

Abstract
Pharmacological studies have suggested that oligodendroglial NMDA glutamate receptors (NMDARs) mediate white matter injury in a variety of CNS diseases, including multiple sclerosis (MS). We tested this hypothesis in experimental autoimmune encephalomyelitis (EAE), a model of human MS, by timed conditional disruption of oligodendroglial NR1, an essential subunit of functional NMDARs, using an inducible proteolipid protein (Plp) promoter-driven Cre-loxP recombination system. We found that selective ablation of oligodendroglial NR1 did not alter the clinical severity of EAE elicited in C57BL/6 mice by immunization with myelin oligodendrocyte glycoprotein peptide 35–55 (MOG-peptide), nor were there significant differences between the oligodendroglial NR1 KO and non-KO mice in numbers of axons lost in spinal cord dorsal funiculi or severity of spinal cord demyelination. Similarly, constitutive deletion of NR3A, a modulatory subunit of oligodendroglial NMDARs, did not alter the course of MOG-peptide EAE. Furthermore, conditional and constitutive ablation of NR1 in neonatal oligodendrocyte progenitor cells did not interrupt their normal maturation and differentiation. Collectively, our data suggest that oligodendroglial lineage NMDARs are neither required for timely postnatal development of the oligodendroglial lineage, nor significant participants in the pathophysiology of MOG-peptide EAE.
Introduction
NMDA receptors (NMDARs) are composed of different combinations of the subunits NR1, NR2A-D, and NR3A-B. NR1 is essential for functional NMDAR assembly, whereas NR3A subunits modulate susceptibility of oligodendroglial lineage NMDARs to Mg2+ block and to glycine or d-serine activation (Stys and Lipton, 2007; Piña-Crespo et al., 2010).
Oligodendroglial lineage NMDARs have been hypothesized to contribute to the pathophysiology of ischemic CNS white matter (WM) disease and multiple sclerosis (MS) (Káradóttir et al., 2005; Salter and Fern, 2005; Micu et al., 2006). In support of this hypothesis, NMDAR antagonist, memantine, has been reported to ameliorate experimental autoimmune encephalomyelitis (EAE) (Wallström et al., 1996) and models of periventricular leukomalacia (PVL) (Manning et al., 2008), but other studies demonstrated no protection by NMDAR inhibitors in EAE (Basso et al., 2008) or ischemia (McCarran and Goldberg, 2007; Tekkök et al., 2007). Because pharmacological NMDAR antagonists are likely to have off-target effects (e.g., on neurons), we decided to address this issue by genetic manipulations that specifically perturb oligodendroglial lineage NMDARs.
We found that conditional ablation of oligodendroglial NR1 did not alter the clinical course of EAE nor severities of spinal cord demyelination and axonal loss. Constitutive deletion of NR3A also had no demonstrable effect on MOG-peptide (myelin oligodendrocyte glycoprotein peptide 35–55) EAE. Furthermore, conditional or constitutive ablation of NR1 from oligodendroglial lineage cells in neonate mice did not alter telencephalic oligodendrocyte progenitor cell (OPC) development. Nonradioactive dual-color in situ hybridization (ISH) for NR1 and plp mRNAs showed that NR1 mRNA expression by oligodendrocytes is very low, consistent with prior OPC electrophysiological and microarray studies (Ziskin et al., 2007; Cahoy et al., 2008; De Biase et al., 2010). Though our study did not exclude a role for oligodendroglial NMDARs in CNS ischemia, we concluded that NMDARs do not have a major influence on oligodendroglial development or EAE.
Materials and Methods
Animals and tamoxifen treatment.
Plp-Cre-ERT2, Rosa26-STOP-EYFP reporter, and Grin1flox/flox (gene encoding NR1 subunit) mice were purchased from The Jackson Laboratory, maintained on a C57BL/6 background, and crossed to produce Plp-Cre-ERT2/Rosa26-STOP-EYFP/Grin1flox/flox triple transgenic (PRG) and Plp-Cre-ERT2/Rosa26-STOP-EYFP double transgenic (PR) controls. CNP-Cre mice were provided by Klaus-Armin Nave (Max Planck Institute of Experimental Medicine, Göttingen, Germany) via Judith Grinspan (CHOP, Philadelphia, PA). Cspg4(NG2)-Cre mice were from The Jackson Laboratory. For EAE studies, 2-month-old PRG or PR mice were injected intraperitoneally with tamoxifen (TM) or vehicle (V) for 5 d (Guo et al., 2010). For developmental studies, postnatal day 7 (P7) neonates were injected intraperitoneally with 2 doses of TM 6–8 h apart (Guo et al., 2009). Three-month-old NR3A−/− (MGI accession ID, 3609573), NR3A−/+ and NR3A+/+ littermates were tested for EAE susceptibility. Both male and female mice were used in this study. All animal procedures were performed according to University of California, Davis Institutional Animal Care and Use Committee guidelines.
MOG-peptide EAE and clinical deficits.
MOG-peptide EAE and clinical assessment were performed according to our previous study (Guo et al., 2011).
