

Glutathione S-transferases (GSTs) represent a large group of enzymes found in organisms ranging from prokaryotes to mammals (for a primary review of GSTs, see ref. 1). GSTs perform important functions in the detoxification of both endogenous and exogenous xenobiotic compounds and in protection against oxidative stress, cancer, and other degenerative diseases, including many diseases associated with aging. In addition to their enzymatic functions, GSTs have been found to bind a large number of compounds including steroids and carcinogens, to act as regulatory proteins, and even to serve in structural roles (S-crystallins). The fundamental chemistry performed by GSTs involves the peptide glutathione (GSH, γ-glutamylcysteinylglycine) in a nucleophilic attack on an electrophilic substrate (Scheme S1). Their protective roles in the cell derive from the fact that the resulting GSH conjugates are more soluble than the original substrates and hence more easily exported from the cell. In some organisms such as prokaryotes, GST superfamily enzymes use the basic GST chemistry to perform more complex reactions associated with biodegradation of persistent environmental pollutants, including halogenated aromatic compounds (2).
Scheme 1.

Over 400 different GST sequences have now been determined [Pfam, release 5.4, June, 2000 (3)] representing at least 25 divergent classes (4, 5). High-resolution structures, both unliganded and bound to relevant ligands or inhibitors, are also available for the best studied classes and provide a wealth of information important to understanding structure–function relationships. All known GST structures exhibit a two-domain fold consisting of an N-terminal thioredoxin fold domain and a more divergent four-helix bundle fold domain (Fig. 1). The thioredoxin fold domain contains the principal determinants for GSH binding, whereas the C-terminal domain provides the primary structural elements associated with the second substrate specificity. The different classes of GSTs are divergent, exhibiting only ≈20–30% sequence identity across the most structurally similar of the mammalian enzymes, the α, μ, and π classes. Important class-specific idiosyncrasies include a so-called “μ loop” common to all of the μ class enzymes and an extra helix, the α9 helix, common to all α class GSTs. It has been suggested that the location of these structural elements adjacent to the substrate-binding sites contributes to formation of somewhat constricted active-site architectures for the α and μ classes compared with the π, σ, and τ classes (1).
Figure 1.
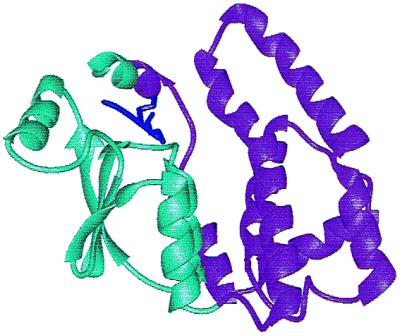
Structure of the A4–4 α-class GST showing the GST-binding domain (cyan) and the second substrate-binding domain (violet). The α carbon trace of the GSH moiety of the inhibitor, 2-(2-iodobenzyl) GSH is shown in dark purple [adapted from Protein Data Bank accession no. 1GUL (13)].
Because of their versatility for reacting with literally thousands of compounds (6), GSTs have been the subject of intense scrutiny aimed at understanding the design paradigms involved. During the last decade, delineation of important structural differences among the classes (1, 7–9) has laid the groundwork for experiments involving both rational and arational† redesign of these enzymes to create new biocatalysts of differing specificities. Although these attempts have been useful in identifying structural elements that appear to be important for GST function and/or specificity, none have approached the success that has been achieved in reengineering other superfamily scaffolds [see Tobin et al. (10) and Cedrone et al. (11) for recent reviews].
