

Abstract
Background/Aims
Chemokine signaling from airway epithelium regulates macrophage recruitment to the lung in inflammatory diseases such as asthma. This study investigates the mechanism by which the α-melanocyte stimulating hormone-derived tripeptide, KPV, and the agonist of the dominant melanocortin receptor in airway epithelium (MC3R), γ-melanocyte stimulating hormone (γ-MSH), suppress inflammation in immortalised human bronchial airway epithelium.
Methods
TNFα and rhino syncitial virus (RSV)-evoked nuclear factor-κB (NFκB) signaling was measured in immortalised human bronchial epithelial cells (16HBE14o-) in response to KPV and γMSH. Cellular and systemic inflammatory signaling was measured by NFκB reporter gene and chemokine (IL8, eotaxin) secretion, respectively.
Results
KPV and γMSH evoked a dose-dependent inhibition of NFκB, matrix metalloproteinase-9 activity, IL8 and eotaxin secretion. The KPV effect was associated with its nuclear import, IκBα stabilisation and suppressed nuclear translocation of YFP-tagged p65RelA. Competition assays revealed an interaction between KPV and the Imp-α3 binding site on p65RelA which may involve blockade of the importin-α armadillo domain 7 and 8. In contrast, the γMSH anti-inflammatory effect required MC3R whose apical expression occurred in epithelium distributed along the length of the respiratory tree in vivo.
Conclusion
KPV and γMSH respectively suppress NFκB signalling in airway epithelium by: i) inhibition of p65RelA nuclear import and, ii) epithelial MC3R activation. Melanocortin peptides therefore provide a robust mechanism for targeting airway inflammation in lung disease.
Keywords: α-melanocyte stimulating hormone-derived tripeptide, KPV, melanocortin receptor, MC3R, γ-melanocyte stimulating hormone (γ-MSH), airway epithelium, chemokine signaling, asthma, inflammation
Introduction
α-Melanocyte stimulating hormone (αMSH) is a member of the melanocortin peptide family which are derived as cleavage products of proopiomelanocortin, a precursor protein synthesised in the pituitary gland. Although αMSH was originally described as a regulator of skin pigmentation it is now widely reported to suppress NFκB-dependent inflammatory signalling induced by bacterial endotoxins and proinflammatory cytokines such as TNFα and interleukins 1 and 6. This anti-inflammatory effect is mediated through a family of 7-transmembrane G-protein coupled receptors (designated MC1… 5R) which regulate skin pigmentation/inflammation (MC1R), adrenal corticosteroid synthesis (MC2R), macrophage activity (MC3R), appetite and obesity (MC4R) and exocrine gland secretion (MC5R). Thus, melanocortin receptors are attractive therapeutic targets in inflammatory, metabolic and cancerous disease states [1].
Amino acid deletion studies have established that C-terminal truncations of αMSH also possess anti-inflammatory properties with the minimum-effective sequence confined to the last 3 residues, K-P-V [2-6]. Several analogues of this sequence have also been tested as well as disulfide bridge linked homodimers (eg (CKPV)2 [7]) which yield improvements in the peptide’s ability to suppress NFκB activity (reviewed [8]). Despite these well documented anti-inflammatory effects, the mechanism of KPV action is poorly understood. Work by Moustafa et al [6] showed that the anti-inflammatory effect of KPV extends over a range of concentrations which would exceed the kinetics of receptor-mediated effects and recent work demonstrates an apparent requirement for PEPTL1-mediated membrane transport of KPV [9] raising the possibility that it mediates its effects by interacting with intracellular targets. Ostensibly, this could occur in two ways. Firstly, the KPV residues may individually or collectively bind polar or non-polar amino acid sequences exposed on the surface of key signalling proteins. The theory of amino acid complimentary binding predicts that K will favour interactions with L or F residues, P with W,G and R and V with Y, H, D or N [10] though alternative algorithms predict some more conservative variations on this theme [11]. As the proline residue places an angle in the KPV peptide, complimentary interactions may occur between one, two or all three residues in several possible orientations. This model cannot, however, explain the apparent specificity of KPV for the NFκB pathway as the short peptide sequence would presumably favour multiple non-specific targets. An alternative hypothesis predicts that the sequence of KPV will specify its actions to a molecule in the NFκB activation pathway. KPV displays the fundamental features of a minimal nuclear localisation sequence (NLS). Although NLS sequences are variable, key features include a cluster of positively charged residues (e.g. K-K/R-X-K/R for monopartite NLSs; [12]), often preceded by a helix breaking residue such as P. For example, the monopartite NLS of the SV40 large T antigen includes K,P and V residues in the critical DNA binding sequence, 126PKKKRKV132 [13] whereas the NLS of lymphoid enhancer factor-1 (LEF-1) contains a KPV-like sequence where the V is substituted for another hydrophobic residue, L, which interacts with the DNA minor groove. Once free from its inhibitor, IκBα, the p50 and p65RelA subunits of the NFκB heterodimer migrate to the nucleus by binding to the N and C-termini of importin-α3 (Imp-α3) respectively [14]. Analysis of the Imp-α3 armadillo (arm) 3 domain which binds p65RelA shows that the critical interacting sequence is rich in amino acids which are complementary for KPV suggesting that a competitive interaction could occur at this site which would disrupt NFκB nuclear import.
To determine how KPV inhibits NFκB-driven inflammatory signalling in airway epithelium, the current study looked for evidence that the peptide might function in one of 3 ways: 1) by promoting the stability of IκBα, 2) by occupying the DNA minor groove or 3) by interfering with the nuclear import of p65RelA. In addition, the expression of melanocortin receptor isoforms in airway epithelial tissue was investigated to establish the potential for receptor-mediated anti-inflammatory effects. The results show that KPV translocates to the nucleus in human bronchial epithelial cells and that it competitively blocks the interaction between Imp-α3 and the p65RelA of NFκB. Wider examination of the role of melanocortin receptor expression demonstrates that MC3R is the dominant receptor expressed in airway epithelium and that its agonist, γMSH, suppresses cellular and systemic inflammation in response to pro-inflammatory stimuli. It is concluded that melanocortin peptides can repress inflammatory signaling in airway epithelial cells either by direct repression of NFκB nuclear transport or through receptor-mediated signalling pathways. As such, the melanocortins and their derivatives represent robust targets for the treatment of inflammatory diseases of the lung.
