

Abstract
As a model organism Saccharomyces cerevisiae has greatly contributed to our understanding of many fundamental aspects of cellular biology in higher eukaryotes. More recently, engineered yeast models developed to study endogenous or heterologous proteins that lay at the root of a given disease have become powerful tools for unraveling the molecular basis of complex human diseases like neurodegeneration. Additionally, with the possibility of performing target-directed large-scale screenings, yeast models have emerged as promising first-line approaches in the discovery process of novel therapeutic opportunities against these pathologies. In this paper, several yeast models that have contributed to the uncovering of the etiology and pathogenesis of several neurodegenerative diseases are described, including the most common forms of neurodegeneration worldwide, Alzheimer's, Parkinson's, and Huntington's diseases. Moreover, the potential input of these cell systems in the development of more effective therapies in neurodegeneration, through the identification of genetic and chemical suppressors, is also addressed.
1. Introduction
The budding yeast Saccharomyces cerevisiae has long been used as an eukaryotic model organism mostly due to its ease of manipulation and amenability to genetic modifications. This can also explain the forefront position of yeast in the development of large scale screening approaches, like DNA and protein microarrays [1–3], two-hybrid analysis [4, 5], and whole-genome deletion and overexpression libraries [6, 7]. Additionally, this contributed to the large amount of easily accessible online dataset for yeast which include genetic interactions, transcriptional changes, protein interactions, and localization (reviewed in [8]).
The use of yeast as a model organism was recently expanded to the dissection of the molecular mechanisms of human diseases, either by directly studying an endogenous protein orthologue of a human counterpart involved in the disease or through the heterologous expression of human disease-associated proteins. Though several aspects of the disease are beyond the reach of a unicellular organism like yeast, many processes and pathways are highly conserved in this organism, namely, mitochondria biogenesis, protein quality control, vesicular trafficking, and autophagic pathways. Actually, the high simplicity of yeast, when compared to human cells, has been widely explored to accelerate the discovery of new drugs and therapeutic targets in human diseases. In fact, when complemented with more physiologically relevant models where the hits discovered will be validated, yeast can be seen as a powerful first-line screening system for large genetic and chemical libraries.
With the increase in life expectancy, neurodegenerative disorders such as Alzheimer's, Parkinson's, and Huntington's diseases became major health problems in our society for which treatments are critically required [9].
The yeast models herein described, based on both classical and high-throughput methodologies, have widely contributed to the uncovering of the molecular mechanism underlying neurodegenerative disorders. Since many of them were adapted to target-directed screening approaches, a promising contribution of yeast to the identification of novel therapeutic opportunities against these pathologies can be envisaged.
2. Yeast Models of Neurodegeneration- Associated Proteins
2.1. Yeast Models Based on Orthologues
Neurodegenerative diseases are hereditary and sporadic conditions, which are characterized by a progressive nervous system dysfunction. Sporadic diseases like Parkinson's disease (PD) also have rare familial forms that have been linked to mutations in several genes, providing research opportunities for pathogenic mechanisms [10]. Because yeast encodes orthologues of many disease-causing proteins, several disease models based on the study of protein functions in yeast were established (Table 1). For example, Yfh1p is the yeast orthologue of human frataxin whose decreased expression and/or function is associated with Friedreich's ataxia (FRDA), a neuro- and cardiodegenerative disorder [11]. Studies with Yfh1p were decisive in determining the function of frataxin. Absence of Yfh1p, likewise of its human orthologue, results in mitochondrial iron accumulation, mitochondrial dysfunction, and oxidative stress [12, 13]. Recently, the yeast model was applied to the high-throughput screening (HTS) of compounds able to rescue mitochondrial function [14]. This was possible because yeast is one of the rare eukaryotes with a good fermenting capacity allowing the analysis of commonly disease-associated mitochondrial defects that would be lethal in other systems [15]. Other proteins that were directly studied in yeast are associated with Batten's [16] and Niemann-Pick's [17, 18] diseases, Ataxia telangiectasia [19, 20], and Hereditary Spastic Paraplegia [21]. Though yeast has no true orthologues of the human prion protein (PrP), responsible in its prion form for the Creutzfeldt-Jakob disease, it has prions, with at least three forms [URE3], [PSI+], and [PIN+], that show similarities concerning transmission of phenotype in a protein-only mode [22]. The yeast prion system has been useful, for example, for the screening of inhibitors with promising activity against mammalian prions [23].
Table 1.
Proteins associated with human neurodegenerative disorders studied in yeast.
| Disease | Protein(s) involved | Yeast orthologue | References |
|---|---|---|---|
| Batten's disease | CLN3 | YHC3/BTN1 | [16] |
| Friedreich's Ataxia | Frataxin | YFH1 | [12, 13] |
| Ataxia-telangiectasia | ATM | TEL1, MEC1 | [19, 20] |
| Niemann-Pick disease | NPC1 | NCR1 | [17, 18] |
| Hereditary Spastic Paraplegia | mAAA-proteases | mAAA proteases | [21] |
| (Afg312 and paraplegin) | (Yta10 and Yta12) | ||
| Creutzfeldt-Jakob disease | PrP | — | [23] |
| Amyotrophic lateral sclerosis | SOD1 | SOD1 | [25] |
| TDP-43 | — | [27] | |
| FUS/TLS | — | [29, 30] | |
| Parkinson's disease | α-synuclein | — | [31] |
| Lrrk2 | — | [32] | |
| Alzheimer's disease | Amyloid β | — | [33, 34] |
| tau | — | [35] | |
| Huntington's disease | Huntingtin | — | [41] |
—: No orthologue.
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the selective degeneration of motor neurons. With the recognition of the involvement of copper-zinc superoxide dismutase (CuZnSOD1) gene in familial ALS [24], the respective yeast orthologue was studied in an attempt to clarify the role of disease-associated mutations [25]. More recently, TDP-43 (TAR DNA binding protein) and FUS/TLS (fused in sarcoma/translocated in liposarcoma) were recognized as major pathological proteins in sporadic cases of ALS. TDP-43 and FUS/TLS are both RNA-binding proteins that are found in ubiquitinated protein aggregates in the cytoplasm of ALS and in frontotemporal lobar degeneration patients [26].
2.2. Yeast Models Based on Heterologous Expression
In many cases, the human gene encoding the neurodegeneration-causing protein has no orthologue in yeast. As such, its heterologous expression in this organism (the so-called “humanized yeast”) has been widely used (see Table 1). For example, yeast cells provided a useful system for investigating ALS through TDP-43 and FUS/TLS expression. Similarly to that observed in ALS patients, yeast expressing human TDP-43 exhibit cytoplasmic TDP-43 aggregates that correlate with toxicity [27, 28]. Likewise, expression of FUS/TLS in yeast was recently described to form protein aggregates and to induce cytotoxicity, with two ALS-associated mutants showing increased cytotoxicity [29, 30]. Several other proteins involved in neurodegeneration, namely, α-synuclein and Lrrk2 in PD, tau and amyloid-β (Aβ) peptide in Alzheimer's disease (AD), and Huntingtin with expanded polyglutamine (polyQ) tracts in Huntington's disease (HD), have been studied in yeast through heterologous expression [31–35]. Expression of these human proteins in yeast recapitulated important aspects related to protein pathology, such as the aggregation phenotype and toxicity.
Recombinant human proteins most thoroughly addressed in yeast include Aβ peptide, α-synuclein, and Huntingtin with expanded polyQ tracts, causally associated with the three most common neurodegenerative disorders worldwide, AD, PD, and HD, respectively. As such, these proteins will be discussed in more detail in the next sections.
3. Amyloid-β Peptide
Amyloid-β disorders include several progressive neurodegenerative disorders characterized by the oligomerization and deposition into plaques of an amyloid peptide, known as Aβ. These disorders include not only AD, that is the most recognized, but also Down syndrome, inclusion body myositis, and cerebral amyloid angiopathy [36].