Tissue preparation, immunohistochemistry and confocal microscopy.
These were conducted as in our previous studies (Guo et al., 2009, 2010).
cRNA probe preparation.
Targeted sequences were generated by RT-PCR. The forward (F) and reverse (R) primers (Table 1) were linked to T7 promoter and SP6 promoter sequences, respectively. After purification, RT-PCR products were used as templates to transcribe into cRNA using in vitro labeling kit. DIG-UTP or FITC-UTP was used to generate DIG- or FITC-conjugated cRNA probes.
Table 1.
Primer sequence used in this study
| Primer name | Sequence (5′-3′) | Notes |
|---|---|---|
| NR1 | F, TGTGACGGCTCTGCTGATGGAA | For NR1 probe 1 |
| R, GGGCTTGACATACACGAAGGGTT | ||
| NR1 | F, ACGGGGCCTAATGACACATCC | For NR1 probe 2 |
| R, TGGCCTCAGCTGCACTCTCA | ||
| NR1 | F, TTAAGGTGAACAGCGAGGAGGAG | For NR1 probe 3 |
| R, TCTTCCTCCACACGTTCACGG | ||
| NR1 | F, TGGAAGCAAAATGTGTCCCTG | For NR1 probe 4 |
| R, GAGCAGCAGGACTCATCAGTGT | ||
| PLP | F, CTGGCTGAGGGCTTCTACAC | For PLP probe |
| R, GTCTTCAGGAGATGCTTGC | ||
| F1 | TCACTTTACAAGCGGTGAGCC | For detection of NR1 deletion at genomic DNA level |
| R1 | GAGTGAAGTGGTCGTTGGGAGT | |
| F2 | AACAAACAATGCTGAGGTGGTAGG | |
| R2 | TGTCTCGGACAAATCAGGGAGCAG | |
| F3 | CTTGGGTGGAGAGGCTATTC | |
| R3 | AGGTGAGATGACAGGAGATC | |
| NR1 intron 4 | F, TGCAGGGTAAGAACGGAATC | For qPCR detection of Grin1 intron 4 |
| R, TGTCCTAAGTGCGTGAGGTG | ||
| NR1 intron 12 | F, TTTGGCACACAGGAGCGGGT | For qPCR detection of Grin1 intron 12 |
| R, AAAGCCCAGCCCAACCACCA | ||
| NR1 | F, AGAGCCCGACCCTAAAAAGAA | For RT-qPCR detection of NR1 or NR2A in both optic nerve and spinal cord |
| R, AGAGCCCGACCCTAAAAAGAA | ||
| NR3A-1 | F, ACCGTAGTGGTACAAGGGGTT | |
| R, AGTCCAGCTCCATCATTTCGC | ||
| NR3A-2 | F, GCATAGTGCGCCACGAGTT | |
| R, GGTCAGGATTGAGACAGTGACAT |
F, Forward; R, reverse.
Dual fluorescence ISH.
Fourteen to 20 μm frozen sections were used. Probe concentrations were 100 ng/100 μl hybridization buffer. Hybridization incubation was at 65°C for 18–20 h. After hybridization, sections were treated with 10 μg/ml RNase A to eliminate nonspecific binding. AP-conjugated anti-DIG and HRP-conjugated anti-FITC were applied simultaneously. After 4°C overnight, sections were first developed for FITC signals by TSA Fluorescein Tyramide kit, and then for DIG signals by NBT/BCIP.
Genomic DNA preparation and PCR.
Spinal cord DNA was prepared using Qiagen kits (#51304), and treated with RNaseA to eliminate RNA contamination. PCR primers were designed to detect the deletion of Grin1 sequence between two loxP sites (Fig. 1A, Table 1).
Figure 1.

Specificity of Cre-mediated NR1 ablation in oligodendroglial lineage cells. A, Strategy of PCR design to detect Cre-mediated excision of floxed Grin1 (gene encoding NR1 subunit) sequence. Primer sets F1 and R1 serve as internal controls for PCR, generating a 770 bp product. Primer sets F2 and R2 generate a 1.3 kb product in WT Grin1 allele and 1.4 kb product in Grin1 floxed allele. Primer sets F3 and R3 target neomycin (neo) transgene in Grin1 floxed allele and Rosa26-STOP-EYFP transgene, and generate 280 bp product. A 1.3 kb PCR product is generated using primers F2 and R3 upon Cre-mediated Grin1 DNA deletion. B, PCR results using spinal cord genomic DNA, prepared 30 d after treatment (TM or V) showing deletion of floxed Grin1 sequence only in PRG+TM mice. Blank ctrl means PCR without DNA template. C, Breeding strategy for triple transgenic PRG mice showing EYFP as a surrogate for Grin1 disruption. D, E, Cellular specificity of Cre-mediated Grin1 deletion in spinal cords, revealed by EYFP expression (green). Note EYFP Sox10+ oligodendroglial lineage cells, including CC1+ mature oligodendrocytes and NG2+ OPCs (arrowheads), but not in GFAP+ astrocytes, Iba1+ microglia nor NeuN+ neurons (arrows). Blue, DAPI. F, EYFP reporter does not turn on in PRG+V mice. PRG+TM, PRG treated with TM; PRG+V, PRG treated with vehicle only. Scale bars, 50 μm.