Materials and methods
Chemicals
Custom synthesised KPV and histidine tagged H6-KPV were from Activotec (Cambridge, UK), Tumour necrosis factor-α, interleukin 8 (IL8) ELISA, recombinant matrix metalloprotease (MMP) 2 and 9 standards and luciferase assay reagents were from R&D systems (Abingdon, UK), MG-132 (carbobenzoxy-L-leucyl-L-leucyl-L-leucinal) was from Merck Biosciences Ltd. (Nottingham, UK), anti rabbit IκBα antibody (9242), Phospho-IκBα (Ser32/36) (5A5) mouse mAb, anti-rabbit IKKα antibody, phospho-IKK-α/β (Ser176/180) antibody, anti-rabbit p70 S6K and Phospho p70 S6K (T389) mouse mAb and secondary detection antibodies were from Cell Signalling Technologies (Hertfordshire, UK). Anti -rabbit IgG p65NFκB (sc109) was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-V5 mouse monoclonal antibody was from Invitrogen (Paisley, UK). DRAQ5 was from Biostatus Ltd, Shepshed, UK). All other chemicals were from Sigma-Aldrich (Poole, UK).
Plasmids and molecular biology
In vitro-translated human IκBα protein, pDNA3-IκBα-V5C and pDNA3-IκBα-(S32A, S36A)-V5C were kind gifts from R. Hay (Dundee University, UK). pDNA3-IκBα (G33A, L34A, D35A)-V5C was generated from pDNA3-IκBα-V5C using standard point mutation techniques. pCMV-N-Flag- RelA was generated by amplifying RelA from reverse transcribed RNA isolated from HBE cells and cloning the RelA gene product into pCR2.1 (Invitrogen). A BamH1-Xho1 fragment containing the RelA insert was then sub-cloned in-frame into pCMV-Tag2 (Stratagene) to generate N-terminally Flag-tagged RelA. YFP-tagged RelA was kindly gifted by N. McTavish (Dundee University). GST-tagged Imp-α3 was a generous gift from R. Fagerlund (National Public Health Institute, Helsinki, Finland). Mammalian expression of a Myc-tagged Imp-α3 was achieved by subconing into pDUAL-GC. The NFκB luciferase reporter gene, κB4-luc was generated by cloning four copies of the κ enhancer element, GGGAATTTCC, ahead of the thymidine kinase promoter into pGL3 (Promega). The same vector lacking the κ enhancer element was used as a negative control.
Cell culture and transfection
The immortalised human bronchial epithelial cell line (16HBE14o- (HBE; [15]) was cultured in RPMI medium supplemented with 8.5% fetal calf serum (Invitrogen, Paisley, UK) and antibiotics (Sigma-Aldrich, Poole, UK). Airway and alveolar epithelial adenocarcinoma cell lines, H441 and A549, plus the human embryonic lung fibroblast cell line, HEL12469, were maintained in DMEM/F12 containing 8.5% fetal calf serum. Cell stocks were maintained in 75cm2 vented culture flasks (Nunc, Roskilde, Denmark) in a humidified atmosphere supplemented with 5% CO2 at 37°C. For experiments, cells were transferred into 6 or 12-well Costar culture plates (Corning, New York, USA) and grown to within 90% confluence. Cells were transfected using Lipofectamine 2000 (Invitrogen, Paisley, UK) according to the manufacturer’s instructions.
Effect of KPV on cell growth
Cells were grown in 16-well coverslips for 3h in serum free medium containing 0-10μg.ml-1 KPV with or without TNFα (5ng.ml-1). At the 3h time point, the medium was replaced with identical medium containing 10μM 5-bromo-2’-deoxyuridine (BrdU). Cells were allowed to incorporate the BrdU for 30 minutes after which they were fixed, the DNA was denatured, then probed using 2μg.ml-1 anti-BrdU-Fluorescein antibody (Roche). The nuclei were counterstained with 5μM DRAQ5 and the slides were imaged using an LSM 510 confocal microscope. The number of S-phase cells in each field of view was determined by calculating the number of BrdU positive nuclei as a percentage of the total number of nuclei in the field of view. Translational activity was inferred from the phosphorylation of ribosomal S6 kinase at threonine 389, a specific substrate of the growth regulating and nutrient sensitive kinase complex, mTORC1.
NFκB activity and p65RelA nuclear translocation
NFκB activity was measured using a luciferase reporter gene containing four copies of the NFκB response element upstream from a minimal thymidine kinase promoter as described before [16]. Cells were transfected with 0.8μg of κB4-luc per 1x106 cells of for 4h in serum free RPMI and then left to recover for 18h in complete medium with serum. NFκB activity was induced with 5 or 10ng.ml-1 TNFα in combination with other treatments for 6h after which the medium was removed for IL8 ELISA and the cells were disrupted in kinase lysis buffer containing Complete Protease Inhibitor Cocktail (Roche) and phosphatase inhibitors. For MC3R-mediated anti-inflammatory responses in HBE, cells were exposed for 6h to 106cfu.ml-1 of rhinosyncitial virus (RSV; generously provided by M Gallacher, Dundee) or 10μg.ml-1 of Pseudomonas aeruginosa lipopolysaccharide (LPS) prior to lysis as above. Luciferase assays were performed using a BMG Labtech Fluostar Optima plate reader and the results were corrected for protein content of the sample.
Interleukin 8 and eotaxin ELISA
Secreted IL8 and eotaxin were detected using the human IL8 or eotaxin DuoSet ELISA Development System (R&D Systems, Abingdon, UK) according to the manufacturer’s instructions. The quantity of cytokine secreted into the medium for each treatment was expressed as a fraction of the respective cellular protein content.
Matrix metalloprotease activity
MMP 2 and 9 activities were determined by gelatin zymography using human MMP-2 and MMP-9 from HT1080 cells as positive controls (R&D systems, Abingdon, UK). Cells were treated with TNFα and KPV in serum free medium for 6h after which the medium was collected and the cells lysed for determination of protein content. 30μls of medium from each sample was applied to a 7.5% non-denaturing polyacrylamide gel containing 2.8mg.ml-1 gelatin. After electrophoresis, gels were washed three times each in reaction medium containing (in mM): 50 Tris-HCl (pH 7.5), 10 CaCl2, 2, PMSF, 0.001 ZnCl2 and 1% Triton X-100. After the last wash, gels were placed into 50mls of reaction medium and incubated overnight at 37°C. The reaction was stopped by adding Ca2+-free reaction medium containing 20mM EDTA and the gels were then stained with Coomassie brilliant blue R250. Gels were then photographed and gelatinolytic activity, observed as clear bands against a blue background, was quantified by densitometry.
