

Abstract
Dysfunction of basal forebrain cholinergic neurons (BFCNs) is an early pathological hallmark of Alzheimer's disease (AD). Numerous studies have indicated that nerve growth factor (NGF) supports survival and phenotypic differentiation of BFCNs. Consistent with a potential link to AD pathogenesis, TrkA, a NGF receptor, is expressed in cholinergic forebrain neuronal populations including those in BF and striatum, and is markedly reduced in individuals with mild cognitive impairment (MCI) without dementia and early-stage AD. To investigate the role of TrkA in the development, connectivity, and function of the BF cholinergic system and its contribution to AD pathology, we have generated a forebrain-specific conditional TrkA knock-out mouse line. Our findings show a key role for TrkA signaling in establishing the BF cholinergic circuitry through the ERK pathway, and demonstrate that the normal developmental increase of choline acetyltransferase expression becomes critically dependent on TrkA signaling before neuronal connections are established. Moreover, the anatomical and physiological deficits caused by lack of TrkA signaling in BFCNs have selective impact on cognitive activity. These data demonstrate that TrkA loss results in cholinergic BF dysfunction and cognitive decline that is reminiscent of MCI and early AD.
Introduction
The in vivo role of nerve growth factor (NGF) as a target-derived survival factor for sensory and sympathetic neurons is well established (Goedert et al., 1984; Crowley et al., 1994; Chen et al., 2005). Studies with mice lacking both Bax and NGF or TrkA, the NGF high-affinity receptor, have shown that NGF/TrkA signaling plays a key role in peripheral target field innervation (Patel et al., 2000). Still, the functions of NGF and its receptors in the CNS are poorly understood. TrkA mRNA and protein expression in CNS is restricted to limited neuronal populations in the forebrain that include cholinergic neurons in basal forebrain (BF) and striatum (Sobreviela et al., 1994). Most studies on NGF signaling have focused on BF cholinergic neurons (BFCNs) because of their important role in cognition and attention behaviors, which have important implications in aging and Alzheimer's disease (AD) (Holtzman et al., 1995).
One of the earliest pathological events in AD is dysfunction of BFCNs (Mufson et al., 2008); however, the molecular and cellular mechanisms underlying this pathology have not been elucidated. Retrograde transport of NGF to the BF is critical for its neurotrophic effects (Schwab et al., 1979). Notably, BFCN survival is supported, in part, by NGF (Honegger and Lenoir, 1982; Hefti, 1986), which is synthesized in the target tissues of cholinergic neurons such as the cortex and hippocampus. In addition, there is a marked reduction in TrkA-positive BFCNs and decreased levels of TrkA mRNA and protein in postmortem brains of AD patients (Salehi et al., 1996; Mufson et al., 1997), and in individuals with mild cognitive impairment (MCI) without dementia (Chu et al., 2001; Ginsberg et al., 2006). This is not accompanied by a decrease in the pan-neurotrophin receptor p75, indicating specificity for TrkA downregulation in association with cognitive decline.
Whether TrkA function is indeed relevant in AD pathogenesis and in the development or function of BFCNs remains unclear. Studies with homozygous null ngf and TrkA mice have implicated NGF/TrkA signaling in regulating normal maturation of BFCNs. However, no definitive conclusions could be drawn about the extent of BFCN survival, function, and dependency on NGF/TrkA signaling because of the poor health and perinatal mortality of these mice (Crowley et al., 1994; Fagan et al., 1997). To bypass these issues, we used a conditional knock-out strategy and generated mice lacking TrkA expression specifically in forebrain cholinergic neurons (TrkAcKO). These mice develop normally with no gross abnormalities. Interestingly, while striatal cholinergic neurons displayed no apparent signaling or morphological defects, BFCNs exhibited decreased choline acetyltransferase (ChAT) expression and ERK signaling, which was accompanied by severe innervation defects. TrkAcKO mice also demonstrated selective attention and working memory impairments. These phenotypes are reminiscent of those observed in MCI and early AD (Levey et al., 2006; Mufson et al., 2008). In this study, we thus provide evidence that TrkA plays a role in the development, connectivity, and function of the BF cholinergic circuitry and discuss its possible implications in disease.
Materials and Methods
Generation and genotyping of TrkA conditional knock-out mice
The TrkA targeting vector was constructed with a loxP site within the promoter region and another in the first intron of TrkA to remove 0.25 kb of promoter sequence immediately 5′ to the transcriptional start site of TrkA and exon 1, which includes the translation initiation site upon Cre recombination (Fig. 1A). Embryonic stem cells derived from 129/SvJ mice were transfected with the targeting construct, and recombinant clones were obtained by neomycin resistance (neoR)-positive selection, and then injected into C57BL/6 blastocysts to generate TrkAflox mice. Upon successful homologous recombination, correctly targeted TrkA allele (trkAneo) had a 43 bp oligonucleotide containing a loxP site inserted into the promoter region and a PGKneoR fragment flanked by FRT and FRT-loxP sequences, respectively, which were inserted into the first intron. Mice heterozygous for the TrkAneo allele were mated with FlpE transgenic mice that express Flp recombinase to bring about Flp-mediated deletion of the neoR cassette and generate animals carrying the TrkAflox allele (Rodríguez et al., 2000). The dlx5/6i-cre mice used in this study were generated in the laboratory of M. Ekker (University of Ottawa, Ottawa, ON, Canada). These transgenic mice express Cre recombinase driven by the enhancer fragment derived from the zebrafish intergenic region between the dlx4 and dlx6 genes, the orthologs of mammalian dlx5 and dlx6. This enhancer has previously been shown to direct Cre expression specifically to the subcortical telencephalon, which later gives rise to cells in the ventral telencephalon, including the BFCNs and the striatum (Zerucha et al., 2000; Kohwi et al., 2007). These transgenic mice were crossed with TrkAflox mice to generate TrkAcKO mice. The TrkA mutant mice were routinely genotyped by PCR using a mix of oligos: TrkA-wt-5′: 5′-TGTACGGCCATAGATAAGCAT-3′; TrkA-wt-3′: 5′-TGCATAACTGTGTATTTCAC-3′; 3202: 5′-CGCCTTCTTGACGAGTTCTTCTG-3′. The PCR conditions are as follows; preheat 94°C for 2 min (94°C for 30 s, 55°C for 45 s, and 72°C for 30 s) × 35 cycles. All mouse protocols were approved by the University of California, San Francisco Institutional Animal Care and Use Committee (IACUC) and the University of Texas Southwestern Medical School IACUC. Both male and female mice were used in all experiments, except for behavioral tests in which male mice were used.
Figure 1.

Generation of TrkAcKO mice. A, Targeting strategy for conditional deletion of TrkA in the forebrain. Diagram shows the TrkA locus and targeted alleles. B, Dlx5/6i-cre-mediated activation of Rosa26R (R26R), as identified by the expression of β-galactosidase (β-gal) from the activated R26R allele. The Dlx5/6i-cre mice were crossed with R26R-LacZ reporter mice. Sagittal section from E16.5 embryonic brain showing LacZ staining in the subcortical region. Sagittal and coronal sections of brains at P30 showing all the cells in the forebrain. Acb, Nucleus accumbens; CPu, striatum. C, Double immunostaining of β-galactosidase and TrkA in MS of Dlx5/6i-cre;Rosa26R. Most of the cells are positive for LacZ staining. Only a few cells are positive for TrkA, all of which are also positive for LacZ. D, H&E-stained coronal brain sections from P30 wild-type and mutant mice showing that TrkAcKO brain is normal in size and has no gross histological defects. E, Quantitative evaluation of septal, striatal, and cortical volume of P30 control and TrkAcKO mice. n = 4. F, Golgi impregnation of P30 control and TrkAcKO coronal brain sections revealed no apparent defects in cortical lamination. Scale bars: C, 100 μm; F, 1 mm.
Histology
Mice were intracardially perfused with ice-cold PBS followed by 4% (w/v) ice-cold paraformaldehyde (PFA) in PBS. For hematoxylin-eosin (H&E) staining and immunohistochemistry, the dissected brains were then post-fixed overnight with 4% PFA at 4°C.
DiI tracing
DiI crystal (total diameter of ∼0.5 mm; Invitrogen) was inserted into the dissected brain at the depth of the medial septum (stereotaxic coordinates:1.2 mm anterior to bregma, 0.0 mm medial lateral, 4.5 mm ventral from dura) with forceps such that the ventral edges of basal forebrain covered the crystal. The brain was then placed in the dark in 2% PFA in PBS at 37°C for 5 weeks. The vibratome sagittal sections (50 μm) were examined for DiI labeling using a fluorescence microscope (Olympus BX50) equipped with a Coolsnap CCD camera (Roper Scientific), and the images were analyzed using ImageJ software (National Institutes of Health).
Golgi staining
Golgi staining was performed as previously described (Glaser and Van der Loos, 1981; Luikart et al., 2005). In brief, mice were perfused with PBS, and the dissected brains were incubated with Golgi-Cox solution for 12 d. Vibratome coronal sections (100 μm) were processed and examined under an Olympus optical microscope. Images were analyzed with ImageJ software.
Immunohistochemistry and antibodies
For DAB staining, the fixed brains were paraffin embedded, coronally sectioned (10 μm), and analyzed using an Olympus BX50 microscope equipped with a color CCD camera (DXM1200; Nikon). For the immunofluorescent staining, 40 μm vibratome sections were analyzed by confocal microscopy (LSM-510; Zeiss). X-gal, DAB, and immunofluorescent staining were performed as previously described (Lei et al., 2005; Luikart et al., 2005). Antibodies used for immunostaining were as follows: anti-TrkA (1:1000; Advanced Targeting Systems); anti-p75 (1:500; Promega); anti-ChAT (1:400; Millipore Bioscience Research Reagents); anti-phospho-ERK (pERK; 1:200; Cell Signaling Technology); anti-phospho-AKT (pAKT; 1:200; Cell Signaling Technology); anti-β-galactosidase (1:1000; Millipore Bioscience Research Reagents); and anti-DARPP-32 (1:200; BD Biosciences). Primary antibodies were visualized by secondary antibodies conjugated with horseradish peroxidase (HRP; 1:200; Santa Cruz Biotechnology), Cy2, and Cy3 (1:400; Jackson Laboratory).