Genomic DNA preparation from laser-captured white matter and qPCR.
QIAamp DNA Micro Kit (#56304) was used for genomic DNA isolation from laser-captured spinal white matter. RT-qPCR primers used for detection of Grin1 intron 4 and 12 are shown in Table 1.
Optic nerve RNA isolation and reverse transcriptase quantitative PCR.
RNeasy Micro-Kit (#74004) was used for optic nerve RNA preparation. RNA was digested with DNaseI. cDNA was prepared using QuantiTect Reverse Transcription Kit (#205311). NR1 and NR3A primer sets for reverse transcriptase quantitative (RT-q)PCR are shown in Table 1.
Axon counting and MBP intensity measurement.
Semiautomated axon counting was performed according to a published protocol (Marina et al., 2010). MBP intensity was measured using ImageJ software. Images of single confocal optical slices were used for both axonal counting and MBP intensity measurement. To quantify immunoreactivity, ventral white matter tracts of individual spinal cords were outlined manually and the total integrated density (IntDen) was used. IntDen provides a measure of intensity proportional to total volume and is expressed here on a logarithmic scale.
Protein extraction and Western blotting.
Protein extraction and immunoblotting were conducted as in our previous study (Soulika et al., 2009).
Antibody-peptide neutralization.
NR1 antibody (Santa Cruz Biotechnology, sc-1467) was preadsorbed with NR1 peptide or a control peptide (doublecortin peptide) before immunohistochemistry to evaluate specificity (Guo et al., 2010).
Statistics.
For data with normal distribution, unpaired two-tail Student's t test was used, and expressed as mean ± SD. At least 3 mice per group were included for quantification. For clinical scores and cumulative disease index, nonparametric Kruskal–Wallis test was used for multicomparison, and expressed as mean ± SEM. p < 0.05 is considered as significant difference.
Results
Deletion of NR1 in oligodendroglial lineage cells
Removal of sequence between exons 11 and 22 of Grin1 has been shown to disrupt functions of NMDARs (Tsien et al., 1996). To demonstrate Cre-mediated Grin1 deletion, we used genomic PCR using different primer sets (Fig. 1A). A 1.3 kb PCR product, which appears upon NR1 deletion, was present in PRG mice treated with TM, but not in the other groups (Fig. 1B, bottom row), suggesting that NR1 disruption required both the Plp-Cre-ERT2 transgene and TM treatment. DNA sequencing confirmed the 1.3 kb product as Grin1 sequence (data not shown). The ratio of Grin1 intron 12 (situated between two loxP sites) to intron 4 (outside the loxP sites) in PRG+TM mice, quantified by qPCR with DNA template isolated from laser-captured micro-dissected spinal cord white matter, was 65% of that in PRG+V mice. RT-qPCR using optic nerve RNA, in which NR1 expression is confined to the oligodendroglial lineage (Salter and Fern, 2005; Micu et al., 2006), showed significantly less NR1 mRNA than in controls (61% decrease, p = 0.008).
We used a Rosa-STOP-EYFP reporter to confirm the cellular specificity of NR1 ablation in PRG mice (Fig. 1C). Immunohistochemistry showed that in adult spinal cord WM, 94.8% of CC1+ mature oligodendrocytes and 89.2% of Sox10+ pan-oligodendroglial lineage cells were EYFP+, whereas no GFAP+ astrocytes, Iba1+ microglia, nor NeuN+ neurons were positive for EYFP (Fig. 1D,E). Furthermore, the percentage of EYFP+ cell among total DAPI+ nucleated cells in spinal cord WM was 43.6 ± 7.3%, approximately equivalent to the decrease of Grin1 DNA in the spinal WM. Consistent with the PCR data (Fig. 1B), no EYFP+ cells were observed in PGR mice treated with vehicle (Fig. 1F). Immunohistochemical results using NR1 antibody showed barely detectable NR1 signals in WM of PRG mice treated with vehicle (see Fig. 3E–G), and no detectable NR1 signals in WM of PRG mice treated with tamoxifen (data not shown), suggesting that tamoxifen treatment further diminished the extremely low level of NR1 expression in the WM. Collectively, these data demonstrated specific deletion of NR1 in oligodendroglial lineage cells, including virtually all mature oligodendrocytes.
Figure 3.