DRAQ5 competition assays
DRAQ5 is a high affinity fluorescent dye which binds to the minor groove of DNA and which is compatible for use with living cells. Prior to the competition assays, cells grown in chambered coverslips were pre-conditioned in serum free medium that contained different concentrations of KPV. Competition assays were performed on a Zeiss LSM510 confocal microscope (excitation λ = 633nm; emission λ = 650nm) by removing the medium and immediately replacing it with serum-free medium containing the same concentration of KPV to which the cells had been pre-treated plus 1μM DRAQ5. Images were scanned at 1.5 second intervals for a total of 6 minutes. The effect of each concentration of KPV was compared with a control treatment in which KPV was omitted.
KPV interaction with DNA was determined using the fluorescence competition assay [17,18]. Reactions were set up in reaction buffer containing (in mM): 10, HEPES (pH 7.5), 50 NaCl, 1 EDTA, 0.0024 herring sperm DNA and concentrations of DRAQ5 ranging from 0 to 15μM. Each reaction series was carried out in the presence of 0, 0.1, 1 or 10μg.ml-1 KPV and the resulting change in DRAQ5 fluorescence was taken as an indicator of competitive interaction between KPV and DNA.
In vitro protein binding competition assays
In vitro-translated, purified human IκBα was generously provided by R. Hay (Dundee University). Lysates containing in vitro-translated GSTtagged Imp-α3 were generated from E. coli BL21 cells by inducing expression of 6h (20°C) with 0.2mM isopropyl-1-thio-β-D-galactopyranoside (IPTG). Control lysates were generated from parallel cultures by omitting the IPTG treatment and bacterial lysis was performed as described elsewhere [14]. Nunc Immunosorp plates were coated with 10μg.ml-1 of the in vitro-translated protein or the same concentration of non-induced lysate protein overnight at 4°C. Plates were then washed three times in Tris-buffered saline containing 0.01% Tween 20 (TBST) before being blocked for 1h in 10μg.ml-1 BSA. After a further series of washes, 1μg of protein obtained from HBE cells expressing FLAG-p65RelA was added to each well in combination with KPV at concentrations ranging from 0.01→30μg.ml-1. Plates were incubated overnight at 4°C and then washed as before in TBST. Each well was then incubated with HRP-conjugated mouse anti-FLAG IgG for 1h at room temperature followed by 3 final washes in TBST. The amount of p65RelA bound to the target protein coating the well was visualised by adding TMB reagent (Sigma-Aldrich) and quantified by measuring absorbance at 450nm with background correction at 595nm.
Immunohistochemistry
Cellular distribution of histidine tagged KPV, p65RelA, IκBα and MC3R was observed in methanol-fixed HBE with secondary fluorescent detection as indicated in the figure legends. MC3R expression in mouse lung was determined in paraformaldehyde fixed tissue counterstained with DAPI as described previously [19]. Fluorescent images were obtained using a Zeiss LSM-510 confocal microscope.
Western blotting
Protein lysates were western blotted for phosphorylated and total IκBα (S32, S36), ribosomal s6 kinase (T389), p65RelA, MC3R and actin using methods described by us previously [19,20]. Blots were quantitated by performing densitometry using Un-Scan-It software.
Melanocortin receptor expression and siRNA knockdown
A nested RT-PCR protocol was used to determine the expression of melanocortin receptors as described before [21]. Knockdown of MC3R was achieved using Gene Silencer siRNA from Santa Cruz Biotechnology (sc40111) and compared to cells treated with a randomised oligonucleotide control.
Statistics
Values are represented as mean ± standard error and the number of independent experiments is given in the figure legend. Statistical significance was assessed by one way analysis of variance with the level of significance determined using Tukey’s test.
Results
The first aim of this study was to determine if KPV could suppress intracellular (NFκB) and systemic chemokine (IL8) pro-inflammatory signalling as well as the activation of metalloproteases responsible for lung remodelling in inflammatory lung diseases. Using immortalised human bronchial epithelial cells as a model, KPV was found to suppress TNFα-evoked NFκB-driven luciferase reporter gene activity in a dose dependent fashion and this was mirrored by a decrease in IL8 release at concentrations of KPV 3 1 μg.ml-1 (Figure 1A). Zymography performed on the same samples revealed that HBE cells spontaneously secrete MMP-9 gelatinolytic activity which was raised two fold by the addition of TNFα; again, the same concentration range of KPV was found to inhibit NFκB activity reduced secreted MMP-9 activity to the nonstimulated level (Figure 1B).
Figure 1.

KPV inhibits NFκB signalling and related signals of systemic inflammation and lung remodelling in HBE cells. A. Dose dependent suppression of NFκB activity (lower histogram) and interleukin 8 secretion (upper histogram) induced by TNFα (5ng.ml-1). Values are mean ± SEM, n=4. *P<0.05 relative to TNFα administered without KPV. B. Zymograph showing dose-dependent suppression of secreted MMP-9 activity in H441 cells treated with 5ng.ml-1 TNFα. The position of MMP-2 and 9 standards is shown in the far left lane. Histogram shows the change in secreted MMP-9 activity (as average band intensity) for four independent experiments. Values are mean ± SEM, *P<0.05 relative to untreated control.
The effect of KPV on cell cycle activity was measured by bromodeoxyuridine (BrdU) incorporation into S-phase cells. KPV did not alter the proportion of cells in S-phase, however, it reversed the inhibition of cell growth caused by TNFα (Figure 2A). This was accompanied by activation of mTORC1, a branch point regulator of transcription and translation (Figure 2B).
Figure 2.