In a recent issue of PNAS, Nilsson, Gustafsson, and Mannervik describe the successful redesign of an α-class GST for a new specificity (12). The goal was to reengineer the human α-class A1–1 GST, which shows high catalytic activity (kcat/Km (M−1⋅s−1) = 1.6 × 105) with 1-chloro-2–4,dinitrobenzene (CDNB) but much lower efficiency with alkenal substrates, e.g., kcat/Km (M−1⋅s−1) ≤ 5 × 103 for conjugation with the alkenal, nonenal. By using structural comparisons with an alkenal-specific enzyme in the α class, A4–4 GST, these investigators altered the A1–1 active site to make it more A4–4 like. The resulting mutants exhibit a greatly increased activity with alkenals, including one variant in which kcat/Km (M−1⋅s−1) = 1.52 × 106 for nonenal. This activity is ≈3-fold better than the catalytic efficiency for this substrate of the wild-type A4–4 GST itself. The reengineered A1–1 alkenal activity is also specific, with the most successful mutant exhibiting a 20-fold drop in catalytic efficiency in the CDNB reaction. The change in substrate specificity is accompanied by the concomitant change in chemical mechanism (Scheme S2), from an aromatic nucleophilic substitution reaction (A1–1 with CDNB) to a Michael addition [A4–4 with alkenals, shown using 4-hydroxynon-2-enal as the substrate in Scheme S2]. Although many previous attempts with both rational and arational design strategies have been made to tailor the active sites of GSTs for new specificities, none have achieved such striking success.
Scheme 2.

The rational design approach developed for this experiment was based on a detailed analysis of structure–function relationships in these two well-studied GSTs. Comparisons of the A1–1 (7) and A4–4 structures (13) with each other and with other classes of GSTs reveal some of nature's design principles that appear to be especially important for understanding specificity in the GSTs in general and the α class in particular. (i) The GST superfamily represents an example of nature-designed combinatorics. Here, the architectural paradigm is represented by a two-domain structure in which the first domain provides the most conserved elements of the active site responsible for common chemistry across the superfamily, whereas the second domain provides the variations within the second substrate-binding site required to provide versatility (Fig. 1). A superposition of the A1–1 and A4–4 GST structures is provided in Fig. 2 Left. The α carbons of the regions of the proteins that superimpose best (≤1.0 Å rms deviation), shown in red, include the GSH-binding site (“below” the α carbon trace of GSH) and portions of the α9 helix (directly “above” GSH in the figure). The regions that superimpose less well, shown in green (A1–1) and yellow (A4–4), respectively, include the majority of the residues that were replaced in the A1–1 scaffold with the analogous residues from A4–4. Analysis of several superfamilies in other fold classes mirror this general theme, that nature has evolved a wide range of structural scaffolds to be similar with respect to their delivery of common elements of chemistry and to be different, sometimes substantially, with respect to substrate specificity and the overall chemical functions they can mediate [see Gerlt and P.C.B. (14) for a short review of some of these superfamilies]. (ii) At first approximation, the GST scaffold can be viewed as having been designed by nature as a modular architecture in which specificity determinants can be varied without disruptive distortions in GSH binding and catalytic function. For the GSTs, this observation is supported by extensive sequence and structural analysis across highly divergent classes and by the recognition that the fold type represented by the GST-binding domain module has apparently been used in many other superfamilies of the thioredoxin fold, including thioredoxins, glutaredoxins, protein disulfide isomerases, and GSH peroxidase (15). (iii) In GSTs, the fairly straightforward modification of first sphere interactions alone can achieve variation in specificity. Although the authors caution that their results should not be generalized to infer that second-sphere or more remote mutations will not be required to achieve similar results in other superfamily architectures, it is noteworthy that recent redesign involving primarily first-sphere structural elements in an α/β barrel fold enzyme also achieved highly successful results (16).
Figure 2.

(Left) α-carbon superposition of A1–1 GST (7) and A4–4 (GST) (13). Shown in red are the regions of the trace in which the α carbons from each structure superimpose at ≤1.0 Å rms deviation. Regions of the two structures that deviate more than this are shown in yellow (A4–4) and green (A1–1). The position of the GSH moiety as generated from the coordinates of the A4–4 structure liganded with 2-(2-iodobenzyl) GSH (13) is shown in cyan. The regions of the two structures that were targeted for substitution in the engineering experiment, the α9 helix, the α4 helix, and the β1-α1 loop, are labeled. The superposition was generated by using the minrms algorithm developed at the University of California, San Francisco, Computer Graphics Laboratory. (Right) Closeup view of the superposition shown on the Left. The side chains of residues considered to be most important for the rational design of an alkenal-conjugating A1–1 GST mutant are shown.