Aβ is a cleavage product derived from the amyloid precursor protein (APP), by the action of specific proteases known as secretases. APP is usually delivered to the cell surface membrane where it is subjected to proteolytic processing by α-secretase. APP molecules that fail to be cleaved by α-secretase can be internalized into endocytic compartments and subsequently cleaved by β-secretase (identified as BACE-1) and γ-secretase, a multimeric complex containing the presenilins. γ-Secretase produces the C-terminal at the end of the Aβ peptide and it can generate the Aβ40 peptide or more rarely the longer Aβ42. The Aβ42, which is more aggregating and cytotoxic, is considered the pathologically important form, responsible for the formation of amyloid plaques [37]. Despite its toxicity, recent evidence suggests that increased amounts of Aβ peptide may have a normal function as an antimicrobial peptide [38]. Mutations in either APP or presenilin genes associated with early-onset AD have been noted to increase the relative production of Aβ42. Therefore, the modulation of the activity of secretases responsible for APP processing can be a potential therapeutic strategy for AD.
One of the initial contributions of S. cerevisiae to the understanding of Aβ disorders has been in the study of APP processing. In an early study, it was found that when APP is expressed in yeast, it gets cleaved at the C-terminus of the α-proteolysis site. This indicated that yeast possessed α-secretase activity [33, 39]. Additionally, the responsible proteases for the α-secretase-like activity in yeast were identified as being two aspartyl proteases, Yap3 and Mkc7 [39], though Mkc7 alone is apparently sufficient to cleave APP [40]. Since the secretases responsible for APP processing in humans are still not completely identified, the discovery of secretases in yeast may help in the identification of new human secretases by homology.
Though a possible β-secretase-like enzymatic activity was also reported in yeast [42], this finding was not supported by other works. Overcoming the lack of β-secretase-like activity, a fragment derived from the APP cleaved by β-secretase (C99) was expressed in yeast [43]. In an attempt to clarify the role of the proteasome in the C99 proteolytic processing, authors used proteomic analysis to compare the proteolysis fragments obtained in the presence and in the absence (using proteasome mutants) of the proteasome. In agreement with the concept that APP proteasomal degradation may be beneficial in mammalian cells [44], in yeast it was observed that the proteasome is involved in C99 processing and that most of the C99 cleavage products are mainly nonamyloidogenic [43]. Interestingly, it was reported the existence in yeast of a compensatory mechanism to overcome the absence of proteasomal activity [43]. This may be relevant for AD since proteasomal function is known to decline with age [45]. With the study of APP processing in yeast, it was found that secondary modifications, like O-glycosylation, are also important for protein solubility and processing in this organism [46].
One of the advantages of using yeast models to study human APP processing is the possibility to individually express human secretase complex subunits. This allowed dissecting the contribution of each subunit, a difficult goal to achieve in mammals due to the high redundancy of these proteases. As such, the γ-secretase activity was reconstituted in yeast by expressing four components of the γ-secretase multimeric complex, which were therefore established as the minimal components required for γ-secretase activity [47]. More recently, reconstitution of γ-secretase activity in yeast was used to produce microsomes. This made possible the study of the enzymatic properties of this complex in vitro upon the addition of its substrates, C99 and C55 [48].
Due to the amenability of yeast to HTS, several systems were developed to search for new proteases. One of those systems took advantage of the yeast GAL reporter system to screen for proteases that cleaved at the C-terminal of APP. When APP was processed, the Gal4 transcription factor was released activating the GAL-reporter genes. Among other hits obtained, the screening of a library of human cDNAs led to the discovery of caspase-3 and -8 [49]. This finding validated this approach for the screening of APP processing proteases since these caspases cleave APP generating a fragment also implicated in the pathogenesis of AD [50]. Another novel approach to identify APP processing secretases used an engineered APP (fused to invertase) that only allows the growth of invertase-null yeast upon processing of APP [51]. The efficacy of this approach was demonstrated by the absence of yeast growth upon the deletion of proteases Yap3 and Mkc7 that, as referred above, cleaved APP in yeast at the α-proteolysis site. Also, expression of human BACE-1, responsible for the β-secretase activity in humans, restored the growth in the absence of yeast proteases, indicating processing of APP. This system, initially developed for the identification of novel secretases, was also adapted for the screening of compounds that inhibit BACE-1 activity [52].
Additionally to the different approaches and tools developed in yeast to study APP processing, yeast has been used as a cell system to study the toxicity of the amyloidogenic APP fragment Aβ42. In humans, Aβ peptide was first identified as a component of extracellular amyloid plaques, but the existence of intracellular Aβ is now recognized [53]. The intracellular Aβ was found in several cellular locations such as cytosol, secretory pathway, autophagosomes, and mitochondria [54], thus potentially affecting diverse processes. In yeast, the effects of both extra- and intracellular Aβ were assessed. Since most of the Aβ is secreted, the addition of APP end products to the culture medium is the most straightforward way of investigating their cellular effects. As in mammals, the physical form in which these products were provided to S. cerevisiae (oligomeric or fibrillar) was important for the obtained effects [34, 55]. While an early study found no toxicity in adding soluble Aβ to yeast cells [34], another group performed the studies in water to better control the physical form of Aβ. Using this approach, authors reported a dose-dependent cytotoxicity for the oligomeric Aβ, while the fibrillar form had a lower toxicity [55]. This observation was consistent with studies in mammalian cells, suggesting the soluble oligomeric Aβ42 as the major determinant of neuronal dysfunction in AD [56]. Unlike the exogenously provided Aβ42, its intracellular expression caused only a mild toxicity, as evidenced by the slight reduction on yeast cell growth [57]. Besides the growth stress, it was observed that Aβ42 activated the yeast heat shock response [57]. This may explain the unexpected report that Aβ42 protected yeast from oxidative stress [58]. However, it was recently reported that directing Aβ42 to the secretory pathway in order to mimic its trafficking in human cells leads to a higher toxicity [59] (Figure 1(a)). This phenotype allowed authors to perform a genome-wide overexpression screen for toxicity modifiers, which resulted in the discovery of several toxicity suppressors. Interestingly, in this study, authors uncovered a previously unknown connection between this peptide and the phosphatidylinositol binding clathrin assembly protein (PICALM), also involved in AD [59].
Figure 1.
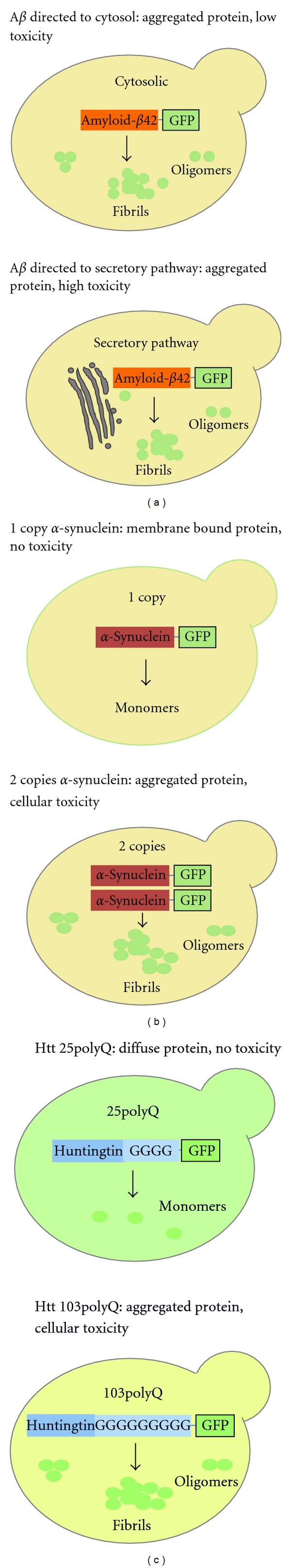
Yeast models of (a) AD, based on amyloid-β42 expression; (b) PD, based on α-synuclein expression; (c) HD, based on Huntingtin's polyQ tract expression. (a) The toxic Aβ42 peptide expressed in yeast causes aggregation and moderate stress, but when expression is directed to the secretory pathway, the toxicity is increased. (b) At low expression levels, α-synuclein associates with the plasma membrane with no effect on cell viability. At doubling expression levels, α-synuclein redistributes from the cell surface into aggregates and induces toxicity. (c) Expression of a Huntingtin polyQ tract with 25 glutamines (normal length) does not lead to aggregation or toxicity. Expression of a polyQ tract with 103 glutamines (HD-associated expansion) leads to the formation of aggregates and toxicity (dependent on the presence of active prions).
