

Abstract
Down syndrome (DS) is caused by the overexpression of genes on triplicated regions of human chromosome 21 (Hsa21). While the resulting physiological and behavioral phenotypes vary in their penetrance and severity, all individuals with DS have variable but significant levels of cognitive disability. At the core of cognitive processes is the phenomenon of synaptic plasticity, a functional change in the strength at points of communication between neurons. A wide variety of evidence from studies on DS individuals and mouse models of DS indicates that synaptic plasticity is adversely affected in human trisomy 21 and mouse segmental trisomy 16, respectively, an outcome that almost certainly extensively contributes to the cognitive impairments associated with DS. In this review, we will highlight some of the neurophysiological changes that we believe reduce the ability of trisomic neurons to undergo neuroplasticity-related adaptations. We will focus primarily on hippocampal networks which appear to be particularly impacted in DS and where consequently the majority of cellular and neuronal network research has been performed using DS animal models, in particular the Ts65Dn mouse. Finally, we will postulate on how altered plasticity may contribute to the DS cognitive disability.
1. Introduction
Down syndrome (DS) results from the triplication of genes on human chromosome 21 (Hsa21) and is associated with a range of phenotypes including craniofacial changes [1, 2], cardiac defects [3], susceptibility to leukemia but with reduced occurrence of solid cancers [4, 5], and intellectual disability [6, 7]. While the presence and severity of these individual phenotypes vary among DS individuals, every individual with DS has some degree of cognitive impairment. These impairments limit the independence of DS subjects and adversely impact their quality of life. Consequently, understanding the genetic causes of cognitive dysfunction in DS has been the focus of much research in this field.
The phenomenon of synaptic plasticity has been strongly linked to cognitive processes, such as learning and memory [8, 9]. Synaptic plasticity refers to the dynamic nature of synapses, sites of communication between neurons, in which the structure, composition, or function of the synapse changes in response to network activity. Depending on the timing and strength of pre- and postsynaptic activity, synapses can either be strengthened or weakened providing a potential mechanism for memory formation and storage [10]. Structurally, synaptic connections on excitatory neurons are typically formed on the heads of dendritic spines [11]. The morphology of the spines enables compartmentalization of signaling cascades and facilitates manipulation of the structure and composition of the cell membrane by second messenger systems [12, 13]. Thus, not only is the number of spines important, as individual locations for excitatory synaptic transmission, but the shape of the individual spines also has a critical functional role.
The link between synaptic plasticity and cognitive processes such as learning and memory is frequently studied within the hippocampus, a structure involved in diverse cognitive processes such as those related to acquisition, coding, storing, and recalling information in physical or perceived spatial environments [14–16]. Multiple lines of evidence indicate that long-lasting up- or downregulation of functional synaptic strengths, referred to as long-term potentiation (LTP) and long-term depression (LTD), respectively, are fundamental synaptic mechanisms underlying hippocampal contributions to these processes. Thus, dendritic and synaptic abnormalities in the hippocampus, either morphological or functional, would be expected to significantly impact spatial cognition. Indeed, neuropsychological investigations requiring the use of spatial information in problem solving indicate that deficits in hippocampal-mediated learning and memory processes are hallmarks of DS [17, 18]. In this paper, we will provide an overview of the morphological and behavioral evidence for altered synaptic plasticity in DS with a focus on the hippocampus and discuss the insights provided by mouse models of this neurodevelopmental disorder into the potential molecular mechanisms contributing to these deficits.
2. Evidence for Altered Synaptic Plasticity in DS: A Neurodevelopmental Impact
The basis for altered synaptic plasticity in DS can be found in changes in the physical structure of the dendrites. Alterations in the shape and densities of dendrites would be expected to adversely affect the information storage capacity of neural networks by reducing the number of potential sites for plasticity to occur. Consistent with this idea and the observed deficits in cognition associated with DS, examination of postmortem brain tissue from DS individuals reveals profound alterations in dendritic and neuronal densities and morphology across many regions of the brain beginning in utero and persisting throughout life. The neocortical development of DS fetuses appears normal up to at least gestational week 22 [19–21]. By 40 weeks gestation, less discrete lamination is observed in the neocortex of DS fetuses with lower and higher cell densities observed in the visual cortex and superior temporal neocortex, respectively [19, 20]. In the hippocampus, deficits begin to appear slightly earlier as DS fetuses (17 to 21 weeks of gestation) show altered morphology, reduced neuron numbers, enhanced apoptosis, and reduced cell proliferation [22–24]. These changes may result, in part, from reductions in serotonin, dopamine, and GABA levels in the fetal DS cortex [25] since, during development, neurotransmitters such as these can act as neurotrophic factors assisting with neuronal migration, axon guidance, and neurite development [26].
During the early postnatal period, significant deficits in brain weight and gross morphology as well as myelination and neuronal densities and morphology appear [27]. Initially, dendritic expansion is enhanced in DS infants, but, by the first to second year of life, this trend reverses to become a deficit [19, 28] which persists into adulthood [19, 29]. Dendritic spine numbers are reduced, and morphology altered in DS [30, 31]. Consistent with adverse changes in dendrite morphology, synaptogenesis is also aberrant in DS fetuses [19, 32, 33] and remains deficient in adulthood [34]. MRI studies reveal that DS children and young adults have smaller overall brain volumes [35, 36] with particular deficits noted in the hippocampus [36, 37]. Hippocampal volume, that continues to decrease with age in DS individuals [38], was found to be inversely correlated with the degree of cognitive impairment [36]. Cognitive tests such as the Cambridge Neuropsychological Testing Automated Battery (CANTAB) and the Arizona Cognitive Test Battery (ACTB), the latter specifically tailored to address DS deficits, indicate that hippocampal function is particularly impacted by the DS genetic condition [17, 39].
These morphological and cognitive deficits are consistent with aberrant synaptic plasticity, and, indeed, while difficult to measure directly in human subjects, evidence suggests that plasticity is reduced at least in the motor cortex of DS individuals [40]. Additionally, functional MRI (fMRI) during cognitive processing tasks reveals abnormal neural activation patterns in DS children and young adults [41, 42]. Examination of resting glucose metabolism in the cerebral cortex of DS individuals found enhanced uptake in brain regions associated with cognition suggesting cellular hyperactivity in those areas [43]. To better understand the functional consequences resulting from altered network morphologies as well as investigate potential alterations in intracellular signaling cascades contributing to aberrant plasticity, it was necessary to develop and then examine animal models of DS.
3. Modeling DS Cognitive Impairment
Over the past few decades, several mouse models of Down syndrome have been developed to further our understanding of the link between enhanced gene dosage and DS phenotypes such as altered plasticity and cognition. The Tc1 mouse model carries an almost complete, freely segregating copy of Hsa21, but the chromosome is present in only approximately 50% of cells making this a mosaic model of DS [46]. Interestingly, some genes have been deleted from the “inserted” Hsa21 [47]. It is important to note that, in spite of the mosaicism and gene deletions, many DS phenotypes have been replicated in this model [46, 48, 49]. Other mouse models have taken advantage of the homology between regions of Hsa21 and mouse chromosomes 10, 16, and 17 (Mmu10, 16, 17) making models in which these genes are triplicated highly useful in understanding the genetic basis of DS phenotypes [50, 51]. A mouse model trisomic for all Hsa21 homologous segments was recently developed and holds great promise for furthering our understanding of DS [52]. As this is a relatively new model, however, most research has been conducted using the Ts65Dn segmental trisomic mouse [53–55] which is trisomic for more than 50% genes of Hsa21 homologs [56, 57] and has well-documented DS-like deficits in behavioral tasks such as those relying upon declarative memory (novel object recognition and spontaneous alternation tasks) and the proper encoding and recollection of spatial information (radial arm and Morris water mazes) [58–63]. While the Ts65Dn mouse is the only mouse model of DS to have a freely segregating supernumerary chromosome, they are also trisomic for 60 genes that do not have Hsa21 homologs [64], and the impact of overexpression of these genes on Ts65Dn phenotypes remains to be determined.
Similar to the Ts65Dn mouse but with smaller triplicated Mmu16 segments are the Ts1Cje and Ts1Rhr mouse models. These mice display phenotypes similar to Ts65Dn mice including hippocampal dysfunction; however, the severity of the deficits is reduced [65–69]. The reduced severity of DS-like deficits in mice with fewer trisomic genes highlights one of the powerful aspects of mouse models: the ability to control expression of certain HSA21 homologs to assess their contribution to specific DS phenotypes. Those deficits associated with the hippocampus, whose function is notably altered in DS individuals [17, 39], will be the focus of the remainder of this paper.
