

Abstract
Bacterial artificial chromosome (BAC) libraries are critical for identifying full-length genomic sequences, correlating genetic and physical maps, and comparative genomics. Here we describe the utilization of the Fluidigm access array genotyping system in conjunction with KASPar genotyping technology to identify individual BAC clones corresponding to specific single-nucleotide polymorphisms (SNPs) from an Amplicon Express seven-plate super pooled Amaranthus hypochondriacus BAC library. Ninety-six SNP loci, spanning the length of A. hypochondriacus linkage groups 1, 2, and 15, were simultaneously tested for clone identification from four BAC super pools, corresponding to 28 384-well plates, using a single Fluidigm integrated fluidic chip (IFC). Forty-six percent of the SNPs were associated with a single unambiguous identified BAC clone. PCR amplification and next-generation sequencing of individual BAC clones confirmed the IFC clone identification. Utilization of the Fluidigm Dynamic array platform allowed for the simultaneous PCR screening of 10,752 BAC pools for 96 SNP tag sites in less than three hours at a cost of ~$0.05 per reaction.
1. Introduction
Sequence-based molecular markers (e.g., SNPs), genetic linkage maps, and expressed sequence tagged libraries are important molecular tools needed for advanced genomic studies. One genomic tool of particular importance has been BAC libraries (large insert DNA libraries). BAC libraries are critical for identifying full-length genomic sequences, correlating genetic and physical maps [1], comparative genomics [2] and traditionally were the first step towards whole genome sequencing projects [3]. BAC libraries have been successfully developed for numerous species, including economically important crop species, secondary/emerging crop species, and model organisms [4–8].
Traditionally, the identification of specific BAC clones corresponding to specific DNA sequences (aka BAC library screening) was accomplished by probing high-density nylon membranes (single or double) spotted with individual BAC clones representing the entire or portions of the BAC library. This hybridization method, while reliable, requires the problematic use of radioactively labeled probes. If many probes are required to be screened, for example, to establish a connection between a linkage map and a physical map, either multiple copies of the spotted library are required for simultaneous screening (increased cost) or the library must be probed serially (significant time requirement), taking care that the blots are not exhausted before all probes have been screened—normally 3–5 hybridization events. Moreover, the presence of repeat elements in the labeled probe themselves often confounds the hybridization results [9].
PCR-based screening of BAC libraries is an attractive alternative to hybridization-based screening, since the PCR screening can be significantly cheaper, and the efficiency of the PCR-based screening can be significantly enhanced by dimensional pooling of the BAC library [1]. In the dimensional pooling scheme, clones are often pooled by plate, then by row and lastly by column. PCR screening of the different pools allows for the identification of specific BAC addresses (plate, row, and column) containing the corresponding sequence tag site (STS) [10].
The genus Amaranthus (Caryophyllales: Amaranthaceae) encompasses about 60 species of worldwide distribution [11]. The grain amaranths (A. hypochondriacus L., A. cruentus L., and A. caudatus L.) produce edible seeds and are an important food crop in several areas of Latin America and Africa [12]. Amaranth seed protein has an exceptional balance of amino acids and an average seed protein content (15% on a dry matter basis) that is notably higher than most cereal grains [13, 14]. Despite the relative minor status of the grain amaranths as an alternative crop, important genomic tools are being developed that should aid in the genetic improvement of the species. These tools include the development of (i) molecular markers (RAPDs, AFLPs, microsatellites, and SNPs) used to resolve taxonomic questions [15–20], (ii) a 10X BAC library utilized for genomic sequencing of herbicide target genes [21], (iii) a densely populated SNP-based linkage map needed to facilitate quantitative trait loci (QTL) discovery experiments [22], and (iv) a deeply sequenced transcriptome generated from stress response leaf and stem tissues [23].
In this study, we describe a proof-of-concept approach that utilizes the Fluidigm IFC technology (96.96 Dynamic Array) with an Amplicon Express seven-plate super pooled dimensional library of an A. hypochondriacus BAC library [21] to accomplish a high-throughput screening of SNP loci from the recently released A. hypochondriacus linkage map [22].