KPV rescues TNFα-evoked growth arrest. A. Percentage of S-phase cells (Brdu positive nuclei) in HBE treated with (Black Circles) or without (Grey Circles) TNFα at increasing concentrations of KPV. *P<0.05 with respect to no TNFα treatment at the same concentration of KPV. Values are mean ± SEM, n=4. B. KPV activates mTOR, a key regulator of protein synthesis. A representative blot shows activation of mTOR as an increase in phosphorylation of the T389 residue on ribosomal S6 Kinase 1. Histogram shows compiled densitometry data from 4 independent experiments. The compiled data shows that KPV rescues TNFα-evoked inhibition of mTOR. *P<0.05 with respect to no TNFα treatment at the same concentration of KPV. Values are mean ± SEM.
KPV can enter the cell via the PEPTL1 transporter raising the possibility that it may interact directly with inflammatory signalling molecules. A FITC tagged anti-histidine antibody was used to follow the uptake and intracellular distribution of histidine tagged KPV (H6KPV) over time. Figure 3 shows that H6KPV was detected in the cells after 3h incubation and became almost exclusively nuclear after 5h. Chromatin competition assays demonstrated that KPV did not compete with the interaction between the minor groove binding dye DRAQ5 and DNA in vitro, however, it did suppress DRAQ5 entry into the nucleus in living cells (Figure 4). This was not associated with a statistical difference in the rate of DRAQ5 passage over the plasma membrane.
Figure 3.

Uptake of histidine tagged KPV in HBE cells. Cells were incubated for the indicated times in 10μg.ml-1 H6-KPV then fixed and probed with FITC-conjugated anti-histidine IgG. Nuclei were counterstained with propidium iodide. FITC was detected using a 505-530nm bandpass filter whereas propidium iodide was detected with a 630nm long pass filter. Note nuclear localisation of staining by 5h of incubation. Bar = 40μm.
Figure 4.

Effect of KPV on DRAQ5 DNA binding in vitro and in intact cells. A. Interaction of DRAQ5 with herring sperm DNA (0.2mg.ml-1) in the presence of 0 (closed circles), 1 (grey squares) or 10 (grey circles) μg.ml-1 KPV. Each data point was determined as the mean of four independent readings. B. Rate of development of DRAQ5 fluorescence in the nuclei of live cells was monitored in the presence of 0, 0.1, 1 or 10μg.ml-1 KPV included in the medium. Each data point represents the Mean ± SEM of four independent experiments.
KPV has been reported to suppress NFκB activity by favouring the interaction between the p65RelA/p50 heterodimer and the inhibitor, IκBα. This involves IκBα translocation to the nucleus where it interacts with the p65RelA subunit, thus competing with the p65RelA-DNA interaction. Given that KPV enters the nucleus, the next possibility examined was that KPV might favour IκBα interaction with NFκB by blocking its phosphorylation by the IKK complex or, alternatively, blocking IκBα degradation. Treatment of cells for 3h with 1 or 10μg.ml-1 KPV in the presence of TNFα caused a statistically significant increase in the cellular abundance of IκBα, however, the ratio of IKK-phosphorylated (S32, S36) IκBα-to-total IκBα remained unchanged (Figure 5A). Investigation of IκBα stability at the higher KPV concentration in cells treated with TNFα showed that the rise in IκBα abundance was half maximal at 65.8±14.9 mins after the addition of KPV and reached a statistically significant maximum at 120mins. Immuno-histochemistry showed no evidence of co-localisation between his tidine tagged KPV and IκBα (Figure 6); moreover, neither wild-type nor constitutively stable mutants of IκBα (S32A/S36A or G33A/L34A/D35A) eluted with H6KPV on nickel agarose beads (data not shown).
Figure 5.

KPV stabilises IκBα in the presence of TNFα. A. Treatment of cells with increasing concentrations of KPV for 3h in the presence of TNFα induces the accumulation of IKK phosphorylated IκBα (closed circles) as well as total IκBκ abundance (open circles). The ratio of phosphorylated to total IκBα remained unaffected (grey circles). A representative blot is shown beneath the graph. Cells were pre-treated for 3h with 10ng.ml-1 TNFα before exposure to KPV at the concentrations shown. The concentration of TNFα remained at 10ng.ml-1 throughout the KPV treatment. Values are mean ± SEM, n=5. *P<0.05 relative to the lowest KPV concentration. B. Rate of IκBα stabilisation induced by KPV in the presence of TNFα Cells were pre-treated for 3h with 10ng.ml-1 TNFα then exposed to medium containing the same concentration of TNFα plus 10μg.ml-1 KPV. The relative abundance of total IκBα was determined by comparing the IκBα band intensity to actin. Values are mean ± SEM, n=5, *P<0.05. A representative experiment is shown beneath the graph. C. Effect of increasing KPV concentration upon p65RelA and IκBα binding in vitro. Nunc Immunosorp plates were coated with either recombinant IκBα or lysate from uninduced BL21 cells (10μg.ml-1), blocked, then exposed overnight to recombinant FLAG-p65RelA in the presence of increasing concentrations of KPV. The amount of FLAG-p65RelA remaining bound in each well was determined as described in the Materials and Methods section. Values are Mean ± SEM, n=9. ANOVA revealed no significant differences between KPV concentrations.
Figure 6.

KPV does not co-localise with IκBα. Distribution of IκBα in cells treated with or without histidine tagged-KPV in HBE cells in the presence of 10ng.ml-1 TNFα for 3h. IκBα was visualised with anti-rabbit IgG-TRITC and H6KPV with anti-histidine IgG-FITC. Images are representative of three independent experiments.
Given the failure of KPV to alter IKK-dependent phosphorylation of IκBα, the relatively slow rate of IκBα stabilisation and the lack of interaction with the DNA minor groove, the next possibility tested was that this peptide might interfere with the nuclear import of p65RelA. Immunofluorescence revealed that KPV abolished TNFα-evoked nuclear import of p65RelA (Figure 7). Real-time experiments in HBE transfected with YFP-p65RelA confirmed this effect in living cells and also revealed a tendency for KPV to suppress YFP-p65RelA fluorescence below the control (i.e. pre-stimulation) level (Figure 7B). TNFα activates NFκB by inducing degradation of IκBα thereby exposing the NLS of p65RelA to Impα3. In vitro competition assays confirmed that KPV suppressed the binding of GST-Imp-α3 with FLAG-p65RelA (Figure 8).
Figure 7.