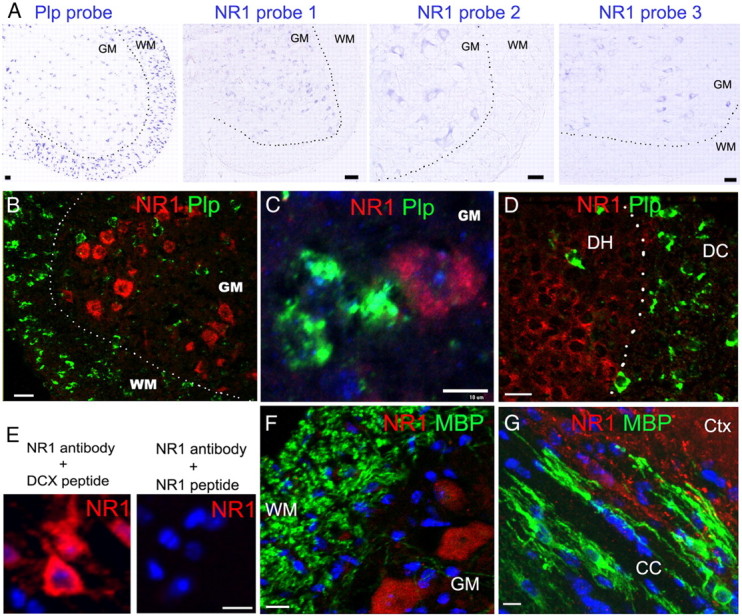
mRNA ISH and immunohistochemistry showing barely detectable NR1 transcript and protein in oligodendrocytes. A, Single mRNA ISH in P8∼P10 spinal cord, revealed by alkaline phosphatase-mediated color development, shows NR1 transcripts in GM, but barely detectable in WM (right three panels), despite presence of many Plp+ oligodendrocytes (left panel). B–D, Confocal images of dual fluorescent mRNA ISH demonstrating NR1 (pooled four NR1 probes) and Plp transcripts in P8∼P10 spinal WM and GM (B, C), and in dorsal column (DC) and dorsal horn (DH) (D). Note that Plp+ oligodendrocytes express barely detectable NR1 transcripts. E, NR1 peptide-antibody neutralization experiments demonstrating the specificity of NR1 immunostaining signals in cortical neurons. DCX, Doublecortin. F, G, NR1 and MBP double immunohistochemistry showing barely detectable NR1 signals in MBP+ myelin and oligodendrocytes from PRG mice treated with vehicle. Note the strongly positive NR1 signals in spinal GM (F) and forebrain cortex (Ctx) (G). CC, Corpus callosum. Scale bars, 10 μm, applied to all.
Disruption of NR1 in oligodendrocytes does not alter their susceptibility to EAE
Because the clinical severity of EAE was diminished if MOG-peptide immunization took place within 10 d after TM treatment (Fig. 2A), in subsequent experiments MOG-peptide immunization was delayed till 35∼40 d after TM to minimize this effect (Guo et al., 2011). During this tamoxifen clearance time, the NR1-ablated PRG mice did not show any neurological deficits (video available on request), suggesting that oligodendroglial NMDARs are dispensable for short-term myelin maintenance.
Figure 2.
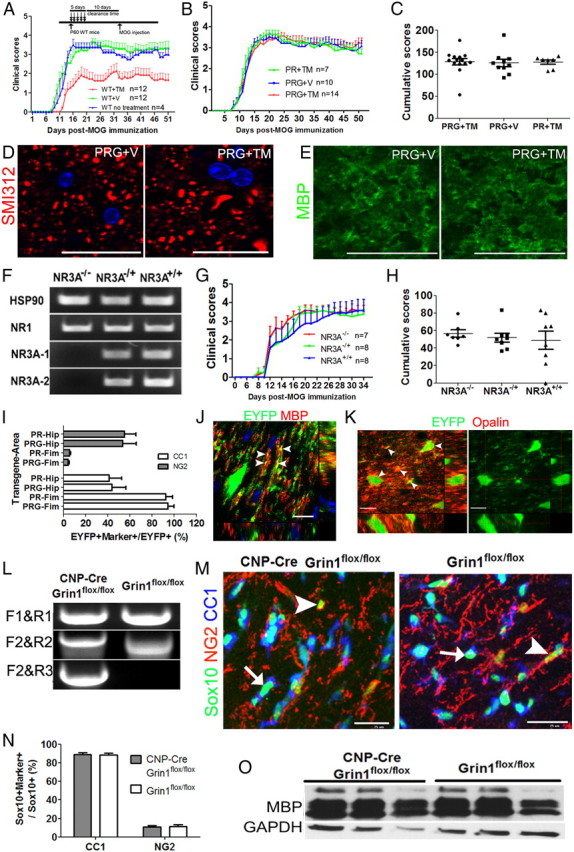
Disruption of NR1 in oligodendroglial lineage cells does not alter EAE disease course, nor interrupt neonatal OPC maturation and differentiation. A, Five days of TM treatment reduces severity of EAE if MOG-peptide immunization is only 10 d post-TM (insert). B, Mean clinical scores of PRG+TM mice did not differ from those in PRG+V and PR+TM mice. C, Cumulative disease index (summed clinical score) on day 51 post-MOG immunization is similar in PRG+TM, PRG+V and PR+TM mice. D, E, Representative confocal images showing SMI312+ axons in dorsal column and MBP+ myelin in PRG+TM and PRG+V mice on day 51 post-MOG immunization. F, RT-qPCR showing the absence of NR3A mRNA in NR3A−/− mice, and that NR1 mRNA levels were similar in these three groups. Note that two primer sets targeting NR3A confirmed the lack of mRNA transcripts in NR3A KO mice. G, H, NR3A KO mice show similar EAE course (G), and similar cumulative disease index (H) to their littermates. I, Percentage of EYFP+ cells that are CC1+ mature oligodendrocytes and NG2+ OPCs in forebrain fimbria (Fim) and hippocampus (Hip) on P15 of PR and PRG mice that received TM on P7. J, K, Orthogonal confocal images showing colocalization (arrowheads) of EYFP and MBP (J) and Opalin (K), a protein expressed in myelinating oligodendrocytes, in fimbria on P15 PRG mice that received TM on P7. Note MBP is mainly restricted to EYFP+ processes, whereas Opalin is