

Abstract
Heat shock protein 27 (HSP27) (or HSPB1) exerts cytoprotection against many cellular insults, including cerebral ischemia. We previously identified apoptosis signal-regulating kinase 1 (ASK1) as a critical downstream target of HSP27 conferring the neuroprotective effects of HSP27 against neuronal ischemia. However, the function of HSP27 is highly influenced by posttranslational modification, with differential cellular effects based on phosphorylation at specific serine residues. The role of phosphorylation in neuronal ischemic neuroprotection is currently unknown. We have created transgenic mice and viral vectors containing HSP27 mutated at three critical serine residues (Ser15, Ser78, and Ser82) to either alanine (HSP27-A, nonphosphorylatable) or aspartate (HSP27-D, phosphomimetic) residues. Under both in vitro and in vivo neuronal ischemic settings, overexpression of wild-type HSP27 (HSP27) and HSP27-D, but not HSP27-A, was neuroprotective and inhibited downstream ASK1 signaling pathways. Consistently, overexpressed HSP27 was phosphorylated by endogenous mechanisms when neurons were under ischemic stress, and single-point mutations identified Ser15 and Ser82 as critical for neuroprotection. Using a panel of inhibitors and gene knockdown approaches, we identified the upstream kinase protein kinase D (PKD) as the primary kinase targeting HSP27 directly for phosphorylation. PKD and HSP27 coimmunoprecipitated, and inhibition or knockdown of PKD abrogated the neuroprotective effects of HSP27 as well as the interaction with and inhibition of ASK1 signaling. Together, these data demonstrate that HSP27 requires PKD-mediated phosphorylation for its suppression of ASK1 cell death signaling and neuroprotection against ischemic injury.
Introduction
Overexpression of heat shock protein 27 (HSP27) provides robust cellular protection against a variety of neurological insults and diseases, including cerebral ischemia (An et al., 2008; Stetler et al., 2008; Badin et al., 2009; van der Weerd et al., 2010). While the neuroprotective effect has been well documented and critical downstream targets of HSP27 involved in mediating cell survival have been identified, the overall upstream regulation of the actions of HSP27 is not well understood in neural contexts. Specific identification of the mechanism(s) controlling HSP27 function is particularly relevant under different disease and cellular states.
Characterized in non-neuronal cell types, regulation of HSP27 function depends in large part upon its phosphorylation—and hence, oligomeric—state (Stetler et al., 2009, 2010). Typically, HSP27 exists in high-molecular-weight oligomers, but in response to cellular stimuli, such as oxidative stress, HSP27 is phosphorylated at several distinct serine residues (Ser15 and Ser86 in rodents; and Ser15, Ser78, and Ser82 in humans). In particular, phosphorylation at Ser82, localized in the region of dimerization, induces a strong negative charge that presumably leads to dissociation to lower-molecular-weight oligomers (Thériault et al., 2004). In turn, the cellular function of HSP27 shifts correspondingly to its phosphorylation state. The chaperone function of HSP27 is associated with the nonphosphorylated, high-molecular-weight oligomeric structure (Knauf et al., 1994). However, HSP27 is capable of direct suppression of cell death-signaling cascades that are similar to cascades activated following cerebral ischemia (Bruey et al., 2000; Concannon et al., 2001; Benn et al., 2002; Rane et al., 2003; Lee et al., 2005; Voss et al., 2007). Several of these reports indicate that cell death suppression necessitates the phosphorylation of HSP27 (Benn et al., 2002). Given that both protein denaturation and cell death signaling can be induced by cerebral ischemia, it remains unknown how the neuroprotective functions of HSP27 against cerebral ischemia are related to its phosphorylation state. Furthermore, although several families of upstream kinases have been implicated in targeting HSP27 for phosphorylation (Kostenko and Moens, 2009), the potential endogenous activators of HSP27 phosphorylation are currently unknown in neuronal and ischemic contexts. Determination of the induction of HSP27 as a neuroprotective molecule against cerebral ischemia may help to determine the mechanistic targets for ischemic intervention.
Having previously determined that exogenous overexpression of HSP27 confers significant neuroprotection via suppression of cell death signaling, we wanted to determine the mechanism and upstream regulation of HSP27 in the context of neuronal ischemia. We hypothesized that endogenously mediated phosphorylation of HSP27 is necessary for neuroprotection against ischemic injury and that inhibition of upstream kinase(s) that target HSP27 for phosphorylation would abrogate neuroprotection.
Materials and Methods
Creation of mutant HSP27 transgenic mice.
The methods for the creation of the HSP27 transgenic mouse were previously described (Stetler et al., 2008). The HSP27-A and HSP27-D transgenic mice were created using identical procedures, with the exception that the sequence encoding Ser15, Ser78, and Ser82 were mutated to encode either alanines (for HSP27-A) or aspartates (for HSP27-D). Microinjection and ES screening procedures were performed at the University of Pittsburgh Transgenic Core. Independent transgenic lines were established by breeding wild-type F1 hybrid mice to founder mice obtained following microinjection. All lines were backbred for at least seven generations to the C57/B6 background to minimize any influence of genetic heterogeneity in sensitivity to the stroke model. Cerebral vasculature was examined by injection of India ink/gelatin to ensure that no statistical differences between animal groups existed (Table 1). To reduce possible sex-related differences (Liu et al., 2009), only male adult (25–30 g) mice were used.
Table 1.
Evaluation of surface cerebral blood vessels in MCA territory and PcomA plasticity in mice
| MCA distance from the midlinea at the specified coronal plane ofb |
Number of hemispheres with PcomA scores ofc |
||||||
|---|---|---|---|---|---|---|---|
| 2 mm | 4 mm | 6 mm | 0 | 1 | 2 | 3 | |
| Wild type | 2.35 (0.04) | 2.10 (0.05) | 2.46 (0.08) | 1 | 7 | 2 | 0 |
| Tg-HSP27 | 2.36 (0.07) | 2.28 (0.03) | 2.40 (0.08) | 1 | 6 | 3 | 0 |
| Tg-HSP27-D | 2.31 (0.05) | 2.15 (0.06) | 2.44 (0.10) | 0 | 6 | 3 | 1 |
| Tg-HSP27-A | 2.33 (0.07) | 2.14 (0.08) | 2.46 (0.06) | 2 | 6 | 2 | 0 |
aValues shown are mean (SE) in millimeters (n = 5/group).
bA coronal plane is specified based on its distance from the frontal pole in millimeters.
cPosterior communicating artery (PcomA) plasticity scoring criteria: 0, no anastomoses between posterior cerebral artery (PCA) and superior cerebellar artery (SCA); 1, anastomoses between PCA and SCA in capillary phase; 2, small truncal anastomoses between PCA and SCA; 3, truncal anastomoses between PCA and SCA.
Murine model of transient focal ischemia and measurement of blood flow and infarct.
All animal studies were performed under protocols approved by the University of Pittsburgh Institutional Animal Care and Use Committee, and in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Adult male transgenic mice (2–4 months of age) or wild-type littermates were anesthetized with 1.5% isoflurane in 30% O2/68.5% N2O mixture under spontaneous breathing. Transient focal cerebral ischemia (tFCI) was induced as described previously (Stetler et al., 2008) by intraluminal occlusion of the left middle cerebral artery (MCA). Rectal temperature was monitored and controlled at 36.8–37.2°C by a temperature-regulated heating pad throughout the course of the experiment. Arterial blood pressure was monitored through a tail cuff (XBP1000 Systems; Kent Scientific Corporation). Arterial blood gas was analyzed 15 min after induction of ischemia and 15 min after reperfusion. tFCI was induced for 60 min and subsequent reperfusion was permitted for the indicated durations. Following recovery from anesthesia, the animals were maintained in an air-conditioned room at 20°C. Regional cerebral blood flow (rCBF) was evaluated before, during, and after MCA occlusion by laser-Doppler flowmetry (Stetler et al., 2008) to ensure successful induction of ischemia and reperfusion. Real-time two-dimensional microcirculation imaging during tFCI was obtained in a separate cohort of animals by using the laser speckle technique (Ayata et al., 2004). Briefly, a CCD camera (PeriCam PSI System; Perimed) was positioned above the head, and a laser diode (785 nm) illuminated the intact skull surface to allow penetration of the laser in a diffuse manner through the brain. Speckle contrast—defined as the ratio of the SD of pixel intensity to the mean pixel intensity—was used to measure CBF as it is derived from the speckle visibility relative to the velocity of the light-scattering particles (blood). This was then converted to correlation time values, which are inversely and linearly proportional to the mean blood velocity. Laser speckle perfusion images were obtained every 7.5 s, starting 10 min before tFCI and continuing throughout the ischemic period and 15 min into the reperfusion. CBF was analyzed two-dimensionally over time by quantifying the area of cortex (in square millimeters) with either severe (0–20% residual CBF, representing the core of the infarct and illustrated by the black bars in Fig. 2H) or moderate CBF reduction (20–35% residual CBF, representing the penumbral region and illustrated by the gray bars in Fig. 2H) using a thresholding paradigm.
Figure 2.
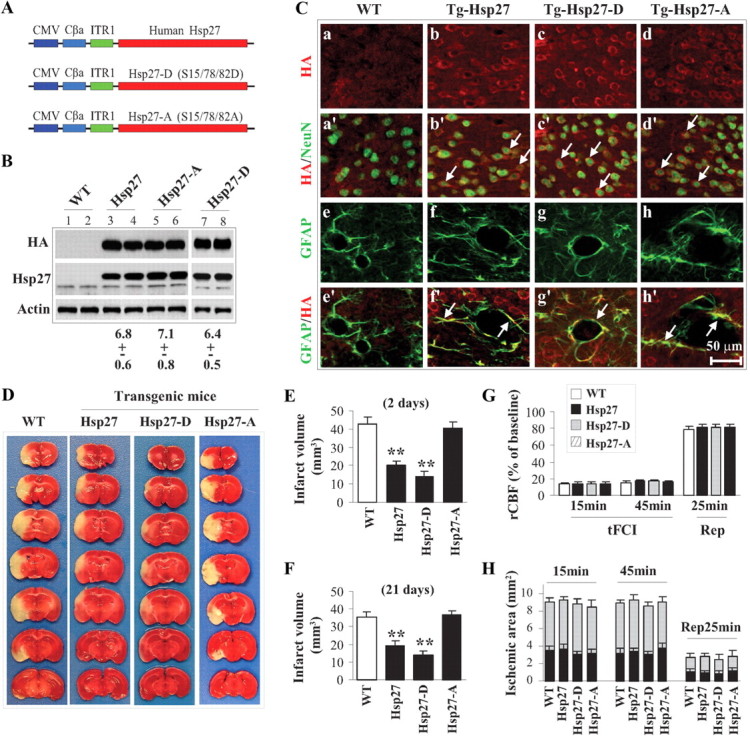
The nonphosphorylatable HSP27 mutant is incapable of conferring neuroprotection against focal ischemia. A, Diagram of transgenic constructs used for the creation of HSP27, HSP27-D, and HSP27-A transgenic mice. B, Western blotting detection of transgenic HSP27 gene products in mouse lines. The lower band visible with the HSP27 antibody (middle panel) represents the endogenously expressed rodent HSP25. The mean ± SEM of fold increases of transgenic HSP27 gene products over endogenous HSP25 in brain extracts are presented below the blots (n = 4/group). C, Representative double-label immunofluorescence images showing neuronal (NeuN) and glial (GFAP) localization of the HSP27 transgene product (HA) in brain. D–F, Transgenic expression of HSP27 or the phosphomimetic HSP27-D reduced infarct volume at both 2 d (D, E; n = 8–9/group) and 21 d (F; n = 7/group) after 60 min of tFCI as measured by 2,3,5-triphenyltetrazolium chloride (TTC) staining and MAP-2 immunohistochemistry, respectively. G, rCBF measured using laser Doppler flowmetry demonstrates no differences between wild-type and transgenic mice during or after tFCI (n = 8–9/group). H, Blood flow was measured by using two-dimensional laser speckle imager during and after tFCI (n = 6/group). The gray bars represent areas with 65–80% blood flow reduction, and the black bars represent areas with >80% blood flow reduction, calculated against preischemic baseline levels. All data are presented as mean ± SEM. **p < 0.01 compared with wild-type mice.