2. Materials and Methods
2.1. BAC Library
The amaranth BAC library utilized consisted of 36,864 clones and was constructed with a HindIII partial digestion of the A. hypochondriacus cultivar “Plainsman” [21]. The average insert size of the library was 125 Kb and the genome coverage was estimated at 10.6X. In this proof-of-concept experiment, a subset consisting of the first 10,752 clones (28 384-plates) was used in the pooling strategy, providing coverage of 3.4X genome equivalents.
2.2. BAC DNA Isolation
BAC DNA isolation and pools were produced by Amplicon Express (Pullman, WA). Each BAC clone was grown independently in 2X YT broth for 16 h at 37°C with 12.5 ug/mL chloramphenicol. Following the growth period, equal quantities of culture for each of 7 plates were pooled as described below. BAC plasmid DNA was isolated using an optimized alkaline lysis method and resuspended in TE and then pooled to create the super pools as described by Bouzidi et al. [24], with the minor modification that the DNA was suspended at high concentration (20 ng/uL).
2.3. Pooling Strategy
Twenty-eight 384-well plates were pooled into four distinct super pools (SP1-4). For each super pool, BAC DNA was pooled into five pools corresponding to each of seven 384-well plates, 8 pools corresponding to each of the 384-well plate rows of all seven plates, and 10 pools corresponding to each of the 384-well plate columns of all seven plates (Figure 1). For each super pool, each plate of the seven plates in the super pool is found in two wells, each row of the seven plates is found in two wells and each column of the 7 plates is found in two wells. The final 96-well plate consisted of four super pools (Figure 1).
Figure 1.
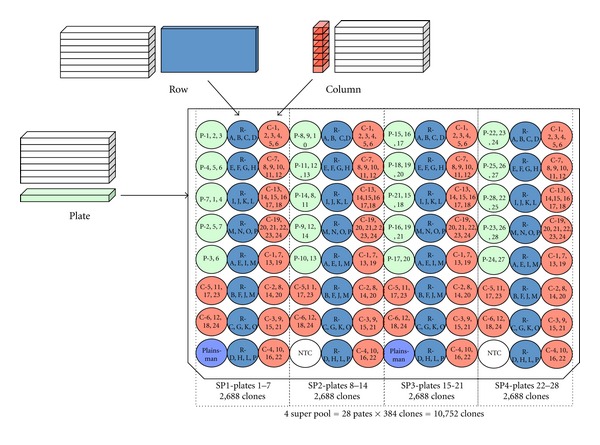
BAC pooling design. A total of 10,752 BAC clones were arranged into four super pools, each consisting of seven BAC plates (384 wells) pooled by plate, row, and column. Each of the plate, row, and column pools was present in two wells of the super pool to allow for target address determination.
2.4. Screening of the BAC Library
The BAC pooled library was screened simultaneously with 96 PCR-based SNP assays. The 96 SNP assays, including primer sequences and genetic locations, were described by Maughan et al. [22]. The assays are based on competitive allele-specific PCR KASPar chemistry (KBioscience Ltd., Hoddesdon, UK) and were performed on a Fluidigm (Fluidigm Corp., South San Francisco, CA) nanofluidic 96.96 dynamic array [25]. For PCR on the 96.96 IFC using the KASPar chemistry, a 5 μL sample mix, consisting of 2.25 μL BAC DNA (20 ng/μL), 2.5 μL of 2X KASP reagent Mix (KBioscience Ltd.) and 0.25 μL of 20X GT sample loading reagent (Fluidigm Corp., South San Francisco, CA) was prepared for each BAC DNA sample. Similarly, a 4 μL 10X KASP Assay, containing 0.56 μL of the KASP assay primer mix (allele-specific primers 12 μM, common reverse primer 30 μM), 2 μL of 2X Assay Loading Reagent (Fluidigm Corp., South San Francisco, CA), and 1.44 μL DNase-free water were prepared for each SNP assay. The assay mix and sample mix were then loaded onto a 96.96 dynamic array chip, mixed, and thermal-cycled using an IFC Controller HX and FC1 thermal cycler (Fluidigm Corp., South San Francisco, CA) according to the manufacturer's protocol. Thermal cycling consisted of an initial thermal mix cycle (70°C-30 min; 25°C-10 min) a hot-start Taq polymerase activation step (94°C-15 min), followed by a touchdown amplification protocol as follows: 10 cycles of 94°C for 20 sec, 65°C for 1 min (decreasing 0.8°C per cycle), 26 cycles of 94°C for 20 sec, 57°C for 1 min; hold at 20°C for 30 sec.