Quantification of cholinergic cell number and size
To quantify cholinergic cell numbers in BF and striatum, six coronal vibratome sections per animal (240 μm apart, bregma: 0.38–1.34 mm) were immunostained for ChAT, TrkA, or p75. Cell counts in BF were restricted to the medial septum nucleus (MS)/vertical nucleus of diagonal band (VDB) region. The MS/VDB area encompasses the triangular region containing ≥95% of cholinergic neurons, is devoid of dopamine and cyclic AMP regulated phosphoprotein of molecular weight 32 kDa (DARPP-32) immunostaining, and starts at the midline, 200 μm below the corpus callosum. It ends at a line drawn 310 μm below and parallel to the anterior commissures. The area of sections that contained MS, but not VDB, were defined as the triangular region beginning at midline, 200 μm below the corpus callosum and ending in a line drawn across the tops of the anterior commissures. The striatum was identified and defined by DARPP-32 immunostaining and by morphological criteria (superior boundary: corpus callosum; lateral boundary: external capsule; medial boundary: lateral ventricle and corpus callosum). Images were analyzed with ImageJ software using the cell counter macro to quantify the ChAT-immunoreactive (IR) cells, and quantification was expressed as cells/area. A minimum of 200 cells were counted per section. To measure cell size of cholinergic neurons, pictures were taken of ChAT-IR neurons at 100× magnification. Two measurements were recorded for each neuronal soma (length and width) and were averaged using ImageJ software.
Quantification of MS/VDB, striatum, and cortex volume
Serial Golgi-stained sections were used to measure the areas of the MS/VDB, striatum, and cortex regions with ImageJ software, from which the volume was calculated. The aforementioned defined morphological criteria were used to evaluate the area of the MS/VDB and striatum. The cortex was evaluated within the same sections and was defined as follows: medial boundary, corpus callosum; lateral boundary, corpus callosum and external capsule; and inferior boundary, anterior commissures. The volume of the striatum and cortex was analyzed in both hemispheres.
Retrograde tracing of BFCNs
The BFCNs were retrogradely traced in vivo by stereotactic injection of 50.6 nl of 20% (w/v) Texas Red-conjugated dextran amine (3000 molecular weight; Invitrogen) in distilled water into the frontal cortex (bregma 0.26, 1.0 mm lateral from midline, 1.0 mm below dura) using a Nanoliter 2000 nanoinjector (World Precision Instruments) at postnatal day 21 (P21). The mice were intracardially perfused at 2 weeks after the injection. Dissected brains were coronally sectioned (40 μm) and analyzed by confocal microscopy.
Western blotting
The basal forebrain including the medial septum, nucleus basalis and diagonal band, as well as the striatum were dissected under a stereoscopic microscope (Leica M420), immediately frozen in liquid nitrogen, and homogenized in lysis buffer containing 1% Triton X-100, 20 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, protease inhibitor mixture (Roche), and 50 mm sodium fluoride. Tissues were then sonicated using Vibra Cell (Sonics & Materials), and insoluble material was removed from the protein extracts by centrifugation at 10,000 rpm for 20 min at 4°C. Protein content in the supernatant was quantified with the BCA assay (ThermoFisher Scientific). Equivalent amounts of total protein (10–25 μg) were separated in either 10% or 4–20% gradient SDS-PAGE gels (Bio-Rad) and transferred to nitrocellulose membranes. Membranes were blocked with Tris-buffered saline containing 0.1% Tween 20 and 5% nonfat milk for 1 h, then incubated with primary antibody to TrkA (1:4000), ChAT (1:1000), p75 (1:1000, rabbit polyclonal; Millipore), DARPP-32 (1:1000; Cell Signaling Technology), phospho-Thr34-DARPP-32 (1:500; Cell Signaling Technology), phospho-ERK1/2 (1:2000, Cell Signaling Technology), ERK1/2 (1:400; Santa Cruz Biotechnology), phospho-AKT (1:1000; Cell Signaling Technology), AKT (1:1000; Cell Signaling Technology), or β-actin (1:5000; Sigma), and were then visualized with appropriate HRP-conjugated secondary antibodies (1:10,000; Santa Cruz Biotechnology) followed by Chemiglow West reagents (Cell Biosciences). Blots were stripped using Re-Blot Plus stripping solution (Millipore) and reprobed with the indicated primary antibodies as indicated in figure legends. Protein levels were determined by band densitometry and normalized to the band intensity values of β-actin for TrkA, ChAT, and DARPP-32; phosphoproteins were normalized to intensity values of the corresponding total protein bands. Band densitometry in blots was performed using ImageJ software.
Behavioral procedures
Morris water maze.
A circular pool (142 cm diameter) was filled with room temperature water to a depth of ∼30 cm. A platform (10 cm diameter) was placed in one quadrant of the pool with the top of the platform ∼2 cm below the water level. White nontoxic paint was added to enhance the contrast with the animal and to hide the location of the platform. Each day the mice were placed in the pool and allowed to swim for 1 min to find the platform. The swim path and time until locating the platform were recorded via video camera and computer using EthoVision (Noldus). If the mouse did not find the platform within 1 min, it was gently guided or placed on the platform for 10 s, then removed from the pool and returned to its home cage. Each animal was placed in the pool for a total of four times each day for 10 d. Immediately following the training days, a probe test was conducted in which the platform was removed from the pool and each mouse was allowed to swim for 1 min to determine whether the animal had learned the location of the platform. To control for visual problems, the mice were given four trials a day for 3 d using the same pool and platform; however, a large black block was placed on top of the platform to clearly mark the location. This control test found no visual problems in any of the mice examined.
Fear conditioning.
The mice were placed in a chamber (31 × 25 × 25 cm; Med Associates) possessing aluminum side walls and Plexiglass rear and front walls with a dim light and constant white noise. The presentation of tone and shock stimuli in all training and testing sessions was controlled by FreezeFrame software (Coulbourn Instruments). Training day: mice were allowed to explore for 120 s, and then a tone [conditioned stimulus (CS)] of 25 kHz and 75 dB was presented for 30 s and coterminated in the last 2 s with a mild footshock (0.5 mA, constant current) [unconditioned stimulus (US)]. Another CS–US pairing was presented twice with 30 s intervals each time, for a total of three CS–US pairings for 5 min. In contextual and cued testing, to test for contextual memory mice were placed in the same chamber 24 h after training and allowed to explore for 4 min without CS or US. Freezing behavior, defined as the complete absence of voluntary movements except for respiratory movement, was observed. Twenty-four hours after the contextual test, the mice were tested for cue memory by returning them to the chamber, which was modified with a plastic floor and a vanilla scent without white noise. Freezing was scored for 2 min without the tone and another 2 min with the tone.
Novel object recognition.
The mice were allowed to explore an empty chamber (70 × 70 × 30 cm) for 10 min per day for 3 d to minimize the disruption, stress, and novelty of handling, and also to become familiarized with the testing environment. In the sample phase, the mice were allowed to interact with the sample objects in the back left and right corner in the chamber. Exploration of an object was defined as directing the nose to the object at a distance of <2 cm and/or touching it with the nose. Turning around or sitting on the object was not considered exploratory behavior. The sample phase ended when the mouse had explored the identical objects for a total of 30 s. Immediately after a 1 h interval, mice were allowed to explore in the chamber with the previously familiarized object in one back corner and the novel object in the other back corner for 3 min. The time spent exploring each object was recorded via video camera to calculate the discrimination ratio b/(a + b), where a is the exploration time of the familiar object, and b is the exploration time of the novel object.
The same cohort of mice was used for all behavioral tests, with littermates used as controls.
Statistical analyses
All data values are presented as the mean ± SEM. At least three littermate mice per genotype were used per experiments, as indicated in figure legends. Student's t tests were applied to data with two groups of samples. One-way ANOVAs were used for comparisons of data with more than two groups and were followed by the Tukey–Kramer post hoc test for significance. A p value of <0.05 was considered statistically significant. Data were analyzed using GraphPad Prism, version 5.04 for Windows.
Results
Generation of forebrain-specific TrkA conditional knock-out mice
A TrkA conditional allele was generated using the Cre/LoxP site-specific recombination system. To ensure complete TrkA inactivation upon exposure to Cre, we placed a loxP site within the promoter region and another in the first intron such that Cre-mediated recombination would remove 0.25 kb of promoter sequence immediately 5′ of the transcriptional start site of TrkA and exon 1, which includes the translation initiation site (Fig. 1A). The targeting construct also contained a neomycin resistance (neoR)-positive selection cassette flanked by two Flp-recombinase target (FRT) sites in the first intron. After correct targeting, the NeoR cassette was removed by germline recombination using FlpE recombinase (Rodríguez et al., 2000), resulting in a TrkA allele modified by insertion of a single loxP site that is inserted 158 bp upstream of the transcription start site and contains single loxP and Frt sites in the first intron without any sequence deletion from the TrkA locus (Fig. 1A).
To investigate TrkA function in BFCN development, we turned to the Dlx5/6i-cre transgenic mouse line (Monory et al., 2006). The enhancer fragment of the Dlx5/6i-cre driver is derived from the zebrafish intergenic region between the Dlx4 and Dlx6 genes, the orthologs of mammalian Dlx5 and Dlx6, respectively. This fragment has been previously shown to direct expression specifically in Dlx expression domains of the forebrain, including parts of the diencephalon and the telencephalon, which contain the BFCNs, the septum, diagonal band, striatum, nucleus accumbens, and cortical and olfactory bulb interneurons that tangentially migrate from the subcortical telencephalon (Zerucha et al., 2000). To verify the Cre expression pattern in Dlx5/6i-cre transgenic mice, they were intercrossed with Rosa26 reporter mice that, upon Cre-mediated recombination, express LacZ. By E16.5, LacZ expression was observed in most of the cells within the subcortical telencephalon (Fig. 1B), long before TrkA expression begins in forebrain cholinergic neurons. In adult brain, LacZ expression was retained in the entire ventral telencephalon and in interneurons of the cortex and olfactory bulb (Fig. 1B). LacZ-expressing cells were also observed in the cortex, representing the cortical interneuron population, and a few scattered LacZ-positive cells were seen in the adult midbrain. Virtually no recombination was detected in the cerebellum or brainstem. Outside the brain, no LacZ expression was detected in the spinal cord, dorsal root ganglia, or sympathetic ganglia (data not shown). To confirm that recombination had occurred in the BF progenitors of TrkA-positive neurons, we performed double-immunofluorescence staining using antibodies against β-galactosidase and TrkA. We found that all TrkA-positive cells also stained for LacZ (Fig. 1C). As previously described (Holtzman et al., 1992; Sobreviela et al., 1994), virtually all of the TrkA-positive neurons colocalized with the cholinergic neuronal marker ChAT (data not shown). Therefore, Dlx5/6icre;TrkAflox transgenic (TrkAcKO) mice undergo Cre-mediated TrkA ablation specifically in forebrain cholinergic neurons. Consequently, these analyses indicate that Dlx5/6i-cre is a suitable Cre-expressing transgenic line to study the role of TrkA in forebrain cholinergic neuron development.