For quantification of infarct volume, animals were killed after 2 or 21 d of reperfusion following tFCI, brains were removed and subjected to 2,3,5-triphenyltetrazolium chloride staining and MAP-2 immunohistochemical straining, respectively. Infarct volume was determined using MCID imaging with correction for edema as previously described (Stetler et al., 2008). Surgeons were blinded to the subjects' genotypes when performing all surgical procedures.
Neurobehavioral testing.
Both sensorimotor deficits and long-term cognitive deficits were assessed during the light phase of the circadian cycle, beginning ∼4 h after the onset of the light phase, by a researcher who was blinded to genotypes of the mice. Sham-operated animals were used as control groups. Because no differences were found between genotypes of sham-operated animals (Stetler et al., 2008) (data not shown), roughly equivalent numbers from each genotype were combined to form the sham-operated group to maximize animal usage. Four neurofunctional tests were performed: the rotarod test, the corner test, the cylinder test, and the Morris water maze (Schallert et al., 2000; Stetler et al., 2008). Corner tests were analyzed based on the direction of each turn recorded over 10 trials for each test. Ischemic mice preferentially turn to the nonimpaired left side. Rotarod training began 2 d before surgery, with five trials per day. On the day of surgery, the mice were subjected to five trials, the mean of which was used as the presurgery baseline value for that particular animal. Following surgical procedures, the animals underwent five trials per day for 7 d. The data are expressed as the percentage of the mean duration spent on the rotarod over the number of trials per day compared with the presurgery baseline value. Cylinder tests determined the presence of paw preferences that can be observed in ischemic-injured mice (Schallert et al., 2000). Briefly, mice were placed in a clear Plexiglas cylinder (9 cm in diameter, 15 cm in height) for 5 min, and the number of times the mouse placed its right, left, or both forepaws on the sides of the cylinder was recorded. The test was repeated for a total of four trials per day of testing. Mice were prescreened before surgery to ensure equal use of forepaws, and subjects that exhibited fewer than five rears over the course of the trial were excluded. The data presented represents the percentage of both-paw use, as it is this outcome that has been most correlated with ischemic injury (Schallert et al., 2000). Data are expressed as the mean of all trials for each test day. A modified Morris water maze was performed to assess spatial reference memory as described previously (Stetler et al., 2008). Briefly, a hidden platform was placed underwater in a large swimming pool, and the latency for the mice to find the platform as an escape from a forced swimming task was recorded. Mice were acclimated to the pool without the platform on the 14th day after tFCI, and then tested daily on days 15–21 following tFCI. Each day of testing included four trials, and data are expressed as the time in seconds to successfully reach the platform (latency), averaged per day. During the final two trials, the platform was removed to allow the animal to swim freely for 30 s. This allowed for the recording of the time spent in the goal quadrant where the platform had previously been located and the overall swim speed.
Primary neuronal cultures and in vitro model of ischemia.
Cortical neurons with different genetic phenotypes, including Tg-HSP27, Tg-HSP27-A, Tg-HSP27-D, and wild type, were derived from embryos of mice and maintained 10–12 d in vitro before experiments, as described previously (Stetler et al., 2008). To model ischemia-like conditions in vitro, primary cultures were exposed to transient oxygen and glucose deprivation (OGD) for 45 or 60 min as described previously (Stetler et al., 2008; Zhao et al., 2009). Control glucose-containing cultures were incubated for the same periods of time in humidified 95% air and 5% CO2. Fluorescence of Alamar blue, an indicator that changes from blue to red and fluoresces when reduced by cellular metabolic activity, was used to measure the viability of the cultured neurons at 24–48 h after OGD. One-half of the culture medium was replaced with MEM-Pak containing 10% (v/v) Alamar blue (AccuMed International), and cultures were incubated for 1.5 h at 37°C in humidified 95% air and 5% CO2. Fluorescence was determined in a Millipore CytoFluor 2300 automated plate-reading fluorometer (Millipore), with excitation at 530 nm and emission at 590 nm. OGD-induced cell death was quantified by measuring lactate dehydrogenase (LDH) release from damaged cells into the culture medium as described previously (Stetler et al., 2008). In selective experiments, cell death was also evaluated after OGD using Hoechst 33258 nuclear staining or Klenow-mediated DNA nick-end labeling. The percentages of cells showing chromatin condensation or DNA damage were quantified by counting at least 3000 cells under each experimental condition (three randomly selected fields per well, four to six wells per condition per experiment, and three independent experiments). Kinase inhibitors used in culture experiments included N-[2-[[3-(4-bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide dihydrate dihydrochloride (H89) and 5,6,7,13-tetrahydro-13-methyl-5-oxo-12H-indolo[2,3-a]pyrrolo[3,4-c]carbazole-12-propanenitrile (Go6976) (Tocris Bioscience), 4-[5-(4-fluorophenyl)-2-[4-(methylsulfonyl)phenyl]-1H-imidazol-4-yl]pyridine (SB203580) (Cell Signaling), and 5-(5-ethyl-2-hydroxy-4-methoxyphenyl)-4-(4-methoxyphenyl)isoxazole (KRIBB3) (Sigma-Aldrich).
Gene transfection in primary neurons by viral vectors and in HEK293 cells using Lipofectamine.
Neuronal cultures were infected with the adenoassociated virus (AAV)-HSP27, AAV-HSP27-A, AAV-HSP27-D, or the control vector (AAV-GFP or empty AAV) for 6 h as previously described (Stetler et al., 2008) and then incubated in vector-free normal media for 72 h. The overexpression of HSP27 in neurons was confirmed by immunocytochemistry and Western blot, respectively, using the anti-hemagglutinin (HA) antibody. For gene transfection using lentivirus vectors, the neuronal cultures were infected for 3 d with the lentiviral vectors encoding shRNA constructs (Lenti-PKDt, Lenti-PKDsc) or encoding the human protein kinase D (PKD) cDNA (Lenti-PKDh), or the control vector (Lenti-GFP). Knockdown of PKD1 and PKD3 in neurons by lentivirus vector was assessed using Western blot.
cDNA isolation, site-directed mutagenesis, and generation of recombinant proteins.
The human full-length HSP27 and PKD1 cDNAs were obtained using PCR from a human kidney cDNA library. Point mutations for HSP27 at Ser78, Ser82, and Ser15 or for PKD1 at Ser738, Ser742, and Lys612 were engineered by site-directed mutagenesis using the Gene Editor system (Promega). The sequences of all cDNAs were verified at the University of Pittsburgh Gene Service Facility by sequencing reactions on both strands. Recombinant proteins of HSP27 and PKD1 were generated by fusing the cDNA into the glutathione S-transferase (GST) gene in PGEX-2T vector (GE Healthcare). The GST fusion protein was expressed in Escherichia coli BL21 cells, absorbed to a glutathione-Sepharose 4B column, and then cleaved by thrombin to remove GST.
Construction of viral vectors.
AAV vectors carrying either the human full-length (AAV-HSP27) or mutated (AAV-HSP27-D, AAV-HSP27-A) HSP27 cDNA were constructed using the expressing vector plasmid described previously (Stetler et al., 2008). Large-scale production of the AAV vector was performed using the adenovirus-free triple-plasmid cotransfection method (Xiao et al., 1998). A total of 50 μg of plasmid mixture was cotransfected into human HEK293 cells with the assistance of 0.25 m CaCl2, and the cells were grown in DMEM containing 10% fetal bovine serum for 48 h and then harvested. AAV vectors were purified using a fast protein liquid chromatography system in conjunction with HiTrap Heparin columns (GE Healthcare).
To construct lentiviral vectors overexpressing human PKD1 (Lenti-PKD1), its dominant-negative form (Lenti-PKDdn, K612W), or its constitutively active form (PKDac, S738/742E), the HA-tagged cDNA was inserted into the lentiviral transfer vector FSW under the control of neuron-specific Synapsin I promoter. To construct lentiviral vectors expressing short hairpin interfering RNA (shRNA) against murine PKD1/3 (targets both isoforms), the gene-specific targeting sequence (PKDt, 5′-GGAAGGGTGGATGGTCCAC-3′ and 5′-GGGTGGATGGTCCACTACA-3′) or its counterpart scramble sequence was inserted into the transfer vector FSW under the control of U6 promoter. The constructed transfer vectors were transformed into Stbl3 Escherichia coli, and then isolated using the EndoFree Plasmid Maxi Kit (QIAGEN). For large-scale production of the virus, a slightly modified protocol (Ramezani and Hawley, 2002) was used. In brief, a plasmid mixture containing 435 μg of pCMVΔR8.9 (packaging construct), 237 μg of pVSVG (envelope plasmid), and 675 μg of FSW (transfer vector) was suspended in 34.2 ml of CaCl2 (250 mm) and then added volume to volume into 2× BES buffer, pH 6.95. The DNA-CaCl2 precipitate was added into human kidney 293 FT cells (on 15 cm plates at the density of 1.1 × 107/plate) drop by drop (1.125 ml each plate) and allowed to incubate for 12 h before switching to fresh culture medium. The supernatant was collected 72 h after transfection, filtered through the 0.45 μm filter flask, and centrifuged at 21,000 rpm for 2 h using the SW28 rotor (Beckman Coulter). Viruses were further purified by sucrose gradient ultracentrifuge. The pellet was suspended in 3 ml of PBS, loaded on the top of 2 ml of 20% sucrose solution, and centrifuged at 22,000 rpm for 2 h using the SW50.1 rotor (Beckman Coulter). The resulting pellet was resuspended in 200 μl of DMEM, aliquoted, and stored at −70°C. The titer of vector stock was determined using ELISA. The average titer typically is ∼5–10 × 1010 particle units per milliliter.
Sucrose gradient.