Along with the fact that Aβ42 is found in yeast predominantly in the insoluble protein fraction, Aβ42 formed lipidic patches, indicating aggregated protein [57]. Taking advantage of the fact that, like in humans, Aβ aggregates in yeast, this model was used to screen for compounds that reduce the aggregation/oligomerization of Aβ peptide. One of such studies used a system consisting of Aβ fused to the green fluorescence protein (GFP), in which Aβ misfolding contributes to GFP misfolding and consequent loss of fluorescence. Using this system, folate was validated as an inhibitor of Aβ misfolding [60]. Bagriantsev and Liebman [61] used an interesting approach to monitor the formation of Aβ42 low-n oligomers formed at the early stage of Aβ42 oligomerization and considered the most toxic Aβ species. Authors used a fusion of Aβ42 with a Sup35p (a translational termination factor) reporter that becomes inactive when low-n oligomers are formed, resulting in a robust phenotype (growth and colony color) that can be easily scored. In a following work, authors adapted this system to (HTS) of small molecule inhibitors of the early oligomerization, since they are potentially more effective than inhibitors of later steps of oligomerization (e.g., fibrillization) [62]. After validation of the approach, by screening several known antiaggregation compounds, authors expanded the screening to a large library of small molecules, resulting in the identification of two new promising antioligomeric compounds: AO-11 (2-(4-methoxyphenyl)-1,3-benzoxazol-5-amine) and AO-15 (1-(2,3-dimethylphenyl)-4-(2-furoyl) piperazine) [62]. Another strategy developed to study aggregation of Aβ peptides took advantage of the same Sup35 protein as a reporter (that is also a prion protein) and consisted in replacing the prion domain in the Sup35 protein by the Aβ42 peptide [60]. Authors found that Aβ42 peptide can restore the Sup35p ability to aggregate and that the Aβ42 aggregates have different properties compared to the original prion aggregates. This model also offers the possibility for genetic and chemical screenings of suppressors of aggregation.
The yeast studies described above allowed better understanding the processing of APP and, more importantly, resulted in a series of high-throughput models for the genetic screening of secretases and genetic/chemical screenings of modulators of Aβ42 aggregation and toxicity with potential therapeutic applicability.
4. α-Synuclein
α-Synuclein plays a crucial role in PD, the second most common neurodegenerative disorder after AD [63]. Though the majority of PD cases arise sporadically, there are some rare (around 10%) familial cases. α-Synuclein was the first gene to be associated with the familial cases, with several missense mutations (A53T, A30P, E46K) as well as locus duplication and triplication causing PD [64, 65].
α-Synuclein is a presynaptic protein with a role in vesicle dynamics. α-Synuclein exhibits a nonclassical chaperone activity that in conjunction with the cysteine-string protein-α (CSPα) helps to promote SNARE complex assembly, crucial for dopamine neurotransmission, vesicle recycling, and synaptic integrity [66, 67]. In both sporadic and familial cases of PD, α-synuclein in the form of amyloid fibrils is the main constituent of Lewy bodies, proteinaceous aggregates that are the major feature of this disease [68]. The first attempt to study α-synuclein in yeast was performed by Outeiro and Lindquist [31]. Three levels of α-synuclein expression were tested in yeast, low (one integrated copy), intermediate (two integrated copies), and high (several copies using a multicopy vector). In addition to the wild-type (wt) α-synuclein, two of the PD-associated mutations, A53T and A30P, were also studied. At low levels, α-synuclein associated with the plasma membrane with no effect on the cell viability [31]. At intermediate expression levels, α-synuclein redistributed from the cell surface into cytoplasmic foci and became cytotoxic (Figure 1(b)). At high expression levels, α-synuclein cytotoxicity was increased, with inhibition of phospholipase D, retardation of endocytosis, and accumulation of cytosolic lipid droplets [31]. This dependence of α-synuclein toxicity on its expression levels in yeast resembled the gene dosage-dependent premature onset of PD found in humans [31]. Despite being PD-associated mutations, and as such we would expect a higher toxicity, A53T and A30P had similar and lower toxicities, respectively, than wt [31]. Following this first study, several other groups used yeast to study α-synuclein pathobiology. The α-synuclein yeast cytotoxicity was shown to be mediated by increased levels of reactive oxygen species (ROS), activation of the heat shock response, endoplasmic reticulum (ER) stress, decreased proteasome function, and altered lipid metabolism [69–76]. In addition, it was shown that α-synuclein triggers an apoptotic cell death, involving ROS accumulation and cytochrome c release from mitochondria [71], which was decreased in the absence of functional mitochondria (lacking mitochondrial DNA) [77]. Supporting an important role of mitochondria on α-synuclein toxicity, microarray data also revealed that mitochondrial stress is an early feature of α-synuclein expression in yeast [78]. The association of α-synuclein with mitochondria is particularly interesting since other PD-associated proteins, for example, Parkin and Pink, have been associated with mitochondrial function/dysfunction [79]. An important feature of α-synuclein expression in yeast is a defect in ER-to-Golgi complex trafficking [73, 80–82]. In addition, other steps of the vesicular trafficking (e.g., vacuolar fusion) were also disrupted by direct interference of α-synuclein with the yeast transport machinery [81–83]. Also, as a result of this vesicular defect, cytoplasmic vesicles that are unable to dock or fuse due to the presence of α-synuclein formed aggregates [80, 81, 83]. Human orthologues of vesicular trafficking yeast proteins involved in α-synuclein toxicity, namely, Rab1, Rab3A, and Rab8A, were found to also suppress α-synuclein toxicity in neuronal models of PD [74, 81].
Aggregation of α-synuclein is the major disease-associated feature of this protein [68], and as such it has been often addressed in the yeast-based α-synuclein model. Using this cell system, sequence determinants for fibrillogenesis were uncovered [84] as well as risk factors for increased α-synuclein fibrillation [85, 86]. In spite of this, the contribution of α-synuclein fibrillation to its toxicity is still dubious since in most studies a correlation between fibrillation rate and α-synuclein toxicity was not found [87]. Rather, through the screening of several random point mutants, the N-terminal amino acids were shown to contribute to α-synuclein cytotoxicity as well as membrane affinity [87, 88]. Amino acid residues in the hydrophobic central core of α-synuclein also contributed to the cytotoxicity, but not interfered with the protein solubility. This further indicated that harmful α-synuclein species were not the insoluble aggregates [89]. In agreement, the defibrillation of α-synuclein fibers with dequalinium, an amphipattic molecule described as a modulator of α-synuclein fibrillation [90], increased its toxicity [41]. This suggests that early misfolded forms of α-synuclein are the toxic species, as has been reported for the Aβ peptide (discussed in the previous section). Since they are not aggregated, this can be explained by the availability of these forms to interact with cell membranes. This membrane interaction-dependent toxicity of α-synuclein is supported by the observation that A30P, with poor membrane binding affinity, is not toxic to yeast cells [31, 73]. In addition, while some yeast strains exhibited α-synuclein inclusions without showing a significant growth defect [41], in other strains α-synuclein proved to be extremely toxic in the absence of visible aggregates [31, 72]. Together, these data indicated that the aggregation or fibrillation of α-synuclein per se is not toxic. Instead, it may be the interference of α-synuclein with membranes and the obstruction of the vesicular trafficking the culprit for its pathogenicity.
Yeast was also used to clarify the association of α-synuclein with other PD-related genes. Coexpression of α-synuclein with Dj-1, Parkin, Pink1, ubiquitin C-terminal hydrolase-L1 (UCH-L1), or Synphilin-1 did not significantly alter the α-synuclein toxicity in exponentially growing yeast cells [87]. Yet, in aged cultures, likewise in mammalian cells, Synphilin-1 increased α-synuclein aggregation and cytotoxicity [91]. Similarly, coexpression of α-synuclein with tau, a protein involved in AD and occasionally associated with α-synuclein inclusions [92], caused synergistic toxicity [93]. Moreover, overexpression of the yeast orthologue of the human ATP13A2, a risk factor for PD, was shown to suppress α-synuclein toxicity in yeast, an effect confirmed in animal and neuronal PD models [94].