2.5. BAC Clone Deconvolution and Verification
End-point fluorescent images of the 96.96 IFC were acquired on an EP-1 imager (Fluidigm Corp., South San Francisco, CA). The data was analyzed with Fluidigm SNP genotyping Analysis Software. Amplification patterns were used to identify candidate BAC clones from the positive amplification data. Verification of candidate BAC clones was accomplished using standard PCR technology and through sequencing using standard protocols for 454-pyrosequencing as a service at the Brigham Young University DNASC (Provo, UT) using a Roche-454 GS FLX instrument, MID barcoding for each BAC clone, and Titanium reagents (Branford, CT). DNA for PCR and 454-pyrosequencing was obtained using a Sigma PhasePrep BAC DNA kit Sigma-Aldrich, St. Louis, MO) according to the manufacturer's protocol.
3. Results and Discussion
3.1. Screening Strategy
The high-throughput screening method employed here incorporates a dimensional pooled BAC library from A. hypochondriacus screened with the Fluidigm 96.96 Dynamic Array. We chose a seven-plate super pooling strategy (as opposed to a deeper pooling strategy of 10 or 12 plates) based on the need for maximizing genome coverage, while minimizing the complexity of the pools to facilitate deconvolution of candidate BAC clone addresses. The A. hypochondriacus BAC library used was described by Maughan et al. [21] and consists of 36,864 clones with an average insert size of 125 kb, 6.9% contamination with extranuclear DNA (chloroplast and mitochondrial), and approximately 1.8% of the clones containing empty vectors. Taking this information into account, we calculated that a 3.4X genome equivalent sublibrary, consisting of 10,752 clones, would result in a 92.8% probability that any specific DNA target sequence screened would be present in the sublibrary (P = 1 − e((N∗ln(1−(i/GS)))), where P is the probability; N is the number of clones; i is the average insert size of clones and GS is the haploid genome size). The 3.4X genome equivalency also suggested that smany of the positive clones could be deconvoluted to specific BAC clone addresses. We note that if a single BAC super pool contains more than one positive clone for the target sequence, the exact address of the positive candidate BAC clone cannot be unambiguously ascertained.