A sucrose gradient was prepared in buffer containing 500 mm Tris-HCl, pH 8.0, 100 mm NaCl, 5 mm MgCl2, and 2 mm DTT for a final concentration gradient of 5, 10, 15, 20, 25, 30, 35, 40, 45, and 50% in 10 ml total volume in a Beckman ultracentrifuge tube. Using crude cellular extracts derived from primary neuronal cultures overexpressing HSP27, HSP27-A, or HSP27-D, 500 μg of cell extract was layered on the top of the sucrose gradient and spun in a prechilled rotor at 36,000 rpm for 21 h at 4°C. Following centrifugation, the fractions were transferred to chilled microcentrifuge tubes and assayed by 12% SDS-PAGE gel and subsequent immunoblotting for the HA tag.
Immunohistochemistry and Western blot.
Animals were killed by carbon dioxide exposure, and the brains were extracted and prepared for frozen coronal sectioning (15 μm thick). Immunocytochemistry was performed as described previously (Stetler et al., 2008), using the appropriate secondary fluorescent antibodies. For cell-specific identification, sections were subjected to a second round of immunohistochemistry using mouse anti-NeuN antibody (1:500; Millipore Bioscience Research Reagents) followed by a fluorescent-conjugated secondary antibody (1:3000). Alternate sections from each experimental condition were incubated in the absence of the primary antibody to assess nonspecific staining. Immunostaining for phosphorylated HSP27 was performed with the rabbit anti-p-HSP27 (Ser15) (1:80; Abcam). Images were acquired using either an inverted Nikon Diaphot-300 fluorescence microscope equipped with a SPOT RT slider camera and Meta Series Software 5.0 (Molecular Devices), or the Olympus Fluoview FV1000 confocal microscope with the FV10-ASW 2.0 software (Olympus America).
Western blot was performed using the standard method and enhanced chemiluminescence detection reagents (GE Healthcare). The following antibodies were used: rabbit monoclonal antibodies against JNK3 (clone 55A8; 1:1000), active caspase-9 (1:1000), p-JNK (Thr183/Tyr185) (clone 81E11; 1:1000), and p-c-Jun (Ser63) (clone 54B3; 1:1000), purchased from Cell Signaling; rabbit polyclonal antibodies against p-MKK4 (Ser257/Thr261) (1:1000), p-MKK7 (Ser271/Thr275) (1:1000), p-ASK1 (Thr845) (1:1000), p-HSP27 (Ser82) (1:500), and p-PKD (Ser916) (1:1000) from Cell Signaling; rabbit polyclonal antibodies against HSP27 (1:1000), Bax (1:1000), β-Actin (1:2000), cytochrome c (1:1000), HA (1:1000), and Flag (1:1000) from Santa Cruz Biotechnology; mouse monoclonal anti-cytochrome c oxidase IV antibody (1:1000) from Invitrogen; rabbit polyclonal anti-p53 upregulated modulator of apoptosis (PUMA) antibody (a gift from Dr. Jian Yu, Pittsburgh Cancer Institute, Pittsburgh, PA); and a rabbit polyclonal anti-PKD3 (a gift from Dr. Qiming Wang, University of Pittsburgh, Pittsburgh, PA).
Protein kinase assays.
Cell lysates were prepared under non-denaturing conditions as described previously (Stetler et al., 2008), and 150 μg of protein was used for each kinase assay. To assay for JNK activity (JNK1 or JNK3), a capture JNK assay was performed using a nonradioactive kinase assay kit according to the manufacturer's instructions (Cell Signaling). Briefly, the cell lysates were first subjected to JNK1 capture using the specific anti-JNK1 antibody (clone F-3; Santa Cruz), and then the immunoprecipitates were incubated with recombinant GST-c-Jun (1–79) in the presence of ATP, and subsequently immunoblotted using the anti-phospho-c-Jun (Ser63) antibody. To assay for the JNK3-specific kinase activity, cell lysates were immunoprecipitated with a mixture of a monoclonal antibody that recognizes JNK1/2 (clone G151-666; BD Biosciences Pharmingen) and a monoclonal antibody that recognizes JNK1 (clone F-3; Santa Cruz) to remove both JNK1 and JNK2 from the lysates (Gao et al., 2005). The remaining kinase activity (JNK3) in the supernatant was assayed by the capture JNK assay as described above. To assay for apoptosis signal-regulating kinase 1 (ASK1) kinase activity, the kinase in cell lysates was captured using the anti-ASK1 antibody (clone sc-7931; Santa Cruz) and then incubated with recombinant myelin basic protein (MBP) in the presence of [γ-32P]ATP, and MBP phosphorylation was detected using autoradiogram. To assay for PKD kinase activity directed toward recombinant HSP27, PKD1 was immunoprecipitated from cell lysates and incubated with recombinant HSP27 (wild type or containing one or more serine-to-alanine mutations) in the presence of [γ-32P]ATP, and visualized via electrophoresis and autoradiogram.
Statistical analysis.
All data are presented as the mean ± SEM. The significance of the difference between means was analyzed using either a t test comparison for single comparisons, or ANOVA followed by Bonferroni's post hoc test for comparisons between more than two groups, with p < 0.05 considered statistically significant.
Results
Serine phosphorylation is critical for HSP27-mediated neuroprotective effects against neural ischemia in vitro and in vivo
Three human HSP27 AAV constructs were generated: wild-type HSP27 (HSP27); a “nonphosphorylatable” HSP27 (HSP27-A) wherein the critical serine residues at Ser15, Ser78, and Ser82 were mutated to alanine; and a “phosphomimetic” HSP27 (HSP27-D) wherein the three serine residues were mutated to aspartate, lending a negatively charged extension similar to phosphorylated serine. When transduced into cortical cultures, all three constructs were recognized by both the HA tag and the HSP27 antibody (Fig. 1A). A faint band corresponding to the endogenous rat HSP25 was detected, demonstrating the low level of HSP25 present in rat cortical neurons.
Figure 1.
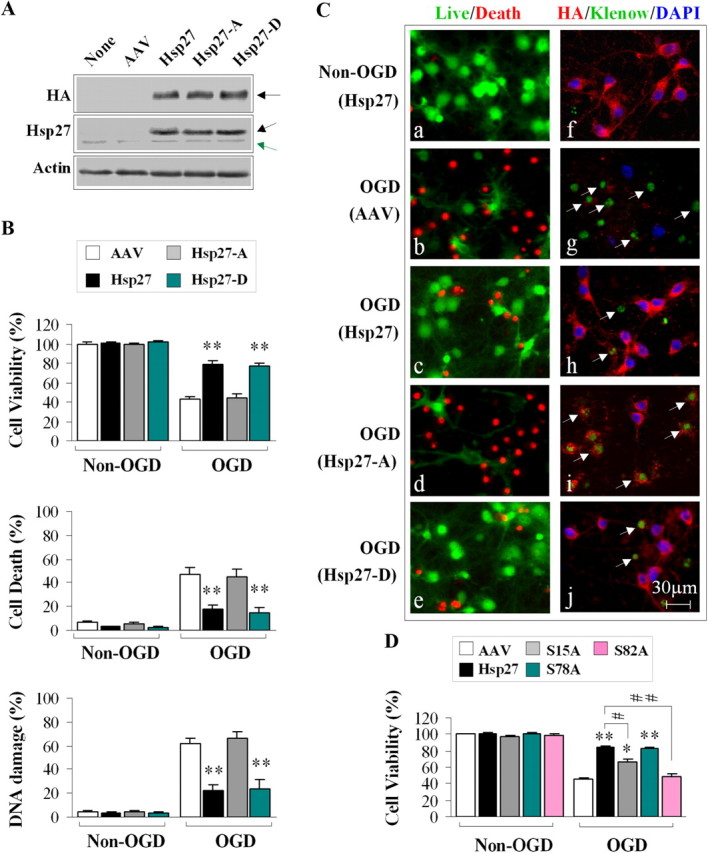
HSP27 phosphorylation is required for neuroprotection against neuronal ischemia in vitro. A, AAV-mediated expression of HA-tagged HSP27 constructs in cortical neuronal cultures. The black arrows indicate the exogenous HSP27 protein product, whereas the green arrow indicates the endogenous rat HSP25. B, Transduction of HSP27 or the phosphomimetic HSP27-D effectively promotes cell viability and decreases cell death and DNA damage following OGD. Viability and death were measured by Alamar blue uptake and LDH release, respectively, and DNA damage was assessed by histological evidence of Klenow fragment labeling and subsequent cell counting. C, Representative images demonstrating that transduction with HSP27 or HSP27-D, but not HSP27-A, promoted cell survival following OGD. a–e were stained using the live/dead kit (calcein labeling in green; dead cells in red). f–j were triple-labeled for HA (red), Klenow fragments (green), and DAPI (blue). D, Ser82 and Ser15 are critical for neuroprotection against OGD. Cultures were transduced with AAV-HSP27 containing single mutations at Ser15, Ser78, or Ser82 to alanine. Mutation of Ser82 or Ser15 significantly decreased the neuroprotective effect of HSP27 against OGD. Data are presented as mean ± SEM; n = 9/condition from 3 independent experiments. *p < 0.05, **p < 0.01 compared with empty vector controls (B, D), and #p < 0.05 and ##p < 0.01 compared with transfection of wild-type HSP27 (D).
Overexpression of either HSP27 or the phosphomimetic HSP27 (HSP27-D) was protective against cellular damage following OGD in cortical neuronal cultures (Fig. 1B,C). However, the nonphosphorylatable mutant was ineffective at preventing cell death. These results suggest that HSP27 phosphorylation is necessary to exert neuroprotection. To determine the contribution of the specific serine residues toward neuroprotection, we transduced cultures with AAV constructs containing HSP27 mutated at only one of the three serine residues. We found that alanine mutation at either Ser82 or Ser15 significantly blocked neuroprotection against OGD afforded by HSP27 overexpression (Fig. 1D). However, mutation of Ser78 to alanine had no significant effect on HSP27-mediated neuroprotection. Together, these results indicate that phosphorylation of HSP27 on Ser82 and Ser15 is critical for the full neuroprotective effect of HSP27 against neuronal ischemia.
Using the phosphomimetic HSP27-D and nonphosphorylatable HSP27-A constructs, we created transgenic mouse lines overexpressing human HSP27 under the control of a cytomegalovirus (CMV) promotor (Fig. 2A). Western blot and immunohistochemical analyses confirmed the presence of the HSP27 transgene protein products in neural tissue, including in neuronal subtypes (Fig. 2B,C). Consistent with our in vitro results, we found that overexpression of the phosphomimetic HSP27-D transgene was equally or more effective at reducing infarct volume following tFCI compared with wild-type HSP27 transgenic mice, even when assessed 21 d following ischemia (Fig. 2D–F), whereas overexpression of nonphosphorylatable HSP27-A was ineffective at decreasing infarct volume. Importantly, cerebral blood flow did not differ between groups when measured by either traditional laser Doppler or two-dimensional laser speckle (Fig. 2G,H). Brain surface vascular anatomy appeared unaffected by transgene expression (Table 1), and physiological variables, such as blood pressure, blood gases, and glucose, did not differ significantly between groups (data not shown).