In the absence of high-resolution structural information for the reengineered A1–1 enzyme, it is difficult to predict how either the active site or the overall structural scaffold may have been altered by these changes. However, some simple observations can be made by using the structural superposition of the A1–1 and A4–4 wild-type enzymes (Fig. 2). The mutations made to generate an alkenal-active GST A1–1 enzyme involve three primary changes in the substrate-binding site (Fig. 2 Right). First, three residues at the end of the α4 helix, which lines the outermost portion of the “right” side of the active site, L107, L108, and V111 of the A1–1 structure (green), were substituted with the analogous residues from the A4–4 structure, I107, M108, F111 (yellow). These substitutions would likely result in a constriction of that side of the active-site relative to the wild-type A1–1 template. A second set of substitutions involves replacement of the α9 helix and the two residues N-terminal of that helix, residues 208–222 of A1–1, with the corresponding residues from the A4–4 sequence. Important changes associated with this substitution include M208P, S212Y, and possibly, A216V (not shown). These changes are also likely to result in further constriction of the active site, pushing the top of the active site “box” down toward the GSH-binding position. The third change in the A1–1 active site, A12G, is required to compensate for the introduction of the longer and bulkier Y212 for S212. Mutagenesis and structural studies of the A4–4 had previously shown that Y212 is required for activity toward alkenals and that it is positioned appropriately in the structure to polarize the alkenal substrate, thus aiding in the Michael addition of GSH (13). In addition to making room for the introduction of the bulky Y212, this A12G substitution also could result in a shift of the active-site “box” downward relative to the wild-type A1–1 structure.
Nilsson et al. note that the activity of the mutant A1–1 is approaching the highest values known for any GST with any substrate and therefore is unlikely to be substantially improved by further reengineering. Thus, the simple message from their study suggests that these few first sphere changes are sufficient to achieve major, predictable alterations in specificity and catalysis. The substitutions that were made represent only ≈6% of the wild-type A1–1 molecule and were chosen by using a rational design strategy that recognized the critical need for a tyrosine at position 212 and a remodeled active-site “box” likely to be slightly more constricted and “closer in” to the GSH-binding position than that of the wild-type A1–1. Because the natural divergence between the wild-type A1–1 and A4–4 enzymes is considerable (only 53% sequence identity), the success of this strategy might seem surprising. The surprise is lessened somewhat, however, when the results are viewed in the context of another recent enzyme engineering experiment detailed by Altamirano et al. (16) in which phosphoribosylanthranilate isomerase activity was grafted into the indoleglycerol-phosphate synthase scaffold with only 10% of the amino acids changed.
The success of both the Nilsson et al. and the Altamirano et al. experiments raises interesting questions for understanding protein engineering in more general terms. For example, is the simple answer sufficient or will structural characterization of the evolved mutants reveal important consequences of these mutations remote from the active site? It is interesting to note, as shown in Fig. 2 Left, substantial structural differences between wild-type A1–1 and A4–4 GSTs outside of the active site (colored yellow and green in Fig. 2). Are these differences important to function, including the determinants of specificity and chemical mechanism, or are they simply the result of neutral drift?
In addition to structural characterization, answers to these questions may require additional arational engineering by using the A1–1/A4–4 systems. Especially pertinent to this issue is the ongoing debate over the relative contributions of rational vs. arational (or irrational) design for understanding the architectural principles used by nature to design new functions. In a relevant review, Tobin et al. cite some examples in which directed evolution approaches have resulted in improved proteins containing multiple substitutions, some far from the active site, which act synergistically to achieve altered function (10). Further, they assert that such multiple changes could not have been predicted a priori from detailed structural information and rational design strategies. Important examples that support these conclusions include reengineering of aspartate aminotransferase (17) and p-nitrobenzyl esterase (18). In both of these cases, structural characterization of the winning mutants shows that mutations remote from the active sites play a role in the observed substantial reorganization of the active sites relative to the wild-type enzymes. If and when such experiments are performed on the A1–1/A4–4 systems, it will be interesting to see how the results of the rational and arational design efforts may differ and whether other equally successful structural solutions will be found.