in both cell body and processes (arrowheads). L, PCR data confirming the Grin1 deletion in CNP-Cre, Grin1flox/flox NR1 KO but not in Grin1flox/flox non-KO mice (same strategy as in Fig. 1A). M, N, Sox10+/NG2+ OPCs and Sox10+/CC1+ mature oligodendrocytes in the corpus callosum on P22 mice. Arrows and arrowheads point to Sox10+/CC1+/NG2− mature oligodendrocytes and Sox10+/CC1−/NG2+ OPCs, respectively. O, Western blotting showing MBP expression in P22 forebrain between NR1 KO and non-KO groups. Scale bars: D, E, J, K, 10 μm; M, 25 μm.
The clinical scores (Fig. 2B) and cumulative disease index (Fig. 2C) of PRG mice treated with TM did not differ from those of control groups, PRG+V and PR+TM (Fig. 2B,C) up to day 51 post-MOG immunization, nor were there significant differences between oligodendroglial NR1-ablated and non-ablated mice in numbers of axons in spinal cord dorsal funiculi (80,700 ± 13,800/mm2 in PRG+TM vs 81,700 ± 13,700 mm2 in PRG+V, p = 0.90, n = 6) or MBP expression in ventral white matter (7.65 ± 0.01 in PRG+TM vs 7.63 ± 0.09 in PRG+V, p = 0.96, n = 3) (Figs. 2D,E).
It has been shown that, unlike most adult neuronal NMDARs, which lack NR3A subunits (Wong et al., 2002), oligodendroglial NMDARs include NR3A subunits, which decrease the amplitude, Ca2+ permeability, and Mg2+ sensitivity of NMDA-induced currents (Stys and Lipton, 2007; Piña-Crespo et al., 2010). We therefore compared the clinical score and cumulative disease index of adult C57BL/6 NR3A−/−, NR3A−/+ and NR3A+/+ littermates (Fig. 2F) during EAE, and observed no differences between the homozygous or heterozygous NR3A knock-out and control mice (Fig. 2G,H). Also, SMI312+ pan-axonal number did not differ between NR3A KO and WT groups (p = 0.58). Collectively, these results suggested that NMDARs in oligodendrocytes do not play a major role in influencing severity of EAE.
Disruption of NR1 in neonate OPCs does not alter their normal development
OPCs receive synaptic inputs from unmyelinated axons (Ziskin et al., 2007) that may regulate their lineage progression and differentiation (Gallo et al., 2008; Etxeberria et al., 2010; Kukley et al., 2010). To determine whether OPC NMDARs influence their differentiation/maturation, we treated P7 PRG mice with TM, using TM-treated P7 PR mice as controls. Fimbria and hippocampus were chosen as representative WM and GM regions for analysis, respectively, because at P7 almost all recombined cells in these regions were NG2+ OPCs (Guo et al., 2009). By P15, we did not observe differences between NR1 KO PRG and control PR mice in percentages of EYFP+CC1+ mature oligodendrocytes and EYFP+NG2+ OPCs in either fimbria or hippocampus (Fig. 2I). The presence of many parallel EYFP+ processes and colocalization with MBP (Fig. 2J, arrowheads) and opalin (Fig. 2K, arrowheads), which marks myelinating oligodendrocytes (Golan et al., 2008), in the PRG mice, as in TM-treated PR mice, indicated successful OPC maturation, differentiation, and myelin formation. Furthermore, NR1 KO PRG mice developed indistinguishably compared with PR mice and did not show neurological deficits (video available on request).