3.1. Morphological Changes
Mouse models of DS, including the Ts65Dn strain, show similar detrimental changes in neuronal and dendritic morphologies observed in humans. The neocortex of Ts65Dn mice contains fewer excitatory neurons but an increased number of a subset of inhibitory neurons relative to euploid controls, a phenotype that was reversed by normalizing the expression levels of Olig1/2 [70]. Additionally, regions both in the neocortex and hippocampus have decreased spine densities with larger spine volumes [71]. In the dentate granule cells of the hippocampus, there is a shift of inhibitory synaptic connections away from the dendritic shafts and onto the necks [71]. Such a change would be expected to increase the efficacy of inhibitory synaptic transmission given the significantly reduced volume of the spine neck compared to the shaft. At a finer resolution, symmetric (presumed inhibitory synapses) have greater opposition lengths in Ts65Dn while asymmetric synapses were unaltered [72], again supporting a shift towards excess inhibition in these mice. Similar but less severe changes are observed in Ts1Cje mice [67]. Beyond suppressing excitatory synaptic activity, the altered spine morphology and shift towards excess inhibition in trisomy would be expected to suppress plasticity-related signaling cascades that frequently rely on depolarization-mediated calcium influx into the postsynaptic structural domains.
3.2. Functional Changes
Synaptic plasticity in the hippocampus is often investigated in the context of long-term potentiation (LTP) in which high-frequency activation of specific inputs in the hippocampus results in a long-lasting potentiation of synaptic responses along the excited afferent pathway. First described in the anesthetized rabbit [73], this phenomenon is believed to be a fundamental mechanism underlying memory formation [8, 74] and is suppressed in Ts65Dn (depicted in Figure 1 for the CA1 region of the hippocampus) [44, 75, 76] and Ts1Cje [66, 67] but not in Ts1Rhr mice [69] (however, see [68]) as well as mice trisomic for Hsa21 syntenic regions of Mmu16 and Mmu17 [77] or those carrying an almost complete copy of Hsa21 [46, 48].
Figure 1.
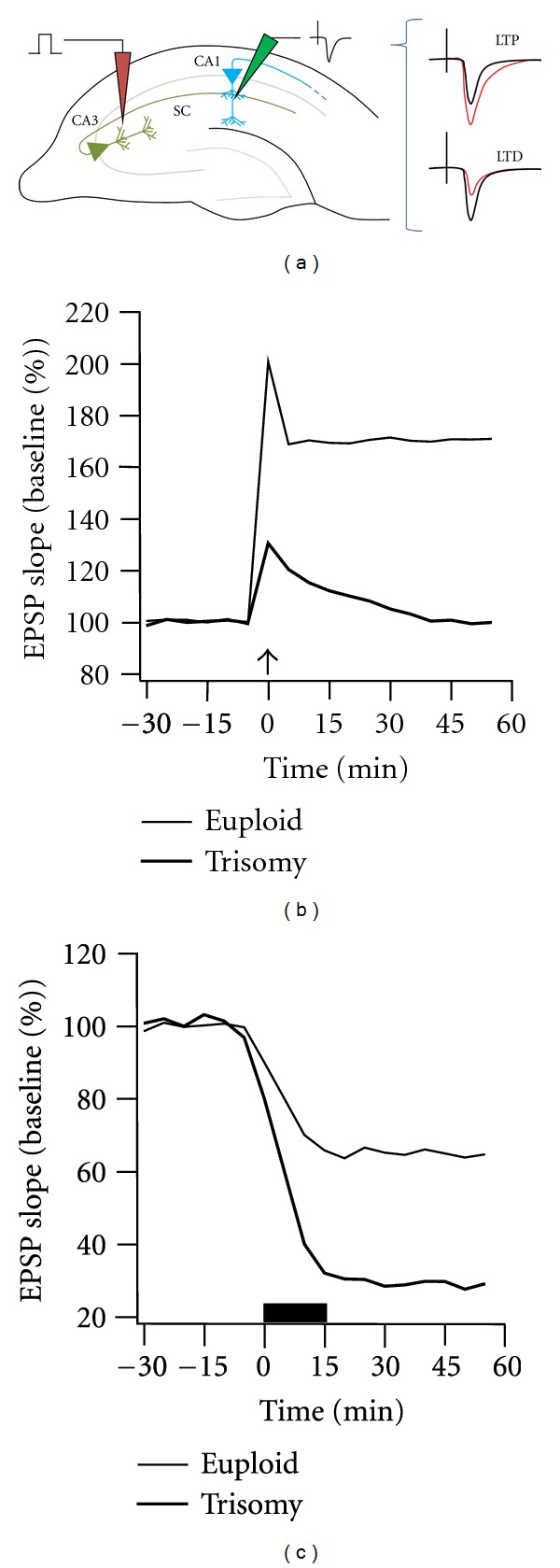
Depiction of altered CA1 hippocampal plasticity in Ts65Dn mice. (a) Diagram indicating electrode placement for stimulating Schaffer collaterals arising from CA3 and recording the evoked field excitatory postsynaptic potential (EPSP) in CA1. Traces to the right indicate the typical change in evoked responses (red) following LTP and LTD. (b) Simulated data depicting suppressed LTP in Ts65Dn mice. After high-frequency stimulation of SC (at arrow head), the field EPSP increases and remains enhanced in euploid mice but fails to remain elevated in Ts65Dn mice. (c) Simulated LTD data depicting exaggerated depression of evoked EPSPs following low-frequency stimulation of SC in Ts65Dn mice. (Traces in B and C based on data from [44, 45].)
As outlined above, structural changes suggest that inhibition is exaggerated in the trisomic hippocampus. Consistent with this idea is the observation that LTP in the dentate gyrus and CA1 regions of Ts65Dn hippocampal slices, induced by high-frequency stimulation and theta burst protocols, respectively, can be rescued by the GABAA antagonists picrotoxin [75, 76]. Blockade of GABAA receptors in hippocampal slices from Ts1Cje and Ts1Rhr mice rescues LTP deficits in the dentate gyrus in these DS mouse models as well [67, 68]. A similar treatment in Ts65Dn mice leads to an enhancement in cognitive performance [60].
In addition to suppressed LTP, hippocampi from Ts65Dn mice show enhanced long-term depression (LTD) in response to sustained activation of excitatory synapses [45, 78]. This latter effect can be reversed with the uncompetitive NMDA receptor antagonist memantine [78] and also improves the cognitive performance of Ts65Dn mice [79–81]. These results draw a clear link between altered synaptic plasticity in the hippocampus and cognitive performance in the Ts65Dn mouse model of Down syndrome.
3.3. Synaptic-Plasticity-Related Signaling Cascades
Changes in intracellular calcium concentrations are important triggers for many intracellular signaling cascades including those underlying LTP and LTD [82]. For example, the presence of a calcium chelator that buffers intracellular calcium levels in postsynaptic neurons prevents the induction of LTP [83] consistent with the hypothesis that a postsynaptic rise in intracellular calcium levels is necessary for LTP [8]. When strongly depolarized, the magnesium block of NMDA channels is lifted providing the main (but not exclusive) mechanism for calcium entry into the postsynaptic cell. Elevated intracellular calcium levels trigger a cascade of intracellular messengers that ultimately lead to the induction and maintenance of synaptic plasticity (both LTP and LTD depending on the kinetics). An excellent overview of this process can be found in several reviews [82, 84], and only key components known to be affected by trisomy (Figure 2) will be discussed here.
Figure 2.
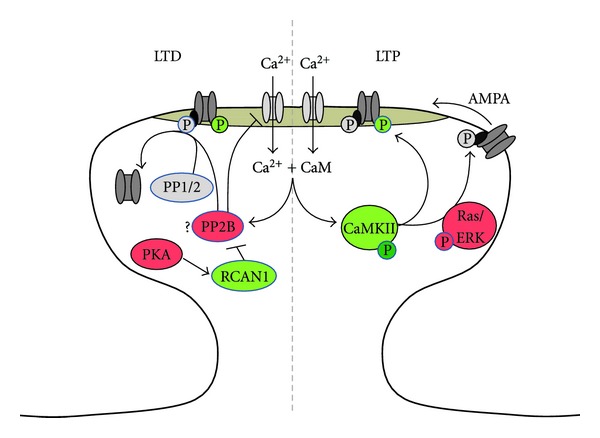
Alterations in intracellular signaling cascades affecting postsynaptic AMPAR response in Ts65Dn hippocampus. Green indicates elevated levels/activity at baseline, while red indicated diminished activity. During LTP (right), enhanced CaMKII and GluR1 subunit phosphorylation in Ts65Dn synapses may result in a saturated condition incapable of additional potentiation. Reduced ERK activity may reduce migration of new AMPARs into the PSD. In LTD, overexpression of RCAN1 should reduce the activity of PP2B (calcineurin) resulting in reduced internalization of AMPA receptors and potential reduction of NMDAR mean open times. Rescue of LTD in Ts65Dn mice by NMDAR antagonists suggests enhanced NMDAR activity contributes to altered LTD through yet unidentified mechanisms.