TrkAcKO cholinergic neurons do not express TrkA
Conventional TrkA homozygous knock-out mice die in utero or soon after birth (Smeyne et al., 1994). In contrast, TrkAcKO mice are fertile, have normal growth and weight gain, and do not exhibit any gross abnormalities. The size and weight of TrkAcKO brains are comparable to those of brains from wild-type mice (data not shown). Comparison of H&E-stained coronal brain sections from P30 wild-type and TrkAcKO mice revealed no obvious histological abnormalities in the mutant brains (Fig. 1D). Evaluation of the volume of the MS, striatum, and cortex from P15, P30, and P60 control and mutant mice showed that the volume of these brain regions is unaffected in TrkAcKO mice (Fig. 1E and data not shown). In addition, the cortical lamination of P30 TrkAcKO brains appeared similar to that of control mice (Fig. 1F).
To confirm that TrkA expression in TrkAflox mice is not affected by the presence of the loxP and Frt sites, and that Cre-mediated homologous recombination effectively inactivated TrkA in the forebrain of TrkAcKO mice, coronal brain sections from P10 wild-type, TrkAflox, and TrkAcKO mice were immunostained with a TrkA-specific antibody (Fig. 2A). Brain sections from TrkAflox mice exhibited a wild-type-like TrkA expression pattern in the MS, the VDB, and the horizontal diagonal band (HDB) region. In contrast, TrkA expression in these regions is not detectable in TrkAcKO mice. Examination of TrkA-positive cells indicated that both the neuronal cell numbers and size in the MS/VDB region of TrkAflox mice were indistinguishable from wild type (Fig. 2B). In addition, immunofluorescence staining for TrkA confirmed the absence of TrkA expression in ChAT-positive neurons of TrkAcKO MS sections (Fig. 2C). Similarly, Western blot analysis showed the absence of TrkA protein in the BF and hippocampus of TrkAcKO mice (Fig. 2D,E).
Figure 2.
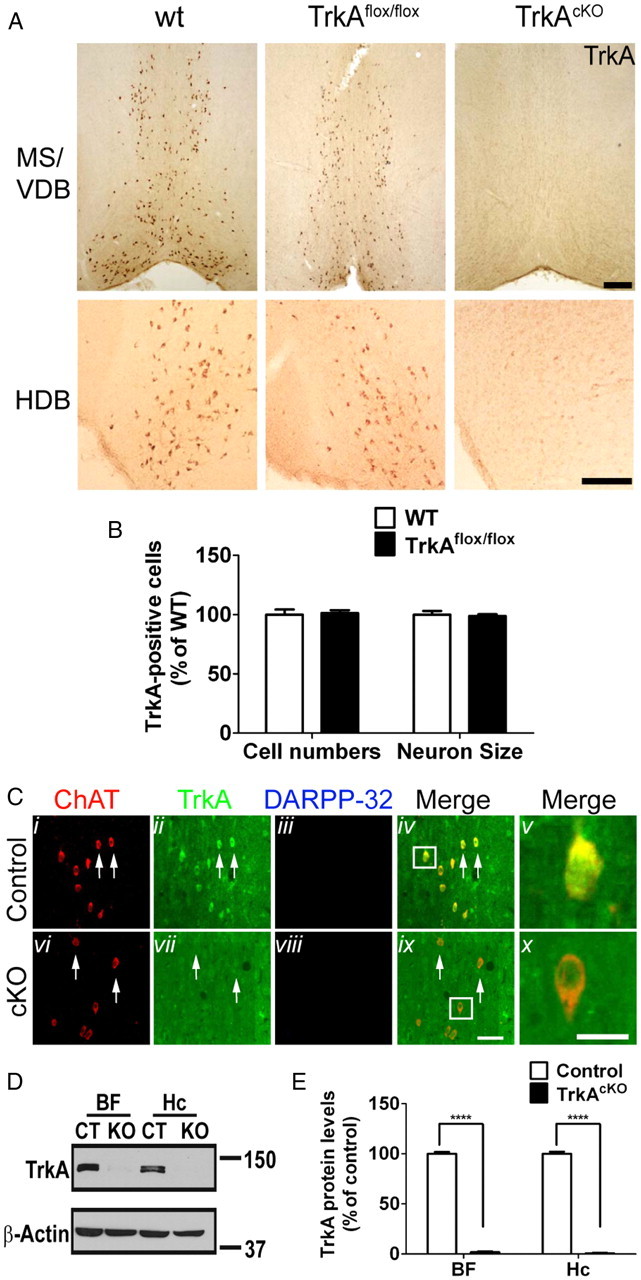
TrkA expression in BF of TrkAcKO mice. A, TrkA expression in wild-type, TrkAflox, and TrkAcKO mice. Neurons in BF of P10 mice were stained with anti-TrkA antibody. B, Analysis of TrkA-positive neuronal cell numbers and size in MS/VDB region. n = 4 mice in cell numbers; n = 17–21 cells in neuron size. C, TrkA is ablated in BFCNs. P30 mice were processed for immunofluorescence using indicated antibodies. Arrows, TrkA-expressing (ii) and TrkA-depleted (vii) BFCNs; in merged pictures, Cv and Cx show enlargement of cells in the boxed areas in Civ and Cix. D, E, Quantitative Western blot analysis of TrkA expression in BF and hippocampus of P30 mutant mice. Molecular weight markers (kDa) are shown. Hc, Hippocampus. ****p < 0.0001 when compared with control; n = 6 per genotype in BF and n = 3 per genotype in Hc group. Scale bars: A, 400 μm; Ci–iv,vi–ix, 50 μm; Cv,x, 25 μm.
In addition to its robust expression in BFCNs, TrkA mRNA and protein have been reported to be expressed in cholinergic interneurons of the striatum (Holtzman et al., 1992; Sobreviela et al., 1994); however, the quality of photomicrographs provided as evidence in those studies was marginal. Thus, we sought to confirm TrkA expression in striatum of wild-type mice by immunofluorescence, using antibodies against ChAT, TrkA, and DARPP-32 in vibratome sections throughout several developmental stages (Fig. 3A). TrkA expression was observed in neurons throughout the striatum and was expressed exclusively in cholinergic, ChAT-positive interneurons, and not in dopaminergic, DARPP-32-positive neurons. This striatal expression pattern of TrkA confirms previous reports (Holtzman et al., 1992, 1995; Steininger et al., 1993; Sobreviela et al., 1994). In addition, TrkA protein levels appear to peak by 2 weeks of age, coinciding with the peak of ChAT protein levels, then start to slowly decrease over time, as opposed to ChAT levels, which remain steady (Fig. 3B–D). Consequently, given the specificity of the Dlx5/6i-Cre expression (Fig. 1B), we examined whether TrkA is ablated in the striatum. In contrast to controls, striatal cholinergic neurons in the TrkAcKO animals contained no detectable TrkA protein as assessed by immunofluorescence studies (Fig. 3E) and Western blot analysis (Fig. 3F,G), indicating that TrkA is also ablated in the striatum of TrkAcKO mice. Therefore, Cre recombinase activity is sufficient to ablate TrkA expression in cholinergic neurons in the striatum as well as in BF.
Figure 3.
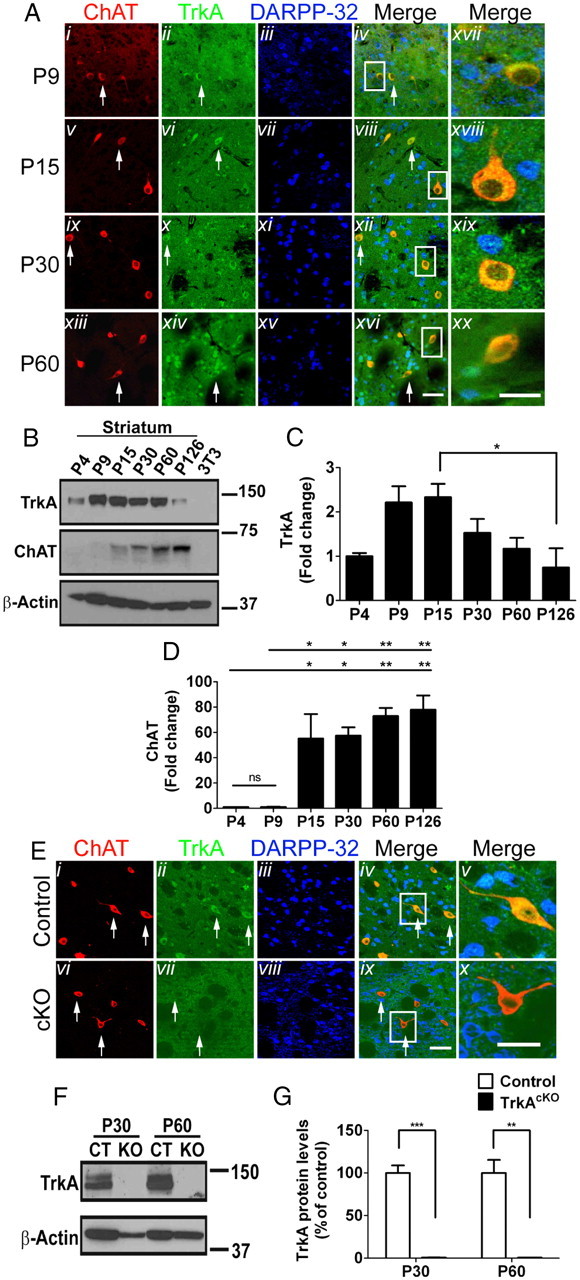
TrkA expression in striatum of wild-type and TrkAcKO mice. A, TrkA expression in striatal cholinergic neurons. Mice were processed for immunofluorescence at the indicated ages. Merged pictures: Axvii–xx show enlargements of cells in boxed area in Aiv, vii, xii, and xvi. B–D, Western blot (B) and quantitative analysis of TrkA (C) and ChAT (D) expression in striatum. 3T3, NIH3T3 lysates were used as negative controls; n = 3 per group. E, TrkA ablation in striatal cholinergic neurons of TrkAcKO mice. At P30, control or mutant animals were processed for immunofluorescence with the indicated antibodies. Arrows, TrkA-expressing (Eii) and TrkA-depleted (Evii) striatal cholinergic neurons; in merged pictures, Ev and Ex show enlargement of cells in Eiv and Eix. F, G, Immunoblot (F) and quantitative analysis (G) of TrkA protein levels in striatal tissue lysates of control or TrkAcKO mice. n = 3 per genotype in each group. *p < 0.05, **p < 0.01, ***p < 0.001. Scale bars: Ai–xvi, 50 μm; Axvii–xx, 25 μm; Ei–iv,vi–ix, 50 μm; Ev,x, 25 μm.