KPV inhibits Nuclear Translocation of p65RelA. A. Distribution of p65RelA in fixed HBE. Cells had been treated with histidine tagged-KPV and 10ng.ml-1 TNFα for 3h as indicated. p65RelA was visualised with anti-rabbit IgG-TRITC and H6KPV with anti-histidine IgG-FITC. Yellow infers co-localisation between the FITC and TRITC fluors. Images are representative of three independent experiments. B. Inhibition of p65RelA nuclear translocation in living HBE transfected with p65RelA-YFP vector. Cells were pre-incubated for 3h in the presence or absence of KPV (10μg.ml-1) and then exposed to TNFα (5ng.ml-1). Confocal images were obtained at 30 second intervals and the change in nuclear fluorescence was determined using Zeiss LSM510 Image Examiner software. Each line represents data from a single nucleus and data was collected from 4 independent experiments.
Figure 8.

p65RelA interaction with Importin-α3 is competitively inhibited by KPV. A. Representative dot blot experiment. BL21 E. coli transformed with Imp-α3 were induced for 6h with 0.2mM IPTG at 25°C. Cells were lysed and then equal concentrations of lysate protein were spotted onto nitrocellulose. A bacterial lysate from identically treated BL21 cells which had not been transformed with the Imp-α3 vector served as a negative control. Dots were incubated in tris buffered saline containing 10μg.ml-1 protein of a lysate from HBE cells expressing pCMV-FLAG-p65RelA and KPV at the concentrations shown. Dots were visualised using Anti-FLAGHRP. B. Competitive ELISA assay for the p65RelA:Imp-α3 interaction. Open circles: Control (plate coating = bacterial lysate not expressing Imp-α3); Closed Circles: Imp-α3-p65RelA interaction (plate coating = Imp-α3). Values are mean ± SEM, n=10. *P<0.05.
As no crystal structure is currently available for Imp-α3, we performed Pepsite peptide binding analysis [22] on the NLS-interacting domains of the closely related isoform, importin-α2 (3L3Q Mouse Impα2 nuclear localisation sequence). Pepsite analyses residue charge, hydrophobicity and protein structure to produce a statistical probability of interaction between peptide and protein sequences. The analysis is compared to the likelihood that a random sequence of residues will bind to the same region. A probability cut-off value of 0.3 was taken to represent statistical significance. With this criterion KPV was found to interact in several possible orientations with arm domains 7 and 8 of Imp-α2 (Figure 9). No significant interaction was found within the available crystal structures of p65RelA or IκBα.
Figure 9.

Predicted KPV interaction with Imp-α2. Pepsite [22] analysis showing predicted interaction with residues found in Armadillo domains 7 and 8 of murine Importin-α2 (PDB: 3L3Q). P values are listed for each residue in the most favoured orientation.
The second general aim of this study was to determine if receptor-mediated melanocortin signalling can also modulate the inflammatory response in airway epithelium. A nested PCR protocol [21] was used to screen for MC receptor expression in two bronchial airway epithelium-derived cell lines (HBE, H441), one alveolar epithelium-derived cell line (A549) and the human embryonic lung fibroblast cell line, HEL12469 (Figure 10). All epithelial cell lines expressed MC3R exclusively whereas HEL12469 cells expressed MC2R, MC3R and MC5R. Immunohistochemistry revealed that MC3R protein was located in the membrane of HBE cells and was also evident in the apical membrane of airway epithelium in the murine lung in vivo (Figure 11). Cellular (NFκB activation) and systemic (eotaxin, IL8 secretion) inflammatory signalling in HBE induced by either rhino syncitial virus (RSV) or lipopolysaccharide (LPS) were suppressed by either α or γMSH (Figure 12). This effect was mediated by MC3R as siRNA knockdown of this receptor abolished the inhibition of NFκB signalling by α and γ MSH (Figure 13).
Figure 10.

Melanocortin receptor expression in lung-derived cell lines. A nested RT-PCR protocol was used to screen for MC receptors 1-5 in human airway (HBE, H441) and alveolar (A549) epithelial cell lines and in human embryonic lung fibroblasts (HEL12469). Anticipated product length for each receptor is shown to the right of each image. Gels are representative of at least 3 independent experiments.
Figure 11.
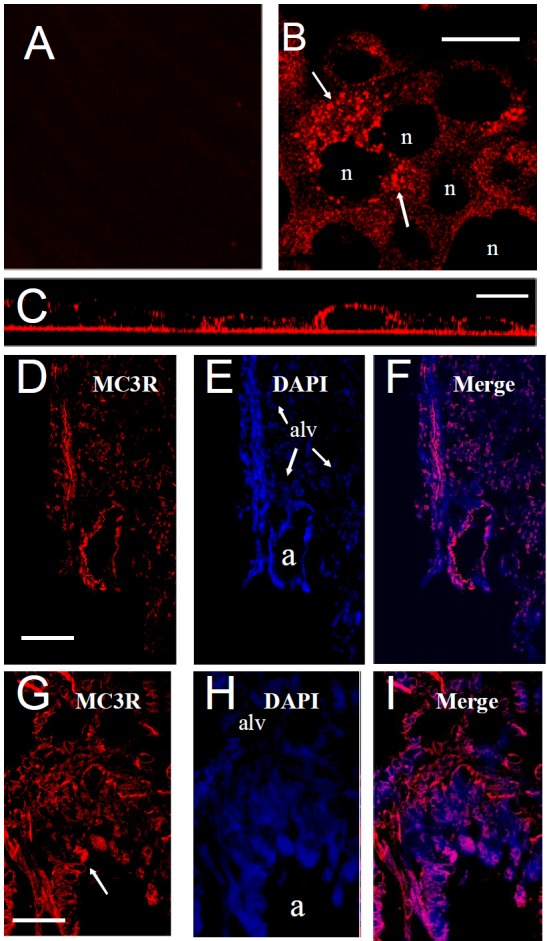
A→I. Distribution of MC3R protein expression in human bronchial epithelial cells and mouse lung. A. Control (2° antibody only), B. MC3R displays punctate expression (arrow) in HBE cells. “n” = nucleus. Bar=15μm. C. Side view Z-stacked image showing predominantly apical MC3R receptor expression. Bar=15μm. D-F. MC3R receptor expression in mouse lung. MC3R, DAPI (blue) staining and merged image respectively, “a” = airway, “alv” = alveoli. Bar = 100μm G-I. Higher resolution image showing apical distribution of MC3R receptor in airway epithelium. “a” = airway lumen. Bar = 30μm.