In addition to decreasing infarct volume, our previous data demonstrated that overexpression of the HSP27 transgene improved neurofunctional recovery following ischemic injury (Stetler et al., 2008). Likewise, the phosphomimetic HSP27-D transgene improved neurofunctional recovery following ischemic injury compared with wild-type, nontransgenic mice, as assessed by the rotarod, corner, cylinder, and Morris water maze tests (Fig. 3A–E). However, the nonphosphorylatable HSP27-A transgenic showed no difference compared with wild-type nontransgenic mice following ischemic injury. No differences among any groups were detected in swim speed, indicating that the observed decrease in latency and increased time spent in the target quadrant were not due to physical differences. Together, these data indicate that serine phosphorylation of HSP27 is necessary for the reduction of ischemic infarct and improved neurofunctional recovery.
Figure 3.

Transgenic overexpression of the phosphomimetic HSP27-D improves neurofunctional recovery following focal ischemia. “WT” represents wild-type mice containing no exogenous transgene, and “sham” represents mice undergoing sham operation without induction of ischemia. The same cohort was used for all long-term studies (C–G). A, Mice were pretrained on the rotarod [establishing the control (100%) value] before the induction of tFCI. HSP27-D transgenic mice performed significantly better than wild-type or HSP27-A transgenic mice following tFCI. B, The number of left body turns made over 10 trials was recorded. Ischemic-injured mice turn predominantly toward the nonimpaired (left) side. HSP27-A transgenic and WT mice demonstrated predominant left turn behavior, whereas HSP27-D transgenic mice performed significantly fewer left turns, indicating reduced injury. C–G, Long-term neurofunctional assessments. Mice were placed in a cylinder, and the percentages of rearings using both paws on the sides of the cylinder were calculated. Ischemic-injured mice use both paws significantly less often than nonischemic (sham) mice, as was noted from ischemic wild-type and HSP27-A transgenic mice. Ischemic-injured HSP27-D, however, performed significantly better than wild-type or HSP27-A transgenic mice, indicating improved functional outcomes following ischemia. D, HSP27-D transgenic mice have significantly improved outcomes in the corner test, as described above, when assessed 21 d following tFCI. E–G, Morris water maze assessments in ischemic animals demonstrate improved performance in time to find the platform (E) as well as time spent in the platform-containing quadrant (F) in HSP27-D transgenic animals following ischemia compared with HSP27-A transgenic or wild-type animals. Swim speed was consistent among all groups (G). Data are presented as mean ± SEM, with n = 8–9/group for short-term studies (A, B) and n = 7/group for long-term studies (C–G). *p < 0.05, **p < 0.01, and ***p < 0.001 compared to nonischemic animals (sham), and ##p < 0.01 compared to wild-type (nontransgenic) ischemic mice.
Exogenous HSP27 is phosphorylated by endogenous neural mechanisms and is required for ASK1 inhibition
Given that overexpression of the phosphomimetic HSP27-D was equally or more effective at conferring ischemic neuroprotection compared with overexpression of the wild-type HSP27, we wanted to determine whether endogenous cellular components phosphorylate overexpressed wild-type HSP27 following ischemia. Indeed, transduction of wild-type HSP27 led to a sustained phosphorylation of HSP27 at both Ser15 and Ser82, beginning at early time points and extending at least 6 h following OGD (Fig. 4A) (data not shown). Conversely, overexpression of HSP27-A was unable to be phosphorylated following OGD (Fig. 4A) or tFCI injury (Fig. 4E). Consistent with the literature, the inability to be phosphorylated correlated with the sustained presence of HSP27 in high-molecular-weight oligomers, even following OGD, whereas wild-type HSP27 shifted to low oligomeric structures (Fig. 4B). Similar to the in vitro data, transgenic overexpression of wild-type HSP27 resulted in a majority of neurons immunopositive for phosphorylated HSP27 (pSer15) following tFCI, whereas the HSP27-A transgenic displayed very few neurons that were immunopositive for pSer15 (Fig. 4E).
Figure 4.

Serine phosphorylation of HSP27 is essential for its interaction with and suppression of ASK1 activity in vitro (A–D) and in vivo (E–G). A, AAV-mediated overexpression of HSP27, but not HSP27-A, leads to phosphorylation at both Ser15 and Ser82 following OGD. The lower arrows represent phosphorylation of endogenous rat HSP25. B, HSP27-A is incapable of shifting to low-molecular-weight oligomers following OGD, whereas wild-type HSP27 and HSP27-D are found predominantly in low-molecular-weight structures following OGD. Oligomeric state was determined by sucrose gel filtration, and the images are representatives of three experiments with similar results. C, Exogenously expressed HSP27 and HSP27-D, but not HSP27-A, coimmunoprecipitate with ASK1. ASK1 was immunoprecipitated from ischemic lysates and then immunoblotted for HSP27 gene products with an antibody against the HA tag; similar results were obtained with immunoprecipitation using the anti-HA antibody. D, HSP27-A is incapable of suppressing OGD-induced ASK1 activity. ASK1 was immunoprecipitated from neuronal lysates, which had been transduced with AAV constructs and exposed to OGD. In vitro ASK1 kinase activity assays were performed with MBP and radiolabeled ATP as substrates. Overexpression of HSP27 or the phosphomimetic HSP27-D effectively inhibited ASK1 activity. E, Immunohistochemistry indicates that HSP27 (a–c), but not HSP27-A (d), transgene product is phosphorylated (pSer15) in neurons (NeuN-positive cells) in the cortex following tFCI. c1–c3 and d1–d3 are high-power images from the boxes indicated in c and d demonstrating NeuN (red) and HSP27-pSer15 (green) immunostaining and subsequent overlay. F, G, HSP27 and HSP27-D, but not HSP27-A, coimmunoprecipitate with ASK1 (F) and effectively inhibit ASK1 activity (G) following tFCI. All graphs represent semiquantitative relative band intensity optical measurements from Western blots. Data are presented as mean ± SEM, with n = 3–4/group (A–D) and n = 5/group (F, G). *p < 0.05, **p < 0.01, and ***p < 0.001 compared with control, non-OGD cultures (A, D), or compared with sham-operated wild-type mice (F, G).
Having established that phosphorylation is required for HSP27-mediated neuroprotection and functional recovery following ischemic insults, and that overexpression of wild-type HSP27 can be phosphorylated by endogenous mechanisms, we wanted to determine whether phosphorylation is required for the interference with cell death signaling that confers HSP27-mediated neuroprotection. Previously, we demonstrated that HSP27 directly interacts with and suppresses apoptosis-signal regulating kinase (ASK1) activity and that the N-terminal region of HSP27 was required for this function. This region encompasses the critical serine residues, leading to the hypothesis that phosphorylation of HSP27 may be involved in its association with and suppression of ASK1 activity following OGD. Using coimmunoprecipitation as well as an in vitro phosphorylation activity assay, we found that the nonphosphorylatable HSP27-A was unable to associate with or inhibit the activity of ASK1 (Fig. 4C,D). Conversely, the phosphomimetic HSP27-D robustly interacted with and suppressed ASK1 activity. Using the transgenic mice, we found that coimmunoprecipitation of endogenous ASK1 and the HSP27 transgene protein following tFCI was evident in both the wild-type and the phosphomimetic HSP27 transgenics (HSP27 and HSP27-D), but absent in the nonphosphorylatable HSP27 transgenic HSP27-A (Fig. 4F). The finding that the phosphomimetic HSP27-D can coimmunoprecipitate with ASK1 indicates that association with ASK1 can occur on a structure that is seemingly already phosphorylated. In addition to the inability to coimmunoprecipitate with ASK1, transgenic overexpression of the nonphosphorylatable HSP27-A was ineffective at decreasing ASK1 activity (Fig. 4G). These data support the concept that association between HSP27 and ASK1 is downstream of HSP27 phosphorylation (i.e., HSP27 must be phosphorylated to associate with and inhibit ASK1).
Others have demonstrated that, following neural ischemic injury, activation of JNK signaling pathways is critical for ischemic cell death (Kuan et al., 2003). Indeed, we previously found that HSP27 overexpression and inhibition of ASK1 activation are associated with suppression of the canonical downstream MKK4/JNK signaling (Stetler et al., 2008). Overexpression of the phosphomimetic HSP27-D, but not the nonphosphorylatable HSP27-A, suppressed MKK4/JNK signaling (Fig. 5A,B) and led to a decreased presence of phosphorylated c-Jun in the nuclei (Fig. 5C). Further downstream cell death signaling, including Bax translocation to the mitochondria, release of cytochrome c into the cytosol, and upregulation of PUMA, was suppressed by overexpression of the wild-type HSP27 and the phosphomimetic HSP27-D following OGD (Fig. 5D). Conversely, the nonphosphorylatable mutant HSP27-A was ineffective at suppression of cell death signaling. Again, these results were recapitulated in the HSP27 transgenic mice. Following tFCI, the nonphosphorylatable HSP27-A transgenic and the nontransgenic (wild-type) mice were incapable of suppressing c-Jun phosphorylation (Fig. 5E), JNK activity (Fig. 5F), or activation of downstream cell death signaling, such as Bax translocation to the mitochondria, release of cytochrome c from the mitochondria, and activation of caspase-9 (Fig. 5G). In contrast, overexpression of the phosphomimetic HSP27-D transgene effectively inhibited both JNK activity and downstream cell death signaling (Fig. 5E–G). These results indicate that phosphorylation of HSP27 is both necessary and sufficient to suppress ASK1 cell death signaling.
Figure 5.