The α-synuclein model was also adapted to genetic large-scale screenings, which allowed the identification of several factors that influence the toxicity/aggregation of α-synuclein. A genetic screening of a collection of deleted nonessential genes led to the identification of several mutants with enhanced α-synuclein toxicity [69]. This approach was also used to identify genes that alter the α-synuclein localization or aggregate formation [82]. Not surprisingly, in both screens many of the hits found were mutants affected in lipid metabolism, vesicular transport, and vacuolar degradation. This reinforced the idea of the importance of these processes to α-synuclein cytotoxicity. With these data, a targeted screen using strains deleted on genes involved in lipid metabolism was recently performed. With this approach, ER lipid elongases were identified as suppressors of α-synuclein toxicity [95]. Using a gene overexpression screening approach, proteins involved in vesicular trafficking were also identified as suppressors of α-synuclein toxicity [74]. Remarkably, with this approach, it was identified the protein Ypt1, whose mammalian homologue Rab1 rescued the loss of dopaminergic neurons in animal models of PD [72]. Other two gene overexpression screens were performed for suppressors of the lethality of wt α-synuclein and A30P in response to hydrogen peroxide [96, 97]. In this study, little overlap was found between the suppressors identified for the wt α-synuclein and the two familial mutations, pointing for distinct mechanisms of toxicity.
A new mathematic algorithm (ResponseNet), allowing the combination of data obtained from microarray expression profiles with genetic screens, was applied to the α-synuclein yeast model [98]. This algorithm revealed new potential cellular responses involved in the toxicity of α-synuclein, including ergosterol biosynthesis and the target of rapamycin (TOR) pathway [98].
Finally, the use of the yeast-based α-synuclein cell model as a drug screening tool also allowed the identification of several toxicity suppressors from large libraries, such as two cyclic peptides and four 1,2,3,4-tetrahydroquinolinones, with promising therapeutic potential [78, 89, 99].
Altogether, the yeast model has provided several insights into the α-synuclein cytotoxicity. The ease to perform HTS allowed the identification of genetic and chemical suppressors of α-synuclein, many of them already validated in higher eukaryotes, with promising applicability in PD therapy.
5. Polyglutamine-Containing Proteins
Inclusions of proteins containing polyglutamine (polyQ) expanded tracts are causatively associated with a group of about 9 fatal hereditary neurodegenerative disorders globally referred as polyglutamine disease, from which HD is the most recognized one [100]. PolyQ expansion in different proteins induces different pathologies; however, these pathologies share the fact that the longer the polyQ expansion, the more severe the disease and the earlier the onset. Huntingtin is a multi-domain protein essential for cell survival and it is thought to play a role in several processes, including transcriptional regulation, vesicular trafficking, metabolism, and synaptic function. It is believed that underlying HD is not only the loss-of-function of the Huntingtin protein due to the expansion of the polyQ tract, but also a new toxic gain-of-function associated with the expansion [101]. Despite the intensive research in this area, the mechanisms underlying the neurodegeneration induced by polyQ expansions remain unclear. Based on this, several research groups have developed yeast models to study the mechanisms of aggregation and toxicity of proteins with expanded polyQ tracts as well as to identify molecular regulators that could serve as therapeutic targets.
Most of the studies performed in yeast used the N-terminal of Huntingtin (which includes the polyQ tract) as a surrogate for proteins with expanded polyQ tracts. When first expressed in yeast, polyQ constructs with different lengths, namely, Q25 (corresponding to a normal polyQ length) and Q47, Q72 and Q103 (representing expanded HD-related polyQ lengths) were analyzed [102]. Authors showed that the Q25 protein did not aggregate. However, the formation of aggregates increased with the polyQ length (Figure 1(c)). With this, it was possible to recapitulate in yeast the dependency of the Huntingtin aggregation on the expansion of the polyQ tract. Despite this, the expression of polyQ in yeast exhibited minimal toxicity, as further confirmed by other authors [103]. However, the presence of the protein Rnq1 in its prion conformation [Pin+] was sufficient to convert the polyQ tract into a toxic protein to yeast, establishing an unexpected link between prions and HD [104, 105]. Toxicity was proposed to be partially due to sequestration of essential proteins, like Sup35 and Sup45, in the polyQ aggregates [106]. Yeast prions are responsible for the transmission of a phenotype but, unlike mammalian prions, they are not harmful to the cell (reviewed in [107]). The connection between polyQ and yeast prions was strengthened by the fact that the polyQ domain facilitated the appearance of prion states [108] and that both proteins can coaggregate [105, 109].
As for yeast prions, the aggregating ability of the expanded polyQ tracts was strongly dependent on chaperones, such as Hsp70 and its cochaperone Hsp40, Hsp26, Hsp104, and the chaperonin TRiC [69, 102, 103, 110–112]. Chaperones bind aggregates in an orderly fashion, with Hsp70 and Hsp90 binding in an early phase and Hsp26 and Hsp104 in a late phase of polyQ aggregate expansion [112]. Chaperones can decrease polyQ toxicity either by decreasing prion propagation or by directly affecting the size of the polyQ aggregates [111]. Besides chaperones, other cellular factors affected the aggregation of Huntingtin polyQ in yeast, including the microtubule cytoskeleton [113, 114], proteins like Cdc48 and cofactors [114], and components of the late endocytic complex [115]. Making clear that unfolded protein aggregation is not a spontaneous process, as initially assumed, the uncovering of aggregation pathways reveals novel opportunities for intervention [116]. Although most studies point to the formation of aggregates as beneficial, whether aggregation should be hindered or promoted to relieve polyQ toxicity is still controversial [113, 114, 117].
In addition to the polyQ length, the sequences flanking the polyQ domain also modulate toxicity. In particular, the polyQ flanking proline-rich region of Huntingtin has an important role in the toxicity [114, 117, 118]. The presence of this domain strongly reduced the polyQ toxicity, and along with the N-terminal N17 domain was proposed to be a sequence determinant for the addressing of the polyQ to the large protective perinuclear aggregates [114]. The proline-rich region can also operate in trans, that is, the coexpression of the polyQ containing the proline-rich domain converted the toxic polyQ without the proline-rich region into benign variants [105]. Also operating in trans and affecting polyQ toxicity were other glutamine rich proteins, such as yeast prions [105].
While many studies addressed the properties of polyQ aggregates, other authors tried to understand the cellular dysfunction underlying toxicity. Most of the cellular effects of polyQ observed in yeast were consistent with reports for HD animal models and HD patients, which validated the yeast model for the study of polyQ toxicity. For instance, it was reported that polyQ tracts led to transcriptional dysregulation [119], caused impairment in ER-protein homeostasis [120] and in endocytosis [115, 121]. Huntingtin polyQ also altered mitochondrial morphology and compromised mitochondrial function, with early alterations in complexes II and III of the respiratory chain associated with an increase in ROS production [122, 123]. Stimulation of mitochondrial biogenesis was able to rescue mitochondrial dysfunction and cellular toxicity, indicating that mitochondria contribute significantly to polyQ toxicity [124]. In addition to mitochondrial dysfunction, the occurrence of DNA fragmentation and caspase activation indicated the activation of an apoptotic pathway by Huntingtin polyQ tracts [123]. The same authors reported a disturbance in the cell cycle that was also involved in polyQ cytotoxicity. This effect on cell cycle was attributed to the accumulation of cell cycle substrates likely due to a hindrance in protein degradation caused by the accumulation of polyQ [123, 125]. Another consequence of the polyQ expression in yeast is the alteration of the cellular concentration of several metabolites, namely, alanine, glycerol, glutamine, and valine. Alterations in these metabolites were proposed as promising biomarkers for HD [126]. Genome-wide screens in yeast identified several modifiers of mutant Huntingtin aggregation and toxicity, which could potentially serve as therapeutic targets. The proteins identified as loss-of-function suppressors were majorly associated with stress, protein folding, vesicular transport, vacuolar degradation, and transcription [69, 127]. An interesting suppressor was the mitochondrial protein kynurenine 3-monooxygenase involved in the kynurenine pathway of tryptophan degradation [127]. The kynurenine pathway was found to be activated in HD patients and in animal models of this disorder and is regulated by histone deacetylase enzymes [128]. Notably, a previous work had shown that the transcriptional profile of yeast expressing a polyQ tract was similar to those of yeast strains deleted for components of the histone acetyltransferase complex [119]. Transcriptional deregulation was rescued by treating the cells with a histone deacetylase inhibitor [119]. Inhibition of histone deacetylase also suppressed the kynurenine pathway in yeast, microglia, and mice expressing the Huntingtin polyQ [129]. This pointed out the kynurenine pathway as a promising therapeutic target for HD. Consistent with the previously established connection between polyQ and yeast prions, the prion form of Rnq1 and proteins involved in prion-like aggregation were also amongst the hits obtained. In one of the screenings, the major functional categories obtained for Huntingtin polyQ was compared to the ones obtained for the PD-associated protein α-synuclein. The overlap obtained for both proteins was minimal. This indicated that despite the coalescence of both proteins into aggregates, they had distinct pathogenic mechanisms [69].