3.2. PCR Screening
The high-throughput screening of the pooled library employed a Fluidigm 96.96 Dynamic Array genotyping platform using KASPar genotyping chemistry. The Fluidigm platform uses an integrated fluidic chip to create 9,216 9.6 nL PCR reactions. Thus four super pool libraries, each consisting of 23 pooled samples (Figure 1), could be screened simultaneously with 96 STS targets. We also included two positive controls (genomic DNA from the cultivar “Plainsman”) and two no template controls (NTCs). Setup of the IFC can be accomplished with an 8-channel multichannel pipettor in less than 1 hour. The use of the fluorescence KASPar genotyping chemistry eliminates the need for detection of the amplified PCR product using radiography or electrophoresis, since successful amplification is detected by a fluorescent signal. Thus in our proof-of-concept experiment we screened 10,752 BAC clones, arrayed in four super pools, with 96 SNP assays using a single IFC. The SNP assays utilized represented SNP loci that were distributed across three linkage groups of a recently published A. hypochondriacus linkage map, specifically linkage groups 1, 2, and 15 (Figure 2). The complete list of SNP markers utilized, along with their GenBank accession number, SNP type, and primer sequences can be found in Supplemental Table S1 in Supplementary Material available online at doi: 10.1155/2012/405940. Of the 96 SNP targets, 5 (5.2%) failed to amplify in any of the BAC DNA pools or with the positive genomic DNA control, while 23 (23.9%) amplified in only the positive genomic DNA control sample (no amplification in the BAC pools). This was somewhat surprising since a 3.4X library should have approximately 92% representation of all the genome sequences, suggesting that the reported genome size of A. hypochondriacus may be slightly larger than originally reported (466 Mb/C; Bennett and Smith [26]) or that the BAC library, developed through partial HindIII digestion, may be underrepresented in some genomic regions. Sixty-eight (70.8%) of the clones amplified in one or more of the BAC pool samples, of which 44 (64.7%) could be unambiguously assigned to specific BAC clone library addresses. The remaining 24 (35.3%) could not be assigned to an unambiguous BAC clone address, since two or more positive clones were present in a single super pool. We note that while specific BAC clone addresses could not be identified for these 24 SNP assays, specific plates, and often, specific columns and/or rows could be determined. Representative images of the BAC pool screening results are shown in Figure 3.
Figure 2.
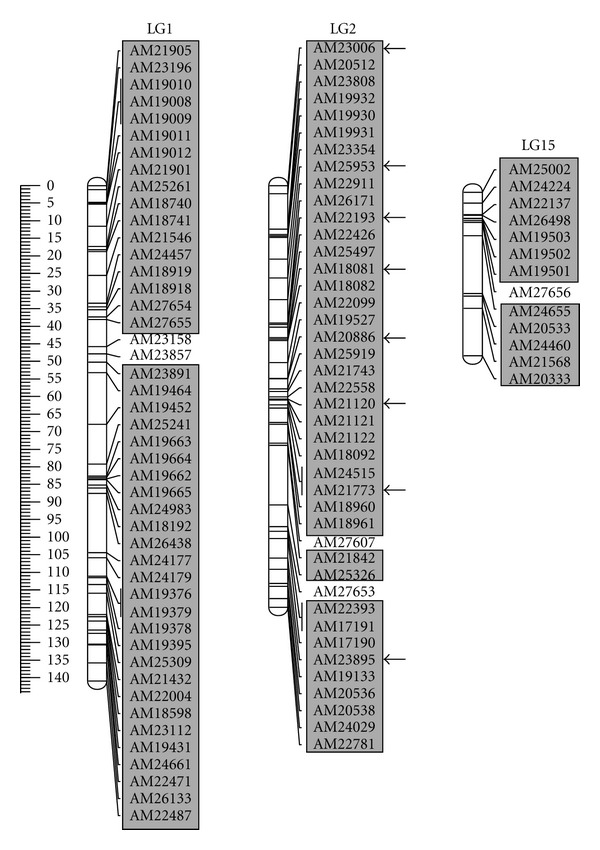
Distribution of the SNP markers screened on linkage groups 1, 2, and 15 [22]. Map distances are in cM, corrected with the Kosambi function. SNP markers in the grey boxes were screened on the BAC library (n = 96). BAC clones corresponding to SNP markers from LG2, indicated with a black arrow, were sequenced using 454-pyrosequencing.
Figure 3.