To confirm the lack of role of OPC NMDARs in differentiation to oligodendroglia, and to exclude the effect of TM itself on normal development, we used a knock-in 2′,3′-cyclic nucleotide phosphodiesterase (CNP)-Cre driver (Lappe-Siefke et al., 2003) to constitutively delete oligodendroglial lineage NR1. CNP-Cre, Grin1flox/flox (NR1 KO) mice (Fig. 2L) developed normally (video available on request). Proportions of NG2+/Sox10+ OPCs and CC1+/Sox10+ mature oligodendrocytes (Fig. 2M) were indistinguishable between NR1 KO and WT mice (Fig. 2N). Furthermore, MBP expression, assessed by Western blotting, was similar in NR1 KO and WT mice (1.56 ± 0.67 in KO Vs 1.11 ± 0.35 in WT, normalized to GAPDH, p = 0.37, n = 3) (Fig. 2O). Similarly, OPC differentiation in Cspg4 (NG2)-Cre, Grin1flox/flox mice, in which NR1 was deleted in NG2+ OPCs, was comparable to that in Grin1flox/flox littermates (data not shown). Together, these findings indicate that neonatal OPC NMDARs do not have a major role in oligodendroglial maturation and differentiation.
NR1 expression in oligodendrocytes revealed by dual mRNA ISH and immunohistochemistry
Transcriptome studies using gene chips showed that acutely isolated oligodendrocytes express extremely low levels of NR1 mRNA (Cahoy et al., 2008; De Biase et al., 2010). To determine whether this was also the case for oligodendroglia in vivo, we designed four NR1 probes targeted to different regions of NR1 (Table 1). Each of these probes demonstrated NR1 signals in spinal GM (Fig. 3A) but not in WM, where many proteolipid protein-positive (Plp+) oligodendrocytes were present (Fig. 3A). To determine the identity of NR1 mRNA+ cells, we conducted dual fluorescence mRNA ISH with the Plp probe and with pooled four NR1 probes. Confocal microscopy showed that Plp+ WM oligodendrocytes had barely detectable NR1 transcripts (Fig. 3B), while in GM NR1+ signals did not colabel with Plp transcripts (Fig. 3C). Similarly, although many NR1+/Plp− cells were present in dorsal horn, no Plp+ cells in dorsal column WM had detectable NR1 transcripts (Fig. 3D). Thus, the expression of NR1 mRNA is extremely low in oligodendrocytes. Consistent with this result, immunohistochemistry showed that NR1 immunoreactivity was barely detectable in MBP+ oligodendroglial cell bodies and processes, compared with an abundant expression in neurons (Fig. 3E–G).
Discussion
NMDARs in oligodendrocytes are activated during CNS ischemia, leading to intracellular Ca2+-dependent injuries to myelin and oligodendroglia (Stys and Lipton, 2007; Alix and Fern, 2009), and to impaired axonal function (Bakiri et al., 2008), although some studies have failed to demonstrate protective roles of NMDAR antagonists against oligodendroglial death and axonal damage (Tekkök et al., 2007; Basso et al., 2008; Baltan, 2009). Since CNS extracellular glutamate homeostasis is impaired in MS and in EAE (Bolton and Paul, 2006), blocking oligodendroglial NMDARs might ameliorate disease severity and axonal damage in these autoimmune demyelinative disorders; the amelioration of EAE obtained with the NMDAR antagonist memantine (Wallström et al., 1996) seemed to support this hypothesis. Unexpectedly, however, we found that specific genetic disruption of NR1 in oligodendrocytes did not alter the clinical or pathological severity of EAE.
Inclusion of NR3A in the NMDARs of neonatal cortical neurons protects them against Ca2+-mediated excitotoxicity (Nakanishi et al., 2009). Adult oligodendrocytes, unlike most adult neurons, express NR3A-containing NMDARs (Stys and Lipton, 2007). We found, however, that there was no difference in EAE severity between wild-type mice and littermate controls in which one or both copies of NR3A gene was constitutively disrupted (Fig. 2G,H), thus further weakening support for a role of oligodendroglial NMDARs (composed of NR1, NR2 and NR3A subunits) in WM injury in demyelinating disease.
Previous studies showed that NMDAR antagonists alleviated the severity of PVL and EAE (Wallström et al., 1996; Manning et al., 2008), but these ameliorative effects might have been due to their effects on neuronal (Stys and Lipton, 2007) or microglial (Murugan et al., 2011) NMDARs. Furthermore, intravitreal injection of NMDA caused axonal loss without myelin alterations in adult optic nerves (Kuribayashi et al., 2010) suggesting that NMDARs in neuronal somas or axons, rather than in oligodendrocytes, were responsible for this axonopathy. Neither genetic ablation of oligodendroglial lineage NR1, a subunit essential for NMDAR function, nor constitutive ablation of NR3A, an NMDAR modulatory subunit in oligodendroglia, altered susceptibility to EAE. These results do not support a role for oligodendroglial NMDARs in the EAE model of multiple sclerosis.