In addition to genetic screenings, yeast polyQ models have also been widely used as a cell-based HTS system of small molecules with potential therapeutic application. The compounds found have already shown promising results to ameliorate polyQ toxicity in higher eukaryotes [130–133].
Overall, yeast models of polyQ-containing proteins provided new insights into the molecular determinants for polyQ aggregation. Importantly, studies in yeast uncovered promising therapeutic compounds and new molecular targets in HD, namely, chaperones and kynurenine 3-monooxygenase.
6. Concluding Remarks
Though this study has been focused on S. cerevisiae, other yeast species like the fission Schizosaccharomyces pombe and the methylotrophic Pichia pastoris have also been used as model organisms of neurodegeneration. Likewise, only proteins strictly involved in the most common neurodegenerative disorders have been addressed. However, other human proteins recognized as central players in apoptotic diseases as neurodegeneration, including several members of the caspase, Bcl-2, p53, and protein kinase C family, have also been widely studied in yeast. Supported by the valuable contribution of yeast as a model organism towards the understanding of the molecular basis underlying neurodegenerative disorders, it is expectable that many other disease-associated proteins will be studied in this unicellular eukaryote. Even though yeast has no orthologues of the majority of these proteins, most of them exhibit some assessable phenotype that can be explored in the development of target-specific approaches easily adapted to HTS analysis. In fact, an enormous input in the identification of new therapeutic opportunities against these pathologies has been given through the development of fast and reliable yeast target-based HTS approaches. Though the obvious limitations of using a microorganism to address human diseases, when used in the early phase and with complementary mammalian systems, yeast may greatly contribute to the identification of novel targets and to the discovery of novel agents against neurodegeneration. Therapeutic options for many patients with neurodegenerative disorders have changed little. A typical delay between the early steps in the discovery of a lead compound, in which yeast participate, and its clinical use make us believe that many great contributions of the yeast model to the discovery of new therapeutic agents will be realized in the near future.
Acknowledgment
This work was supported by FCT (Fundação para a Ciência e a Tecnologia) through REQUIMTE (Grant no. PEst-C/EQB/LA0006/2011) and the fellowships of M. Leão (SFRH/BD/64184/2009) and J. Soares (SFRH/BD/78971/2011), FEDER funds through the COMPETE program under Project FCOMP-01-0124-FEDER-015752 (ref FCT PTDC/SAU-FAR/110848/2009), and by U.Porto/Santander Totta.
References
- 1.Cho RJ, Campbell MJ, Winzeler EA, et al. A genome-wide transcriptional analysis of the mitotic cell cycle. Molecular Cell. 1998;2(1):65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- 2.Zhu H, Bilgin M, Bangham R, et al. Global analysis of protein activities using proteome chips. Science. 2001;293(5537):2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 3.Giaever G, Chu AM, Ni L, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 4.Uetz P, Glot L, Cagney G, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403(6770):623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 5.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(8):4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tong AHY, Evangelista M, Parsons AB, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294(5550):2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 7.Jones GM, Stalker J, Humphray S, et al. A systematic library for comprehensive overexpression screens in Saccharomyces cerevisiae. Nature Methods. 2008;5(3):239–241. doi: 10.1038/nmeth.1181. [DOI] [PubMed] [Google Scholar]
- 8.Khurana V, Lindquist S. Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker’s yeast? Nature Reviews Neuroscience. 2010;11(6):436–449. doi: 10.1038/nrn2809. [DOI] [PubMed] [Google Scholar]
- 9.Forman MS, Trojanowski JQ, Lee VMY. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nature Medicine. 2004;10(10):1055–1063. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- 10.Saiki S, Sato S, Hattori N. Molecular pathogenesis of Parkinson's disease: update. Journal of Neurology, Neurosurgery & Psychiatry. 2012;83:430–436. doi: 10.1136/jnnp-2011-301205. [DOI] [PubMed] [Google Scholar]
- 11.Pandolfo M, Pastore A. The pathogenesis of Friedreich ataxia and the structure and function of frataxin. Journal of Neurology. 2009;256(1):9–17. doi: 10.1007/s00415-009-1003-2. [DOI] [PubMed] [Google Scholar]
- 12.Babcock M, De Silva D, Oaks R, et al. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science. 1997;276(5319):1709–1712. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- 13.Wilson RB, Roof DM. Respiratory deficiency due to loss of mitochondrial dna in yeast lacking the frataxin homologue. Nature Genetics. 1997;16(4):352–357. doi: 10.1038/ng0897-352. [DOI] [PubMed] [Google Scholar]
- 14.Cotticelli MG, Rasmussen L, Kushner NL, et al. Primary and secondary drug screening assays for friedreich ataxia. Journal of Biomolecular Screening. 2012;17(3):303–313. doi: 10.1177/1087057111427949. [DOI] [PubMed] [Google Scholar]
- 15.de Moura MB, dos Santos LS, Van Houten B. Mitochondrial dysfunction in neurodegenerative diseases and cancer. Environmental and Molecular Mutagenesis. 2010;51(5):391–405. doi: 10.1002/em.20575. [DOI] [PubMed] [Google Scholar]
- 16.Pearce DA, Sherman F. A yeast model for the study of Batten disease. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(12):6915–6918. doi: 10.1073/pnas.95.12.6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang S, Ren J, Li H, et al. Ncr1p, the yeast ortholog of mammalian Niemann Pick C1 protein, is dispensable for endocytic transport. Traffic. 2004;5(12):1017–1030. doi: 10.1111/j.1600-0854.2004.00241.x. [DOI] [PubMed] [Google Scholar]
- 18.Berger AC, Hanson PK, Nichols JW, Corbett AH. A yeast model system for functional analysis of the Niemann-Pick type C protein 1 homolog, Ncr1p. Traffic. 2005;6(10):907–917. doi: 10.1111/j.1600-0854.2005.00327.x. [DOI] [PubMed] [Google Scholar]
- 19.Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science. 1996;271(5247):357–360. doi: 10.1126/science.271.5247.357. [DOI] [PubMed] [Google Scholar]
- 20.Fritz E, Friedl AA, Zwacka RM, Eckardt-Schupp F, Meyn MS. The yeast TEL1 gene partially substitutes for human ATM in suppressing hyperrecombination, radiation-induced apoptosis and telomere shortening in A-T cells. Molecular Biology of the Cell. 2000;11(8):2605–2616. doi: 10.1091/mbc.11.8.2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nolden M, Ehses S, Koppen M, Bernacchia A, Rugarli EI, Langer T. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell. 2005;123(2):277–289. doi: 10.1016/j.cell.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 22.Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264(5158):566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 23.Bach S, Talarek N, Andrieu T, et al. Isolation of drugs active against mammalian prions using a yeast-based screening assay. Nature Biotechnology. 2003;21(9):1075–1081. doi: 10.1038/nbt855. [DOI] [PubMed] [Google Scholar]