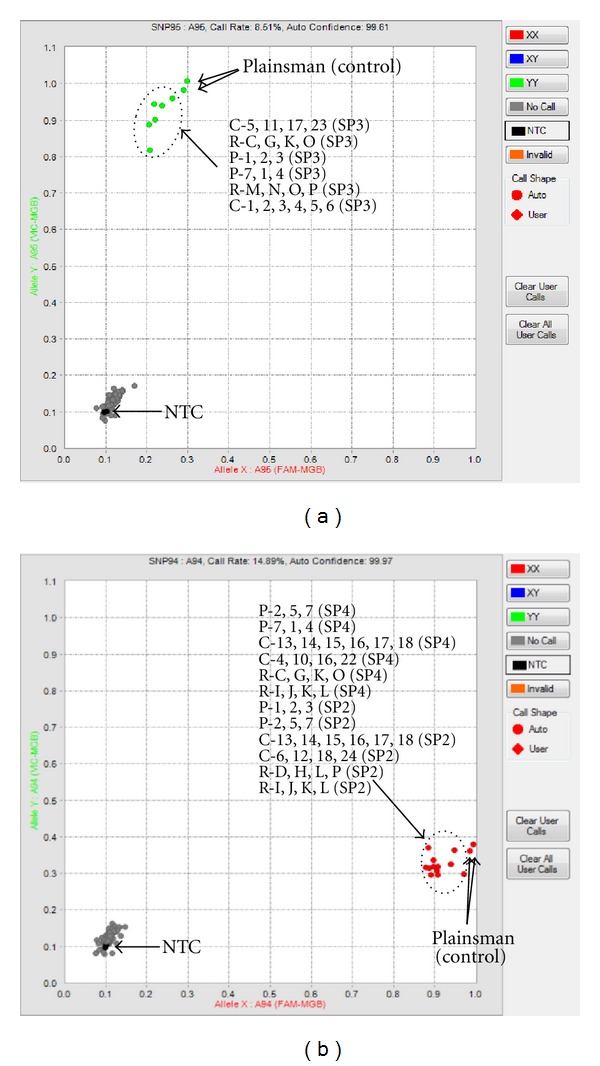
BAC pool screening using the Fluidigm Access Array. (a) shows assay AM25002 which was detected at a single BAC library address, specifically in Plate 1, row O, column 5 of Super Pool x no. 3 (SP3-1O5). (b) shows assay AM24655 which was detected at two library addresses in Super Pools no. 2 (SP2-2L18) and no. 4 (SP4-7K16). No template controls (NTC) are identified at the origin of each Cartesian graph. Genomic DNA of the amaranth cultivar “Plainsman” was used as positive control samples. Successfully amplified pooled BAC clones are circled with a dashed line and are identified as being either Column (C-), Row (R-) or Plate (P-) pools followed by the specific columns, rows or plates utilized in those pools. Pools without the target sequence fail to amplify and are found near the NTC.
3.3. BAC Clone Verification
Standard PCR amplification and next-generation sequencing using 454-pyrosequencing were used to verify the Fluidigm IFC screening of the BAC sublibrary. Of the 44 clones that could be unambiguously assigned to specific BAC clone addresses, all produced specific amplification products of predicted lengths using standard PCR and gel electrophoresis (data not shown), indicating that BAC clones with the correct target sequence had been identified. Furthermore, we 454-pyrosequenced eight BAC clones targeted by eight SNP loci distributed across linkage group 2 as further evidence of the successful identification of targeted BAC clones. Based on the BAC library average insert size, the selected eight BAC clones represented an estimated 1.0 Mb of the A. hypochondriacus genome. The BAC clones, together with their corresponding SNP assay, can be found in Table 1. DNA from each clone was MID-barcoded and pooled in equimolar amounts and sequenced on a quarter portion of a 454-pyrosequencing picotiter plate. A total of 218,786 reads were obtained, producing 82 Mb of total sequence with an average read length of 376 bp. Newbler assembler (v. 2.6) was used to partition reads into their respective barcode pools and to remove the MID-barcode and to trim the sequences of any contaminating BAC vector sequences (pAGIBAC1). Contigs, specific to each BAC, were constructed using the Newbler, and only large contigs (≥500 bp; 81% of all contigs) were used in all subsequent analyzes. The number of contigs assembled for each BAC clone ranged from a low of 4 to a high of 15, with an average N50 Contig size across all BACs of 60,364 Kb. The largest contig assembled was for the BAC clone detected with SNP AM25953, which spanned greater than 147 Kb. The final assembly statistics for each BAC clone are given in Table 1. Verification of the SNP target sequence in the targeted BAC clone was determined using Basic Local Alignment Search Tool (BLASTn), where the SNP marker and its flanking sequence (200 bp) were used as the query sequence and the large contigs from all clones as the search database. For each SNP sequence query, the only significant (E-value < 1E − 10) hit was with a contig corresponding to the specific BAC clone identified via the IFC-based PCR screen of the super pools (Table 1), further verifying the screening methodology. All significant hits spanned the SNP itself, as well as the entire flanking sequence with 100% nucleotide identity. All hits had E-values < 1E − 100 (Table 1).