NMDA-evoked currents have been detected in a subpopulation of OPCs (Ziskin et al., 2007); current amplitude was significantly downregulated after OPC differentiation (De Biase et al., 2010; Kukley et al., 2010). We used two color nonradioactive ISH to visualize NR1 transcripts in Plp+ oligodendrocytes (Fig. 3A–D). Our results, which were consistent with prior reports using 35S-labeled and 33P-labeled (South et al., 2003) NR1 probes, showed that NR1 transcript signals were barely detectable in WM tracts, where many Plp+ oligodendrocytes were present (Fig. 3A–D). Our NR1 immunostaining results were consistent with the barely detectable ISH transcript expression (Fig. 3E–G), supporting previous microarray observations (Cahoy et al., 2008; De Biase et al., 2010). It is possible that the extremely low-level expression of oligodendroglial NR1, previously estimated by qRT/PCR to be only 1–2% of the abundance in neurons (Salter and Fern, 2005), is still sufficient to generate physiologically relevant NMDA-evoked currents in the oligodendroglial lineage (Burzomato et al., 2010), and sufficient Ca2+ influx to be pathologically significant (Káradóttir et al., 2005). Thus, our data do not negate the concept that oligodendroglial lineage NMDARs contribute to CNS WM ischemic injury. However, our findings do strongly suggest that oligodendroglial NMDARs are not major players in EAE, and likely also in multiple sclerosis, and also argue against a major role for OPC NMDARs in regulating OPC maturation and differentiation.
Footnotes
This work was supported by Shriners Hospitals for Children (F.G., D.P.), California Institute for Regenerative Medicine (F.G., D.P.), NIH Grant RO1NS025044 (D.P.), and National Multiple Sclerosis Society Grant RG 4397-A-5 (D.P.).
References
- Alix JJ, Fern R. Glutamate receptor-mediated ischemic injury of premyelinated central axons. Ann Neurol. 2009;66:682–693. doi: 10.1002/ana.21767. [DOI] [PubMed] [Google Scholar]
- Bakiri Y, Hamilton NB, Káradóttir R, Attwell D. Testing NMDA receptor block as a therapeutic strategy for reducing ischaemic damage to CNS white matter. Glia. 2008;56:233–240. doi: 10.1002/glia.20608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S. Ischemic injury to white matter: an age-dependent process. Neuroscientist. 2009;15:126–133. doi: 10.1177/1073858408324788. [DOI] [PubMed] [Google Scholar]
- Basso AS, Frenkel D, Quintana FJ, Costa-Pinto FA, Petrovic-Stojkovic S, Puckett L, Monsonego A, Bar-Shir A, Engel Y, Gozin M, Weiner HL. Reversal of axonal loss and disability in a mouse model of progressive multiple sclerosis. J Clin Invest. 2008;118:1532–1543. doi: 10.1172/JCI33464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton C, Paul C. Glutamate receptors in neuroinflammatory demyelinating disease. Mediators Inflamm. 2006;2006:93684. doi: 10.1155/MI/2006/93684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzomato V, Frugier G, Pérez-Otaño I, Kittler JT, Attwell D. The receptor subunits generating NMDA receptor mediated currents in oligodendrocytes. J Physiol. 2010;588:3403–3414. doi: 10.1113/jphysiol.2010.195503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Biase LM, Nishiyama A, Bergles DE. Excitability and synaptic communication within the oligodendrocyte lineage. J Neurosci. 2010;30:3600–3611. doi: 10.1523/JNEUROSCI.6000-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etxeberria A, Mangin JM, Aguirre A, Gallo V. Adult-born SVZ progenitors receive transient synapses during remyelination in corpus callosum. Nat Neurosci. 2010;13:287–289. doi: 10.1038/nn.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Mangin JM, Kukley M, Dietrich D. Synapses on NG2-expressing progenitors in the brain: multiple functions? J Physiol. 2008;586:3767–3781. doi: 10.1113/jphysiol.2008.158436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan N, Adamsky K, Kartvelishvily E, Brockschnieder D, Möbius W, Spiegel I, Roth AD, Thomson CE, Rechavi G, Peles E. Identification of Tmem10/Opalin as an oligodendrocyte enriched gene using expression profiling combined with genetic cell ablation. Glia. 2008;56:1176–1186. doi: 10.1002/glia.20688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Ma J, McCauley E, Bannerman P, Pleasure D. Early postnatal proteolipid promoter-expressing progenitors produce multilineage cells in vivo. J Neurosci. 2009;29:7256–7270. doi: 10.1523/JNEUROSCI.5653-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Maeda Y, Ma J, Xu J, Horiuchi M, Miers L, Vaccarino F, Pleasure D. Pyramidal neurons are generated from oligodendroglial progenitor cells in adult piriform cortex. J Neurosci. 2010;30:12036–12049. doi: 10.1523/JNEUROSCI.1360-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Maeda Y, Ma J, Delgado M, Sohn J, Miers L, Ko EM, Bannerman P, Xu J, Wang Y, Zhou C, Takebayashi H, Pleasure D. Macroglial plasticity and the origins of reactive astroglia in experimental autoimmune encephalomyelitis. J Neurosci. 