3.3.1. CaMKII
Activation of postsynaptic NMDA receptors (NMDARs) concomitant with the depolarization of the postsynaptic membrane is sufficient to relieve the magnesium block of NMDA channels leading to an influx of calcium into the intracellular postsynaptic space. In the case of LTP, the rise of intracellular calcium leads to the activation of calcium calmodulin-dependent protein kinase II (CaMKII), a necessary step for initiating NMDAR-dependent LTP [82]. Blocking CaMKII prevents induction of LTP, [82, 85, 86], while constitutively active forms can induce LTP [87]. During all phases of LTP (induction, early, and late), levels of phosphorylated CaMKII are increased in the hippocampus [88]. CaMKII phosphorylated at threonine 286 (Thr286) can become constitutively active providing a potential switch for initiating and then maintaining potentiation [89]. Alternatively, phosphorylation of Thr305/306 can inhibit the expression of LTP by interfering with the binding of calcium/calmodulin [90, 91]. Indeed, cognitive deficits associated with Angelman syndrome were reversed in a mouse model of the disorder by reducing the levels of CaMKII phosphorylated at Thr305/306 [92]. Thus, depending on the site of phosphorylation, CaMKII can facilitate or suppress initiation and maintenance of LTP. In Ts65Dn mice, we found that baseline levels of CaMKII phosphorylated at Thr286 are elevated in the hippocampus [93]. Excessive basal phosphorylation of the CaMKII site leading to constitutive activation could leave the DS modeling trisomic network in a saturated state unable to shift to more potentiated levels.
One of the substrates targeted by CaMKII during the initial expression of LTP is the serine 831 residue on GluR1 subunits of AMPA receptors [94, 95]. This phosphorylation leads to an increase in conductance of the AMPA channel [96] providing a rapid mechanism for enhancing glutamatergic synaptic strength. In Ts65Dn mice, we find that baseline levels of phosphorylated serine 831 in synaptically located GluR1 receptors are elevated [93]. This apparent increase in AMPA channel conductance appears not to have any significant effect on baseline excitatory synaptic transmission which is normal in the Ts65Dn hippocampus [45, 75, 76, 93]. However, it could also partially occlude the initiation of LTP in these mice by leaving Ts65Dn hippocampal excitatory synapses with fewer AMPA channels available for potentiation. This finding would be consistent with our observation of increased CaMKII in Ts65Dn hippocampus noted above [93] and the suggestion that some components of the LTP network are in an apparent saturated state in these mice.
3.4. PKA, RCAN1, Calcineurin
Protein kinase A (PKA) also plays a critical role in establishing LTP. In particular, evidence suggests that it is involved in initiating the protein synthesis required for the late phase of LTP [97, 98]. Blocking PKA activity suppresses the late phase of LTP (lasting beyond 3 hours) while leaving the early phase of LTP (less than 3 hours) unaffected [99, 100]. Transgenic mice in which PKA activity is reduced have significantly decreased late-phase LTP in CA1 but normal early LTP and perform poorly on tasks requiring long- but not short-term memory formation [101].
PKA plays a role in LTD where its substrates, such as GluR1, show increased dephosphorylation following induction [102, 103]. Dephosphorylation of GluR1 subunits should reduce the conductance levels of affected AMPA receptors [96] resulting in a reduction of synaptic strength. PKA also enhances the activity of RCAN1 [104], an inhibitor of calcineurin which contributes to AMPA receptor internalization [105] and reductions in NMDA receptor mean open time [106]. In Ts65Dn mice, we found that PKA activity is reduced in the hippocampus [93], which should adversely affect LTP by reducing protein expression required for the late phase. With respect to LTD, reduced PKA activity would result in more AMPA receptors remaining in a high conductance state and less facilitation of RCAN1 activity. This latter effect is offset, however, by the overexpression of the gene encoding RCAN1 in DS and Ts65Dn mice [4]. How these factors contribute to the enhancement of LTD in Ts65Dn hippocampi [45, 78] remains to be determined.
3.5. Extracellular Receptor Kinase (ERK)
In addition to GluR1 subunits, both CaMKII and PKA converge on another common effector, the mitogen-activated protein kinase (MAPK/ERK), that is associated with a host of synaptic-plasticity-related cellular processes [107]. In the case of hippocampal LTP, there is a rapid increase in the amount of phosphorylated ERK following induction [108] and blocking ERK activation prevents expression of LTP [109]. Cultured hippocampal neurons undergo phosphorylated ERK-dependent spine generation following LTP conditioning stimuli implicating this pathway in spine formation [110]. Additionally, it is believed that lateral diffusion of extrasynaptic AMPA receptors containing GluR1 subunits into the postsynaptic density (PSD) is a major contributor to LTP expression [111]. This process is assisted by Ras/Erk phosphorylation of stargazin on extrasynaptic AMPA receptors enabling them to be structurally secured at the synapse to PSD95 [112]. In the Ts65Dn hippocampus, ERK phosphorylation is decreased [93] suggesting decreased activity. This would be expected to adversely affect the insertion of new AMPA receptors into the PSD as well as morphological restructuring of synaptic spines observed after LTP in normal mice [113, 114].
3.6. BDNF Pathway
Brain-derived neurotrophic factor (BDNF) contributes to LTP by stimulating protein synthesis. In activating postsynaptic TrkB receptors, BDNF stimulates the PI3K pathway [115] which can initiate translation through mammalian target of rapamycin (mTOR) thereby enhancing synthesis of proteins such as CaMKIIα, GluR1, and NMDA receptor subunit 1 [116]. In Ts65Dn mice, we found that PI3K phosphorylation failed to increase following an LTP induction protocol suggesting this pathway is perturbed by trisomy [93]. Consistent with this notion, in DS individuals, BDNF blood plasma levels are approximately 5 times higher than in age-matched controls [117]. As BDNF readily crosses the blood-brain barrier [118], these levels likely reflect those present in the CNS as well.
Examination of BDNF levels in DS mouse models presents a complex picture. In Ts65Dn mice, levels of BDNF in the frontal cortex are diminished [119]. In the hippocampus, both no difference [81] and a reduction [120] compared to control have been reported. In the latter case, the reduction in BDNF levels was associated with decreased neurogenesis and was reversible through treatment with fluoxetine [120]. In the Ts1Cje mouse model of DS, BDNF is overexpressed in the hippocampus, particularly in the dentate gyrus and CA1 regions and in the dendrites of dissociated hippocampal neurons grown in culture [121]. Increased BDNF levels in Ts1Cje mice hippocampi were associated with greater levels of phosphorylated Akt-mTOR and expression of GluR1 protein which could not be further enhanced with exogenous supplemental BDNF suggesting this pathway related to synaptic plasticity is saturated in these mice preventing further contributions to LTP [121].
The discrepancies between observations in Ts65Dn and Ts1Cje BDNF levels may reflect how BDNF expression is distributed in these structures, elevated in some subregions or subcellular compartments while diminished in others, resulting in an increased functional effect despite reduced global levels. Conversely, the differences in observed BDNF levels could be related to the different numbers of genes overexpressed in these two mouse lines [53, 65, 122] or, as mice of differing age groups were used in the studies, may reflect differences in expression levels as a function of age. Further investigation is necessary to fully align these observations. However, the observation that rapamycin has a restorative effect on phosphorylated Akt-mTOR levels in Ts1Cje suggests a potential therapeutic mechanism for improving cognition in DS individuals [121] possibly by normalizing a pathway involved in synaptic plasticity.
3.7. GABAB-GIRK2 Attenuation of Synaptic Plasticity
As mentioned above, postsynaptic calcium influx is critical for LTP and LTD in the hippocampus. This initiating step relies heavily upon depolarization of the postsynaptic membrane to relieve the voltage-dependent magnesium block of NMDA channels. Any phenomenon that reduces the ability of the postsynaptic membrane to depolarize would thus be expected to adversely affect plasticity. Through its coupling to GABA B receptors, the type 2 G-protein-activated inward rectifying potassium (GIRK2) channel may act to dampen the expression of LTP in Ts65Dn hippocampus through a shunting mechanism.
