/cell, as already noted in Figure 2.
ATP deficiency may affect intercellular signaling and neurotransmission
The patients of both cohorts have reduced ATP compared to the control group. As a result they have reduced access to energy which is needed for all processes within the body. However, ATP has a second important and essential role which is not so widely appreciated. It is a critical signaling molecule and neurotransmitter for a wide variety of processes [27-29]. How much this role is compromised in patients with ME/CFS and what the consequences are have not yet been investigated.
Discussion and conclusions
Diagnosis of ME/CFS
The ATP profile is a test which provides numerical values of 6 biomedical quantities regarding energy provision by the mitochondria in neutrophils, the main effectors of the innate immune system. The ATP Profile is an objective test of ME/CFS and clearly shows that this illness has a physical basis. Individually and collectively the biomedical quantities select patients whose symptoms are the direct result of mitochondrial dysfunction. These quantities also reflect the severity of the illness and, together with one or more additional tests such as Cell-free DNA they demonstrate that it is not just neutrophils that are dysfunctional but also other biological systems.
In some cases there may be a co-morbid psychiatric disorder but it hardly seems necessary to perform a psychiatric diagnosis as a matter of course, as has been proposed [30]. Many accompanying mental symptoms can be explained as resulting from the long period of physical disability endured by many sufferers.
Pathophysiology of ME/CFS
Our results clearly show mitochondrial dysfunction in all 138 patients of Cohort 2 and also the 61 patients of Cohort 1 [3]. A major factor in the dysfunction is partial blocking of the translocator protein TL, and this has not been investigated in any other study of ME/CFS or in most other studies of illnesses with mitochondrial dysfunctions of various types [9].
We also find that lack of substrate or essential co-factors contributes to the mitochondrial dysfunction of some patients, particularly those with super-normal values of TL IN (sub-group A2).
Another feature that we have uncovered is that there are at least two alternative processes that cells and their mitochondria use in order to partially compensate for the dysfunction. This division into 2 distinct groups, A and B, appears to correlate with the 2 groups observed in some exercise studies [21,23]. Future exercise studies coupled with tests like the ATP Profile are needed to confirm this correlation.
Our measurements of Cell-free DNA show that ME/CFS patients have abnormally high levels of damaged and necrotic cells and that there is strong correlation with the measured mitochondrial dysfunction. Taken together, these measurements show that ME/CFS is a serious illness which may affect every cell in the body.
Implications for the treatment of ME/CFS
Here we have emphasized the use of biomedical tests to aid in the diagnosis and to vastly improve our knowledge of the pathophysiology involved in this illness. In addition, these biomedical tests can act as a valuable guide for medical and therapeutic interventions. These will be discussed in a paper which is in preparation.
Acknowledgment
We thank the patients and controls for their written permission to use their test results anonymously.
References
- 1.Acheson ED. The clinical syndrome variously called benign myalgic encephalomyelitis, Iceland disease and epidemic neuromyasthenia. Am J Med. 1959;26:569–595. doi: 10.1016/0002-9343(59)90280-3. [DOI] [PubMed] [Google Scholar]
- 2.Carruthers BM, van de Sande MI, De Meirleir KL, Klimas NG, Broderick G, Mitchell T, Staines D, Powles ACP, Speight N, Vallings R, Bateman L, Baumgarten-Austrheim B, Bell DS, Carlo-Stella N, Chia J, Darragh A, Jo D, Lewis D, Light AR, Marshall-Gradisbik S, Mena I, Mikovits JA, Miwa K, Murovska M, Pall ML, Stevens S. Myalgic encephalomyelitis: International Consensus Criteria. J Intern Med. 2011;270:327–338. doi: 10.1111/j.1365-2796.2011.02428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Myhill S, Booth NE, McLaren-Howard J. Chronic fatigue syndrome and mitochondrial dysfunction. Int J Clin Exp Med. 2009;2:1–16. [PMC free article] [PubMed] [Google Scholar]
- 4.Bell DS. The Doctor’s Guide to Chronic Fatigue Syndrome. New York: Da Capo Press; 1994. [Google Scholar]
- 5.Fukuda K, Straus SE, Hickie I, Sharpe MC, Dobbins JG, Komaroff A. The chronic fatigue syndrome: a comprehensive approach to its definition and study. Ann Intern Med. 1994;121:953–959. doi: 10.7326/0003-4819-121-12-199412150-00009. [DOI] [PubMed] [Google Scholar]