- 24.Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(59):p. 62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 25.Nishida CR, Gralla EB, Valentine JS. Characterization of three yeast copper-zinc superoxide dismutase mutants analogous to those coded for in familial amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(21):9906–9910. doi: 10.1073/pnas.91.21.9906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Current Opinion in Neurobiology. 2011;21:904–919. doi: 10.1016/j.conb.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson BS, McCaffery JM, Lindquist S, Gitler AD. A yeast TDP-43 proteinopathy model: exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(17):6439–6444. doi: 10.1073/pnas.0802082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP−43 intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. The Journal of Biological Chemistry. 2009;284(37):20329–20339. doi: 10.1074/jbc.M109.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fushimi K, Long C, Jayaram N, Chen X, Li L, Wu JY. Expression of human FUS/TLS in yeast leads to protein aggregation and cytotoxicity, recapitulating key features of FUS proteinopathy. Protein and Cell. 2011;2(2):141–149. doi: 10.1007/s13238-011-1014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ju S, Tardiff DF, Han H, et al. A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biology. 2011;9(4) doi: 10.1371/journal.pbio.1001052.e1001052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science. 2003;302(5651):1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiong Y, Coombes CE, Kilaru A, et al. GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genetics. 2010;6(4) doi: 10.1371/journal.pgen.1000902.e1000902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H, Komano H, Fuller RS, Gandy SE, Frail DE. Proteolytic processing and secretion of human β-amyloid precursor protein in yeast. Evidence for a yeast secretase activity. Journal of Biological Chemistry. 1994;269(45):27799–27802. [PubMed] [Google Scholar]
- 34.Chacińska A, Woźny W, Boguta M, Misicka A, Brzyska M, Elbaum D. Effects of beta-amyloid on proliferation and morphology of yeast Saccharomyces cerevisiae. Letters in Peptide Science. 2002;9(4-5):197–201. [Google Scholar]
- 35.Vandebroek T, Terwel D, Van Helmont T, Winderickx J, Van Leuven F. Phosphorylation and aggregation of protein tau in humanized yeast cells and in transgenic mouse brain. New Trends in Alzheimer and Parkinson Related Disorders. 2005;256:15–19. [Google Scholar]
- 36.Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314(5800):777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 37.Jarrett JT, Berger EP, Lansbury PT. The C-terminus of the β protein is critical in amyloidogenesis. Annals of the New York Academy of Sciences. 1993;695:144–148. doi: 10.1111/j.1749-6632.1993.tb23043.x. [DOI] [PubMed] [Google Scholar]
- 38.Soscia SJ, Kirby JE, Washicosky KJ, et al. The Alzheimer’s disease-associated amyloid β-protein is an antimicrobial peptide. PLoS ONE. 2010;5(3) doi: 10.1371/journal.pone.0009505.e9505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang W, Espinoza D, Hines V, Innis M, Mehta P, Miller DL. Characterization of β-amyloid peptide precursor processing by the yeast Yap3 and Mkc7 proteases. Biochimica et Biophysica Acta. 1997;1359(2):110–122. doi: 10.1016/s0167-4889(97)00082-7. [DOI] [PubMed] [Google Scholar]
- 40.Komano H, Seeger M, Gandyl S, Wang GT, Krafft GA, Fuller RS. Involvement of cell surface glycosyl-phosphatidylinositol-linked aspartyl proteases in α-secretase-type cleavage and ectodomain solubilization of human Alzheimer β-amyloid precursor protein in yeast. Journal of Biological Chemistry. 1999;273(48):31648–31651. doi: 10.1074/jbc.273.48.31648. [DOI] [PubMed] [Google Scholar]
- 41.Park JW, Lee IH, Hahn JS, Kim J, Chung KC, Paik SR. Disintegration of amyloid fibrils of α-synuclein by dequalinium. Biochimica et Biophysica Acta. 2008;1780(10):1156–1161. doi: 10.1016/j.bbagen.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 42.Greenfield JP, Xu H, Greengard P, Gandy S, Seeger M. Generation of the amyloid-β peptide N terminus in Saccharomyces cerevisiae expressing human Alzheimer’s amyloid-β precursor protein. Journal of Biological Chemistry. 1999;274(48):33843–33846. doi: 10.1074/jbc.274.48.33843. [DOI] [PubMed] [Google Scholar]
- 43.Sparvero LJ, Patz S, Brodsky JL, Coughlan CM. Proteomic analysis of the amyloid precursor protein fragment C99: expression in yeast. Analytical Biochemistry. 2007;370(2):162–170. doi: 10.1016/j.ab.2007.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song S, Jung YK. Alzheimer’s disease meets the ubiquitin-proteasome system. Trends in Molecular Medicine. 2004;10(11):565–570. doi: 10.1016/j.molmed.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 45.Keller JN, Gee J, Ding Q. The proteasome in brain aging. Ageing Research Reviews. 2002;1(2):279–293. doi: 10.1016/s1568-1637(01)00006-x. [DOI] [PubMed] [Google Scholar]
- 46.Murakami-Sekimata A, Sato K, Sato K, Takashima A, Nakano A. O-Mannosylation is required for the solubilization of heterologously expressed human β-amyloid precursor protein in Saccharomyces cerevisiae. Genes to Cells. 2009;14(2):205–215. doi: 10.1111/j.1365-2443.2008.01263.x. [DOI] [PubMed] [Google Scholar]
- 47.Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of γ-secretase activity. Nature Cell Biology. 2003;5(5):486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- 48.Yagishita S, Futai E, Ishiura S. In vitro reconstitution of γ-secretase activity using yeast microsomes. Biochemical and Biophysical Research Communications. 2008;377(1):141–145. doi: 10.1016/j.bbrc.2008.09.090. [DOI] [PubMed] [Google Scholar]
- 49.Gunyuzlu PL, White WH, Davis GL, Hollis GF, Toyn JH. A yeast genetic assay for caspase cleavage of the Amyloid-β precursor protein. Applied Biochemistry and Biotechnology B. 2000;15(1):29–37. doi: 10.1385/MB:15:1:29. [DOI] [PubMed] [Google Scholar]
- 50.Park SA, Shaked GM, Bredesen DE, Koo EH. Mechanism of cytotoxicity mediated by the C31 fragment of the amyloid precursor protein. Biochemical and Biophysical Research Communications. 2009;388(2):450–455. doi: 10.1016/j.bbrc.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lüthi U, Schaerer-Brodbeck C, Tanner S, Middendorp O, Edler K, Barberis A. Human β-secretase activity in yeast detected by a novel cellular growth selection system. Biochimica et Biophysica Acta. 2003;1620(1–3):167–178. doi: 10.1016/s0304-4165(02)00529-9. [DOI] [PubMed] [Google Scholar]
- 52.Middendorp O, Ortler C, Neumann U, Paganetti P, Lüthi U, Barberis A. Yeast growth selection system for the identification of cell-active inhibitors of β-secretase. Biochimica et Biophysica Acta. 2004;1674(1):29–39. doi: 10.1016/j.bbagen.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 53.Bayer TA, Wirths O. Intracellular accumulation of amyloid-Beta—a predictor for synaptic dysfunction and neuron loss in Alzheimer's disease. Frontiers in Aging Neuroscience. 2010;2, article 8 doi: 10.3389/fnagi.2010.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer’s disease. Nature Reviews Neuroscience. 2007;8(7):499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 55.Bharadwaj P, Waddington L, Varghese J, Macreadie IG. A new method to measure cellular toxicity of non-fibrillar and fibrillar Alzheimer’s Aβ using yeast. Journal of Alzheimer’s Disease. 2008;13(2):147–150. doi: 10.3233/jad-2008-13204. [DOI] [PubMed] [Google Scholar]
- 56.McLean CA, Cherny RA, Fraser FW, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Annals of Neurology. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 57.Caine J, Sankovich S, Antony H, et al. Alzheimer’s Aβ fused to green fluorescent protein induces growth stress and a heat shock response. FEMS Yeast Research. 2007;7(8):1230–1236. doi: 10.1111/j.1567-1364.2007.00285.x. [DOI] [PubMed] [Google Scholar]
- 58.Dubey AK, Bharadwaj PR, Varghese JN, MacReadie IG. Alzheimer’s amyloid-β rescues yeast from hydroxide toxicity. Journal of Alzheimer’s Disease. 2009;18(1):31–33. doi: 10.3233/JAD-2009-1121. [DOI] [PubMed] [Google Scholar]
- 59.Treusch S, Hamamichi S, Goodman JL, et al. Functional links between abeta toxicity, endocytic trafficking, and Alzheimer's disease risk factors in yeast. Science. 2011;334:1241–1245. doi: 10.1126/science.1213210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Macreadie I, Lotfi-Miri M, Mohotti S, Shapira D, Bennett L, Varghese J. Validation of folate in a convenient yeast assay suited for identification of inhibitors of Alzheimer’s amyloid-β aggregation. Journal of Alzheimer’s Disease. 2008;15(3):391–396. doi: 10.3233/jad-2008-15305. [DOI] [PubMed] [Google Scholar]
- 61.Bagriantsev S, Liebman S. Modulation of Aβ42 low-n oligomerization using a novel yeast reporter system. BMC Biology. 2006;4:p. 32. doi: 10.1186/1741-7007-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park SK, Pegan SD, Mesecar AD, Jungbauer LM, LaDu MJ, Liebman SW. Development and validation of a yeast high-throughput screen for inhibitors of Abeta oligomerization. Disease Models & Mechanisms. 2011;4:822–831. doi: 10.1242/dmm.007963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurology. 2006;5(6):525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 64.Eriksen JL, Przedborski S, Petrucelli L. Gene dosage and pathogenesis of Parkinson’s disease. Trends in Molecular Medicine. 2005;11(3):91–96. doi: 10.1016/j.molmed.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 65.Schiesling C, Kieper N, Seidel K, Krüger R. Review: familial Parkinson’s disease—genetics, clinical phenotype and neuropathology in relation to the common sporadic form of the disease. Neuropathology and Applied Neurobiology. 2008;34(3):255–271. doi: 10.1111/j.1365-2990.2008.00952.x. [DOI] [PubMed] [Google Scholar]