GIRK2 is encoded by the gene Kcnj6 which is located on the chromosomal segment triplicated in DS and Ts65Dn mice, and, consequently, elevated expression levels have been found in the Ts65Dn hippocampus [50, 123]. At a cellular level, overexpression of GIRK2 leads to a more hyperpolarized resting potential in cultured hippocampal neurons [124] and CA1 pyramidal neurons in vitro [125]. Selectively reducing the expression level of GIRK2 by crossing euploid and Ts65Dn mice with mice heterozygous for GIRK2 (GIRK2+/−) resulted in a gene dosage-dependent change in the resting membrane potential and facilitation of LTP in GIRK2 knockout mice [50]. Selective overexpression of GIRK2 alone in mice results in cognitive deficits, reduced depotentiation (a functional reversal of potentiation at a synapse), and enhanced LTD [126].
These effects on LTP and LTD could be mediated through GABAB receptors which, in pyramidal neurons, are in closest proximity to GIRK2-contaning potassium channels near glutamatergic synapses on dendritic spines [127]. GABAB receptors are functionally linked to GIRK channels, and, indeed, whole-cell GABAB-mediated potassium currents are exaggerated in Ts65Dn hippocampal neurons [50, 124, 128]. In CA1, these exaggerated currents have a greater functional impact on the distal dendrites of pyramidal neurons as opposed to those located more proximally [128]. A similar enhancement of GABAB-mediated currents is also found in the dentate gyrus where the presynaptic release probability of GABA is increased [129]. Thus, GIRK channels, activated by GABAB and other G-protein coupled receptors, appear to act as a break on synaptic plasticity in the Ts65Dn hippocampus.
4. Potential Impact of Altered Plasticity on Hippocampal Processing
The hippocampus receives major inputs from the entorhinal cortex (EC) which converge on CA1 pyramidal through two main pathways: the perforant pathway (PP) and the temporoammonic (TA) pathway [130]. The PP pathway passes through the dentate gyrus to pyramidal neurons in CA3 before impinging upon the relatively proximal dendrites of CA1 pyramidal neurons in stratum radiatum (SR). Conversely, inputs to CA1 from TA target the distal dendrites located in stratum lacunosum molecular (SLM). In the normal hippocampus, frequency-based synaptic plasticity at the CA3-CA1 synapse, coupled with a feed-forward inhibition loop from stratum oriens alveus interneurons that suppress inputs to distal CA1 dendrites, enables segregation of information flow through these two pathways [131]. During high-frequency synaptic activity, the CA3-CA1 synapse would be expected to undergo LTP, increasing the excitatory drive to CA1 pyramidal neurons and, consequently, enhanced suppression of inputs to distal CA1 dendrites by the feed-forward inhibition loop (Figure 3(a)). Thus, TA inputs that target distal CA1 dendrites would be suppressed, and information flows through the CA3-CA1 pathway enhanced during during high-frequency events. Conversely, during low-frequency synaptic activity, the CA3-CA1 synapse would be expected to undergo LTD and become less effective. Inhibition of distal CA1 synapses would then be decreased and information flow through the TA pathway would likely be enhanced (Figure 3(b)). Diminished LTP resulting from trisomy would then interfere with this frequency-based segregation of information flow through the hippocampus. Without LTP, feed-forward inhibition would cause suppression of information flow through the TA pathway causing inputs from the two pathways to become superimposed upon and interfere with each other (Figure 3(c)). In contrast, the flow of information during low frequency signaling would likely remain intact, or potentially facilitated, since enhanced LTD at CA3-CA1 would prevent interference from this pathway (Figure 3(d)).
Figure 3.

Potential impact of altered synaptic plasticity on hippocampal processing in Ts65Dn mouse model of Down syndrome. Schematic of two main pathways through hippocampus arriving from the entorhinal cortex: temporoammonic (TA)—direct to CA1 distal dendrites; trisynaptic pathway from DG through CA3 to proximal CA1 dendrites. LTP and LTD are proposed to minimize interference between the two pathways [50, 131]. (a) In euploid hippocampi, high-frequency inputs induce LTP in CA1 resulting in enhanced suppression of inputs from TA by feed-forward inhibition arising from interneurons in stratum oriens. (b) Low-frequency inputs depress the trisynaptic pathway releasing distal CA1 dendrites from feed-forward inhibition and allowing information to flow through the TA pathway. (c) In Ts65Dn hippocampi, aberrant LTP in CA1 results in diminished feed-forward inhibition during high-frequency activity allowing TA inputs to become superimposed on those flowing through the trisynaptic pathway. (d) Enhanced LTD would be expected to facilitate flow of low-frequency information through the direct TA pathway in Ts65Dn mice.
Electroencephalogram (EEG) recordings from DS individuals suggest that such a preferential suppression of high-frequency information flow may result from overexpression of Hsa21 genes. Compared to controls, DS individuals have increased power at low EEG frequencies and a corresponding reduction in power at higher frequencies [132]. Similar observations have been reported in Ts65Dn mice [133]. While it is not clear that hippocampal activity is accurately reflected in EEG recordings, abnormal EEGs findings are consistent with aberrant processing of high-frequency information in DS individuals and Ts65Dn mice.
5. Cognitive Therapies Targeting Plasticity
A number of studies using the Ts65Dn mouse model of DS have examined the possibility of pharmacologically reversing cognitive deficits (reviewed in [122]). Of particular note with respect to hippocampal plasticity are those targeting the excess GABAergic inhibitory tone or NMDA receptors whose activation, as outlined above, is a critical step in initiating LTP and LTD.
Application of the GABAA receptor antagonist, picrotoxin to hippocampal slices from Ts65Dn mice rescues LTP in the dentate gyrus [75] and CA1 region [76]. Chronic administration of low doses of picrotoxin or other GABAA receptor antagonists (pentylenetetrazole or bilobalide) improves cognition in Ts65Dn mice suggesting that the efficacy of this class of pharmacological agents could be tested for reversing impaired cognition in DS [60]. However, as overinhibition of GABAA receptors can induce seizures, translating these findings to humans requires great caution. Careful screening of similar drugs or design of pharmacological compounds with similar blocking capabilities but reduced propensities for inducing seizures may prove to be effective treatments. Currently, a small molecule targeting GABAA receptors developed by F. Hoffmann-La Roche Ltd (Pharmaceutical pipeline molecule RG1662 http://www.roche.com/roche_pharma_pipeline.htm) is in clinical trials with the goal of safely improving cognition in DS individuals.
Braudeau et al. [134, 135] are currently investigating a similar promising inhibitor that targets the alpha-5 subunit of GABAA receptors.
Another pharmacological avenue targets aberrant NMDA receptor-mediated signaling apparently present in Ts65Dn mice. The uncompetitive NMDA receptor antagonist memantine improves the cognitive performance of Ts65Dn mice [79] and normalizes hippocampal LTD [78]. Memantine is an FDA approved and fairly well-tolerated drug already in use for treating dementia in Alzheimer disease. Clinical trials assessing the safety, tolerability, and efficacy in alleviating DS cognitive phenotypes are currently underway [57].
In addition to pharmacological approaches, behavioral therapies have been shown to improve cognition in Ts65Dn mice. When housed in enriched environments (larger cage with novel objects such as toys and running wheels), trisomic mice performed as well as euploid littermates in the Morris water maze and had normalized hippocampal LTP [136, 137]. Interestingly, environmental enrichment was effective for trisomic females but not trisomic males potentially due to social and physical factors associated with the new environments [138].
The benefits of environmental enrichment appear to be linked in part to regulation of excess inhibition in the neocortex and hippocampus. Release of GABA from synaptosomes isolated from the hippocampus and neocortex is elevated in Ts65Dn mice, an effect that is reversed by environmental enrichment [137]. In adult rats with amblyopia (via monocular deprivation during critical period), environmental enrichment reversed visual deficits reduced GABA levels in the visual cortex contralateral to the deprived eye while increasing plasticity [137]. It thus appears possible to regulate aberrant levels of inhibition in trisomic mice behaviorally without pharmacological intervention and achieve similar behavioral outcomes without the concerns associated with nonspecific actions or adverse side-effects of drugs.
Deficits in neurogenesis in the dentate gyrus and forebrain subventricular zone in Ts65Dn mice are also reversed following environmental enrichment [139] and may add an additional therapeutic layer to the beneficial effect of a decrease in inhibitory tone. A structural benefit of environmental enrichment appears lacking; however, as, unlike euploid mice, this treatment has failed to significantly increase dendritic branching and spine density in Ts65Dn mice [140].
Early behavioral intervention techniques designed to improve development in DS children show great promise [141, 142] suggesting that this comparatively easily translatable therapeutic approach, either used alone or in combination with pharmacological agents, could potentially increase cognitive capacities in DS individuals.