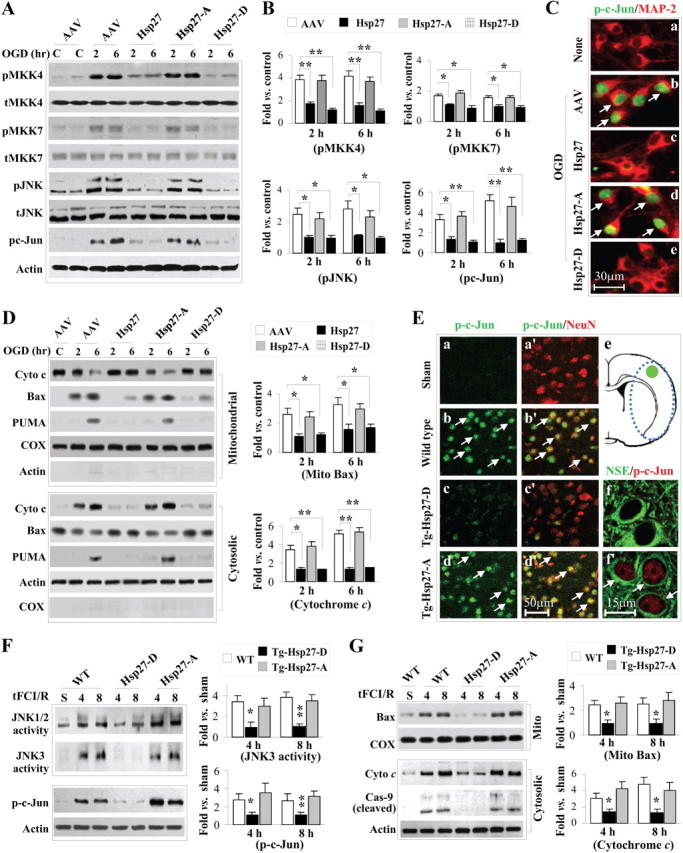
Ischemia-induced activation of ASK1 downstream signaling pathways is inhibited by overexpressed HSP27 or HSP27-D, but not HSP27-A, in both in vitro (A–D) and in vivo models (E–G). A, B, OGD-induced activation of the MKK4,7/JNK signaling pathway is attenuated in neuron cultures transfected with HSP27 or HSP27-D, but not HSP27-A. Western blots (A) were semiquantified and expressed as fold increase over non-OGD controls (B). C, Representative immunofluorescence images demonstrating suppression of c-Jun phosphorylation (green, arrows in b and d) in HSP27- or HSP27-D-transfected neurons 2 h after OGD. D, OGD-induced cytochrome c release, mitochondrial translocation of Bax, and upregulation of PUMA expression are attenuated in neuron cultures transfected with HSP27 or HSP27-D, but not HSP27-A. E, tFCI increases the levels of phosphorylated c-Jun (p-c-Jun) with predominant nuclear localization in neurons of wild-type (nontransgenic) mice and HSP27-A transgenic mice (b, b′, d, d′, f′), whereas HSP27-D transgenic mice demonstrate reduced c-Jun phosphorylation (c, c′, f). The region in e outlined by blue dots indicates the ischemic zone, and the green circle indicates the region from which the images were acquired. F, G, Overexpression of HSP27-D, but not HSP27-A, significantly suppresses JNK activation (increased JNK3-specific kinase activity and levels of p-c-Jun) and downstream prodeath signaling (mitochondrial translocation of Bax, cytochrome c release, and caspase-9 activation) at 4 and 8 h after tFCI. JNK3 activity, p-c-Jun induction, mitochondrial translocation of Bax, and cytochrome c release were semiquantified and expressed as fold increases over sham controls. Data are presented as mean ± SEM, with n = 3–4/group (B, D) and n = 4–5/group (F, G). *p < 0.05 and **p < 0.01 compared with empty vector (B, D) or wild-type mice (F, G).
PKD functions as the primary upstream kinase directed to HSP27 phosphorylation
Having determined that phosphorylation of HSP27 is required for neuroprotection and having demonstrated that overexpressed wild-type HSP27 can be phosphorylated by endogenous mechanisms, we sought to determine the endogenous upstream kinase(s) responsible for HSP27 phosphorylation. Several other model systems have indicated that PKA, PKD, PI3K/PKB, PKC, and p38-mediated MAPKAP kinase can serve as upstream kinases leading to phosphorylation of HSP27. However, the relevance of the particular kinase appears to be highly context dependent. Using a panel of kinase inhibitors, we found that only the PKD inhibitor 2,3,4,5-tetrahydro-7-hydroxy-1H-benzofuro[2,3-c]azepin-1-one (CID755673) (3 μm), which blocks the activation of PKD by inhibiting phosphorylation at serine 916 (Sharlow et al., 2008), effectively blocked phosphorylation of HSP27 at both Ser15 and Ser82 (Fig. 6A). Interestingly, phosphorylation of Ser15 at an early time point (0.5 h) following OGD was unaffected by PKD inhibition despite the abolition of PKD activation (Fig. 6B), suggesting that an alternate kinase may contribute to HSP27 Ser15 phosphorylation in the initial phase of reperfusion. However, the extended suppression of HSP27 phosphorylation at both Ser82 and Ser15 by PKD inhibition suggests that PKD is the primary kinase responsible for continued phosphorylation of HSP27 at these critical residues.
Figure 6.
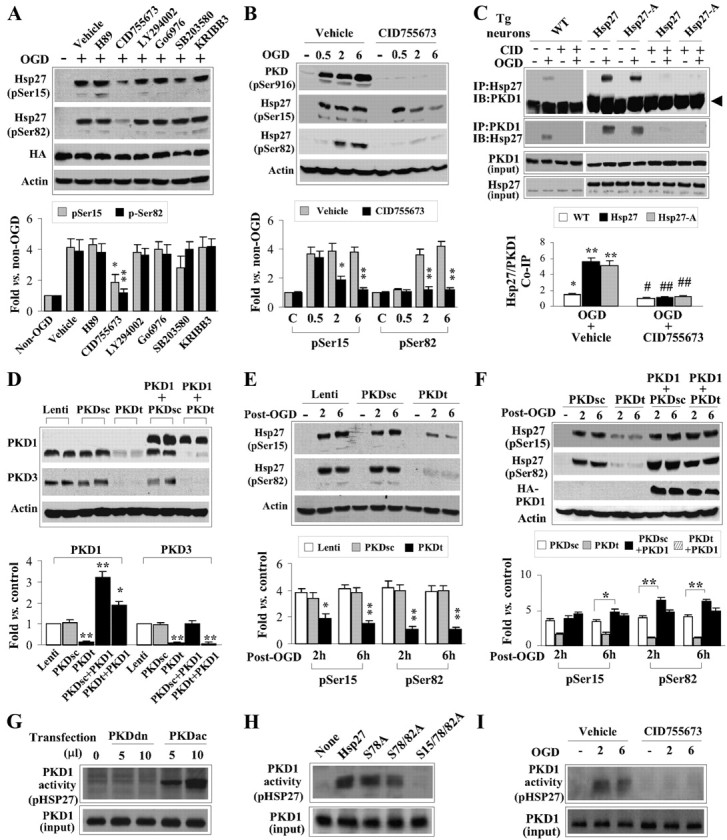
PKD is the major kinase responsible for phosphorylation of HSP27 following neuronal ischemia. A, A panel of kinase inhibitors was applied to cortical neuron cultures derived from Tg-HSP27 mice 30 min before induction of OGD, including the PKA inhibitor H89 (10 μm), the PKD inhibitor CID755673 (3 μm), the PI3K/Akt inhibitor LY294002 (10 μm), the PKC inhibitor Go6976 (3 μm), the p38 inhibitor SB203580 (10 μm), and the PKC inhibitor KRIBB3 (10 μm). Only CID755673 significantly blocked HSP27 phosphorylation at Ser15 and Ser82 at 2 h after OGD. B, Endogenously expressed PKD undergoes robust phosphorylation following OGD in a time frame consistent with phosphorylation of HSP27. The PKD inhibitor CID755673 (3 μm) effectively blocked phosphorylation of PKD as well as significantly prevented HSP27 phosphorylation at Ser15 and Ser82. C, OGD promoted the interaction between HSP27 or HSP27-A and PKD1. The interaction required activation of PKD, as the inhibitor CID755673 significantly blocked HSP27 and PKD1 association. Coimmunoprecipitation was performed using cell extracts from cultures derived from Tg-HSP27 or Tg-HSP27-A mice and wild-type mice. The arrow on the right points to the endogenous IgG. D, Lentivirus shRNA was infected to simultaneously target endogenous PKD1 and PKD3 [PKDt; no PKD2 isoform is expressed in neurons in vitro or in vivo (data not shown)]. Overexpression of human PKD1 (3HA-tagged) cDNA restored protein expression. The scrambled shRNA sequence (PKDsc) had no effect on endogenous expression of PKD1 or PKD3. E, PKD knockdown significantly attenuated OGD-induced HSP27 phosphorylation at Ser15 and Ser82 in cultures derived from Tg-HSP27 mice. F, Overexpression of PKD1 promoted OGD-induced HSP27 phosphorylation in neurons transfected with PKDsc or PKDt. G, Using 293HEK cells transfected with human PKD1 [either a dominant-negative (PKDdn) form or constitutively active (PKDac) form], PKD1 was pulled down and subjected to a cell-free activity assay, with recombinant HSP27 protein as the substrate. PKDac, but not PKDdn, demonstrated robust activity in the direct phosphorylation of HSP27. H, 293HEK cells were transfected with PKDac as described in G, and activity was assessed with recombinant HSP27 protein mutated at the various serine residues. Mutation of Ser78 to alanine had little effect on PKD1-directed HSP27 phosphorylation, whereas mutation of Ser82 and Ser15 to alanines resulted in near-complete abrogation of PKD1-directed HSP27 phosphorylation. I, Endogenously expressed PKD1 was extracted from ischemic neuronal cultures and subjected to the activity assay using recombinant HSP27 as the substrate. Inhibition of PKD with CID755673 effectively blocked the activity of endogenous PKD toward phosphorylation of HSP27. All data are mean ± SEM, with n = 3–4/group. *p < 0.05 and **p < 0.01 compared with vehicle (A, B), non-OGD controls (C), or empty vector or PKDsc (D, E). The blots at the bottom for ASK1 input or actin were used to demonstrate equal loading and were not used in the semiquantification.
We next wanted to determine whether PKD directly associates with HSP27. Indeed, transgenic HSP27 [and endogenous HSP25 as noted by the faint band in nontransgenic (wild-type) mice; Fig. 6C, left panel] was able to be coimmunoprecipitated with PKD1 following OGD in cortical cultures (Fig. 6C). Using cultures derived from HSP27-A transgenic mice, we further determined that the association between HSP27 and PKD1 is independent of HSP27 phosphorylation, as HSP27-A retained the capability of being coimmunoprecipitated with PKD1 following OGD (Fig. 6C). Furthermore, pharmacological inhibition of PKD1 with CID755673 blocked the association between HSP27 and PKD1. These results indicate that HSP27 can physically associate with PKD and that the activation of PKD1 is required for this association. Because the nonphosphorylatable HSP27-A was able to associate with PKD following OGD, these data suggest that phosphorylation of HSP27 occurs downstream of PKD activation and association with HSP27.
To further confirm that PKD is required for HSP27 phosphorylation, we used a targeted knockdown of PKD1 and PKD3. A single siRNA sequence was found that effectively targets both the PKD1 and PKD3 isoforms (PKDt). PKD2 was not found to be expressed in either cortical cultures or brain (data not shown). This sequence or a scrambled control sequence (PKDsc) was constructed into a lentiviral vector for maximal neuronal transduction. PKDt, but not PKDsc, effectively knocked down expression of PKD1 and PKD3 3 d following infection (Fig. 6D). Cotransduction with the human sequence of PKD1 (PKD1h) was insensitive to the rat-targeted PKDt shRNA construct and effectively restored PKD1 expression (Fig. 6D). The targeted knockdown of PKD1/3 (PKDt) significantly decreased HSP27 phosphorylation in wild-type HSP27 transgenic cortical cultures exposed to OGD, whereas the scrambled control shRNA (PKDsc) had no effect (Fig. 6E). Furthermore, cotransfection with the human sequence of PKD1 restored both expression of PKD1 in the presence of the PKDt and phosphorylation of HSP27 following OGD (Fig. 6D,F), confirming that HSP27 phosphorylation requires the presence of PKD.