- 66.Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 67.Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329(5999):1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schulz-Schaeffer WJ. The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathologica. 2010;120(2):131–143. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or α-synuclein. Science. 2003;302(5651):1769–1772. doi: 10.1126/science.1090389. [DOI] [PubMed] [Google Scholar]
- 70.Chen Q, Thorpe J, Keller JN. α-synuclein alters proteasome function, protein synthesis, and stationary phase viability. Journal of Biological Chemistry. 2005;280(34):30009–30017. doi: 10.1074/jbc.M501308200. [DOI] [PubMed] [Google Scholar]
- 71.Flower TR, Chesnokova LS, Froelich CA, Dixon C, Witt SN. Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of Parkinson’s disease. Journal of Molecular Biology. 2005;351(5):1081–1100. doi: 10.1016/j.jmb.2005.06.060. [DOI] [PubMed] [Google Scholar]
- 72.Sharma N, Brandis KA, Herrera SK, et al. α-synuclein budding yeast model: toxicity enhanced by impaired proteasome and oxidative stress. Journal of Molecular Neuroscience. 2006;28(2):161–178. doi: 10.1385/JMN:28:2:161. [DOI] [PubMed] [Google Scholar]
- 73.Dixon C, Mathias N, Zweig RM, Davis DA, Gross DS. α-synuclein targets the plasma membrane via the secretory pathway and induces toxicity in yeast. Genetics. 2005;170(1):47–59. doi: 10.1534/genetics.104.035493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cooper AA, Gitler AD, Cashikar A, et al. α-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313(5785):324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arimon M, Grimminger V, Sanz F, Lashuel HA. Hsp104 targets multiple intermediates on the amyloid pathway and suppresses the seeding capacity of Aβ fibrils and protofibrils. Journal of Molecular Biology. 2008;384(5):1157–1173. doi: 10.1016/j.jmb.2008.09.063. [DOI] [PubMed] [Google Scholar]
- 76.Sere YY, Regnacq M, Colas J, Berges T. A Saccharomyces cerevisiae strain unable to store neutral lipids is tolerant to oxidative stress induced by α-synuclein. Free Radical Biology and Medicine. 2010;49(11):1755–1764. doi: 10.1016/j.freeradbiomed.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 77.Büttner S, Bitto A, Ring J, et al. Functional mitochondria are required for α-synuclein toxicity in aging yeast. Journal of Biological Chemistry. 2008;283(12):7554–7560. doi: 10.1074/jbc.M708477200. [DOI] [PubMed] [Google Scholar]
- 78.Su LJ, Auluck PK, Outeiro TF, et al. Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson’s disease models. DMM Disease Models and Mechanisms. 2010;3(3-4):194–208. doi: 10.1242/dmm.004267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pilsl A, Winklhofer KF. Parkin, PINK1 and mitochondrial integrity: emerging concepts of mitochondrial dysfunction in Parkinson's disease. Acta Neuropathologica. 2012;123:173–188. doi: 10.1007/s00401-011-0902-3. [DOI] [PubMed] [Google Scholar]
- 80.Soper JH, Roy S, Stieber A, et al. α-synuclein-induced aggregation of cytoplasmic vesicles in Saccharomyces cerevisiae. Molecular Biology of the Cell. 2008;19(3):1093–1103. doi: 10.1091/mbc.E07-08-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gitler AD, Bevis BJ, Shorter J, et al. The Parkinson’s disease protein α-synuclein disrupts cellular Rab homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(1):145–150. doi: 10.1073/pnas.0710685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zabrocki P, Bastiaens I, Delay C, et al. Phosphorylation, lipid raft interaction and traffic of α-synuclein in a yeast model for Parkinson. Biochimica et Biophysica Acta. 2008;1783(10):1767–1780. doi: 10.1016/j.bbamcr.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 83.Soper JH, Kehm V, Burd CG, Bankaitis VA, Lee VMY. Aggregation of α-synuclein in S. cerevisiae is associated with defects in endosomal trafficking and phospholipid biosynthesis. Journal of Molecular Neuroscience. 2011;43(3):391–405. doi: 10.1007/s12031-010-9455-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zibaee S, Jakes R, Fraser G, Serpell LC, Crowther RA, Goedert M. Sequence determinants for amyloid fibrillogenesis of human α-synuclein. Journal of Molecular Biology. 2007;374(2):454–464. doi: 10.1016/j.jmb.2007.09.039. [DOI] [PubMed] [Google Scholar]
- 85.Oien DB, Shinogle HE, Moore DS, Moskovitz J. Clearance and phosphorylation of alpha-synuclein are inhibited in methionine sulfoxide reductase a null yeast cells. Journal of Molecular Neuroscience. 2009;39(3):323–332. doi: 10.1007/s12031-009-9274-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fiske M, Valtierra S, Solvang K, et al. Contribution of alanine-76 and serine phosphorylation in alpha-synuclein membrane association and aggregation in yeasts. Parkinson's Disease. 2011;2011:12 pages. doi: 10.4061/2011/392180.392180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Volles MJ, Lansbury PT. Relationships between the sequence of α-synuclein and its membrane affinity, fibrillization propensity, and yeast toxicity. Journal of Molecular Biology. 2007;366(5):1510–1522. doi: 10.1016/j.jmb.2006.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vamvaca K, Volles MJ, Lansbury PT. The first N-terminal amino acids of α-synuclein are essential for α-helical structure formation in vitro and membrane binding in yeast. Journal of Molecular Biology. 2009;389(2):413–424. doi: 10.1016/j.jmb.2009.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Griffioen G, Duhamel H, Van Damme N, et al. A yeast-based model of α-synucleinopathy identifies compounds with therapeutic potential. Biochimica et Biophysica Acta. 2006;1762(3):312–318. doi: 10.1016/j.bbadis.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 90.Lee CH, Hyun JK, Lee JH, et al. Dequalinium-induced protofibril formation of α-synuclein. Journal of Biological Chemistry. 2006;281(6):3463–3472. doi: 10.1074/jbc.M505307200. [DOI] [PubMed] [Google Scholar]
- 91.Büttner S, Delay C, Franssens V, et al. Synphilin-1 enhances a-synuclein aggregation in yeast and contributes to cellular stress and cell death in a sir2-dependent manner. PLoS ONE. 2010;5(10) doi: 10.1371/journal.pone.0013700.e13700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Duda JE, Giasson BI, Mabon ME, et al. Concurrence of α-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathologica. 2002;104(1):7–11. doi: 10.1007/s00401-002-0563-3. [DOI] [PubMed] [Google Scholar]
- 93.Zabrocki P, Pellens K, Vanhelmont T, et al. Characterization of α-synuclein aggregation and synergistic toxicity with protein tau in yeast. The FEBS Journal. 2005;272(6):1386–1400. doi: 10.1111/j.1742-4658.2005.04571.x. [DOI] [PubMed] [Google Scholar]
- 94.Gitler AD, Chesi A, Geddie ML, et al. α-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nature Genetics. 2009;41(3):308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee YJ, Wang S, Slone SR, Yacoubian TA, Witt SN. Defects in very long chain fatty acid synthesis enhance alpha-synuclein toxicity in a yeast model of Parkinson’s disease. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0015946.e15946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Flower TR, Clark-Dixon C, Metoyer C, et al. YGR198w (YPP1) targets A30P α-synuclein to the vacuole for degradation. Journal of Cell Biology. 2007;177(6):1091–1104. doi: 10.1083/jcb.200610071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liang J, Clark-Dixon C, Wang S, et al. Novel suppressors of α-synuclein toxicity identified using yeast. Human Molecular Genetics. 2008;17(23):3784–3795. doi: 10.1093/hmg/ddn276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yeger-Lotem E, Riva L, Su LJ, et al. Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity. Nature Genetics. 2009;41(3):316–323. doi: 10.1038/ng.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kritzer JA, Hamamichi S, McCaffery JM, et al. Rapid selection of cyclic peptides that reduce α-synuclein toxicity in yeast and animal models. Nature Chemical Biology. 2009;5(9):655–663. doi: 10.1038/nchembio.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Katsuno M, Banno H, Suzuki K, et al. Molecular genetics and biomarkers of polyglutamine diseases. Current Molecular Medicine. 2008;8(3):221–234. doi: 10.2174/156652408784221298. [DOI] [PubMed] [Google Scholar]