6. Conclusion
Synaptic plasticity is believed to be the process central to learning and memory. This belief is bolstered by experiments where drugs that normalize aberrant plasticity in hippocampal slices isolated from mouse models of DS also confer improvements in cognition in in intact adult mice. The initiation and maintenance of plastic changes involve structural and compositional modifications of synapses that depend on intracellular signaling cascades. The reduced excitatory neuronal densities and deficits in dendritic morphologies present in individuals with DS diminish the capacity of their neural networks in general to undergo neuroplastic adaptations. Combined with the deficits in signaling pathways reported in Ts65Dn mice, evidence strongly suggests that synaptic plasticity is severely impaired in DS neural networks. By understanding how plasticity is perturbed, we can design therapies to reverse these phenotypes and ultimately improve cognition in DS individuals.
Acknowledgments
This work was supported in part by NIH R01 HD05780 and NS076503, Lejeune Foundation and intramural grant from the Uniformed Services University of the Health Sciences.
References
- 1.Suri S, Tompson BD, Cornfoot L. Cranial base, maxillary and mandibular morphology in Down syndrome. Angle Orthodontist. 2010;80(5):861–869. doi: 10.2319/111709-650.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sureshbabu R, Kumari R, Ranugha S, Sathyamoorthy R, Udayashankar C, Oudeacoumar P. Phenotypic and dermatological manifestations in Down syndrome. Dermatology Online Journal. 2011;17(2):p. 3. [PubMed] [Google Scholar]
- 3.Weijerman ME, van Furth AM, van der Mooren MD, et al. Prevalence of congenital heart defects and persistent pulmonary hypertension of the neonate with Down syndrome. European Journal of Pediatrics. 2010;169(10):1195–1199. doi: 10.1007/s00431-010-1200-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baek KH, Zaslavsky A, Lynch RC, et al. Down’s syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature. 2009;459(7250):1126–1130. doi: 10.1038/nature08062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan I, Malinge S, Crispino JD. Myeloid leukemia in down syndrome. Critical Reviews in Oncogenesis. 2011;16(1-2):25–36. doi: 10.1615/critrevoncog.v16.i1-2.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapman RS, Hesketh LJ. Behavioral phenotype of individuals with Down syndrome. Mental Retardation and Developmental Disabilities Research Reviews. 2000;6:84–95. doi: 10.1002/1098-2779(2000)6:2<84::AID-MRDD2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 7.Haydar TF, Reeves RH. Trisomy 21 and early brain development. Trends in Neurosciences. 2012;35(2):81–91. doi: 10.1016/j.tins.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361(6407):31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 9.Hofer SB, Bonhoeffer T. Dendritic spines: the stuff that memories are made of? Current Biology. 2010;20(4):R157–R159. doi: 10.1016/j.cub.2009.12.040. [DOI] [PubMed] [Google Scholar]
- 10.Dan Y, Poo M-M. Spike timing-dependent plasticity: from synapse to perception. Physiological Reviews. 2006;86(3):1033–1048. doi: 10.1152/physrev.00030.2005. [DOI] [PubMed] [Google Scholar]
- 11.Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annual Review of Biochemistry. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- 12.Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annual Review of Neuroscience. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holcman D, Schuss Z. Diffusion laws in dendritic spines. The Journal of Mathematical Neuroscience . 2011;1, article 10 doi: 10.1186/2190-8567-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nadel L, Bohbot V. Consolidation of memory. Hippocampus. 2001;11:56–60. doi: 10.1002/1098-1063(2001)11:1<56::AID-HIPO1020>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 15.Moser EI, Kropff E, Moser MB. Place cells, grid cells, and the brain’s spatial representation system. Annual Review of Neuroscience. 2008;31:69–89. doi: 10.1146/annurev.neuro.31.061307.090723. [DOI] [PubMed] [Google Scholar]
- 16.Kesner RP. Behavioral functions of the CA3 subregion of the hippocampus. Learning and Memory. 2007;14(11):771–781. doi: 10.1101/lm.688207. [DOI] [PubMed] [Google Scholar]
- 17.Pennington BF, Moon J, Edgin J, Stedron J, Nadel L. The Neuropsychology of Down syndrome: evidence for hippocampal dysfunction. Child Development. 2003;74(1):75–93. doi: 10.1111/1467-8624.00522. [DOI] [PubMed] [Google Scholar]
- 18.Nadel L. Down’s syndrome: a genetic disorder in biobehavioral perspective. Genes, Brain and Behavior. 2003;2(3):156–166. doi: 10.1034/j.1601-183x.2003.00026.x. [DOI] [PubMed] [Google Scholar]
- 19.Becker L, Mito T, Takashima S, Onodera K. Growth and development of the brain in Down syndrome. Progress in Clinical and Biological Research. 1991;373:133–152. [PubMed] [Google Scholar]
- 20.Golden JA, Hyman BT. Development of the superior temporal neocortex is anomalous in trisomy 21. Journal of Neuropathology and Experimental Neurology. 1994;53(5):513–520. doi: 10.1097/00005072-199409000-00011. [DOI] [PubMed] [Google Scholar]
- 21.Schmidt-Sidor B, Wisniewski KE, Shepard TH, Sersen EA. Brain growth in Down syndrome subjects 15 to 22 weeks of gestational age and birth to 60 months. Clinical Neuropathology. 1990;9(4):181–190. [PubMed] [Google Scholar]
- 22.Sylvester PE. The hippocampus in Down’s syndrome. Journal of Mental Deficiency Research. 1983;27(part 3):227–236. doi: 10.1111/j.1365-2788.1983.tb00294.x. [DOI] [PubMed] [Google Scholar]
- 23.Contestabile A, Fila T, Ceccarelli C, et al. Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with down syndrome and in Ts65Dn mice. Hippocampus. 2007;17(8):665–678. doi: 10.1002/hipo.20308. [DOI] [PubMed] [Google Scholar]
- 24.Guidi S, Bonasoni P, Ceccarelli C, et al. Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with Down syndrome. Brain Pathology. 2008;18(2):180–197. doi: 10.1111/j.1750-3639.2007.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whittle N, Sartori SB, Dierssen M, Lubec G, Singewald N. Fetal Down syndrome brains exhibit aberrant levels of neurotransmitters critical for normal brain development. Pediatrics. 2007;120(6):e1465–e1471. doi: 10.1542/peds.2006-3448. [DOI] [PubMed] [Google Scholar]
- 26.Ruediger T, Bolz J. Neurotransmitters and the development of neuronal circuits. Advances in Experimental Medicine and Biology. 2007;621:104–115. doi: 10.1007/978-0-387-76715-4_8. [DOI] [PubMed] [Google Scholar]
- 27.Wisniewski KE. Down syndrome children often have brain with maturation delay, retardation of growth, and cortical dysgenesis. American Journal of Medical Genetics. 1990;(7):274–281. doi: 10.1002/ajmg.1320370755. [DOI] [PubMed] [Google Scholar]
- 28.Becker LE, Armstrong DL, Chan F. Dendritic atrophy in children with Down’s syndrome. Annals of Neurology. 1986;20(4):520–526. doi: 10.1002/ana.410200413. [DOI] [PubMed] [Google Scholar]
- 29.Takashima S, Ieshima A, Nakamura H, Becker LE. Dendrites, dementia and the Down syndrome. Brain and Development. 1989;11(2):131–133. doi: 10.1016/s0387-7604(89)80082-8. [DOI] [PubMed] [Google Scholar]
- 30.Purpura DP. Normal and aberrant neuronal development in the cerebral cortex of human fetus and young infant. UCLA Forum in Medical Sciences. 1975;(18):141–169. doi: 10.1016/b978-0-12-139050-1.50014-8. [DOI] [PubMed] [Google Scholar]
- 31.Marin Padilla M. Pyramidal cell abnormalities in the motor cortex of a child with Down’s syndrome. A Golgi study. Journal of Comparative Neurology. 1976;167(1):63–81. doi: 10.1002/cne.901670105. [DOI] [PubMed] [Google Scholar]
- 32.Petit TL, LeBoutillier JC, Alfano DP, Becker LE. Synaptic development in the human fetus: a morphometric analysis of normal and Down’s syndrome neocortex. Experimental Neurology. 1984;83(1):13–23. doi: 10.1016/0014-4886(84)90041-4. [DOI] [PubMed] [Google Scholar]
- 33.Weitzdoerfer R, Dierssen M, Fountoulakis M, Lubec G. Fetal life in Down syndrome starts with normal neuronal density but impaired dendritic spines and synaptosomal structure. Journal of Neural Transmission, Supplement. 2001;(61):59–70. doi: 10.1007/978-3-7091-6262-0_5. [DOI] [PubMed] [Google Scholar]
- 34.Takashima S, Iida K, Mito T, Arima M. Dendritic and histochemical development and ageing in patients with Down’s syndrome. Journal of Intellectual Disability Research. 1994;38(part 3):265–273. doi: 10.1111/j.1365-2788.1994.tb00394.x. [DOI] [PubMed] [Google Scholar]