Figure 12.

α and γ MSH inhibit systemic inflammatory signalling induced by the NFκB pathway. Human bronchial epithelial cells (HBE) were exposed for 6h to 106cfu.ml-1 of Rhinosyncitial Virus (RSV) or 10μg.ml-1 of Pseudomonas aeruginosa lipopolysaccharide (LPS). α, β, or γ MSH were added at 10μg.ml-1. NFκB activity was measured by luciferase assay. Secreted interleukin 8 (IL8) and eotaxin were measured by ELISA (R&D). (*P<0.05 relative to control; n=4 independent experiments).
Figure 13.
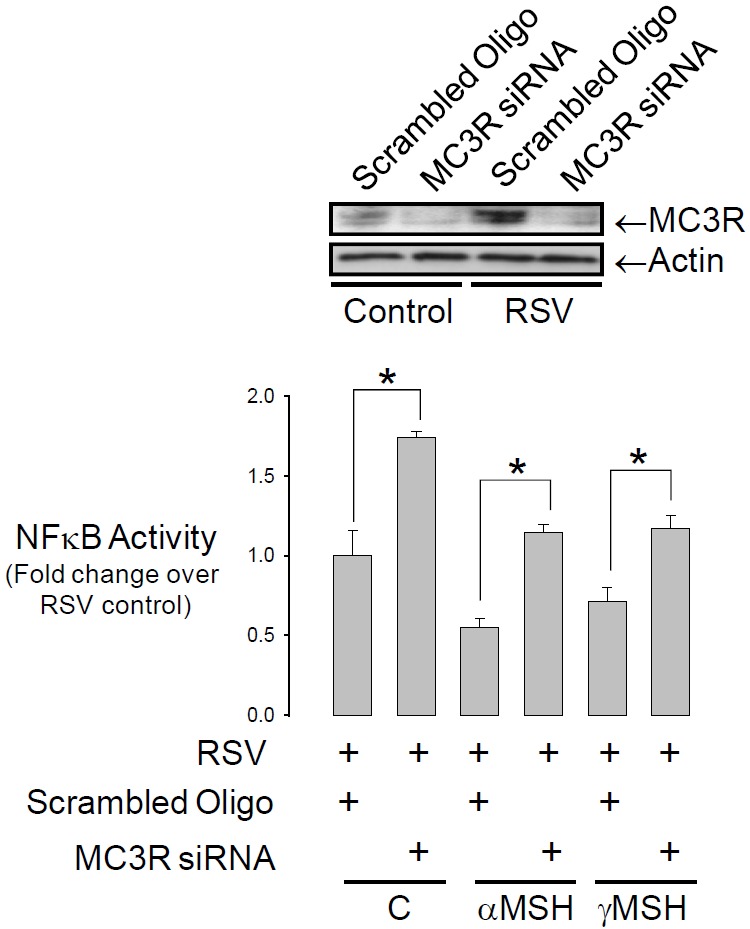
The inhibition of NFκB activity occurs through the MC3R. HBE were co-transfected with siRNA against the MC3R receptor and the NFκB luciferase reporter. Cells were treated with RSV as in Figure 12. siRNA treatment abolished the inhibition of NFκB by α and γ MSH (*P<0.05, n=4). Blot confirms knockout of the MC3R.
Discussion
This study confirms that the melanocortin derived tripeptide, KPV, suppresses both local and systemic immune responses which commonly induce airway damage and remodelling in inflammatory lung disease. These observations are consistent with the widely described ability of KPV and its stereoisomers (dKPV, KPdV, KdPV, dKPdV and KdPT) to act as potent anti-inflammatories and anti-pyretics (reviewed, [1,8]) making them attractive pharmacological targets for the treatment of a wide range of inflammatory diseases.
The purpose of this study was to determine how KPV mediates its anti-inflammatory effects. The H+-coupled oligopeptide transporter, PEPT1, mediates the intracellular uptake of KPV in epithelium and this is necessary for the anti-inflammatory effects [9]. This extends a growing body of evidence which suggests that the antiinflammatory effect of KPV is independent of the melanocortin receptor signalling system and likely occurs through an intracellular mechanism. One of the most consistently reported actions of KPV is that it reduces the duration of NFκB activation, therefore, attention was focussed on its interactions with IκBα, p65RelA and Imp-α3 as critical mediators of NFκB activation and nuclear import. The results corroborate previous observations that KPV promotes IκBα stabilisation in the presence of pro-inflammatory cytokines, however, the major finding is that the predominant site of KPV accumulation is in the nucleus where it competitively inhibits the interaction between p65RelA and Imp-α3. Importantly, this effect occurred without significant interaction with chromatin.
The NLS of p65RelA is located at C-terminal residues 301-304 and contains the consensus sequence KRKR which is flanked by two α-helices, helix three (289-300) and helix four (305-321). Helix four contains critical sites necessary for stable interaction with IκBα which masks the NLS and so blocks interaction with Imp-α3. Phosphorylation and proteolytic degradation of IκBα enables Imp-α3 to bind to the NLS and thus promotes nuclear translocation of the NFκB dimer [14]. The observation that KPV can interfere with the interaction between the p65RelA subunit and Imp-α3 in vitro suggests that this peptide may competitively bind critical sequences in the NLS of either protein. Although crystal structures of Imp-α3 are not yet available, Pepsite binding analysis of KPV interaction with the closely related isoform, mouse Imp-α2, demonstrated multiple possible interactions involving two or more of the KPV residues with amino acids 360-403 spanning arms 7 and 8. Pepsite analysis revealed this to be quite specific to this region of the molecule with no predicted interaction at other sites. Although this study did not demonstrate this interaction in vivo, an interaction between KPV and importin family proteins would explain the tendency for KPV to accumulate in the nucleus in hand with hindered p65RelA nuclear import despite IκBα phosphorylation.