- 101.Caviston JP, Holzbaur ELF. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends in Cell Biology. 2009;19(4):147–155. doi: 10.1016/j.tcb.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(4):1589–1594. doi: 10.1073/pnas.97.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(14):7841–7846. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. Huntingtin toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. Journal of Cell Biology. 2002;157(6):997–1004. doi: 10.1083/jcb.200112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Duennwald ML, Jagadish S, Giorgini F, Muchowski PJ, Lindquist S. A network of protein interactions determines polyglutamine toxicity. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(29):11051–11056. doi: 10.1073/pnas.0604548103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kochneva-Pervukhova NV, Alexandrov AI, Ter-Avanesyan MD, et al. Amyloid-mediated sequestration of essential proteins contributes to mutant huntingtin toxicity in yeast. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0029832.e29832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chien P, Weissman JS, DePace AH. Emerging principles of conformation-based prion inheritance. Annual Review of Biochemistry. 2004;73:617–656. doi: 10.1146/annurev.biochem.72.121801.161837. [DOI] [PubMed] [Google Scholar]
- 108.Derkatch IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL, Liebman SW. Effects of Q/N-rich, polyQ, and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(35):12934–12939. doi: 10.1073/pnas.0404968101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Douglas PM, Summers DW, Ren HY, Cyr DM. Reciprocal efficiency of RNQ1 and polyglutamine detoxification in the cytosol and nucleus. Molecular Biology of the Cell. 2009;20(19):4162–4173. doi: 10.1091/mbc.E09-02-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tam S, Geller R, Spiess C, Frydman J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nature Cell Biology. 2006;8(10):1155–1162. doi: 10.1038/ncb1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gokhale KC, Newnam GP, Sherman MY, Chernoff YO. Modulation of prion-dependent polyglutamine aggregation and toxicity by chaperone proteins in the yeast model. Journal of Biological Chemistry. 2005;280(24):22809–22818. doi: 10.1074/jbc.M500390200. [DOI] [PubMed] [Google Scholar]
- 112.Walter GM, Smith MC, Wisen S, et al. Ordered assembly of heat shock proteins, Hsp26, Hsp70, Hsp90, and Hsp104, on expanded polyglutamine fragments revealed by chemical probes. Journal of Biologial Chemistry. 2011;286:40486–40493. doi: 10.1074/jbc.M111.284448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Muchowski PJ, Ning K, D’Souza-Schorey C, Fields S. Requirement of an intact microtubule cytoskeleton for aggregation and inclusion body formation by a mutant huntingtin fragment. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(2):727–732. doi: 10.1073/pnas.022628699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang Y, Meriin AB, Zaarur N, et al. Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. The FASEB Journal. 2009;23(2):451–463. doi: 10.1096/fj.08-117614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Meriin AB, Zhang X, Alexandrov IM, et al. Endocytosis machinery is involved in aggregation of proteins with expanded polyglutamine domains. The FASEB Journal. 2007;21(8):1915–1925. doi: 10.1096/fj.06-6878com. [DOI] [PubMed] [Google Scholar]
- 116.Nagai Y, Fujikake N, Popiel HA, Wada K. Induction of molecular chaperones as a therapeutic strategy for the polyglutamine diseases. Current Pharmaceutical Biotechnology. 2010;11(2):188–197. doi: 10.2174/138920110790909650. [DOI] [PubMed] [Google Scholar]
- 117.Dehay B, Bertolotti A. Critical role of the proline-rich region in Huntingtin for aggregation and cytotoxicity in yeast. Journal of Biological Chemistry. 2006;281(47):35608–35615. doi: 10.1074/jbc.M605558200. [DOI] [PubMed] [Google Scholar]
- 118.Duennwald ML, Jagadish S, Muchowski PJ, Lindquist S. Flanking sequences profoundly after polyglutamine toxicity in yeast. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(29):11045–11050. doi: 10.1073/pnas.0604547103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hughes RE, Lo RS, Davis C, et al. Altered transcription in yeast expressing expanded polyglutamine. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(23):13201–13206. doi: 10.1073/pnas.191498198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Duennwald ML, Lindquist S. Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes and Development. 2008;22(23):3308–3319. doi: 10.1101/gad.1673408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Meriin AB, Zhang X, Miliaras NB, et al. Aggregation of expanded polyglutamine domain in yeast leads to defects in endocytosis. Molecular and Cellular Biology. 2003;23(21):7554–7565. doi: 10.1128/MCB.23.21.7554-7565.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Solans A, Zambrano A, Rodríguez M, Barrientos A. Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III. Human Molecular Genetics. 2006;15(20):3063–3081. doi: 10.1093/hmg/ddl248. [DOI] [PubMed] [Google Scholar]
- 123.Sokolov S, Pozniakovsky A, Bocharova N, Knorre D, Severin F. Expression of an expanded polyglutamine domain in yeast causes death with apoptotic markers. Biochimica et Biophysica Acta. 2006;1757(5-6):660–666. doi: 10.1016/j.bbabio.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 124.Ocampo A, Zambrano A, Barrientos A. Suppression of polyglutamine-induced cytotoxicity in Saccharomyces cerevisiae by enhancement of mitochondrial biogenesis. The FASEB Journal. 2010;24(5):1431–1441. doi: 10.1096/fj.09-148601. [DOI] [PubMed] [Google Scholar]
- 125.Bocharova NA, Sokolov SS, Knorre DA, Skulachev VP, Severin FF. Unexpected link between anaphase promoting complex and the toxicity of expanded polyglutamines expressed in yeast. Cell Cycle. 2008;7(24):3943–3946. doi: 10.4161/cc.7.24.7398. [DOI] [PubMed] [Google Scholar]
- 126.Joyner PM, Matheke RM, Smith LM, Cichewicz RH. Probing the metabolic aberrations underlying mutant huntingtin toxicity in yeast and assessing their degree of preservation in humans and mice. Journal of Proteome Research. 2010;9(1):404–412. doi: 10.1021/pr900734g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Giorgini F, Guidetti P, Nguyen Q, Bennett SC, Muchowski PJ. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nature Genetics. 2005;37(5):526–531. doi: 10.1038/ng1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schwarcz R. The kynurenine pathway of tryptophan degradation as a drug target. Current Opinion in Pharmacology. 2004;4(1):12–17. doi: 10.1016/j.coph.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 129.Giorgini F, Möller T, Kwan W, et al. Histone deacetylase inhibition modulates kynurenine pathway activation in yeast, microglia, and mice expressing a mutant huntingtin fragment. Journal of Biological Chemistry. 2008;283(12):7390–7400. doi: 10.1074/jbc.M708192200. [DOI] [PubMed] [Google Scholar]
- 130.Ehrnhoefer DE, Duennwald M, Markovic P, et al. Green tea (-)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Human Molecular Genetics. 2006;15(18):2743–2751. doi: 10.1093/hmg/ddl210. [DOI] [PubMed] [Google Scholar]
- 131.Zhang X, Smith DL, Meriin AB, et al. A potent small molecule inhibits polyglutamine aggregation in Huntington’s disease neurons and suppresses neurodegeneration in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(3):892–897. doi: 10.1073/pnas.0408936102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bodner RA, Outeiro TF, Altmann S, et al. Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington’s and Parkinson’s diseases. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(11):4246–4251. doi: 10.1073/pnas.0511256103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sarkar S, Perlstein EO, Imarisio S, et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nature Chemical Biology. 2007;3(6):331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]