- 35.Pinter JD, Eliez S, Schmitt JE, Capone GT, Reiss AL. Neuroanatomy of Down’s syndrome: a high-resolution MRI study. American Journal of Psychiatry. 2001;158(10):1659–1665. doi: 10.1176/appi.ajp.158.10.1659. [DOI] [PubMed] [Google Scholar]
- 36.Śmigielska-Kuzia J, Boćkowski L, Sobaniec W, et al. A volumetric magnetic resonance imaging study of brain structures in children with Down syndrome. Neurologia I Neurochirurgia Polska. 2011;45(4):363–369. doi: 10.1016/s0028-3843(14)60107-9. [DOI] [PubMed] [Google Scholar]
- 37.Pinter JD, Brown WE, Eliez S, Schmitt JE, Capone GT, Reiss AL. Amygdala and hippocampal volumes in children with Down syndrome: a high-resolution MRI study. Neurology. 2001;56(7):972–974. doi: 10.1212/wnl.56.7.972. [DOI] [PubMed] [Google Scholar]
- 38.Teipel SJ, Schapiro MB, Alexander GE, et al. Relation of corpus callosum and hippocampal size to age in nondemented adults with Down’s syndrome. American Journal of Psychiatry. 2003;160(10):1870–1878. doi: 10.1176/appi.ajp.160.10.1870. [DOI] [PubMed] [Google Scholar]
- 39.Edgin JO, Pennington BF, Mervis CB. Neuropsychological components of intellectual disability: the contributions of immediate, working, and associative memory. Journal of Intellectual Disability Research. 2010;54(5):406–417. doi: 10.1111/j.1365-2788.2010.01278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Battaglia F, Quartarone A, Rizzo V, et al. Early impairment of synaptic plasticity in patients with Down’s syndrome. Neurobiology of Aging. 2008;29(8):1272–1275. doi: 10.1016/j.neurobiolaging.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 41.Losin EAR, Rivera SM, O’Hare ED, Sowell ER, Pinter JD. Abnormal fMRI activation pattern during story listening in individuals with Down syndrome. American Journal on Intellectual and Developmental Disabilities. 2009;114(5):369–380. doi: 10.1352/1944-7558-114.5.369. [DOI] [PubMed] [Google Scholar]
- 42.Jacola LM, Byars AW, Chalfonte-Evans M, et al. Functional magnetic resonance imaging of cognitive processing in young adults with Down syndrome. American Journal on Intellectual and Developmental Disabilities. 2011;116(5):344–359. doi: 10.1352/1944-7558-116.5.344. [DOI] [PubMed] [Google Scholar]
- 43.Lengyel Z, Balogh E, Emri M, et al. Pattern of increased cerebral FDG uptake in Down syndrome patients. Pediatric Neurology. 2006;34(4):270–275. doi: 10.1016/j.pediatrneurol.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 44.Siarey RJ, Stoll J, Rapoport SI, Galdzicki Z. Altered long-term potentiation in the young and old Ts65Dn mouse, a model for Down Syndrome. Neuropharmacology. 1997;36(11-12):1549–1554. doi: 10.1016/s0028-3908(97)00157-3. [DOI] [PubMed] [Google Scholar]
- 45.Siarey RJ, Carlson EJ, Epstein CJ, Balbo A, Rapoport SI, Galdzicki Z. Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in Down syndrome. Neuropharmacology. 1999;38(12):1917–1920. doi: 10.1016/s0028-3908(99)00083-0. [DOI] [PubMed] [Google Scholar]
- 46.O’Doherty A, Ruf S, Mulligan C, et al. An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science. 2005;309(5743):2033–2037. doi: 10.1126/science.1114535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reynolds LE, Watson AR, Baker M, et al. Tumour angiogenesis is reduced in the Tc1 mouse model of Down’ s syndrome. Nature. 2010;465(7299):813–817. doi: 10.1038/nature09106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morice E, Andreae LC, Cooke SF, et al. Preservation of long-term memory and synaptic plasticity despite short-term impairments in the Tcl mouse model of down syndrome. Learning and Memory. 2008;15(7):492–500. doi: 10.1101/lm.969608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dunlevy L, Bennett M, Slender A, et al. Down’s syndrome-like cardiac developmental defects in embryos of the transchromosomic Tc1 mouse. Cardiovascular Research. 2010;88(2):287–295. doi: 10.1093/cvr/cvq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cramer NP, Best TK, Stoffel M, Siarey RJ, Galdzicki Z. GABAB-GIRK2-mediated signaling in down syndrome. Advances in Pharmacology. 2010;58:397–426. doi: 10.1016/S1054-3589(10)58015-3. [DOI] [PubMed] [Google Scholar]
- 51.Gotti S, Caricati E, Panzica G. Alterations of brain circuits in Down syndrome murine models. Journal of Chemical Neuroanatomy. 2011;42(4):317–326. doi: 10.1016/j.jchemneu.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 52.Yu T, Li Z, Jia Z, et al. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Human Molecular Genetics. 2010;19(14):2780–2791. doi: 10.1093/hmg/ddq179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davisson MT, Schmidt C, Akeson EC. Segmental trisomy of murine chromosome 16: a new model system for studying Down syndrome. Progress in Clinical and Biological Research. 1990;360:263–280. [PubMed] [Google Scholar]
- 54.Davisson MT, Schmidt C, Reeves RH, et al. Segmental trisomy as a mouse model for Down syndrome. Progress in Clinical and Biological Research. 1993;384:117–133. [PubMed] [Google Scholar]
- 55.Galdzicki Z, Siarey RJ. Understanding mental retardation in down’s syndrome using trisomy 16 mouse models. Genes, Brain and Behavior. 2003;2(3):167–178. doi: 10.1034/j.1601-183x.2003.00024.x. [DOI] [PubMed] [Google Scholar]
- 56.Gardiner K, Costa ACS. The proteins of human chromosome 21. American Journal of Medical Genetics. 2006;142(3):196–205. doi: 10.1002/ajmg.c.30098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Costa ACS. On the promise of pharmacotherapies targeted at cognitive and neurodegenerative components of down syndrome. Developmental Neuroscience. 2011;33(5):414–427. doi: 10.1159/000330861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reeves RH, Irving NG, Moran TH, et al. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nature Genetics. 1995;11(2):177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- 59.Hunter CL, Bimonte HA, Granholm ACE. Behavioral comparison of 4 and 6 month-old Ts65Dn mice: age-related impairments in working and reference memory. Behavioural Brain Research. 2003;138(2):121–131. doi: 10.1016/s0166-4328(02)00275-9. [DOI] [PubMed] [Google Scholar]
- 60.Fernandez F, Morishita W, Zuniga E, et al. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nature Neuroscience. 2007;10(4):411–413. doi: 10.1038/nn1860. [DOI] [PubMed] [Google Scholar]
- 61.Rueda N, Flórez J, Martínez-Cué C. Effects of chronic administration of SGS-111 during adulthood and during the pre- and post-natal periods on the cognitive deficits of Ts65Dn mice, a model of Down syndrome. Behavioural Brain Research. 2008;188(2):355–367. doi: 10.1016/j.bbr.2007.11.020. [DOI] [PubMed] [Google Scholar]
- 62.Blanchard J, Bolognin S, Chohan MO, Rabe A, Iqbal K, Grundke-Iqbal I. Rescue of synaptic failure and alleviation of learning and memory impairments in a trisomic mouse model of down syndrome. Journal of Neuropathology and Experimental Neurology. 2011;70(12):1070–1079. doi: 10.1097/NEN.0b013e318236e9ad. [DOI] [PubMed] [Google Scholar]
- 63.Faizi M, Bader PL, Tun C, et al. Comprehensive behavioral phenotyping of Ts65Dn mouse model of Down Syndrome: activation of β1-adrenergic receptor by xamoterol as a potential cognitive enhancer. Neurobiology of Disease. 2011;43(2):397–413. doi: 10.1016/j.nbd.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duchon A, Raveau M, Chevalier C, Nalesso V, Sharp AJ, Herault Y. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling down syndrome. Mammalian Genome. 2011;22(11-12):674–684. doi: 10.1007/s00335-011-9356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sago H, Carlson EJ, Smith DJ, et al. Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(11):6256–6261. doi: 10.1073/pnas.95.11.6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Siarey RJ, Villar AJ, Epstein CJ, Galdzicki Z. Abnormal synaptic plasticity in the Ts1Cje segmental trisomy 16 mouse model of Down syndrome. Neuropharmacology. 2005;49(1):122–128. doi: 10.1016/j.neuropharm.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 67.Belichenko PV, Kleschevnikov AM, Salehi A, Epstein CJ, Mobley WC. Synaptic and cognitive abnormalities in mouse models of Down syndrome: exploring genotype-phenotype relationships. Journal of Comparative Neurology. 2007;504(4):329–345. doi: 10.1002/cne.21433. [DOI] [PubMed] [Google Scholar]
- 68.Belichenko NP, Belichenko PV, Kleschevnikov AM, Salehi A, Reeves RH, Mobley WC. The “Down syndrome critical region” is sufficient in the mouse model to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of Down syndrome. Journal of Neuroscience. 2009;29(18):5938–5948. doi: 10.1523/JNEUROSCI.1547-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Olson LE, Roper RJ, Sengstaken CL, et al. Trisomy for the Down syndrome “critical region” is necessary but not sufficient for brain phenotypes of trisomic mice. Human Molecular Genetics. 2007;16(7):774–782. doi: 10.1093/hmg/ddm022. [DOI] [PubMed] [Google Scholar]