The data thus far indicate that PKD is required for HSP27 phosphorylation at Ser82 and Ser15, and that these two proteins physically associate. However, the possibility still exists that the mechanism is indirect. To test this, we transfected HEK293 cells with a constitutively active PKD1 (PKDac) containing the S738/742E mutation or a dominant-negative PKD1 (PKDdn) containing the K612W mutation, immunoprecipitated the expressed PKD1 protein, and performed an in vitro kinase activity assay using recombinant HSP27 as the target substrate with [32P]ATP as a radiolabel. Indeed, immunoprecipitated PKDac, but not PKDdn, was able to significantly phosphorylate wild-type HSP27 (Fig. 6G,H). Although mutation of Ser78 to alanine did not diminish the ability of PKD to incorporate 32P into recombinant HSP27, additional mutations of Ser82 and Ser15 to alanines significantly decreased PKD-mediated phosphorylation of recombinant HSP27 (Fig. 6H). Using cortical neuronal cultures exposed to OGD, we were able to recapitulate these results by immunoprecipitating endogenous PKD1 and assessing its ability to phosphorylate recombinant HSP27 in vitro (Fig. 6I). Addition of CID755673 before OGD abolished this activity. These results indicate that PKD1 specifically is capable of HSP27 phosphorylation, particularly at Ser82 and Ser15.
Suppression of PKD activity disinhibits ASK1 activation
Thus far, we have demonstrated that PKD specifically phosphorylates HSP27 and that phosphorylated HSP27 is necessary for neuroprotection against neuronal ischemic injury. We previously reported that HSP27 overexpression directly suppresses ASK1 activity and downstream cell death signaling, and have now demonstrated that phosphorylation of HSP27 is required for ASK1 interaction and inhibition. However, HSP27 is phosphorylated at three distinct serine residues, and the possibility remains that the PKD-targeted phosphorylation of HSP27 may or may not regulate the HSP27-mediated suppression of ASK1 activity. Therefore, we wanted to determine whether PKD-directed phosphorylation of HSP27 confers ASK1 pathway suppression.
Both the PKD inhibitor CID755673 and the targeted shRNA knockdown of PKD1/3 effectively inhibited the interaction between HSP27 and ASK1 (Fig. 7A,C) and subsequently allowed ASK1 to be activated following OGD (Fig. 7B,E). Confirmation of the PKD shRNA was performed by cotransfection of the human PKD1 sequence (PKDh) or a dominant-negative mutant (PKDdn), both of which are resistant to the mouse-directed shRNA PKD1/3 targeting sequence. Expression of PKDh protein, but not the PKDdn, restored interaction between HSP27 and ASK1, and subsequent suppression of ASK1 activity (Fig. 7D,F). As expected, interaction between ASK1 and the phosphomimetic HSP27-D was insensitive to alterations directed at PKD activity (Fig. 7A–E). Given that the aspartate mutation introduces a long, negatively charged arm highly similar to serine phosphorylation, the phosphomimetic structure is thus uncoupled from kinase regulation. This further supports the conclusion that PKD targets wild-type HSP27 for phosphorylation and that it is this posttranslational modification that appears to be required for ASK1 inhibition.
Figure 7.

PKD-mediated phosphorylation of HSP27 is required for inhibition of ASK1 following OGD. A, B, HSP27- or HSP27-D-transfected neurons were subjected to OGD in the presence or absence of CID755673 (CID); at 2 h after OGD, coimmunoprecipitation was performed for ASK1/HSP27 interaction (A) and ASK1 activity was assayed (B). CID inhibits ASK1/HSP27 interaction (A) and the ability of HSP27 to suppress ASK1 activity (B) in OGD-challenged neurons. Overexpressed HSP27-D bypasses the need for PKD-mediated phosphorylation, and thus HSP27-D still interacts with and suppresses ASK1 in OGD neurons. C–F, Neuron cultures derived from transgenic animals overexpressing HSP27 or the phosphomimetic HSP27-D were subjected to PKD inhibition (shRNA or dominant-negative) and assayed for ASK1 activity and interaction with HSP27 following OGD. C, D, Targeted knockdown of PKD (PKDt) but not a scrambled control (PKDsc) significantly inhibited the ability of transgenically expressed HSP27 to coimmunoprecipitate with ASK1 (C) and suppress the activity of ASK1 (D) following OGD. The phosphomimetic HSP27-D was still capable of interaction with and suppression of ASK1 activity with or without the presence of PKD. E, F, Restoration of PKD by lentivirus-mediated transfection of human PKD1 (PKD1-wt) but not PKDdn reversed the PKD knockdown effect, promoting HSP27/ASK1 interaction (E) and suppressing ASK1 activity (F) following OGD. All data are mean ± SEM, with n = 3–4/group. *p < 0.05 and **p < 0.01 compared with vehicle (A, B), empty vector or PKDsc (C, D), or as indicated in the graphs (E, F).
PKD is required for HSP27-mediated neuroprotection against neuronal ischemia
The data thus far support the conclusion that PKD is the primary kinase leading to phosphorylation of HSP27 in neurons and that phosphorylation of HSP27 is required for neuroprotective actions, including the suppression of ASK1 activity. To directly link neuroprotection to PKD-mediated HSP27 phosphorylation, we directly assessed the consequence of PKD inhibition on the neuroprotective activity of HSP27. Pharmacological inhibition of PKD (Fig. 8A), shRNA knockdown (Fig. 8B), or competition by overexpression of PKDdn (Fig. 8C) significantly blocked the neuroprotection against OGD afforded by HSP27 overexpression. Importantly, inhibition of PKD activity did not affect the protection afforded by the phosphomimetic HSP27-D, suggesting that the activity of PKD in neuroprotection is targeted primarily via phosphorylation of HSP27.
Figure 8.

Inhibition of PKD negates HSP27 overexpression-afforded neuroprotection against OGD. Cortical neuron cultures transduced with AAV constructs (A) or derived from transgenic mice (B–D) were exposed to the PKD inhibitor CID755673 for 30 min (A) or transduced with shRNA targeting PKD (PKDt) or a scrambled control (PKDsc) for 3 d (B), and then subjected to OGD. Cell viability (cell live/dead kit) and cell death (LDH release) were assessed 24 h after OGD. A, B, PKD inhibition with CID755673 or PKD knockdown significantly blocked the neuroprotective effects of HSP27, but not of HSP27-D, following OGD. C, Overexpression of human PKD1, but not PKDdn, restored HSP27-afforded neuroprotection against OGD in PKD-knockdown neurons. All quantitative data are mean ± SEM, with n = 9/group from 3 independent experiments. *p < 0.05 and **p < 0.01 compared with OGD cultures either transduced with empty vector (A) or derived from wild-type (nontransgenic) animals (B, C), and #p < 0.05 and ##p < 0.01 as indicated by the horizontal brackets.
To confirm the in vitro results demonstrating the role of PKD in HSP27-mediated neuroprotection against ischemic injury, we retested this mechanism using the mouse model of tFCI. Concurrent phosphorylation of both PKD and HSP27 occurred rapidly following reperfusion (Fig. 9A), and confocal microscopic imaging indicated a high colocalization of phosphorylated PKD (p-PKD) and phosphorylated HSP27 (p-HSP27) (Fig. 9B). Intracerebroventricular injection of the PKD inhibitor CID755673 blocked the phosphorylation of both PKD (Fig. 9C) and HSP27 at Ser15 and Ser82 (Fig. 9D). Additionally, inhibition of ASK1 activity by overexpression of HSP27 was abolished by infusion of the PKD inhibitor CID (Fig. 9E). However, PKD inhibition had no effect on suppression of ASK1 activity in the phosphomimetic HSP27-D transgenic. Thus, mimicking the phosphorylation of HSP27 by HSP27-D decouples PKD from ASK1 inhibition.
Figure 9.
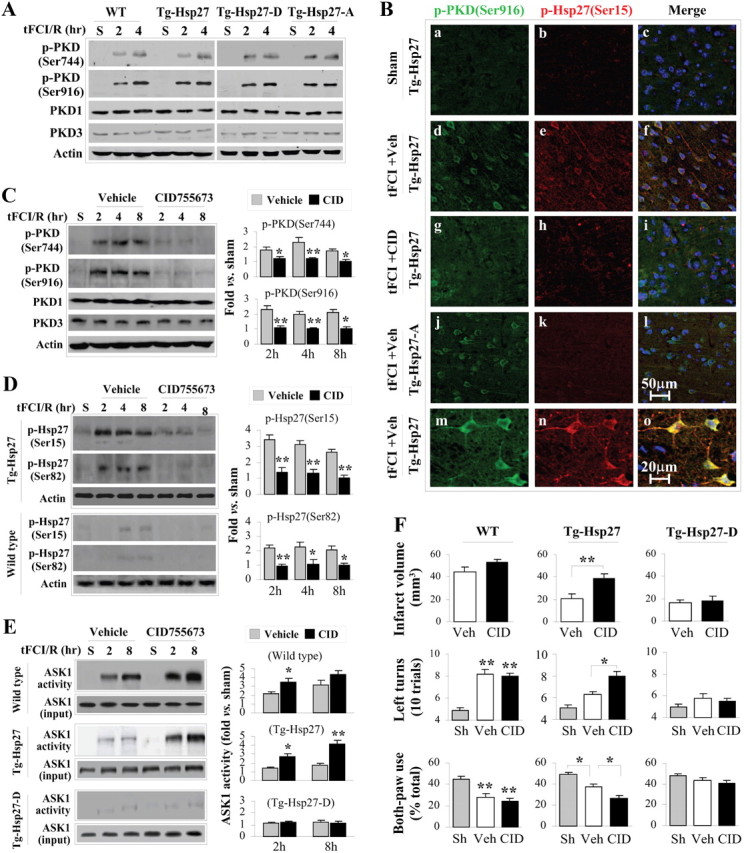
PKD inhibition in vivo abrogates HSP27-mediated neuroprotection and suppression of ASK1 following tFCI. A, Time course of PKD phosphorylation following tFCI and reperfusion. PKD phosphorylation at Ser916 [p-PKD(Ser916)] is closely associated with its kinase activity and is markedly increased within 2 h following post-tFCI reperfusion. B, Representative double-label immunofluorescence images of cerebral cortex at 2 h following post-tFCI reperfusion show colocalization of p-PKD (Ser916) and p-HSP27 (Ser15) in HSP27 transgenic mice (d–f; high-power magnification, m–o), but not in HSP27-A transgenic mice (j–l). Inhibition of PKD with intracerebroventricular infusion of CID755673 attenuated the immunofluorescence of both p-PKD and p-HSP27 (g–i). C, Western blots demonstrate that CID755673 infusion significantly decreased PKD phosphorylation (both Ser744 and Ser916) at 2–8 h after tFCI. D, Western blots demonstrate that CID755673 infusion significantly decreased HSP27 phosphorylation (both Ser15 and Ser82) in Tg-HSP27 mice at 2–8 h after tFCI. E, CID755673 infusion abolished HSP27-afforded suppression of ASK1 activity following tFCI. HSP27-D retains its inhibitory effects on ASK1 activity, as it is a phosphomimetic structure that does not necessitate phosphorylation. F, Improved histological and neurofunctional outcomes observed in HSP27 (wild-type) transgenic mice were abrogated by CID755673 infusion. HSP27-D transgenic mice showed improved outcomes even in the presence of PKD inhibitor. Infarct volume was measured on TTC-stained brain sections at 24 h after tFCI; neurofunctional outcomes were assessed using corner test and cyclinder test. Data are presented as mean ± SEM, with n = 4/group (C–E) and n = 8/group (F). *p < 0.05 and **p < 0.01 compared with vehicle (C–E), sham controls (F), or as indicated in the graphs (F). The blots at the bottom for ASK1 input or actin were used to demonstrate equal loading and were not used in the semiquantification.