Table 1.
Targeted BAC clone sequencing. Assembly statistics, including N50 contig size, max contig, and BLASTn E-value correspondence with SNP query sequence.
| Positive BAC clone address | SNP target | BAC Assembly Statistics | BLASTn E-value | |||||
|---|---|---|---|---|---|---|---|---|
| No. of reads | No. of bases | Contigs (>500 bp) | N50 contig Size (bp) | Max contig (bp) | Number of bases | |||
| 16O4 | AM18081 | 28417 | 11346335 | 14 | 52842 | 58444 | 187937 | 9.65E − 101 |
| 124I | AM20886 | 58578 | 21913991 | 15 | 25282 | 57023 | 181556 | 6.68E − 111 |
| 3L14 | AM21120 | 12523 | 4717010 | 8 | 124794 | 124794 | 235849 | 5.36E − 109 |
| 6M9 | AM21773 | 38937 | 14568922 | 15 | 23276 | 54030 | 125585 | 1.90E − 113 |
| 13G11 | AM22193 | 13529 | 4999229 | 15 | 30565 | 40572 | 206077 | 7.58E − 111 |
| 15O11 | AM23006 | 9887 | 3722293 | 9 | 30149 | 66008 | 184367 | 6.79E − 111 |
| 9N2 | AM23895 | 26614 | 9798881 | 4 | 48808 | 56894 | 181431 | 9.33E − 101 |
| 3L19 | AM25953 | 30301 | 11263931 | 7 | 147194 | 147194 | 164652 | 6.10E − 111 |
|
| ||||||||
| Average | 10.9 | 60364 | 75620 | 183432 | ||||
4. Conclusions
We report the development and verification of a high-throughput method for screening BAC libraries for specific DNA sequence tag sites (STSs). In this method we utilize a Fluidigm Access Array platform combined with KBioscience KASPar genotyping chemistry to screen a super pooled BAC library. A single Fluidigm 96.96 IFC is capable of producing 9,216 PCR reactions in a single run (~3 hours) with little technical expertise, and since each PCR reaction is done on a nanoliter scale (9.6 nL), the consumable reagent costs (i.e., Taq polymerase, primers, and IFC chip) are only ~$0.05 for PCR reaction. The seven-plate pooling strategy of the BAC library allowed for the simultaneous screening of 10,752 BAC clones (four super pools, representing 28 384-well plates from the BAC library) with 96 DNA sequence tags sits (here we used SNP tag sites), with nearly 50% of the STS unambiguously identifying specific BAC clone addresses. A significantly greater proportion of unambiguously identified addresses could be obtained by simply increasing the number of seven-plate pools screened—in this proof-of-concept experiment we screened only 4 of the 14 seven-plate pooled libraries possible from our 10.6X (36,864 clone) BAC library. The approach presented here provides a simple and cost-effective method to rapidly and reliably screen a BAC library with multiple (up to 96 simultaneously) sequence tag site. Such STS may represent SNP loci, which could be used to anchor genetic and physical maps, resolve local marker order, and speed assembly and validation of full genome sequences.
Supplementary Material
Supplemental Table S1: provides the SNP marker name, GenBank dbSNP accession ID, polymorphism type, KASParTM primer sequences (A1, A2 and Common Reverse) for all 96 SNP assays screened against the BAC super pooled library using the Fluidigm Dynamic array platform.