- 6.Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neuro. 2008;4:600–609. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- 7.Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry. 2012;17:290–314. doi: 10.1038/mp.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicolson GL. Metabolic syndrome and mitochondrial function: Molecular replacement and antioxidant supplements to prevent membrane peroxidation and restore mitochondrial function. J Cell Biochem. 2007;100:1352–1369. doi: 10.1002/jcb.21247. [DOI] [PubMed] [Google Scholar]
- 9.Golomb BA. Oxidative Stress and Mitochondrial Injury in Chronic Multisystem Conditions: From Gulf War Illness to Autism Spectrum Disorder. Nature Precedings. 2012 Available from Nature Precedings http://hdl.handle.net/10101/npre.2012.6847.1. [Google Scholar]
- 10.Barrientos A. In vivo and in organello assessment of OXPHOS activities. Methods. 2002;26:307–316. doi: 10.1016/S1046-2023(02)00036-1. [DOI] [PubMed] [Google Scholar]
- 11.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lanza IR, Nair KS. Chapter 20: Functional Assessment of Isolated Mitochondria In Vitro. In: William SA, Anne NM, editors. Methods in Enzymology. Academic Press; 2009. pp. 349–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta. 2008;1778:1978–2021. doi: 10.1016/j.bbamem.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 14.Pieczenik SR, Neustadt J. Mitochondrial dysfunction and molecular pathways of disease. Exp Mol Pathol. 2007;83:84–92. doi: 10.1016/j.yexmp.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Vermeulen R, Kurk R, Visser F, Sluiter W, Scholte H. Patients with chronic fatigue syndrome performed worse than controls in a controlled repeated exercise study despite a normal oxidative phosphorylation capacity. J Transl Med. 2010;8:93. doi: 10.1186/1479-5876-8-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smits B, van den Heuvel L, Knoop H, Küsters B, Janssen A, Borm G, Bleijenberg G, Rodenburg R, van Engelen B. Mitochondrial enzymes discriminate between mitochondrial disorders and chronic fatigue syndrome. Mitochondrion. 2011;11:735–738. doi: 10.1016/j.mito.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 17.Savabi F. Interaction of creatine kinase and adenylate kinase systems in muscle cells. Mol Cell Biochem. 1994;133:145–152. doi: 10.1007/BF01267953. [DOI] [PubMed] [Google Scholar]
- 18.Dzeja PP, Terzic A. Phosphotransfer networks and cellular energetics. J Exp Biol. 2003;206:2039–2047. doi: 10.1242/jeb.00426. [DOI] [PubMed] [Google Scholar]
- 19.Koolman J, Roehm K-H. Color Atlas of Biochemistry. Stuttgart - New York: Thieme; 2005. pp. 336–337. [Google Scholar]
- 20.VanNess JM, Stevens SR, Bateman L, Stiles TL, Snell CR. Postexertional Malaise in Women with Chronic Fatigue Syndrome. J Womens Health. 2010;19:239–244. doi: 10.1089/jwh.2009.1507. [DOI] [PubMed] [Google Scholar]
- 21.Lane RJM, Barrett MC, Taylor DJ, Kemp GJ, Lodi R. Heterogeneity in chronic fatigue syndrome: evidence from magnetic resonance spectroscopy of muscle. Neuromuscul Disord. 1998;8:204–209. doi: 10.1016/s0960-8966(98)00021-2. [DOI] [PubMed] [Google Scholar]
- 22.Barnes PR, Taylor DJ, Kemp GJ, Radda GK. Skeletal muscle bioenergetics in the chronic fatigue syndrome. J Neurol Neurosurg Psychiatry. 1993;56:679–683. doi: 10.1136/jnnp.56.6.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones DEJ, Hollingsworth KG, Jakovljevic DG, Fattakhova G, Pairman J, Blamire AM, Trenell MI, Newton JL. Loss of capacity to recover from acidosis on repeat exercise in chronic fatigue syndrome: a case–control study. Eur J Clin Invest. 2012;42:186–194. doi: 10.1111/j.1365-2362.2011.02567.x. [DOI] [PubMed] [Google Scholar]
- 24.Binzoni T, Cooper CE, Wittekind AL, Beneke R, Elwell CE, Van De Ville D, Leung TS. A new method to measure local oxygen consumption in human skeletal muscle during dynamic exercise using near-infrared spectroscopy. Physiol Meas. 2010;31:1257–1269. doi: 10.1088/0967-3334/31/9/014. [DOI] [PubMed] [Google Scholar]
- 25.Wu T-L, Zhang D, Chia J-H, Tsao K-C, Sun C-F, Wu JT. Cell-free DNA: measurement in various carcinomas and establishment of normal reference range. Clin Chim Acta. 2002;321:77–87. doi: 10.1016/s0009-8981(02)00091-8. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt B, Weickmann S, Witt C, Fleischhacker M. Improved Method for Isolating Cell-Free DNA. Clin Chem. 2005;51:1561–1563. doi: 10.1373/clinchem.2005.051003. [DOI] [PubMed] [Google Scholar]
- 27.Khakh BS. Molecular physiology of P2X receptors and ATP signalling at synapses. Nature Rev Neurosci. 2001:165–174. doi: 10.1038/35058521. [DOI] [PubMed] [Google Scholar]
- 28.Burnstock G. Pathophysiology and therapeutic potential of purinerguc signaling. Pharm Rev. 2006;58:58–86. doi: 10.1124/pr.58.1.5. [DOI] [PubMed] [Google Scholar]
- 29.Khakh BS, Burnstock G. The Double Life of ATP. Sci Amer. 2009;301:84–92. doi: 10.1038/scientificamerican1209-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawn T, Kumar P, Knight B, Sharpe M, White PD. Psychiatric misdiagnoses in patients with chronic fatigue syndrome. J R Soc Med Sh Rep. 2010;1:1–7. doi: 10.1258/shorts.2010.010042. [DOI] [PMC free article] [PubMed] [Google Scholar]