In vivo TrkA is required for normal ChAT expression in BFCNs but not striatal cholinergic neurons
In cell culture, NGF has been shown to support survival and promote ChAT expression in BFCNs (Honegger and Lenoir, 1982; Hartikka and Hefti, 1988). In TrkAcKO mice, the cell size and number of BFCNs were not affected, compared with control mice (Fig. 2C). Consistent with this, Golgi staining in P30 and P60 MS neurons did not reveal any obvious differences in cellular organization, cell morphology, or cell number between control and TrkAcKO mice (Fig. 4A). Immunofluorescence studies revealed that the number of striatal cholinergic neurons of TrkAcKO animals was comparable to those of control mice (Fig. 4B,C). Similarly, the striatal cholinergic cell size appeared unaffected in TrkAcKO mice at the examined ages (cell perikarya measured ∼20 μm) (Fig. 4D,E). Consistent with these data, Golgi staining of neurons in the ventral striatum of TrkAcKO mice showed no obvious morphological deficiencies and no significant differences in number, size, or organization of these neurons compared with neurons of control animals (Fig. 4F). Thus, at the morphological level, in vivo TrkA ablation has no discernible effect on cholinergic neuron survival or size.
Figure 4.
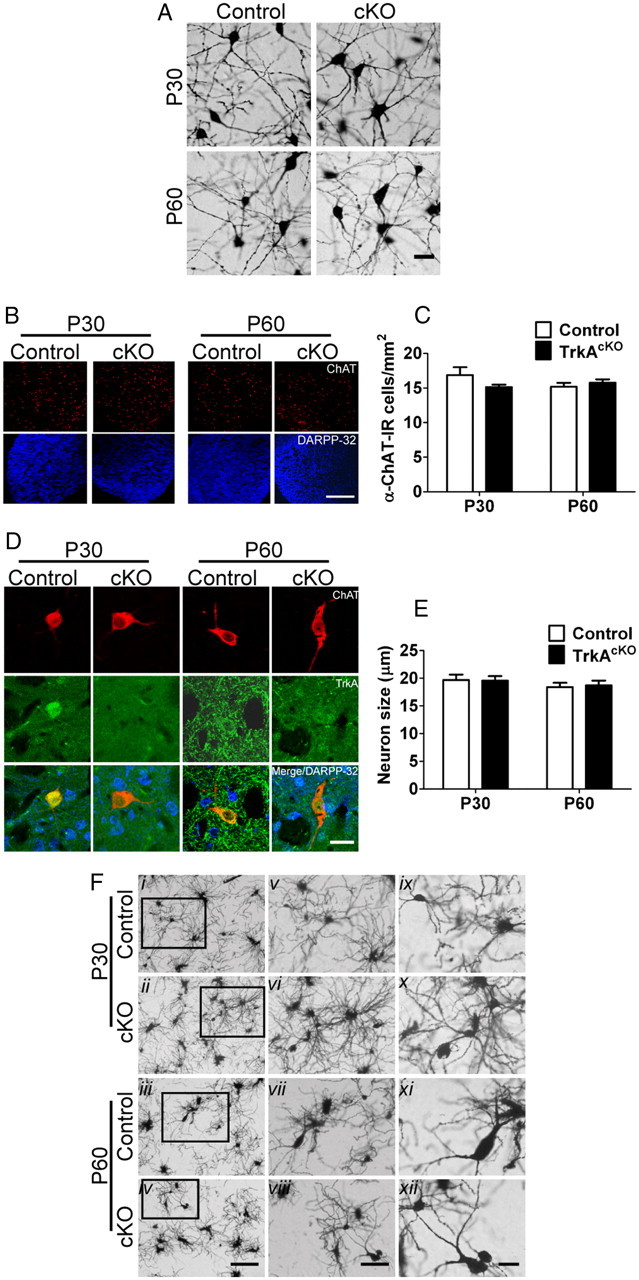
TrkA ablation does not affect morphology of cholinergic neurons in either BF or striatum. A, Golgi staining of basal forebrain neurons. At P30, staining indicates no apparent morphological defects or any deficiency in cell number in medial septum neurons of brains derived from TrkAcKO animals compared with control mice. B, C, Number of striatal cholinergic neurons does not change in TrkAcKO mice. n = 4–7 mice per genotype. D, E, TrkA conditional deletion has no effect on striatal cholinergic cell size. n = 15 neurons per genotype among three mice. F, Golgi staining of ventral striatal neurons shows no obvious morphological differences. Fix,x, Medium spiny neurons; Fxii, cholinergic neurons; Fv–vii,ix–xii, successive magnifications of regions outlined in Fi–iv. Scale bars: A, 10 μm; B, 1 mm; D, 25 μm; Fi–iv, 50 μm; Fv–vii, 25 μm; Fix–xii, 10 μm.
To investigate BFCN differentiation in TrkAcKO mice, we next evaluated ChAT enzyme levels from control and mutant BF tissue (Fig. 5A). Immunostaining of coronal brain sections with ChAT antibody indicated a reduction in ChAT-IR in mutant mice compared with controls beginning at P14 and sustained throughout adulthood (Fig. 5A). Morphometric and quantitative analyses of ChAT-IR neurons revealed no appreciable differences between mutants and controls (Fig. 5B,C), indicating that these parameters are not responsible for the reduction in ChAT intensity. To verify the changes in ChAT expression, Western blot analysis was performed at various ages. Consistent with the immunohistochemistry data, a substantial increase in ChAT levels was observed in control mice from P9 to P14 (∼10-fold) that became more pronounced at P155 (∼30-fold) (Fig. 5D,E). Additional anti-ChAT Western blot analysis using TrkAcKO mice tissue lysates further confirmed a clear reduction in expression by P9 and P14 in the mutants compared with control littermates, as was observed in the immunostained sections (Fig. 5F,G). Therefore, loss of TrkA in BFCNs causes a marked decrease in ChAT levels produced by these neurons without any evidence of morphological alteration or reduced survival.
Figure 5.
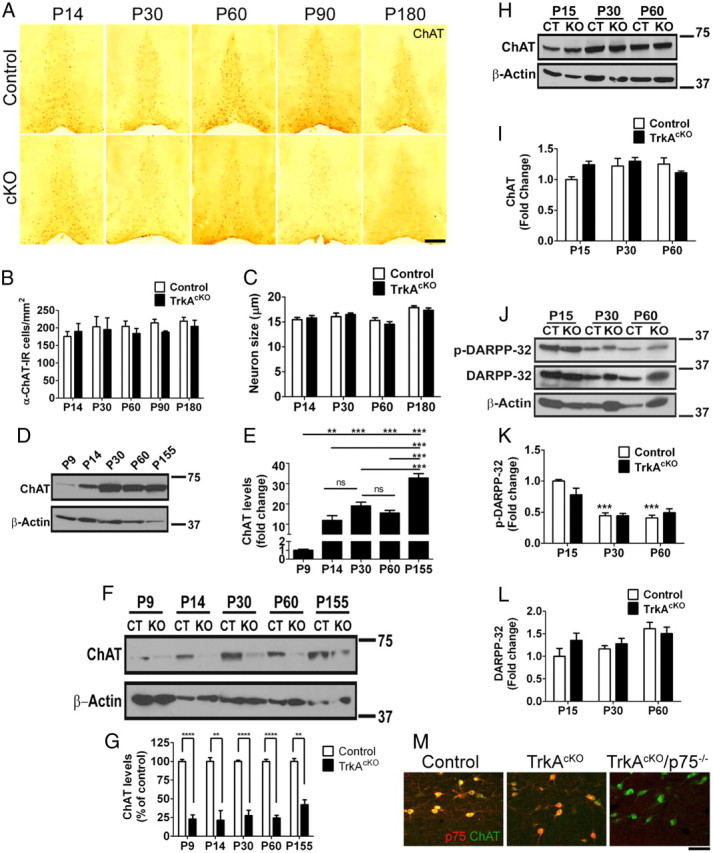
TrkA loss affects ChAT expression in BFCNs, but not striatal neurons. A, Neurons in the medial septum of P14, P30, P60, P90, and P180 control and TrkAcKO mice stained with anti-ChAT antibody. Neurons of mutant mice exhibited decreased ChAT staining compared with control. B, Quantitative analysis of ChAT-IR neuronal cell numbers of control and TrkAcKO mice. n = 3–4 mice per genotype. C, Evaluation of control and TrkAcKO ChAT-IR neuronal cell size. n = 35 cells per genotype analyzed among three mice. D, E, ChAT protein levels in BF of control mice at different ages. n = 4. F, G, Quantitative Western blot of ChAT levels in BF. A reduction in ChAT expression in the TrkAcKO mice compared with WT mice is already seen at P9 and is sustained throughout cholinergic development. n = 4 mice per genotype at P9, P14; n = 7–8 at P30, P60; n = 3 at P155. H–L, Analysis of cholinergic signaling in striatum of TrkAcKO mice. Whole tissue lysates were prepared from striatum of either control or mutant animals at various ages and subjected to Western blotting using the indicated antibodies. H, I, Total protein levels of ChAT remain unaffected in mutant animals. n = 4 mice per genotype. J–L, Unaltered DARPP-32 signaling in striatum of TrkAcKO mice. ***p < 0.001 compared with P15-control; n = 6 mice per genotype. M, Neurons in the medial septum of P30 control, TrkAcKO, and TrkAcKO/p75−/− double-mutant mice stained with anti-ChAT and anti-p75 antibodies. Although there is complete absence of NGF signaling in double-mutant mice (right panel), there is no significant change in BFCN morphology or ChAT expression compared with TrkAcKO littermates. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Scale bars: A, 500 μm; M, 50 μm.
We next examined ChAT levels in TrkA-deficient striatal neurons. Western blot analysis indicated that, in contrast to BF neurons, ablation of TrkA did not affect striatal ChAT expression (Fig. 5H,I). To further investigate possible effects of TrkA loss on striatal cholinergic neurons, we examined the dopaminergic pathway. Dopamine has been shown to regulate striatal cholinergic neurons, in part via dopamine D2 receptor-induced inhibition of acetylcholine release (MacKenzie et al., 1989; DeBoer et al., 1996; Pisani et al., 2007). Phosphorylated DARPP-32 is a pivotal dopamine signaling regulator in striatum that is selectively enriched in medium spiny neurons, which are the main synaptic target of cholinergic interneurons (Nishi et al., 1997; Calabresi et al., 2000). We therefore investigated whether phosphorylated or total DARPP-32 levels were affected by TrkA ablation, as an indicator of striatal dopamine signaling integrity. Upon immunoblot analysis, we found that the levels of neither phosphorylated nor total DARPP-32 were affected in striatal cholinergic neurons of TrkAcKO mice compared with controls at different ages (Fig. 5J–L), further illustrating the absence of defects in striatal dopamine signaling. Based on these cellular and molecular analysis parameters, we conclude that ablation of TrkA in forebrain does not affect maturation of cholinergic neurons in striatum. Thus, this mouse model is suitable for studying the role of TrkA in BFCN development.
We next crossed TrkAcKO mice with p75 mutant mice to determine the effect of the complete absence of NGF receptors in BFCNs. The double-mutant cholinergic neurons displayed no significant changes in number, morphology, or ChAT expression levels (Fig. 5M). Together, these data suggest that TrkA, but not p75, signaling is necessary for the normal developmental increase of ChAT expression that occurs in BFCNs, but is not required for their survival. In addition, these data highlight a difference between NGF function in vitro as a survival factor for BFCNs compared with an in vivo role in ChAT regulation.