Clearly this raises the question as to whether KPV might interfere with the import of other nuclear proteins. The Pepsite analysis suggests that KPV interaction is specific to the Imp-α2 C-terminal NLS arms 7 and 8 and this domain is critically important among other importin isoforms for the nuclear import of HIF-1α and p65relA (by Imp-α3; [14,23]), STAT1 and influenza A virus nuclear protein (NP) (by Imp-α5; [24]). Indeed, this study shows that KPV can ameliorate the growth inhibition caused by TNFα in addition to its anti-inflammatory effect and also induces the activity of mTORC1, an important regulator of cell growth and differentiation. Thus, the intracellular effects of KPV are pleiotropic and likely to involve a range of effector molecules which may explain the unusually protracted dose response curve for this molecule reported in pharmacological studies [6].
This study demonstrates that KPV suppresses inflammatory signalling in lung bronchial epithelial cells and raises the question as to whether KPV and other melanocortin peptides may be of use in combating inflammatory lung diseases. Previous work in lung cells demonstrates that melanocortin peptides can arrest various forms of inflammation in lung. For example, α-MSH suppressed PGE synthesis in fetal human lung fibroblasts stimulated with IL-1 [25] and also TNFα-induced mucin protein expression in cultured human nasal epithelium [26]. In allergic and non-allergic models of lung inflammation both the MC1R and MC3R agonists, αMSH and [D-TRP(8)]-γ-MSH], inhibited leucocyte accumulation in lung and suppressed inflammatory lung remodelling [27,28]. The current study found that both α and γ-MSH can suppress intracellular and systemic inflammatory cues via the MC3R in airway epithelial cells challenged with either RSV or LPS. This compliments work by Getting et al [27] which demonstrated a central role for MC3R in suppressing leucocyte mediated airway inflammation. Importantly, this work did not rule out a role for MC receptor activation in the airway epithelium and the present study affirms this by demonstrating widespread MC3R expression in airway epithelial cells, both in vitro and in vivo, and by demonstrating that melanocortin peptides suppress chemotactic cytokine secretion from the airway epithelium. KPV affords the advantage that its actions do not appear to be receptor mediated and its structure may be potentially modulated to improve its targeting to one particular pathway or another. Moreover its small size and aqueous solubility mean that it may be delivered in nebulised form to the lung where our data shows it is capable of mediating anti-inflammatory effects in lung epithelia. The suppression of MMP activity is particularly notable as this family of proteases plays a major role in immune signalling and lung remodelling in varied forms of inflammatory and cancerous lung diseases [reviewed 29]. Taken as a whole, this work suggests that KPV and MC3R agonists represent ideal candidates for the suppression of the early stages of inflammation in airway epithelia.
In conclusion, the present study demonstrates that the small melanocortin-related tri-peptide, KPV, inhibits inflammatory signalling in human bronchial epithelial cells through a mechanism which involves the disruption of p65RelA nuclear translocation. The data show that this disruption may occur by a competitive interaction between KPV and p65RelA with the arm domains of Imp-α3 and Pepsite analysis suggests that this may be confined to arm 7 and 8 which are involved in the nuclear trafficking of other transcription factors. Thus, KPV mediates its major anti-inflammatory effect via the nuclear transport system and independently of melanocortin receptors. The pharmaceutical development of KPV as an anti-inflammatory therapy may therefore depend on the degree to which it can be targeted to specific interactions between importin molecules and their protein cargo. In addition to the KPV effect, this study demonstrates that melanocortin agonists of the MC3R can repress early phases of cellular and systemic inflammation highlighting melanocortin peptides as a robust tool for the treatment of inflammatory lung disease.
Acknowledgements
I wish to thank Drs J MacInally and M MacLean for assistance with western blotting and nested PCR experiments and also Dr. K. Treharne for several useful discussions and insightful suggestions. SCL is supported by grants from The Wellcome Trust, Anonymous Trust and Tenovus Scotland.
References
- 1.Catania A, Gatti S, Colombo G, Lipton JM. Targeting melanocortin receptors as a novel strategy to control inflammation. Pharmacol Rev. 2004;56:1–29. doi: 10.1124/pr.56.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Wong KY, Rajora N, Boccoli G, Catania A, Lipton JM. A potential mechanism of local anti-inflammatory action of α-melanocyte- stimulating hormone within the brain: modulation of tumor necrosis factor-α production by human astrocytic cells. Neuroimmunomodulation. 1997;4:37–41. doi: 10.1159/000097313. [DOI] [PubMed] [Google Scholar]
- 3.Delgado R, Carlin A, Airaghi L, Demitri MT, Meda L, Galimberti D, Baron P, Lipton JM, Catania A. Melanocortin peptides inhibit production of proinflammatory cytokines and nitric oxide by activated microglia. J Leukoc Biol. 1998;63:740–745. doi: 10.1002/jlb.63.6.740. [DOI] [PubMed] [Google Scholar]
- 4.Galimberti D, Baron P, Meda L, Prat E, Scarpini E, Delgado R, Catania A, Lipton JM, Scarlato G. α-MSH peptides inhibit production of nitric oxide and tumor necrosis factor-α by microglial cells activated with β-amyloid and interferon-γ. Biochem Biophys Res Commun. 1999;263:251–256. doi: 10.1006/bbrc.1999.1276. [DOI] [PubMed] [Google Scholar]
- 5.Barcellini W, Colombo G, La Maestra L, Clerici G, Garofalo L, Brini AT, Lipton JM, Catania A. α-Melanocyte-stimulating hormone peptides inhibit HIV-1 expression in chronically infected promonocytic U1 cells and in acutely infected monocytes. J Leukoc Biol. 2000;68:693–699. [PubMed] [Google Scholar]