- 70.Chakrabarti L, Best TK, Cramer NP, et al. Olig1 and Olig2 triplication causes developmental brain defects in Down syndrome. Nature Neuroscience. 2010;13(8):927–934. doi: 10.1038/nn.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Belichenko PV, Masliah E, Kleschevnikov AM, et al. Synaptic structural abnormalities in the Ts65Dn mouse model of Down syndrome. Journal of Comparative Neurology. 2004;480(3):281–298. doi: 10.1002/cne.20337. [DOI] [PubMed] [Google Scholar]
- 72.Belichenko PV, Kleschevnikov AM, Masliah E, et al. Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of down syndrome. Journal of Comparative Neurology. 2009;512(4):453–466. doi: 10.1002/cne.21895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bliss TVP, Lømo T. Long lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. Journal of Physiology. 1973;232(2):331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neves G, Cooke SF, Bliss TVP. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nature Reviews Neuroscience. 2008;9(1):65–75. doi: 10.1038/nrn2303. [DOI] [PubMed] [Google Scholar]
- 75.Kleschevnicov AM, Belichenko PV, Villar AJ, Epstein CJ, Malenka RC, Mobley WC. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of down syndrome. Journal of Neuroscience. 2004;24(37):8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Costa ACS, Grybko MJ. Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: a model of Down syndrome. Neuroscience Letters. 2005;382(3):317–322. doi: 10.1016/j.neulet.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 77.Yu T, Liu C, Belichenko P, et al. Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Research. 2010;1366:162–171. doi: 10.1016/j.brainres.2010.09.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Scott-McKean JJ, Costa ACS. Exaggerated NMDA mediated LTD in a mouse model of Down syndrome and pharmacological rescuing by memantine. Learning and Memory. 2011;18(12):774–778. doi: 10.1101/lm.024182.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Costa ACS, Scott-McKean JJ, Stasko MR. Acute injections of the NMDA receptor antagonist memantine rescue performance deficits of the Ts65Dn mouse model of Down syndrome on a fear conditioning test. Neuropsychopharmacology. 2008;33(7):1624–1632. doi: 10.1038/sj.npp.1301535. [DOI] [PubMed] [Google Scholar]
- 80.Rueda N, Llorens-Martín M, Flórez J, et al. Memantine normalizes several phenotypic features in the Ts65Dn mouse model of down syndrome. Journal of Alzheimer’s Disease. 2010;21(1):277–290. doi: 10.3233/JAD-2010-100240. [DOI] [PubMed] [Google Scholar]
- 81.Lockrow J, Boger H, Bimonte-Nelson H, Granholm AC. Effects of long-term memantine on memory and neuropathology in Ts65Dn mice, a model for Down syndrome. Behavioural Brain Research. 2011;221(2):610–622. doi: 10.1016/j.bbr.2010.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44(1):5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 83.Lynch G, Larson J, Kelso S. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305(5936):719–721. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- 84.Bliss TVP, Collingridge GL, Morris RGM. Introduction. Long-term potentiation and structure of the issue. Philosophical Transactions of the Royal Society B. 2003;358:607–611. doi: 10.1098/rstb.2003.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Malenka RC, Kauer JA, Perkel DJ, et al. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989;340(6234):554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- 86.Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245(4920):862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 87.Pettit DL, Perlman S, Malinow R. Potentiated transmission and prevention of further LTP by increased CaMKII activity in postsynaptic hippocampal slice neurons. Science. 1994;266(5192):1881–1885. doi: 10.1126/science.7997883. [DOI] [PubMed] [Google Scholar]
- 88.Ahmed T, Frey JU. Plasticity-specific phosphorylation of CaMKII, MAP-kinases and CREB during late-LTP in rat hippocampal slices in vitro. Neuropharmacology. 2005;49(4):477–492. doi: 10.1016/j.neuropharm.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 89.Lai Y, Nairn AC, Greengard P. Autophosphorylation reversibly regulates the Ca2+/calmodulin-dependence of Ca2+/calmodulin-dependent protein kinase II. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(12):4253–4257. doi: 10.1073/pnas.83.12.4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Colbran RJ, Soderling TR. Calcium/calmodulin-independent autophosphorylation sites of calcium/calmodulin-dependent protein kinase II. Studies on the effect of phosphorylation of threonine 305/306 and serine 314 on calmodulin binding using synthetic peptides. Journal of Biological Chemistry. 1990;265(19):11213–11219. [PubMed] [Google Scholar]
- 91.Zhang L, Kirschstein T, Sommersberg B, et al. Hippocampal synaptic metaplasticity requires inhibitory autophosphorylation of Ca2+/calmodulin-dependent kinase II. Journal of Neuroscience. 2005;25(33):7697–7707. doi: 10.1523/JNEUROSCI.2086-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.van Woerden GM, Harris KD, Hojjati MR, et al. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of αCaMKII inhibitory phosphorylation. Nature Neuroscience. 2007;10(3):280–282. doi: 10.1038/nn1845. [DOI] [PubMed] [Google Scholar]
- 93.Siarey RJ, Kline-Burgess A, Cho M, et al. Altered signaling pathways underlying abnormal hippocampal synaptic plasticity in the Ts65Dn mouse model of Down syndrome. Journal of Neurochemistry. 2006;98(4):1266–1277. doi: 10.1111/j.1471-4159.2006.03971.x. [DOI] [PubMed] [Google Scholar]
- 94.Barria A, Derkach V, Soderling T. Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. Journal of Biological Chemistry. 1997;272(52):32727–32730. doi: 10.1074/jbc.272.52.32727. [DOI] [PubMed] [Google Scholar]
- 95.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405(6789):955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 96.Derkach V, Barria A, Soderling TR. Ca2+/calmodulin-kinase II enhances channel conductance of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(6):3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260(5114):1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- 98.Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265(5175):1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- 99.Matthies H, Reymann KG. Protein kinase A inhibitors prevent the maintenance of hippocampal long-term potentiation. NeuroReport. 1993;4(6):712–714. doi: 10.1097/00001756-199306000-00028. [DOI] [PubMed] [Google Scholar]
- 100.Huang YY, Kandel ER. Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learning Memory. 1994;1(1):74–82. [PubMed] [Google Scholar]
- 101.Abel T, Nguyen PV, Barad M, Deuel TAS, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88(5):615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- 102.Kameyama K, Lee H-K, Bear MF, Huganir RL. Involvement of a postsynaptic protein kinase A substrate in the expression of homosynaptic long-term depression. Neuron. 1998;21(5):1163–1175. doi: 10.1016/s0896-6273(00)80633-9. [DOI] [PubMed] [Google Scholar]
- 103.Lee H-K, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21(5):1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- 104.Kim SS, Oh Y, Chung KC, Seo SR. Protein kinase A phosphorylates Down syndrome critical region 1 (RCAN1) Biochemical and Biophysical Research Communications. 2012;418(4):657–661. doi: 10.1016/j.bbrc.2012.01.071. [DOI] [PubMed] [Google Scholar]
- 105.Beattie EC, Carroll RC, Yu X, et al. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nature Neuroscience. 2000;3(12):1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- 106.Lieberman DN, Mody I. Regulation of NMDA channel function by endogenous Ca2+-dependent phosphatase. Nature. 1994;369(6477):235–239. doi: 10.1038/369235a0. [DOI] [PubMed] [Google Scholar]
- 107.Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Current Opinion in Neurobiology. 2004;14(3):311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 108.Davis S, Vanhoutte P, Pagès C, Caboche J, Laroche S. The MAPK/ERK cascade targets both Elk-1 and cAMP response element- binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. Journal of Neuroscience. 2000;20(12):4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. Journal of Biological Chemistry. 1997;272(31):19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 110.Goldin M, Segal M. Protein kinase C and ERK involvement in dendritic spine plasticity in cultured rodent hippocampal neurons. European Journal of Neuroscience. 2003;17(12):2529–2539. doi: 10.1046/j.1460-9568.2003.02694.x. [DOI] [PubMed] [Google Scholar]