Acknowledgments
This research was funded by the Ezra Taft Benson Agriculture and Food Institute. The authors gratefully acknowledge Dr. Edward Wilcox (BYU) for his assistance with 454-pyrosequencing and David Harker (BYU) for his assistance with the BAC clone verification.
Abbreviations
- BAC:
Bacterial artificial chromosome
- SNP:
Single-nucleotide polymorphism
- IFC:
Integrated fluidic chip
- PCR:
Polymerase chain Reaction
- STS:
Sequence tag site.
References
- 1.Yim YS, Moak P, Sanchez-Villeda H, et al. A BAC pooling strategy combined with PCR-based screenings in a large, highly repetitive genome enables integration of the maize genetic and physical maps. BMC Genomics. 2007;8:p. 47. doi: 10.1186/1471-2164-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maughan PJ, Turner TB, Coleman CE, et al. Characterization of salt overly sensitive 1 (SOS1) gene homoeologs in quinoa (Chenopodium quinoa Willd.) Genome. 2009;52(7):647–657. doi: 10.1139/G09-041. [DOI] [PubMed] [Google Scholar]
- 3.Monaco AP, Larin Z. YACs, BACs, PACs and MACs: artificial chromosomes as research tools. Trends in Biotechnology. 1994;12(7):280–286. doi: 10.1016/0167-7799(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 4.Woo SS, Jiang J, Gill BS, Paterson AH, Wing RA. Construction and characterization of a bacterial artificial chromosome library of Sorghum bicolor. Nucleic Acids Research. 1994;22(23):4922–4931. doi: 10.1093/nar/22.23.4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tomkins J, Fregene M, Main D, Kim H, Wing R, Tohme J. Bacterial artificial chromosome (BAC) library resource for positional cloning of pest and disease resistance genes in cassava (Manihot esculenta Crantz) Plant Molecular Biology. 2004;56(4):555–561. doi: 10.1007/s11103-004-5045-7. [DOI] [PubMed] [Google Scholar]
- 6.Stevens MR, Coleman CE, Parkinson SE, et al. Construction of a quinoa (Chenopodium quinoa Willd.) BAC library and its use in identifying genes encoding seed storage proteins. Theoretical and Applied Genetics. 2006;112(8):1593–1600. doi: 10.1007/s00122-006-0266-6. [DOI] [PubMed] [Google Scholar]
- 7.Luo M, Wang YH, Frisch D, Joobeur T, Wing RA, Dean RA. Melon bacterial artificial chromosome (BAC) library construction using improved methods and identification of clones linked to the locus conferring resistance to melon Fusarium wilt (Fom-2) Genome. 2001;44(2):154–162. [PubMed] [Google Scholar]
- 8.Wu C, Sun S, Nimmakayala P, et al. A BAC- and BIBAC-based physical map of the soybean genome. Genome Research. 2004;14(2):319–326. doi: 10.1101/gr.1405004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yim YS, Davis GL, Duru NA, et al. Characterization of three maize bacterial artificial chromosome libraries toward anchoring of the physical map to the genetic map using high-density bacterial artificial chromosome filter hybridization. Plant Physiology. 2002;130(4):1686–1696. doi: 10.1104/pp.013474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruno WJ, Knill E, Balding DJ, et al. Efficient pooling designs for library screening. Genomics. 1995;26(1):21–30. doi: 10.1016/0888-7543(95)80078-z. [DOI] [PubMed] [Google Scholar]
- 11.Sauer JD. Grain amaranths. In: Simmonds NW, editor. Evolution of Crop Plants. London, UK: Longman Group; 1976. pp. 4–7. [Google Scholar]
- 12.Bressani R, Sanchez-Marroquin A, Morales E. Chemical composition of grain amaranth cultivars and effects of processing on their nutritional quality. Food Reviews International. 1992;8(1):23–49. [Google Scholar]