2011;31:11914–11928. doi: 10.1523/JNEUROSCI.1759-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Káradóttir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature. 2005;438:1162–1166. doi: 10.1038/nature04302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukley M, Nishiyama A, Dietrich D. The fate of synaptic input to NG2 glial cells: neurons specifically downregulate transmitter release onto differentiating oligodendroglial cells. J Neurosci. 2010;30:8320–8331. doi: 10.1523/JNEUROSCI.0854-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuribayashi J, Kitaoka Y, Munemasa Y, Ueno S. Kinesin-1 and degenerative changes in optic nerve axons in NMDA-induced neurotoxicity. Brain Res. 2010;1362:133–140. doi: 10.1016/j.brainres.2010.09.053. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Manning SM, Talos DM, Zhou C, Selip DB, Park HK, Park CJ, Volpe JJ, Jensen FE. NMDA receptor blockade with memantine attenuates white matter injury in a rat model of periventricular leukomalacia. J Neurosci. 2008;28:6670–6678. doi: 10.1523/JNEUROSCI.1702-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina N, Bull ND, Martin KR. A semiautomated targeted sampling method to assess optic nerve axonal loss in a rat model of glaucoma. Nat Protoc. 2010;5:1642–1651. doi: 10.1038/nprot.2010.128. [DOI] [PubMed] [Google Scholar]
- McCarran WJ, Goldberg MP. White matter axon vulnerability to AMPA/kainate receptor-mediated ischemic injury is developmentally regulated. J Neurosci. 2007;27:4220–4229. doi: 10.1523/JNEUROSCI.5542-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, Yin X, Trapp BD, McRory JE, Rehak R, Zamponi GW, Wang W, Stys PK. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature. 2006;439:988–992. doi: 10.1038/nature04474. [DOI] [PubMed] [Google Scholar]
- Murugan M, Sivakumar V, Lu J, Ling EA, Kaur C. Expression of N-methyl d-aspartate receptor subunits in amoeboid microglia mediates production of nitric oxide via NF-kappaB signaling pathway and oligodendrocyte cell death in hypoxic postnatal rats. Glia. 2011;59:521–539. doi: 10.1002/glia.21121. [DOI] [PubMed] [Google Scholar]
- Nakanishi N, Tu S, Shin Y, Cui J, Kurokawa T, Zhang D, Chen HS, Tong G, Lipton SA. Neuroprotection by the NR3A subunit of the NMDA receptor. J Neurosci. 2009;29:5260–5265. doi: 10.1523/JNEUROSCI.1067-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piña-Crespo JC, Talantova M, Micu I, States B, Chen HS, Tu S, Nakanishi N, Tong G, Zhang D, Heinemann SF, Zamponi GW, Stys PK, Lipton SA. Excitatory glycine responses of CNS myelin mediated by NR1/NR3 “NMDA” receptor subunits. J Neurosci. 2010;30:11501–11505. doi: 10.1523/JNEUROSCI.1593-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MG, Fern R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature. 2005;438:1167–1171. doi: 10.1038/nature04301. [DOI] [PubMed] [Google Scholar]
- Soulika AM, Lee E, McCauley E, Miers L, Bannerman P, Pleasure D. Initiation and progression of axonopathy in experimental autoimmune encephalomyelitis. J Neurosci. 2009;29:14965–14979. doi: 10.1523/JNEUROSCI.3794-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- South SM, Kohno T, Kaspar BK, Hegarty D, Vissel B, Drake CT, Ohata M, Jenab S, Sailer AW, Malkmus S, Masuyama T, Horner P, Bogulavsky J, Gage FH, Yaksh TL, Woolf CJ, Heinemann SF, Inturrisi CE. A conditional deletion of the NR1 subunit of the NMDA receptor in adult spinal cord dorsal horn reduces NMDA currents and injury-induced pain. J Neurosci. 2003;23:5031–5040. doi: 10.1523/JNEUROSCI.23-12-05031.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Lipton SA. White matter NMDA receptors: an unexpected new therapeutic target? Trends Pharmacol Sci. 2007;28:561–566. doi: 10.1016/j.tips.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Tekkök SB, Ye Z, Ransom BR. Excitotoxic mechanisms of ischemic injury in myelinated white matter. J Cereb Blood Flow Metab. 2007;27:1540–1552. doi: 10.1038/sj.jcbfm.9600455. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Wallström E, Diener P, Ljungdahl A, Khademi M, Nilsson CG, Olsson T. Memantine abrogates neurological deficits, but not CNS inflammation, in Lewis rat experimental autoimmune encephalomyelitis. J Neurol Sci. 1996;137:89–96. doi: 10.1016/0022-510x(95)00339-4. [DOI] [PubMed] [Google Scholar]
- Wong HK, Liu XB, Matos MF, Chan SF, Pérez-Otaño I, Boysen M, Cui J, Nakanishi N, Trimmer JS, Jones EG, Lipton SA, Sucher NJ. Temporal and regional expression of NMDA receptor subunit NR3A in the mammalian brain. J Comp Neurol. 2002;450:303–317. doi: 10.1002/cne.10314. [DOI] [PubMed] [Google Scholar]
- Ziskin JL, Nishiyama A, Rubio M, Fukaya M, Bergles DE. Vesicular release of glutamate from unmyelinated axons in white matter. Nat Neurosci. 2007;10:321–330. doi: 10.1038/nn1854. [DOI] [PMC free article] [PubMed] [Google Scholar]