Finally, inhibition of PKD significantly decreased the neuroprotection against tFCI afforded by HSP27 transgene overexpression, as measured by both infarct volume and neurofunctional outcomes (Fig. 9F). Again, the effects of PKD inhibition were absent in the presence of the phosphomimetic HSP27-D. Together, these data demonstrate that PKD is required for phosphorylation of wild-type HSP27 and that, through this pathway, ASK1 activity is inhibited, leading to suppression of ischemic neuronal injury.
Discussion
The data presented here detail the mechanistic actions required for and leading to the neuroprotection afforded by HSP27 against neural ischemic injury. In particular, overexpression of HSP27 itself requires serine phosphorylation to suppress ASK1-mediated cell death signaling and neuroprotection against ischemic injury. Furthermore, we have identified PKD as the primary kinase present in ischemic neurons responsible for phosphorylation of HSP27, and have presented data to indicate that the endogenous activation of PKD is necessary for neuroprotection afforded by exogenous HSP27 overexpression. These data provide a sound basis for targeting an inherent regulatory mechanism of HSP27 for use as a therapeutic molecule and open the role of PKD in supporting neuroprotection against ischemic injury.
The observation that phosphorylation of HSP27 is required for neuroprotection is novel, which differentiates the effects of HSP27 under neuronal ischemic conditions from other cellular systems. Previous studies in non-neuronal systems have indicated that HSP27 possesses several roles in the cellular context, primarily based on two major functions associated with differential phosphorylation and oligomerization states. In the high-molecular-weight oligomeric, nonphosphorylated form, HSP27 functions primarily as a chaperone (Knauf et al., 1994) and has been best characterized in the targeting of aberrantly folded proteins to inhibit aggregate formation (Jakob et al., 1993). However, upon phosphorylation and dissociation to lower-molecular-weight oligomers, HSP27 can function as an inhibitor of cell death, targeting both upstream and downstream of mitochondrial signaling. Although the presence of the high-molecular-weight, nonphosphorylated form of HSP27 appears to be necessary for cellular protection against cardiac ischemia/reperfusion (Hollander et al., 2004) and under some neural contexts, such as neurite outgrowth (Williams and Mearow, 2011) or against tau fibril formation (Abisambra et al., 2010), differences in cellular phenotype and environment may affect the necessity for the phosphorylated state of HSP27. Given the complex pathogenesis of ischemic injury (Lo et al., 2003), and our previous finding that HSP27 effectively inhibited ASK1 activity in the neural ischemic context, our data are consistent with previous findings that phosphorylation of HSP27 is required for suppression of cell death signaling in neural ischemia. Further studies comparing and contrasting different cellular contexts might further elucidate (and predict) the regulation and actions of HSP27.
Determination of the upstream effectors controlling HSP27 function is critical in developing therapeutic strategies. Several serine/threonine kinases, including PKC, p38, PI3K/Akt, and PKD, are capable of phosphorylating HSP27 but appear to vary widely based on cell phenotype and environment. Under neuronal ischemic conditions, many of these kinases have been demonstrated to be activated and contribute to outcomes (Aronowski et al., 2000; Irving and Bamford, 2002); thus, any could feasibly regulate the oligomeric state and subsequent function of HSP27. However, PKD is a particularly attractive candidate. Although PKD has not been investigated in cerebral ischemia models, HSP27 has been identified as a substrate of PKD (Döppler et al., 2005), and PKD specifically phosphorylates HSP27 in a number of non-neuronal systems, including pancreatic cancer cells (Yuan and Rozengurt, 2008) and endothelial cells (Evans et al., 2008). In a neuronal cell line (SH5Y5), several kinases including PKD were found to be capable of phosphorylating HSP27 in the presence of different agonists (Dokas et al., 2011). The current finding that the endogenous neuronal PKD is required for the phosphorylation—and hence neuroprotection—afforded by HSP27 not only identifies an endogenous regulator of HSP27 in neural settings but also opens the field for further investigation into the role of PKD in protection against cerebral ischemia.
Stemming from our observations that (1) immunoprecipitated PKD1/3 from ischemic lysates phosphorylated recombinant HSP27 in a cell-free system, (2) only the inhibitor of PKD (CID755673) had a significant effect on blocking HSP27 phosphorylation at critical serine residues, and (3) knockdown of PKD1/3 blocked both HSP27 phosphorylation and its neuroprotective effects, we conclude that PKD is the primary kinase responsible for mediating the neuroprotective function of HSP27 via phosphorylation. This finding is novel, as a role for PKD in neuronal ischemia has not before been identified. Interestingly, we further demonstrated that PKD and HSP27 associate in an immunocomplex, implying that the interaction between the kinase and its substrate should be fairly direct. When PKD was immunoprecipitated from HEK293 cell lysates and incubated with HSP27 in the presence of radiolabeled ATP in a purified assay, HSP27 was phosphorylated. These data indicate a close physical interaction between the two proteins. However, the possibility cannot be excluded that other components may have been inadvertently immunoprecipitated with PKD and may also be required to facilitate phosphorylation of HSP27. However, given that the pulldown of the dominant-negative PKD and incubation of ischemic cultures with the PKD inhibitor CID755673 effectively abrogated the in vitro phosphorylation of recombinant HSP27, we conclude that PKD is the critical kinase involved in the directed phosphorylation of HSP27 at critical serine residues.
Identification of Ser15 and Ser82 as critical residues in HSP27-mediated neuroprotection may allow for structural screening of small peptides in the search for potential therapeutic agents. Ser82 lies directly within the oligomeric interacting surface and appears to be the critical residue that shifts the oligomeric state of HSP27 (Thériault et al., 2004). Thus, phosphorylation at Ser82 would likely be the main “effector” step of the shift from the large-molecular-weight multimers to the differentially functional oligomers by incorporating a large negative charge at the face of the dimer surface. Ser15, on the other hand, is localized in the WDPF domain in the N-terminal region of HSP27. Its function is less defined, but it is thought to facilitate the full activity of HSP27 in binding to substrates (Thériault et al., 2004). Although our data suggest that phosphorylation of both of these residues is critical to the full neuroprotective effect of HSP27 against ischemia, it was unclear whether the two residues were phosphorylated by the same upstream kinase. Consistent with the data demonstrating that pharmacological inhibition of PKD did not completely block phosphorylation of HSP27 at Ser15, targeted knockdown of PKD1 and PKD3 also did not completely abrogate HSP27 phosphorylation at Ser15. Therefore, it is possible that another kinase may play an initial role in the process of HSP27 phosphorylation at Ser15, which could contribute to the full phosphorylation state of HSP27. As opposed to Ser15 and Ser82, Ser78 appears to play an insignificant role in the neuroprotective effects of HSP27 under neuronal ischemia. However, it is possible that the lack of perceived involvement of Ser78 may be due to the absence of this residue in the murine HSP25 (Kostenko and Moens, 2009). In this case, the endogenous murine mechanisms explored in our model systems may not target or require Ser78 in exogenously expressed human HSP27. Alternatively, the upstream kinase responsible for Ser78 phosphorylation may not be activated following ischemic injury. The regions surrounding Ser15 and Ser82 contain putative PKD consensus phosphorylation motifs (Kostenko and Moens, 2009) not present around Ser78. Consistently, in vitro PKD activity assays using recombinant HSP27 mutated only at Ser78 did not appear to affect the ability of PKD1 to phosphorylate HSP27. Thus, the activation of PKD may preferentially target these two serine residues over Ser78.
Our data reveal that a very small amount of endogenous HSP25 exists in both neuronal cultures and brain, and that this endogenous HSP25 is phosphorylated by endogenous PKD. This observation then leads to several interesting questions. First, given that the endogenous HSP25 interacts with PKD and can be phosphorylated, we hypothesize that the stoichiometric ratio of endogenous phosphorylated HSP25 to ischemic-activated ASK1 is insufficient to overcome activation of cell death signaling. In this scenario, overexpression of exogenous HSP27 would allow enough HSP27 substrate for PKD to phosphorylate and thus inhibit ASK1-mediated cell death signaling. Second, the failure of neurons to induce endogenous HSP25 expression under ischemic conditions is not understood; however, it is possible that the rapidity of the injury is insufficient to induce enough protein to mount a protective response. Indeed, several studies have identified upregulation of HSP27 in retinal ischemic preconditioning (Whitlock et al., 2005) or in glial cells, which are more resistant to cerebral ischemia (Kato et al., 1994; Nishino and Nowak, 2004). Further investigation into the control of endogenous HSP25 gene induction is necessary to better understand the limited availability of endogenous HSP25 in neurons.
Together, our data describe a newly identified endogenous mechanism for the regulation of HSP27 in conferring neuroprotection against cerebral ischemia (Fig. 10). The presence of HSP27 in its nonphosphorylatable form was insufficient to confer protection or to suppress ASK1-mediated cell death signaling. Instead, we identified the endogenous PKD-directed phosphorylation of HSP27 as a critical upstream step for the subsequent interaction between HSP27 and ASK1 and suppression of downstream cell death signaling. In particular, ischemia-induced phosphorylated PKD directly associates with HSP27, leading to phosphorylation at Ser15 and Ser82. The phosphorylated HSP27 can then associate with ischemia-activated (phosphorylated) ASK1 and inhibit its kinase activity, leading to suppression of MKK4/7 cell death signaling and subsequent cell death. These findings present a discernible model of HSP27-mediated neuroprotection, which should contribute significantly to the identification of small-molecule therapeutics geared toward ischemic neuroprotection.
Figure 10.

HSP27 acts via phosphorylation by PKD1 to suppress ASK1 cell death signaling. Following ischemia, PKD1 is phosphorylated (activated), but without HSP27, fails to suppress ASK1 activity and leads to cell death. In the presence of HSP27, endogenously activated PKD1 phosphorylates HSP27, which can then suppress the activity of ASK1, leading to cell survival.
Footnotes
This work was supported by NIH–NINDS Grants NS43802, NS45048, NS36736, and NS56118, and Veterans Affairs Merit Review grant (J.C.); and Chinese Natural Science Foundation Grants 30670642, 30870794, and 81020108021 (Y.G.). F.Z. and X.H. were supported American Heart Association Grants 10SDG2560122 and 10POST4150028, respectively. P.V. was supported by NIH National Research Service Award Fellowship 1F30NS057886. We thank Dr. Qiming Wang (University of Pittsburgh, Pittsburgh, PA) for providing the PKD inhibitor and the anti-PKD3 antibody. We thank Carol Culver for editorial assistance and Pat Strickler for secretarial support. The contents of this article do not represent the views of the Department of Veterans Affairs or the United States Government.