Disruption of BF cholinergic connectivity in TrkAcKO mice
We next turned our attention from the soma of BFCNs to investigate whether absence of TrkA had consequences on axon development and target innervation. BFCN fiber morphology and patterning in the hippocampus was examined using lipophilic dye tracers and p75 immunostaining. Although p75 immunoreactivity has been reported within some astrocytes and postsynaptic terminals in rodent dentate gyrus, anti-p75 antibody is also a useful reagent to stain axonal projections from BFCNs to their terminal innervation sites (Dougherty and Milner, 1999). Previous studies using conventional TrkA knock-out neonatal mice showed a significant reduction of cholinergic fibers into the hippocampus and a decreased size of the bundle of projections from BFCNs (Smeyne et al., 1994). Our DiI tracing study of BFCNs indicated that projections toward the hippocampus appeared intact in mutant mice at P7 (Fig. 6A,B). However, by P14, control hippocampus exhibited p75-IR fiber density and a mature laminar pattern of innervation, whereas the mutants showed little p75-IR fiber density and no clear laminar pattern of innervation (Fig. 6C,E). Throughout the TrkAcKO mutant hippocampus, we observed a similar cholinergic fiber defect at all stages examined (data not shown) (Fig. 6C). Evidence of impaired cholinergic innervation into the frontal cortex was also observed in mutants as early as P7 (Fig. 6D,E). At P90, there was a complete absence of visible p75-IR fibers in mutant hippocampus, and only a small number of p75-IR fibers was observed in the mutant frontal cortex (Fig. 6D, 6). Retrograde tracing with dextran amine conjugates of Texas Red into frontal cortex at earlier stages failed to label BFCNs from nucleus basalis of TrkAcKO mice (Fig. 6F). These data suggest that the few remaining p75-IR fibers in the mutant frontal cortex likely represent debris from retracting cholinergic fibers. In addition, Western blot analysis of TrkAcKO or control hippocampus tissue lysates showed a significant decrease in ChAT levels in mutant hippocampus relative to control (Fig. 6G,H), confirming the loss of target tissue cholinergic innervation in TrkAcKO animals. The absence of p75 immunoreactivity at the hippocampus and frontal cortex was not accompanied by reduction in number, size, or p75 expression in neuronal somata at the TrkAcKO BF (Fig. 6I–K). Furthermore, analysis of TrkAcKO or control BF tissue lysates by Western blot showed that the p75 levels are unaltered in mutant mice (Fig. 6L,M). These results indicate that BFCN target projection to the hippocampus does not require NGF/TrkA signaling; however, development of a normal pattern of innervation does require this signaling pathway. Thus, loss of TrkA in BFCNs has profound effects on connectivity following its effects on ChAT expression.
Figure 6.
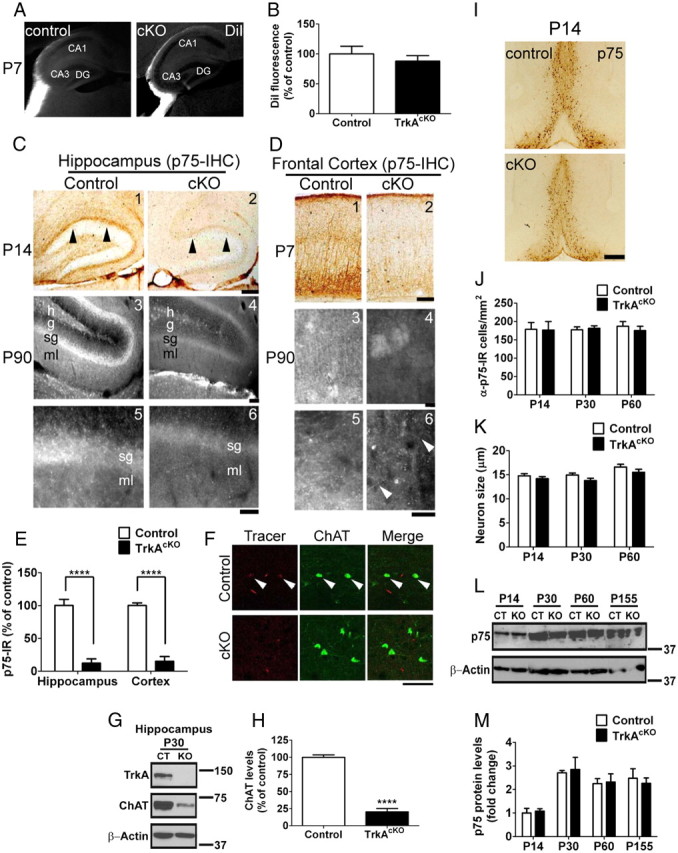
Effect of TrkA ablation on cholinergic input to hippocampus and cortex. A, DiI anterograde tracing of BFCNs. Cholinergic projections around the hippocampus are clearly seen in both brains as early as P7. Analysis at later stages showed a similar projection pattern (data not shown). CA, Cornu ammonis; DG, dentate gyrus. B, Quantification of DiI fluorescence in P7 control and mutant mice brains. C, DAB (1,2) and fluorescence staining (3–6) of cholinergic axonal innervation into hippocampus with anti-p75 antibody. As early as P14, control hippocampus contains p75-IR fibers not observed in TrkAcKO mice (C1,2). This is not due to a delay of innervation as there is no innervation of p75-IR fibers at P90 in TrkAcKO hippocampus (C4,6); a mature pattern of innervation was seen in control (C3,5). h, Hilus; g, granule cell layer; sg, sub granular cell layer; ml, molecular layer. D, DAB (1,2) and fluorescence staining (3–6) of cholinergic axonal innervation to cortex. At P7, frontal cortex from control mice exhibits intense staining of p75-IR fibers, which is dramatically decreased in TrkAcKO mice. Unlike hippocampus, there are some p75-IR fibers in the frontal cortex in TrkAcKO (C6, arrowheads in magnified field). E, Quantification of axonal innervation as indicated by p75-IR in P14 hippocampus and P7 cortex of control and mutant mice. n = 6 in hippocampus; n = 5 in cortex; ****p < 0.0001. F, Retrograde tracing of BFCNs by Texas Red-dextran amine. Texas Red-dextran amine was retrogradely transported from frontal cortex to BFCNs in control (arrowheads), but not in TrkAcKO mice. n = 3 mice per genotype. G,H, Quantitative Western blot showing decreased ChAT levels in hippocampus. n = 4; ****p < 0.0001. I, Despite the dramatic reduction of p75-IR fibers in TrkAcKO hippocampus and cortex, p75-IR neurons in BF are comparable between control and mutant animals. J, Evaluation of cell density in control and TrkAcKO p75-IR neurons. n = 4 mice per genotype. K, Cell size analysis of p75-IR neurons in control and mutant mice. n = 35 neurons per genotype among three mice. L, M, Immunoblot of p75 levels in BF. n = 3 mice per genotype. Scale bars: C, D, 200 mm; F, 50 μm; I, 400 mm.
Loss of ERK activation in TrkAcKO mice BFCNs
To begin to investigate the mechanisms involved in the observed TrkAcKO phenotypes, we examined whether signaling from two major downstream pathways of TrkA, the ERK and AKT pathways, was altered. At P5, no significant ERK activation, as measured by pERK immunostaining, was observed in the BF of control mice (Fig. 7A). By P9, ERK activation was observed in control mice and, to a lesser extent, in mutant mice; however, there appeared to be little colocalization of pERK and ChAT immunoreactivity at this stage. By P14, when BFCN axons are innervating the target fields, increased colocalization of pERK and ChAT-IR neurons was observed in control mice, but not in TrkAcKO mice. Consistent with this, Western blot analysis of BF tissue lysates showed an evident and significant reduction of pERK in TrkAcKO mice (Fig. 7B,C). Given the absence of pERK and ChAT colocalization, it is likely that noncholinergic cells present in BF may account for the residual pERK signal observed in the mutant samples. We also examined the PI3–kinase pathway and observed similar and weak pAKT signal in both control and mutant mice at P14 that did not colocalize with cholinergic neurons (Fig. 7D). Western blots showed no evidence of AKT activation in BF tissue lysates (Fig. 7E). These data support the model that, through retrograde signaling, activation of TrkA results in strong activation of the ERK, but not the AKT, pathway in BFCN cell bodies during development.
Figure 7.

Loss of ERK activation in BFCNs of TrkAcKO mice. A, pERK immunostaining in BF. At P9, weak activation of ERK was observed in both control and TrkAcKO (arrowheads) BF. By P14, stronger ERK activation, mostly limited to ChAT-positive neurons, was observed in control, but not in TrkAcKO MS. B, C, Immunoblot (B) and quantification (C) of ERK activation in BF tissue lysates of control or TrkAcKO mice. n = 4 per genotype in each group; *p < 0.05, **p < 0.01. D, E, Absence of AKT activation in BFCNs as determined by immunofluorescence (D) and Western blot (E) analyses. Although weak activation of AKT was observed in both control and mutant mice MS at P14 as measured by pAKT immunofluorescence, it did not colocalize with ChAT-positive neurons and was no longer present at P30. Consistent with this, pAKT signal was undetectable in BF tissue lysates by Western blots (E). In E, cortex tissue lysates of control mice were used as the positive control; n = 3. Scale bars: A, D, 50 μm.