- 111.Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64(3):381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nature Reviews Neuroscience. 2012;13(3):169–182. doi: 10.1038/nrn3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Matsuzaki M, Honkura N, Ellis-Davies GCR, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429(6993):761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature. 2007;450(7173):1195–1200. doi: 10.1038/nature06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Current Opinion in Neurobiology. 2001;11(3):272–280. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- 116.Schratt GM, Nigh EA, Chen WG, Hu L, Greenberg ME. BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. Journal of Neuroscience. 2004;24(33):7366–7377. doi: 10.1523/JNEUROSCI.1739-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dogliotti G, Galliera E, Licastro F, Corsi MM. Age-related changes in plasma levels of BDNF in Down syndrome patients. Immunity and Ageing. 2010;7, article 2 doi: 10.1186/1742-4933-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pan W, Banks WA, Fasold MB, Bluth J, Kastin AJ. Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology. 1998;37(12):1553–1561. doi: 10.1016/s0028-3908(98)00141-5. [DOI] [PubMed] [Google Scholar]
- 119.Bimonte-Nelson HA, Hunter CL, Nelson ME, Granholm ACE. Frontal cortex BDNF levels correlate with working memory in an animal model of Down syndrome. Behavioural Brain Research. 2003;139(1-2):47–57. doi: 10.1016/s0166-4328(02)00082-7. [DOI] [PubMed] [Google Scholar]
- 120.Bianchi P, Ciani E, Guidi S, et al. Early pharmacotherapy restores neurogenesis and cognitive performance in the Ts65Dn mouse model for down syndrome. Journal of Neuroscience. 2010;30(26):8769–8779. doi: 10.1523/JNEUROSCI.0534-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Troca-Marín JA, Alves-Sampaio A, Montesinos ML. An increase in basal BDNF provokes hyperactivation of the Akt-Mammalian target of rapamycin pathway and deregulation of local dendritic translation in a mouse model of down’s syndrome. Journal of Neuroscience. 2011;31(26):9445–9455. doi: 10.1523/JNEUROSCI.0011-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Das I, Reeves RH. The use of mouse models to understand and improve cognitive deficits in down syndrome. Disease Models and Mechanisms. 2011;4(5):596–606. doi: 10.1242/dmm.007716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Harashima C, Jacobowitz DM, Witta J, et al. Abnormal expression of the G-protein-activated inwardly rectifying potassium channel 2 (GIRK2) in hippocampus, frontal cortex, and substantia nigra of Ts65Dn mouse: a model of Down syndrome. Journal of Comparative Neurology. 2006;494(5):815–833. doi: 10.1002/cne.20844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Best TK, Siarey RJ, Galdzicki Z. Ts65Dn, a mouse model of Down syndrome, exhibits increased GABAB-induced potassium current. Journal of Neurophysiology. 2007;97:892–900. doi: 10.1152/jn.00626.2006. [DOI] [PubMed] [Google Scholar]
- 125.Best TK, Cramer NP, Chakrabarti L, Haydar TF, Galdzicki Z. Dysfunctional hippocampal inhibition in the Ts65Dn mouse model of Down syndrome. Experimental Neurology. 2012;233(2):749–757. doi: 10.1016/j.expneurol.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cooper A, Grigoryan G, Guy-David L, Tsoory MM, Chen A, Reuveny E. Trisomy of the G protein-coupled K+ channel gene, Kcnj6, affects reward mechanisms, cognitive functions, and synaptic plasticity in mice. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(7):2642–2647. doi: 10.1073/pnas.1109099109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kulik A, Vida I, Fukazawa Y, et al. Compartment-dependent colocalization of Kir3.2-containing K+ channels and GABAB receptors in hippocampal pyramidal cells. Journal of Neuroscience. 2006;26(16):4289–4297. doi: 10.1523/JNEUROSCI.4178-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Best TK, Cramer NP, Chakrabarti L, Haydar TF, Galdzicki Z. Dysfunctional hippocampal inhibition in the Ts65Dn mouse model of Down syndrome. Experimental Neurology. 2012;233(2):749–757. doi: 10.1016/j.expneurol.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kleschevnikov AM, Belichenko PV, Gall J, et al. Increased efficiency of the GABAA and GABAB receptor-mediated neurotransmission in the Ts65Dn mouse model of Down syndrome. Neurobiology of Disease. 2012;45(2):683–691. doi: 10.1016/j.nbd.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kajiwara R, Wouterlood FG, Sah A, Boekel AJ, Baks-Te Bulte LTG, Witter MP. Convergence of entorhinal and CA3 inputs onto pyramidal neurons and interneurons in hippocampal area CA1—an anatomical study in the rat. Hippocampus. 2008;18(3):266–280. doi: 10.1002/hipo.20385. [DOI] [PubMed] [Google Scholar]
- 131.Maccaferri G, McBain CJ. Passive propagation of LTD to stratum oriens-alveus inhibitory neurons modulates the temporoammonic input to the hippocampal CA1 region. Neuron. 1995;15(1):137–145. doi: 10.1016/0896-6273(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 132.Babiloni C, Albertini G, Onorati P, et al. Cortical sources of EEG rhythms are abnormal in down syndrome. Clinical Neurophysiology. 2010;121(8):1205–1212. doi: 10.1016/j.clinph.2010.02.155. [DOI] [PubMed] [Google Scholar]
- 133.Colas D, Valletta JS, Takimoto-Kimura R, et al. Sleep and EEG features in genetic models of Down syndrome. Neurobiology of Disease. 2008;30(1):1–7. doi: 10.1016/j.nbd.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Braudeau J, Delatour B, Duchon A, et al. Specific targeting of the GABA-A receptor α5 subtype by a selective inverse agonist restores cognitive deficits in Down syndrome mice. Journal of Psychopharmacology. 2011;25(8):1030–1042. doi: 10.1177/0269881111405366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Braudeau J, Dauphinot L, Duchon A, et al. Chronic treatment with a promnesiant GABA-A α-selective inverse agonist increases immediate early genes expression during memory processing in mice and rectifies their expression levels in a down syndrome mouse model. Advances in Pharmacological Sciences. 2011;2011 doi: 10.1155/2011/153218.153218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Martínez-Cué C, Baamonde C, Lumbreras M, et al. Differential effects of environmental enrichment on behavior and learning of male and female Ts65Dn mice, a model for Down syndrome. Behavioural Brain Research. 2002;134(1-2):185–200. doi: 10.1016/s0166-4328(02)00026-8. [DOI] [PubMed] [Google Scholar]
- 137.Begenisic T, Spolidoro M, Braschi C, et al. Environmental enrichment decreases GABAergic inhibition and improves cognitive abilities, synaptic plasticity, and visual functions in a mouse model of Down syndrome. Frontiers in Cellular Neuroscience. 2011;5, article 29 doi: 10.3389/fncel.2011.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Martínez-Cué C, Rueda N, García E, Davisson MT, Schmidt C, Flórez J. Behavioral, cognitive and biochemical responses to different environmental conditions in male Ts65Dn mice, a model of Down syndrome. Behavioural Brain Research. 2005;163(2):174–185. doi: 10.1016/j.bbr.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 139.Chakrabarti L, Scafidi J, Gallo V, Haydar TF. Environmental enrichment rescues postnatal neurogenesis defect in the male and female Ts65Dn mouse model of down syndrome. Developmental Neuroscience. 2011;33(5):428–441. doi: 10.1159/000329423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dierssen M, Benavides-Piccione R, Martínez-Cué C, et al. Alterations of neocortical pyramidal cell phenotype in the Ts65Dn mouse model of Down syndrome: effects of environmental enrichment. Cerebral Cortex. 2003;13(7):758–764. doi: 10.1093/cercor/13.7.758. [DOI] [PubMed] [Google Scholar]
- 141.Mahoney G, Perales F, Wiggers B, Herman B. Responsive teaching: early intervention for children with Down syndrome and other disabilities. Down's syndrome, research and practice. 2006;11(1):18–28. doi: 10.3104/perspectives.311. [DOI] [PubMed] [Google Scholar]
- 142.Bonnier C. Evaluation of early stimulation programs for enhancing brain development. Acta Paediatrica, International Journal of Paediatrics. 2008;97(7):853–858. doi: 10.1111/j.1651-2227.2008.00834.x. [DOI] [PubMed] [Google Scholar]