- 6.Moustafa M, Szabo M, Ghanem GE, Morandini R, Kemp EH, MacNeil S, Haycock JW. Inhibition of tumor necrosis factor-α stimulated NFκB/p65 in human keratinocytes by α-melanocyte stimulating hormone and adrenocorticotropic hormone peptides. J Invest Dermatol. 2002;119:1244–1253. doi: 10.1046/j.1523-1747.2002.19602.x. [DOI] [PubMed] [Google Scholar]
- 7.Capsoni F, Ongari A, Colombo G, Turcatti F, Catania A. The synthetic melanocortin (CKPV)2 exerts broad anti-inflammatory effects in human neutrophils. Peptides. 2007;28:2016–2022. doi: 10.1016/j.peptides.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Brzoska T, Luger TA, Maaser C, Abels C, Böhm M. Alpha-melanocyte-stimulating hormone and related tripeptides: biochemistry, antiinflammatory and protective effects in vitro and in vivo, and future perspectives for the treatment of immune-mediated inflammatory diseases. Endocr Rev. 2008;29:581–602. doi: 10.1210/er.2007-0027. [DOI] [PubMed] [Google Scholar]
- 9.Dalmasso G, Charrier-Hisamuddin L, Thu Nguyen HT, Yan Y, Sitaraman S, Merlin D. PepT1-mediated tripeptide KPV uptake reduces intestinal inflammation. Gastroenterology. 2008;134:166–178. doi: 10.1053/j.gastro.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blalock JE. Complementarity of peptides specified by 'sense' and 'antisense' strands of DNA. TIBTECH. 1991;6:140–144. doi: 10.1016/0167-7799(90)90159-u. [DOI] [PubMed] [Google Scholar]
- 11.Siemion IZ, Cebrat M, Kluczyk A. The Problem of Amino Acid Complementarity and Antisense Peptides. Curr Prot Peptide Sci. 2004;5:507–527. doi: 10.2174/1389203043379413. [DOI] [PubMed] [Google Scholar]
- 12.Chelsky D, Ralph R, Jonak G. Sequence requirements for synthetic peptide-mediated translocation to the nucleus. Mol Cell Biol. 1989;9:2487–2492. doi: 10.1128/mcb.9.6.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fontes MR, Teh T, Kobe B. Structural basis of recognition of monopartite and bipartite nuclear localization sequences by mammalian importin-α. J Mol Biol. 2000;297:1183–1194. doi: 10.1006/jmbi.2000.3642. [DOI] [PubMed] [Google Scholar]
- 14.Fagerlund R, Kinnunen L, Köhler M, Julkunen I, Melén K. NF-κB is transported into the nucleus by importin-α3 and importin-α4. J Biol Chem. 2005;280:15942–15951. doi: 10.1074/jbc.M500814200. [DOI] [PubMed] [Google Scholar]
- 15.Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH, Gruenert DC. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1994;10:38–47. doi: 10.1165/ajrcmb.10.1.7507342. [DOI] [PubMed] [Google Scholar]
- 16.Hunter MJ, Treharne KJ, Winter AK, Cassidy DM, Land S, Mehta A. Expression of wild-type CFTR suppresses NF-κB-driven inflammatory signalling. PLoS One. 2010;5:e11598. doi: 10.1371/journal.pone.0011598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki M. SPKK, a new nucleic acid-binding unit of protein found in histone. EMBO J. 1989;8:797–804. doi: 10.1002/j.1460-2075.1989.tb03440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reeves R, Nissen MS. The A.T-DNA binding domain of mammalian high mobility group I chromosomal proteins. J Biol Chem. 1990;265:8573–8582. [PubMed] [Google Scholar]
- 19.Land SC, Darakhshan F. Thymulin evokes IL-6-C/EBPbeta regenerative repair and TNF-alpha silencing during endotoxin exposure in fetal lung explants. Am J Physiol Lung Cell Mol Physiol. 2004;286:L473–487. doi: 10.1152/ajplung.00401.2002. [DOI] [PubMed] [Google Scholar]
- 20.Land SC, Tee AR. Hypoxia-inducible factor 1α is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem. 2007;282:20534–20543. doi: 10.1074/jbc.M611782200. [DOI] [PubMed] [Google Scholar]
- 21.Cooper A, Robinson SJ, Pickard C, Jackson CL, Friedmann PS, Healy E. α-melanocyte-stimulating hormone suppresses antigeninduced lymphocyte proliferation in humans independently of melanocortin 1 receptor gene status. J Immunol. 2005;175:4806–4813. doi: 10.4049/jimmunol.175.7.4806. [DOI] [PubMed] [Google Scholar]
- 22.Petsalaki E, Stark A, García-Urdiales E, Russell RB. Accurate prediction of peptide binding sites on protein surfaces. PLoS Comput Biol. 2009;5:e1000335. doi: 10.1371/journal.pcbi.1000335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Depping R, Steinhoff A, Schindler SG, Friedrich B, Fagerlund R, Metzen E, Hartmann E, Köhler M. Nuclear translocation of hypoxia-inducible factors (HIFs): involvement of the classical importin alpha/beta pathway. Biochim Biophys Acta. 2008;1783:394–404. doi: 10.1016/j.bbamcr.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 24.Melen K, Fagerlund R, Franke J, Kohler M, Kinnunen L, Julkunen I. Importin alpha nuclear localization signal binding sites for STAT1, STAT2, and influenza A virus nucleoprotein. J Biol Chem. 2003;278:28193–28200. doi: 10.1074/jbc.M303571200. [DOI] [PubMed] [Google Scholar]
- 25.Cannon JG, Tatro JB, Reichlin S, Dinarello CA. α-Melanocyte stimulating hormone inhibits immunostimulatory and inflammatory actions of interleukin 1. J Immunol. 1986;137:2232–2236. [PubMed] [Google Scholar]
- 26.Lee SN, Ryu JH, Joo JH, Choi YH, Lee HJ, Kim YJ, Kim KB, Yoon JH. α-Melanocyte-stimulating hormone Inhibits TNFα stimulated MUC5AC expression in human nasal epithelial cells. Am J Respir Cell Mol Biol. 2010 doi: 10.1165/rcmb.2009-0420OC. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 27.Getting SJ, Riffo-Vasquez Y, Pitchford S, Kaneva M, Grieco P, Page CP, Perretti M, Spina D. A role for MC3R in modulating lung inflammation. Pulm Pharmacol Ther. 2008;21:866–873. doi: 10.1016/j.pupt.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 28.Raap U, Brzoska T, Sohl S, Path G, Emmel J, Herz U, Braun A, Luger T, Renz H. α-Melanocyte -stimulating hormone inhibits allergic airway inflammation. J Immunol. 2003;171:353–359. doi: 10.4049/jimmunol.171.1.353. [DOI] [PubMed] [Google Scholar]
- 29.Yao H, Rahman I. Current concepts on the role of inflammation in COPD and lung cancer. Curr Opin Pharmacol. 2009;9:375–383. doi: 10.1016/j.coph.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]