Mild cognitive decline in TrkAcKO mice
Our results demonstrate that BFCN ChAT expression becomes dependent upon TrkA signaling before the establishment of cholinergic connections to hippocampus and cortex and, furthermore, that TrkA is required for proper establishment of BFCN innervation into cortex and hippocampus. We therefore wished to assess the impact of these cellular defects on animal behavior. By visual inspection, TrkAcKO mice did not exhibit any obvious behavioral abnormalities and could not be distinguished from littermates. To test cognitive function, 6-month-old mice were examined in fear-conditioning and Morris water maze (MWM) tests; 2-month-old mice were used in the novel object recognition task. We first subjected control and mutant mice to contextual and tone-cued fear-conditioning learning paradigms. TrkAcKO mice displayed lower levels of freezing to tone and context compared with control littermates (Fig. 8A,B). Statistical analysis showed a significant difference (p = 0.015) in the tone-cued test (Fig. 8A), a test that is reported to be exclusively amygdala dependent (Rogan et al., 1997); however, TrkAcKO mice did not display a statistical difference (p = 0.247) in their response to the contextual test (Fig. 8B), which is more dependent on hippocampus. On this basis, the data suggest that loss of TrkA in BFCNs has a profound impact on mnemonic processing within the amygdala, but appears to have less impact on hippocampal function. This is consistent with the result of the MWM test, which tests spatial learning and memory, where we also did not observe a statistically significant difference in performance between the two animal cohorts (Fig. 8C). The velocity (23.75 cm/s control vs 22.85 cm/s mutant; data not shown) and learning curves (Fig. 8D) were similar between controls and mutants. In a probe trial conducted after the 10th trial, both control and mutant mice spent more time in the target quadrant (mean ± SEM: control, 25.72 ± 1.87 s; mutant, 19.96 ± 2.23 s; N = 16 for control; N = 17 for TrkAcKO) than in the other three quadrants. Although the mutant mice spent less time than control mice in the target quadrant, this difference was not statistically significant (p = 0.058) (Fig. 8C). We further assessed the functional impact of TrkA deletion in the BF cholinergic system using the novel object recognition task, which relies on the integrity of working memory and attention system without placing any stress on subjects. Previous studies have reported that rats with damaged amygdalae or perirhinal cortex showed deficient responses to novel objects and object relationships (Moses et al., 2005). Therefore, in this task, we only used 2-month-old male mice since older mice showed a significant drop in visual attention as assessed by the single-novel object task, apparently caused by a reduction of sensitivity in the novel object recognition task (data not shown). We calculated the discrimination ratio as 0.868 ± 0.025 for control versus 0.674 ± 0.049 for mutant mice (Fig. 8E), indicating that TrkAcKO mice still show preference for interacting with a novel object. However, there is a significant decrease in the recognition of novelty compared with control mice (p = 0.002). Although there is a selective difference in the contribution of BFCN input to the function of target regions, in general, based on the reported anatomical correlates for behavior, the behavior test results show a measurable decline in amygdala-dependent, but not hippocampal-dependent, cognitive performance in TrkAcKO mice.
Figure 8.
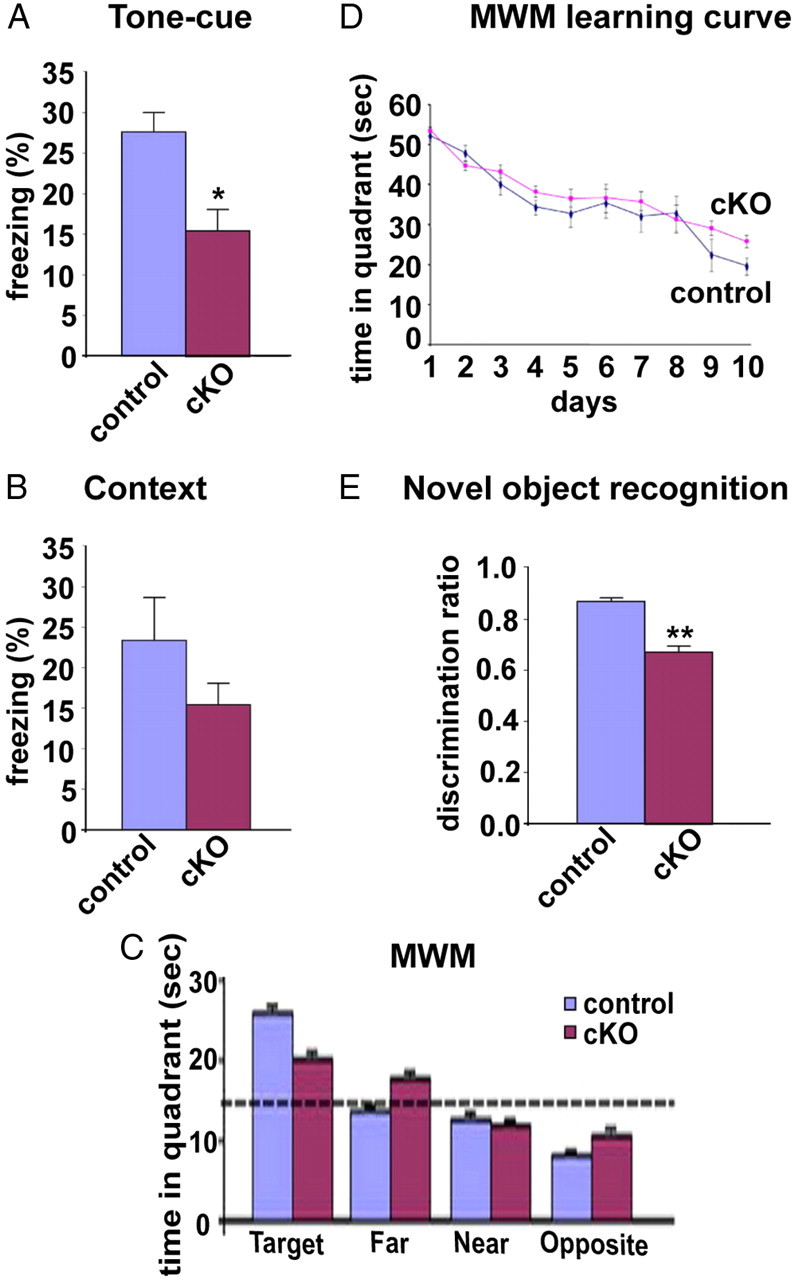
Impact of TrkA loss in BFCNs on cognitive activity. A, Tone-cued fear conditioning. The percentage of freezing of control = 27.19 ± 4.41%; TrkAcKO = 15.24 ± 4.01% (n = 12 per genotype, p = 0.015). B, Contextual fear conditioning. The percentage of freezing of control = 23.13 ± 6.77%; TrkAcKO = 15.28 ± 3.20% (n = 12 per genotype, p = 0.247). C, MWM. The time in the target quadrant of control = 25.72 ± 1.87 s; TrkAcKO = 19.96 ± 2.23 s (n = 16 for control; n = 17 for TrkAcKO, p = 0.058). D, Learning curve from MWM test showed no statistical difference between control and TrkAcKO mice. E, Novel object recognition. The discrimination ratio of control = 0.867 ± 0.025; TrkAcKO = 0.673 ± 0.054 (n = 17 per genotype, **p = 0.002). For the fear-conditioning and MWM tests, male mice were tested at 6 months of age. For the novel object recognition task, male mice were tested at 2 months of age.
Discussion
The TrkA/NGF receptor tyrosine kinase has been intensely studied primarily in the PNS where it undertakes principal roles in survival and target selectivity in the sensory and sympathetic systems. Shortly after its identification as the NGF receptor (Kaplan et al., 1991), evidence of TrkA presence in the BF was discovered (Holtzman et al., 1992). In the intervening years, data have emerged pointing to a role for TrkA in the CNS, but it was limited to tissue culture or to indirect lesion experiments in vivo. No definitive evidence for TrkA function in the CNS has been available.
Development, survival, and connectivity of BFCNs
In the present study, we demonstrate that TrkA is required for appropriate BFCN development. Genetic ablation of TrkA in these cells results in a profound deficit of stable cholinergic innervation to the cortex and hippocampus without affecting survival. Within the hippocampus, the target innervation deficit does not reflect an inability of these neurons to extend processes to the vicinity of their targets, but instead reflects their failure to connect to and innervate these target areas. TrkA loss also leads to reduced ERK activation and ChAT expression within the BF; as a result, mutant mice exhibit mild cognitive deficits in behavioral tests. Despite TrkA ablation in striatal cholinergic neurons, we were unable to identify cellular or molecular changes in this region. Thus, the cortical and hippocampal cellular deficiencies observed in TrkAcKO mice must be considered to be the consequence of TrkA signaling deficits, specifically in BFCNs.
There is extensive indirect evidence supporting a role for NGF, and by inference TrkA, in BFCN function. Exogenous NGF increases ChAT activity in the BF (Gnahn et al., 1983; Mobley et al., 1985) and promotes BFCN survival in culture (Hartikka and Hefti, 1988). Our study indicates that in vivo, TrkA is required for the normal developmental regulation and increase of ChAT expression. It appears that TrkA signaling is not required for many aspects of the phenotypic differentiation of BFCNs, as we observed comparable number and morphology of ChAT-IR neurons in TrkAcKO BF (Fig. 5B,C), indicating that the absence of TrkA signaling does not significantly affect neuronal survival. Our data also indicate that p75 does not influence survival of BFCNs, as the BF of both TrkAcKO/p75 double-knock-out and TrkAcKO mice appear indistinguishable at P30. Our results differ from a previous study that reported 36% fewer ChAT-IR neurons in the septum of TrkA-null mice compared with wild type (Fagan et al., 1997). TrkA-null mice have broad and severe embryonic abnormalities and postnatal growth retardation. Thus, the reduction in ChAT-positive cells in those mice may be an indirect consequence of general developmental deficiencies, either in conjunction with or independent of specific loss of TrkA signaling in these cells. Although our evidence shows that NGF/TrkA signaling is critical for maintaining BFCN ChAT expression, inactivation of TrkA in TrkAcKO mice does not lead to a total loss of ChAT production in BF. In addition, while other neurotrophins (i.e., BDNF, NT-4/5) and growth factors are equally effective in maintaining ChAT expression in vitro (Knusel and Hefti, 1988; Friedman et al., 1993), in vivo, none of these factor/receptor interactions compensated for the lack of normal developmental increase of ChAT expression in the BF of TrkAcKO mutants.
In BFCNs, we found that TrkA was required for the normal activation of ERK without obvious influence on the activation of AKT during target innervation. Although ERK activity is observed in both BF cholinergic and noncholinergic neurons in early neonates (P9), it becomes primarily dependent on NGF/TrkA signaling by P14, and mostly cholinergic neurons exhibit ERK activation at this point. Since target innervation of BFCNs is TrkA dependent and the timing of maximal ERK activation in these neurons correlates with the establishment of target innervation, TrkA-mediated activation of the ERK pathway may play a crucial role in the maintenance of target innervation by BFCNs. The primary ligand for TrkA is NGF; therefore, we presume that this is the factor responsible for the paracrine activation of the receptor. However, receptor promiscuity has been observed for the Trk family in other systems, and this issue will have to be rigorously examined in the future.