References
- Abisambra JF, Blair LJ, Hill SE, Jones JR, Kraft C, Rogers J, Koren J, 3rd, Jinwal UK, Lawson L, Johnson AG, Wilcock D, O'Leary JC, Jansen-West K, Muschol M, Golde TE, Weeber EJ, Banko J, Dickey CA. Phosphorylation dynamics regulate Hsp27-mediated rescue of neuronal plasticity deficits in tau transgenic mice. J Neurosci. 2010;30:15374–15382. doi: 10.1523/JNEUROSCI.3155-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An JJ, Lee YP, Kim SY, Lee SH, Lee MJ, Jeong MS, Kim DW, Jang SH, Yoo KY, Won MH, Kang TC, Kwon OS, Cho SW, Lee KS, Park J, Eum WS, Choi SY. Transduced human PEP-1-heat shock protein 27 efficiently protects against brain ischemic insult. FEBS J. 2008;275:1296–1308. doi: 10.1111/j.1742-4658.2008.06291.x. [DOI] [PubMed] [Google Scholar]
- Aronowski J, Grotta JC, Strong R, Waxham MN. Interplay between the gamma isoform of PKC and calcineurin in regulation of vulnerability to focal cerebral ischemia. J Cereb Blood Flow Metab. 2000;20:343–349. doi: 10.1097/00004647-200002000-00016. [DOI] [PubMed] [Google Scholar]
- Ayata C, Dunn AK, Gursoy-Ozdemir Y, Huang Z, Boas DA, Moskowitz MA. Laser speckle flowmetry for the study of cerebrovascular physiology in normal and ischemic mouse cortex. J Cereb Blood Flow Metab. 2004;24:744–755. doi: 10.1097/01.WCB.0000122745.72175.D5. [DOI] [PubMed] [Google Scholar]
- Badin RA, Modo M, Cheetham M, Thomas DL, Gadian DG, Latchman DS, Lythgoe MF. Protective effect of post-ischaemic viral delivery of heat shock proteins in vivo. J Cereb Blood Flow Metab. 2009;29:254–263. doi: 10.1038/jcbfm.2008.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn SC, Perrelet D, Kato AC, Scholz J, Decosterd I, Mannion RJ, Bakowska JC, Woolf CJ. Hsp27 upregulation and phosphorylation is required for injured sensory and motor neuron survival. Neuron. 2002;36:45–56. doi: 10.1016/s0896-6273(02)00941-8. [DOI] [PubMed] [Google Scholar]
- Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA, Diaz-Latoud C, Gurbuxani S, Arrigo AP, Kroemer G, Solary E, Garrido C. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol. 2000;2:645–652. doi: 10.1038/35023595. [DOI] [PubMed] [Google Scholar]
- Concannon CG, Orrenius S, Samali A. Hsp27 inhibits cytochrome c-mediated caspase activation by sequestering both pro-caspase-3 and cytochrome c. Gene Expr. 2001;9:195–201. doi: 10.3727/000000001783992605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokas LA, Malone AM, Williams FE, Nauli SM, Messer WS., Jr Multiple protein kinases determine the phosphorylated state of the small heat shock protein, HSP27, in SH-SY5Y neuroblastoma cells. Neuropharmacology. 2011;61:12–24. doi: 10.1016/j.neuropharm.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döppler H, Storz P, Li J, Comb MJ, Toker A. A phosphorylation state-specific antibody recognizes Hsp27, a novel substrate of protein kinase D. J Biol Chem. 2005;280:15013–15019. doi: 10.1074/jbc.C400575200. [DOI] [PubMed] [Google Scholar]
- Evans IM, Britton G, Zachary IC. Vascular endothelial growth factor induces heat shock protein (HSP) 27 serine 82 phosphorylation and endothelial tubulogenesis via protein kinase D and independent of p38 kinase. Cell Signal. 2008;20:1375–1384. doi: 10.1016/j.cellsig.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Gao Y, Signore AP, Yin W, Cao G, Yin XM, Sun F, Luo Y, Graham SH, Chen J. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab. 2005;25:694–712. doi: 10.1038/sj.jcbfm.9600062. [DOI] [PubMed] [Google Scholar]
- Hollander JM, Martin JL, Belke DD, Scott BT, Swanson E, Krishnamoorthy V, Dillmann WH. Overexpression of wild-type heat shock protein 27 and a nonphosphorylatable heat shock protein 27 mutant protects against ischemia/reperfusion injury in a transgenic mouse model. Circulation. 2004;110:3544–3552. doi: 10.1161/01.CIR.0000148825.99184.50. [DOI] [PubMed] [Google Scholar]
- Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab. 2002;22:631–647. doi: 10.1097/00004647-200206000-00001. [DOI] [PubMed] [Google Scholar]
- Jakob U, Gaestel M, Engel K, Buchner J. Small heat shock proteins are molecular chaperones. J Biol Chem. 1993;268:1517–1520. [PubMed] [Google Scholar]
- Kato H, Liu Y, Kogure K, Kato K. Induction of 27-kDa heat shock protein following cerebral ischemia in a rat model of ischemic tolerance. Brain Res. 1994;634:235–244. doi: 10.1016/0006-8993(94)91926-7. [DOI] [PubMed] [Google Scholar]
- Knauf U, Jakob U, Engel K, Buchner J, Gaestel M. Stress- and mitogen-induced phosphorylation of the small heat shock protein Hsp25 by MAPKAP kinase 2 is not essential for chaperone properties and cellular thermoresistance. EMBO J. 1994;13:54–60. doi: 10.1002/j.1460-2075.1994.tb06234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostenko S, Moens U. Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell Mol Life Sci. 2009;66:3289–3307. doi: 10.1007/s00018-009-0086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan CY, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ, Dong C, Bao J, Banasiak KJ, Haddad GG, Flavell RA, Davis RJ, Rakic P. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc Natl Acad Sci U S A. 2003;100:15184–15189. doi: 10.1073/pnas.2336254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Lee DH, Cho CK, Bae S, Jhon GJ, Lee SJ, Soh JW, Lee YS. HSP25 inhibits protein kinase C delta-mediated cell death through direct interaction. J Biol Chem. 2005;280:18108–18119. doi: 10.1074/jbc.M501131200. [DOI] [PubMed] [Google Scholar]
- Liu M, Dziennis S, Hurn PD, Alkayed NJ. Mechanisms of gender-linked ischemic brain injury. Restor Neurol Neurosci. 2009;27:163–179. doi: 10.3233/RNN-2009-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Nishino K, Nowak TS., Jr Time course and cellular distribution of hsp27 and hsp72 stress protein expression in a quantitative gerbil model of ischemic injury and tolerance: thresholds for hsp72 induction and hilar lesioning in the context of ischemic preconditioning. J Cereb Blood Flow Metab. 2004;24:167–178. doi: 10.1097/01.WCB.0000100853.67976.8B. [DOI] [PubMed] [Google Scholar]
- Ramezani A, Hawley RG. Generation of HIV-1-based lentiviral vector particles. Curr Protoc Mol Biol Chapter. 2002;16 doi: 10.1002/0471142727.mb1622s60. Unit 16.22. [DOI] [PubMed] [Google Scholar]
- Rane MJ, Pan Y, Singh S, Powell DW, Wu R, Cummins T, Chen Q, McLeish KR, Klein JB. Heat shock protein 27 controls apoptosis by regulating Akt activation. J Biol Chem. 2003;278:27828–27835. doi: 10.1074/jbc.M303417200. [DOI] [PubMed] [Google Scholar]
- Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST. CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology. 2000;39:777–787. doi: 10.1016/s0028-3908(00)00005-8. [DOI] [PubMed] [Google Scholar]
- Sharlow ER, Giridhar KV, LaValle CR, Chen J, Leimgruber S, Barrett R, Bravo-Altamirano K, Wipf P, Lazo JS, Wang QJ. Potent and selective disruption of protein kinase D functionality by a benzoxoloazepinolone. J Biol Chem. 2008;283:33516–33526. doi: 10.1074/jbc.M805358200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler RA, Cao G, Gao Y, Zhang F, Wang S, Weng Z, Vosler P, Zhang L, Signore A, Graham SH, Chen J. Hsp27 protects against ischemic brain injury via attenuation of a novel stress-response cascade upstream of mitochondrial cell death signaling. J Neurosci. 2008;28:13038–13055. doi: 10.1523/JNEUROSCI.4407-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler RA, Gao Y, Signore AP, Cao G, Chen J. HSP27: mechanisms of cellular protection against neuronal injury. Curr Mol Med. 2009;9:863–872. doi: 10.2174/156652409789105561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler RA, Gan Y, Zhang W, Liou AK, Gao Y, Cao G, Chen J. Heat shock proteins: cellular and molecular mechanisms in the central nervous system. Prog Neurobiol. 2010;92:184–211. doi: 10.1016/j.pneurobio.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thériault JR, Lambert H, Chávez-Zobel AT, Charest G, Lavigne P, Landry J. Essential role of the NH2-terminal WD/EPF motif in the phosphorylation-activated protective function of mammalian Hsp27. J Biol Chem. 2004;279:23463–23471. doi: 10.1074/jbc.M402325200. [DOI] [PubMed] [Google Scholar]
- van der Weerd L, Tariq Akbar M, Aron Badin R, Valentim LM, Thomas DL, Wells DJ, Latchman DS, Gadian DG, Lythgoe MF, de Belleroche JS. Overexpression of heat shock protein 27 reduces cortical damage after cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:849–856. doi: 10.1038/jcbfm.2009.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss OH, Batra S, Kolattukudy SJ, Gonzalez-Mejia ME, Smith JB, Doseff AI. Binding of caspase-3 prodomain to heat shock protein 27 regulates monocyte apoptosis by inhibiting caspase-3 proteolytic activation. J Biol Chem. 2007;282:25088–25099. doi: 10.1074/jbc.M701740200. [DOI] [PubMed] [Google Scholar]
- Whitlock NA, Agarwal N, Ma JX, Crosson CE. Hsp27 upregulation by HIF-1 signaling offers protection against retinal ischemia in rats. Invest Ophthalmol Vis Sci. 2005;46:1092–1098. doi: 10.1167/iovs.04-0043. [DOI] [PubMed] [Google Scholar]
- Williams KL, Mearow KM. Phosphorylation status of heat shock protein 27 influences neurite growth in adult dorsal root ganglion sensory neurons in vitro. J Neurosci Res. 2011;89:1160–1172. doi: 10.1002/jnr.22634. [DOI] [PubMed] [Google Scholar]
- Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Rozengurt E. PKD, PKD2, and p38 MAPK mediate Hsp27 serine-82 phosphorylation induced by neurotensin in pancreatic cancer PANC-1 cells. J Cell Biochem. 2008;103:648–662. doi: 10.1002/jcb.21439. [DOI] [PubMed] [Google Scholar]
- Zhao X, Strong R, Zhang J, Sun G, Tsien JZ, Cui Z, Grotta JC, Aronowski J. Neuronal PPARγ deficiency increases susceptibility to brain damage after cerebral ischemia. J Neurosci. 2009;29:6186–6195. doi: 10.1523/JNEUROSCI.5857-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]