- 13.Bressani R, Elias LG, Garcia-Soto A. Limiting amino acids in raw and processed amaranth grain protein from biological tests. Plant Foods for Human Nutrition. 1989;39(3):223–234. doi: 10.1007/BF01091933. [DOI] [PubMed] [Google Scholar]
- 14.Breene WM. Food uses of grain Amaranth. Cereal Foods World. 1991;36(5):426–430. [Google Scholar]
- 15.Transue DK, Fairbanks DJ, Robison LR, Andersen WR. Species identification by RAPD analysis of grain amaranth genetic resources. Crop Science. 1994;34(5):1385–1389. [Google Scholar]
- 16.Mandal N, Das PK. Intra- and interspecific genetic diversity in grain amaranthus using ramdom amplified polymorphic DNA markers. Plant Tissue Culture and Biotechnology. 2002;12(1):49–56. [Google Scholar]
- 17.Chan KF, Sun M. Genetic diversity and relationships detected by isozyme and RAPD analysis of crop and wild species of Amaranthus. Theoretical and Applied Genetics. 1997;95(5-6):865–873. [Google Scholar]
- 18.Xu F, Sun M. Comparative analysis of phylogenetic relationships of grain amaranths and their wild relatives (Amaranthus; Amaranthaceae) using internal transcribed spacer, amplified fragment length polymorphism, and double-primer fluorescent intersimple sequence repeat markers. Molecular Phylogenetics and Evolution. 2001;21(3):372–387. doi: 10.1006/mpev.2001.1016. [DOI] [PubMed] [Google Scholar]
- 19.Mallory MA, Hall RV, McNabb AR, Pratt DB, Jellen EN, Maughan PJ. Development and characterization of microsatellite markers for the grain amaranths. Crop Science. 2008;48(3):1098–1106. [Google Scholar]
- 20.Maughan PJ, Yourstone SM, Jellen EN, Udall JA. SNP discovery via genomic reduction, barcoding and 454-pyrosequencing in amaranth. Plant Genome. 2009;2(2):260–270. [Google Scholar]
- 21.Maughan PJ, Sisneros N, Luo M, Kudrna D, Ammiraju JSS, Wing RA. Construction of an Amaranthus hypochondriacus bacterial artificial chromosome library and genomic sequencing of herbicide target genes. Crop Science. 2008;48(1):S85–S94. [Google Scholar]
- 22.Maughan P, Smith S, Fairbanks D, Jellen E. Development, characterization, and linkage mapping of single nucleotide polymorphisms in the grain amaranths (Amaranthus sp.) Plant Genome. 2011;4(1):92–101. [Google Scholar]
- 23.Delano-Frier JP, Aviles-Arnaut H, Casarrubias-Castillo K, et al. Transcriptomic analysis of grain amaranth (Amaranthus hypochondriacus) using 454 pyrosequencing: comparison with A. tuberculatus, expression profiling in stems and in response to biotic and abiotic stress. BMC Genomics. 2011;12:p. 363. doi: 10.1186/1471-2164-12-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouzidi MF, Franchel J, Tao Q, et al. A sunflower BAC library suitable for PCR screening and physical mapping of targeted genomic regions. Theoretical and Applied Genetics. 2006;113(1):81–89. doi: 10.1007/s00122-006-0274-6. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Lin M, Crenshaw A, et al. High-throughput single nucleotide polymorphism genotyping using nanofluidic Dynamic Arrays. BMC Genomics. 2009;10:p. 561. doi: 10.1186/1471-2164-10-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett MD, Smith JB. Nuclear dna amounts in angiosperms. Philosophical transactions of the Royal Society of London B. 1976;274(933):227–274. doi: 10.1098/rstb.1976.0044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1: provides the SNP marker name, GenBank dbSNP accession ID, polymorphism type, KASParTM primer sequences (A1, A2 and Common Reverse) for all 96 SNP assays screened against the BAC super pooled library using the Fluidigm Dynamic array platform.