BF cholinergic system in cognitive activity
The cellular deficits in hippocampal and cortical innervation compelled us to look for behavioral sequelae. The role of the BF cholinergic system in spatial memory and learning has been studied by inducing lesions with the immunotoxin 192 IgG-saporin, a monoclonal antibody against p75 coupled to saporin. 192 IgG-saporin induces an efficient and specific lesion of neocortical and hippocampal cholinergic afferents (Wiley et al., 1991; Book et al., 1992) and, to a lesser extent, amygdala cholinergic afferents (Heckers et al., 1994), which affects tasks dependent on attention (McGaughy et al., 2002; Dalley et al., 2004) rather than learning and memory (Torres et al., 1994; Baxter et al., 1995; Frielingsdorf et al., 2006). We selectively abolished BF cholinergic innervation in target fields and assessed the effect on certain behavioral tasks. Our results indicate that the BF cholinergic system does not have a prominent impact on spatial learning and memory, data largely consistent with studies using 192 IgG-saporin in rats (Baxter et al., 1995; Kirby and Rawlins, 2003; Frielingsdorf et al., 2006). However, these observations differ from the significant impairment in spatial learning and memory reported in anti-NGF-treated mice (Capsoni et al., 2000; Ruberti et al., 2000). Inflammation, and other complications associated with immune responses may be responsible for the slightly more extensive behavioral impairments observed in anti-NGF-treated mice. Contrary to contextual fear-conditioning and MWM tests, TrkAcKO mice showed a significant impairment in tone-cued fear-conditioning tests, which are reported to depend on many of the same brain regions as those for contextual fear conditioning, including the amygdala, but not the hippocampus (Phillips and LeDoux, 1992). On this basis, our results would indicate that the BF cholinergic system has more impact on amygdala-dependent mnemonic processing. The role of amygdala in formation of emotions and fear memory is modulated by cortical regions such as the piriform cortex (Fujii et al., 2011) and the medial prefrontal cortex (Maren and Quirk, 2004), respectively. Thus, the cholinergic innervation deficit to cortex and hippocampus in TrkAcKO mice could also contribute to the amygdala-dependent behavioral deficits observed in these animals. The significant impairment observed in object discrimination in TrkAcKO mice is reminiscent of the severe visual object recognition deficits caused by 192 IgG-saporin-induced removal of BF cholinergic inputs into perirhinal cortex (Winters and Bussey, 2005). In addition, these deficits observed in TrkAcKO mice are consistent with the improvement reported in severely cognitively impaired 24-month-old rats treated with a TrkA peptidomimetic ligand (Bruno et al., 2004). Collectively, these data support the notion of a central role for TrkA in cognition.
Despite the involvement of striatal cholinergic interneurons in learning behaviors, the selective ablation of those interneurons has been shown to have no effect on the spatial learning in the MWM or in contextual and tone-cued fear-conditioning paradigms (Kitabatake et al., 2003). Our data indicate that TrkA ablation in these interneurons did not affect cellular and molecular aspects of these neurons such as size and morphology, as well as cholinergic and dopamine signaling in striatum. Therefore, ablation of TrkA in striatal cholinergic neurons does not likely influence the selective impairments in attention and working memory exhibited in TrkAcKO mice, indicating that cognitive decline is a consequence of BF cholinergic system disruption triggered by TrkA loss in these neurons.
Phenotypic relevance of TrkAcKO mice to Alzheimer's disease
TrkA expression is downregulated in individuals with MCI and AD (Boissiere et al., 1997; Chu et al., 2001; Ginsberg et al., 2006), while p75 protein levels remain stable during progression of AD (Goedert et al., 1989; Counts et al., 2004). Pro-NGF has also been reported to be increased in AD (Fahnestock et al., 2001; Peng et al., 2004) and to promote apoptosis through the p75/sortilin complex (Nykjaer et al., 2004); therefore, preferential pro-NGF/p75 signaling, together with a reduction of TrkA, may play a key role in the degeneration of BFCNs that occurs in AD. However, TrkAcKO mice exhibited a comparable number and morphology of p75-positive BFCNs as control animals (Fig. 6J,K), arguing against the negative effects of p75 on BFCN survival. An age-dependent slight increase in phosphorylated tau was equally observed in both control and TrkAcKO brains (data not shown), thus failing to establish a direct link between loss of TrkA and amyloid pathology. This is in contrast to the anti-NGF-treated mouse brain, which showed BFCN loss and hyperphosphorylated tau in hippocampal and cortical neurons (Capsoni et al., 2000). The absence of significant cell loss in TrkAcKO/p75 double-knock-out mice, where NGF signaling is selectively ablated in BF, indicates that the BFCN loss observed in anti-NGF-treated mice may result from inflammatory responses or other secondary effects.
In summary, our data indicate that TrkA plays a pivotal role in cholinergic BF dysfunction as well as mild cognitive decline that is reminiscent of MCI and early-stage AD. Consequently, TrkA and ERK pathway components could be suitable targets for the development of novel treatment strategies for these diseases.
Footnotes
This work was supported by NIMH Grants MH049428 (J.L.R.), P01-NS16033 (L.F.R.), and P50MH66172 (L.F.P.). L.F.P. is an American Cancer Society Research Professor. We thank Dr. Shari Birnbaum and Steven McKinnon for their technical support, and Dr. Renée McKay for assistance with the manuscript. J.L.R. thanks Nina Ireland.
Authors declare no financial conflict of interest.
References
- Baxter MG, Bucci DJ, Gorman LK, Wiley RG, Gallagher M. Selective immunotoxic lesions of basal forebrain cholinergic cells: effects on learning and memory in rats. Behav Neurosci. 1995;109:714–722. doi: 10.1037//0735-7044.109.4.714. [DOI] [PubMed] [Google Scholar]
- Boissiere F, Faucheux B, Ruberg M, Agid Y, Hirsch EC. Decreased TrkA gene expression in cholinergic neurons of the striatum and basal forebrain of patients with Alzheimer's disease. Exp Neurol. 1997;145:245–252. doi: 10.1006/exnr.1997.6443. [DOI] [PubMed] [Google Scholar]
- Book AA, Wiley RG, Schweitzer JB. Specificity of 192 IgG-saporin for NGF receptor-positive cholinergic basal forebrain neurons in the rat. Brain Res. 1992;590:350–355. doi: 10.1016/0006-8993(92)91121-t. [DOI] [PubMed] [Google Scholar]
- Bruno MA, Clarke PB, Seltzer A, Quirion R, Burgess K, Cuello AC, Saragovi HU. Long-lasting rescue of age-associated deficits in cognition and the CNS cholinergic phenotype by a partial agonist peptidomimetic ligand of TrkA. J Neurosci. 2004;24:8009–8018. doi: 10.1523/JNEUROSCI.1508-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Pisani A, Bernardi G. Acetylcholine-mediated modulation of striatal function. Trends Neurosci. 2000;23:120–126. doi: 10.1016/s0166-2236(99)01501-5. [DOI] [PubMed] [Google Scholar]
- Capsoni S, Ugolini G, Comparini A, Ruberti F, Berardi N, Cattaneo A. Alzheimer-like neurodegeneration in aged antinerve growth factor transgenic mice. Proc Natl Acad Sci U S A. 2000;97:6826–6831. doi: 10.1073/pnas.97.12.6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ye H, Kuruvilla R, Ramanan N, Scangos KW, Zhang C, Johnson NM, England PM, Shokat KM, Ginty DD. A chemical-genetic approach to studying neurotrophin signaling. Neuron. 2005;46:13–21. doi: 10.1016/j.neuron.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Chu Y, Cochran EJ, Bennett DA, Mufson EJ, Kordower JH. Down-regulation of trkA mRNA within nucleus basalis neurons in individuals with mild cognitive impairment and Alzheimer's disease. J Comp Neurol. 2001;437:296–307. doi: 10.1002/cne.1284. [DOI] [PubMed] [Google Scholar]
- Counts SE, Nadeem M, Wuu J, Ginsberg SD, Saragovi HU, Mufson EJ. Reduction of cortical TrkA but not p75(NTR) protein in early-stage Alzheimer's disease. Ann Neurol. 2004;56:520–531. doi: 10.1002/ana.20233. [DOI] [PubMed] [Google Scholar]
- Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts-Meek S, Armanini MP, Ling LH, McMahon SB, Shelton DL, Levinson AD. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 1994;76:1001–1011. doi: 10.1016/0092-8674(94)90378-6. [DOI] [PubMed] [Google Scholar]
- Dalley JW, Theobald DE, Bouger P, Chudasama Y, Cardinal RN, Robbins TW. Cortical cholinergic function and deficits in visual attentional performance in rats following 192 IgG-saporin-induced lesions of the medial prefrontal cortex. Cereb Cortex. 2004;14:922–932. doi: 10.1093/cercor/bhh052. [DOI] [PubMed] [Google Scholar]
- DeBoer P, Heeringa MJ, Abercrombie ED. Spontaneous release of acetylcholine in striatum is preferentially regulated by inhibitory dopamine D2 receptors. Eur J Pharmacol. 1996;317:257–262. doi: 10.1016/s0014-2999(96)00761-3. [DOI] [PubMed] [Google Scholar]
- Dougherty KD, Milner TA. p75NTR immunoreactivity in the rat dentate gyrus is mostly within presynaptic profiles but is also found in some astrocytic and postsynaptic profiles. J Comp Neurol. 1999;407:77–91. doi: 10.1002/(sici)1096-9861(19990428)407:1<77::aid-cne6>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Garber M, Barbacid M, Silos-Santiago I, Holtzman DM. A role for TrkA during maturation of striatal and basal forebrain cholinergic neurons in vivo. J Neurosci. 1997;17:7644–7654. doi: 10.1523/JNEUROSCI.17-20-07644.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahnestock M, Michalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer's disease. Mol Cell Neurosci. 2001;18:210–220. doi: 10.1006/mcne.2001.1016. [DOI] [PubMed] [Google Scholar]
- Friedman WJ, Ibáñez CF, Hallböök F, Persson H, Cain LD, Dreyfus CF, Black IB. Differential actions of neurotrophins in the locus coeruleus and basal forebrain. Exp Neurol. 1993;119:72–78. doi: 10.1006/exnr.1993.1007. [DOI] [PubMed] [Google Scholar]
- Frielingsdorf H, Thal LJ, Pizzo DP. The septohippocampal cholinergic system and spatial working memory in the Morris water maze. Behav Brain Res. 2006;168:37–46. doi: 10.1016/j.bbr.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Fujii T, Onimaru H, Homma I. Effects of corticotropin releasing factor on spontaneous burst activity in the piriform-amygdala complex of in vitro brain preparations from newborn rats. Neurosci Res. 2011;71:134–139. doi: 10.1016/j.neures.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer's disease. J Neurochem. 2006;97:475–487. doi: 10.1111/j.1471-4159.2006.03764.x. [DOI] [PubMed] [Google Scholar]
- Glaser EM, Van der Loos H. Analysis of thick brain sections by obverse-reverse computer microscopy: application of a new, high clarity Golgi-Nissl stain. J Neurosci Methods. 1981;4:117–125. doi: 10.1016/0165-0270(81)90045-5. [DOI] [PubMed] [Google Scholar]