Acknowledgments
I thank Mr. Walter R. Novak for generating the structural superpositions shown in Fig. 2. Molecular graphics images were produced using midasplus. This work was funded by National Institutes of Health GM60590 to P.C.B.
Footnotes
See companion article on page 9408 in issue 17 of volume 97.
Protein engineering by using combinatorial methods without benefit of detailed structural and functional information about a template enzyme has sometimes been termed “irrational design” to contrast it to so-called rational approaches. Here, we have used the term “arational design” instead of “irrational design,” a term that seems to connote an almost deliberate avoidance of thoughtfulness in the design process.
References
- 1.Armstrong R N. Chem Res Toxicol. 1997;10:2–18. doi: 10.1021/tx960072x. [DOI] [PubMed] [Google Scholar]
- 2.Vuilleumier S. J Bacteriol. 1997;179:1431–1441. doi: 10.1128/jb.179.5.1431-1441.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bateman A, Birney E, Durbin R, Eddy S R, Howe K L, Sonnhammer E L L. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Board P G, Baker R T, Chelvanayagam G, Jermin L S. Biochem J. 1997;328:929–935. doi: 10.1042/bj3280929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snyder M J, Maddison D R. DNA Cell Biol. 1997;16:1373–1384. doi: 10.1089/dna.1997.16.1373. [DOI] [PubMed] [Google Scholar]
- 6.Mannervik B. Adv Enzymol Relat Areas Mol Biol. 1985;57:357–417. doi: 10.1002/9780470123034.ch5. [DOI] [PubMed] [Google Scholar]
- 7.Sinning I, Kleywegt G J, Cowan S W, Reinemer P, Dirr H W, Huber R, Gilliland G L, Armstrong R N, Ji X, Board P G, et al. J Mol Biol. 1993;232:192–212. doi: 10.1006/jmbi.1993.1376. [DOI] [PubMed] [Google Scholar]
- 8.Dirr H, Reinemer P, Huber R. Eur J Biochem. 1994;220:645–661. doi: 10.1111/j.1432-1033.1994.tb18666.x. [DOI] [PubMed] [Google Scholar]
- 9.Wilce M C J, Parker M W. Biochim Biophys Acta. 1994;1205:1–18. doi: 10.1016/0167-4838(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 10.Tobin M B, Gustafsson C, Huisman G. Curr Opin Struct Biol. 2000;10:421–427. doi: 10.1016/s0959-440x(00)00109-3. [DOI] [PubMed] [Google Scholar]
- 11.Cedrone F, Ménez A, Quéméneur E. Curr Opin Struct Biol. 2000;10:405–410. doi: 10.1016/s0959-440x(00)00106-8. [DOI] [PubMed] [Google Scholar]
- 12.Nilsson L O, Gustafsson A, Mannervik B. Proc Natl Acad Sci USA. 2000;97:9408–9412. doi: 10.1073/pnas.150084897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruns C M, Hubatsch I, Ridderström M, Mannervik B, Tainer J A. J Mol Biol. 1999;288:427–439. doi: 10.1006/jmbi.1999.2697. [DOI] [PubMed] [Google Scholar]
- 14.Gerlt J A, Babbitt P C. Curr Opin Chem Biol. 1998;2:607–612. doi: 10.1016/s1367-5931(98)80091-4. [DOI] [PubMed] [Google Scholar]
- 15.Murzin A G, Brenner S E, Hubbard T, Chothia C. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 16.Altamirano M M, Blackburn J M, Aguayo C, Fersht A R. Nature (London) 2000;403:617–622. doi: 10.1038/35001001. [DOI] [PubMed] [Google Scholar]
- 17.Oue S, Okamoto A, Yano T, Kagamiyama H. J Biol Chem. 1999;274:2344–2349. doi: 10.1074/jbc.274.4.2344. [DOI] [PubMed] [Google Scholar]
- 18.Spiller B, Gershenson A, Arnold F H, Stevens R C. Proc Natl Acad Sci USA. 1999;96:12305–12310. doi: 10.1073/pnas.96.22.12305. [DOI] [PMC free article] [PubMed] [Google Scholar]