- Gnahn H, Hefti F, Heumann R, Schwab ME, Thoenen H. NGF-mediated increase of choline acetyltransferase (ChAT) in the neonatal rat forebrain: evidence for a physiological role of NGF in the brain? Brain Res. 1983;285:45–52. doi: 10.1016/0165-3806(83)90107-4. [DOI] [PubMed] [Google Scholar]
- Goedert M, Otten U, Hunt SP, Bond A, Chapman D, Schlumpf M, Lichtensteiger W. Biochemical and anatomical effects of antibodies against nerve growth factor on developing rat sensory ganglia. Proc Natl Acad Sci U S A. 1984;81:1580–1584. doi: 10.1073/pnas.81.5.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Fine A, Dawbarn D, Wilcock GK, Chao MV. Nerve growth factor receptor mRNA distribution in human brain: normal levels in basal forebrain in Alzheimer's disease. Brain Res Mol Brain Res. 1989;5:1–7. doi: 10.1016/0169-328x(89)90011-9. [DOI] [PubMed] [Google Scholar]
- Hartikka J, Hefti F. Development of septal cholinergic neurons in culture: plating density and glial cells modulate effects of NGF on survival, fiber growth, and expression of transmitter-specific enzymes. J Neurosci. 1988;8:2967–2985. doi: 10.1523/JNEUROSCI.08-08-02967.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckers S, Ohtake T, Wiley RG, Lappi DA, Geula C, Mesulam MM. Complete and selective cholinergic denervation of rat neocortex and hippocampus but not amygdala by an immunotoxin against the p75 NGF receptor. J Neurosci. 1994;14:1271–1289. doi: 10.1523/JNEUROSCI.14-03-01271.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefti F. Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J Neurosci. 1986;6:2155–2162. doi: 10.1523/JNEUROSCI.06-08-02155.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Li Y, Parada LF, Kinsman S, Chen CK, Valletta JS, Zhou J, Long JB, Mobley WC. p140trk mRNA marks NGF-responsive forebrain neurons: evidence that trk gene expression is induced by NGF. Neuron. 1992;9:465–478. doi: 10.1016/0896-6273(92)90184-f. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Kilbridge J, Li Y, Cunningham ET, Jr, Lenn NJ, Clary DO, Reichardt LF, Mobley WC. TrkA expression in the CNS: evidence for the existence of several novel NGF-responsive CNS neurons. J Neurosci. 1995;15:1567–1576. doi: 10.1523/JNEUROSCI.15-02-01567.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honegger P, Lenoir D. Nerve growth factor (NGF) stimulation of cholinergic telencephalic neurons in aggregating cell cultures. Brain Res. 1982;255:229–238. doi: 10.1016/0165-3806(82)90023-2. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Hempstead BL, Martin-Zanca D, Chao MV, Parada LF. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- Kirby BP, Rawlins JN. The role of the septo-hippocampal cholinergic projection in T-maze rewarded alternation. Behav Brain Res. 2003;143:41–48. doi: 10.1016/s0166-4328(03)00005-6. [DOI] [PubMed] [Google Scholar]
- Kitabatake Y, Hikida T, Watanabe D, Pastan I, Nakanishi S. Impairment of reward-related learning by cholinergic cell ablation in the striatum. Proc Natl Acad Sci U S A. 2003;100:7965–7970. doi: 10.1073/pnas.1032899100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knusel B, Hefti F. Development of cholinergic pedunculopontine neurons in vitro: comparison with cholinergic septal cells and response to nerve growth factor, ciliary neuronotrophic factor, and retinoic acid. J Neurosci Res. 1988;21:365–375. doi: 10.1002/jnr.490210228. [DOI] [PubMed] [Google Scholar]
- Kohwi M, Petryniak MA, Long JE, Ekker M, Obata K, Yanagawa Y, Rubenstein JL, Alvarez-Buylla A. A subpopulation of olfactory bulb GABAergic interneurons is derived from Emx1- and Dlx5/6-expressing progenitors. J Neurosci. 2007;27:6878–6891. doi: 10.1523/JNEUROSCI.0254-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L, Laub F, Lush M, Romero M, Zhou J, Luikart B, Klesse L, Ramirez F, Parada LF. The zinc finger transcription factor Klf7 is required for TrkA gene expression and development of nociceptive sensory neurons. Genes Dev. 2005;19:1354–1364. doi: 10.1101/gad.1227705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey A, Lah J, Goldstein F, Steenland K, Bliwise D. Mild cognitive impairment: an opportunity to identify patients at high risk for progression to Alzheimer's disease. Clin Ther. 2006;28:991–1001. doi: 10.1016/j.clinthera.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Luikart BW, Nef S, Virmani T, Lush ME, Liu Y, Kavalali ET, Parada LF. TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J Neurosci. 2005;25:3774–3786. doi: 10.1523/JNEUROSCI.0041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie RG, Stachowiak MK, Zigmond MJ. Dopaminergic inhibition of striatal acetylcholine release after 6-hydroxydopamine. Eur J Pharmacol. 1989;168:43–52. doi: 10.1016/0014-2999(89)90631-6. [DOI] [PubMed] [Google Scholar]
- Maren S, Quirk GJ. Neuronal signalling of fear memory. Nat Rev Neurosci. 2004;5:844–852. doi: 10.1038/nrn1535. [DOI] [PubMed] [Google Scholar]
- McGaughy J, Dalley JW, Morrison CH, Everitt BJ, Robbins TW. Selective behavioral and neurochemical effects of cholinergic lesions produced by intrabasalis infusions of 192 IgG-saporin on attentional performance in a five-choice serial reaction time task. J Neurosci. 2002;22:1905–1913. doi: 10.1523/JNEUROSCI.22-05-01905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley WC, Rutkowski JL, Tennekoon GI, Buchanan K, Johnston MV. Choline acetyltransferase activity in striatum of neonatal rats increased by nerve growth factor. Science. 1985;229:284–287. doi: 10.1126/science.2861660. [DOI] [PubMed] [Google Scholar]
- Monory K, Massa F, Egertová M, Eder M, Blaudzun H, Westenbroek R, Kelsch W, Jacob W, Marsch R, Ekker M, Long J, Rubenstein JL, Goebbels S, Nave KA, During M, Klugmann M, Wölfel B, Dodt HU, Zieglgänsberger W, Wotjak CT, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51:455–466. doi: 10.1016/j.neuron.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses SN, Cole C, Driscoll I, Ryan JD. Differential contributions of hippocampus, amygdala and perirhinal cortex to recognition of novel objects, contextual stimuli and stimulus relationships. Brain Res Bull. 2005;67:62–76. doi: 10.1016/j.brainresbull.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Lavine N, Jaffar S, Kordower JH, Quirion R, Saragovi HU. Reduction in p140-TrkA receptor protein within the nucleus basalis and cortex in Alzheimer's disease. Exp Neurol. 1997;146:91–103. doi: 10.1006/exnr.1997.6504. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Counts SE, Perez SE, Ginsberg SD. Cholinergic system during the progression of Alzheimer's disease: therapeutic implications. Expert Rev Neurother. 2008;8:1703–1718. doi: 10.1586/14737175.8.11.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi A, Snyder GL, Greengard P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J Neurosci. 1997;17:8147–8155. doi: 10.1523/JNEUROSCI.17-21-08147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nykjaer A, Lee R, Teng KK, Jansen P, Madsen P, Nielsen MS, Jacobsen C, Kliemannel M, Schwarz E, Willnow TE, Hempstead BL, Petersen CM. Sortilin is essential for proNGF-induced neuronal cell death. Nature. 2004;427:843–848. doi: 10.1038/nature02319. [DOI] [PubMed] [Google Scholar]
- Patel TD, Jackman A, Rice FL, Kucera J, Snider WD. Development of sensory neurons in the absence of NGF/TrkA signaling in vivo. Neuron. 2000;25:345–357. doi: 10.1016/s0896-6273(00)80899-5. [DOI] [PubMed] [Google Scholar]
- Peng S, Wuu J, Mufson EJ, Fahnestock M. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J Neuropathol Exp Neurol. 2004;63:641–649. doi: 10.1093/jnen/63.6.641. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Pisani A, Bernardi G, Ding J, Surmeier DJ. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 2007;30:545–553. doi: 10.1016/j.tins.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Rodríguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF, Dymecki SM. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25:139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- Rogan MT, Stäubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- Ruberti F, Capsoni S, Comparini A, Di Daniel E, Franzot J, Gonfloni S, Rossi G, Berardi N, Cattaneo A. Phenotypic knockout of nerve growth factor in adult transgenic mice reveals severe deficits in basal forebrain cholinergic neurons, cell death in the spleen, and skeletal muscle dystrophy. J Neurosci. 2000;20:2589–2601. doi: 10.1523/JNEUROSCI.20-07-02589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi A, Verhaagen J, Dijkhuizen PA, Swaab DF. Co-localization of high-affinity neurotrophin receptors in nucleus basalis of Meynert neurons and their differential reduction in Alzheimer's disease. Neuroscience. 1996;75:373–387. doi: 10.1016/0306-4522(96)00273-4. [DOI] [PubMed] [Google Scholar]
- Schwab ME, Otten U, Agid Y, Thoenen H. Nerve growth factor (NGF) in the rat CNS: absence of specific retrograde axonal transport and tyrosine hydroxylase induction in locus coeruleus and substantia nigra. Brain Res. 1979;168:473–483. doi: 10.1016/0006-8993(79)90303-2. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature. 1994;368:246–249. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- Sobreviela T, Clary DO, Reichardt LF, Brandabur MM, Kordower JH, Mufson EJ. TrkA-immunoreactive profiles in the central nervous system: colocalization with neurons containing p75 nerve growth factor receptor, choline acetyltransferase, and serotonin. J Comp Neurol. 1994;350:587–611. doi: 10.1002/cne.903500407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steininger TL, Wainer BH, Klein R, Barbacid M, Palfrey HC. High-affinity nerve growth factor receptor (Trk) immunoreactivity is localized in cholinergic neurons of the basal forebrain and striatum in the adult rat brain. Brain Res. 1993;612:330–335. doi: 10.1016/0006-8993(93)91681-h. [DOI] [PubMed] [Google Scholar]
- Torres EM, Perry TA, Blockland A, Wilkinson LS, Wiley RG, Lappi DA, Dunnet SB. Behavioural, histochemical and biochemical consequences of selective immunolesions in discrete regions of the basal forebrain cholinergic system. Neuroscience. 1994;63:95–122. doi: 10.1016/0306-4522(94)90010-8. [DOI] [PubMed] [Google Scholar]
- Wiley RG, Oeltmann TN, Lappi DA. Immunolesioning: selective destruction of neurons using immunotoxin to rat NGF receptor. Brain Res. 1991;562:149–153. doi: 10.1016/0006-8993(91)91199-b. [DOI] [PubMed] [Google Scholar]
- Winters BD, Bussey TJ. Removal of cholinergic input to perirhinal cortex disrupts object recognition but not spatial working memory in the rat. Eur J Neurosci. 2005;21:2263–2270. doi: 10.1111/j.1460-9568.2005.04055.x. [DOI] [PubMed] [Google Scholar]
- Zerucha T, Stühmer T, Hatch G, Park BK, Long Q, Yu G, Gambarotta A, Schultz JR, Rubenstein JL, Ekker M. A highly conserved enhancer in the Dlx5/Dlx6 intergenic region is the site of cross-regulatory interactions between Dlx genes in the embryonic forebrain. J Neurosci. 2000;20:709–721. doi: 10.1523/JNEUROSCI.20-02-00709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]