

Abstract
Objective
The endothelium-dependent dilation of skeletal muscle arterioles is mediated by factors that have not been identified in young rats, and partly mediated by an unidentified hyperpolarizing factor in maturing rats. This study was designed to determine if endogenous hydrogen peroxide (H2O2) contributes to this arteriolar dilation at either of these growth stages.
Methods
Gracilis muscle arterioles were isolated from rats at ages 24-26 days (“weanlings”) and 46-48 days (“juveniles”). We investigated the effects of catalase treatment on the endothelium-dependent dilation of these vessels to simvastatin and ACh. Catalase-sensitive 2′,7′-dichlorofluorescein (DCF) fluorescence also was measured as an index of H2O2 formation, and arteriolar dilation to exogenous H2O2 was pharmacologically probed in each age group.
Results
Responses to simvastatin and ACh were attenuated by catalase in juvenile, but not weanling, arterioles. Juvenile, but not weanling, arterioles also displayed catalase-sensitive DCF fluorescence that was increased by ACh. Exogenous H2O2 could induce dilation in juvenile, but not weanling, arterioles. In juvenile arterioles, this dilation was abolished by the K+ channel inhibitors TEA and glibenclamide, and attenuated by NOS inhibition or endothelial removal.
Conclusion
These findings suggest that endogenous H2O2 contributes to endothelium-dependent arteriolar dilation in juvenile rats but not in younger rats, and that H2O2 acts in juvenile rats by stimulating endothelial NO release and activating smooth muscle K+ channels.
Keywords: arterioles, endothelium, postnatal growth, hydrogen peroxide
Introduction
Although relatively high levels of hydrogen peroxide (H2O2) are sometimes responsible for the vascular dysfunction associated with certain disease states (2, 25), H2O2 at lower concentrations has been recognized as an important signaling molecule that can mediate normal vascular function (5, 19). For example, endothelium-derived H2O2 has been identified as one of the factors that mediate arteriolar dilation to some agonists in the cerebral (35), mesenteric (28) and coronary (27) vascular beds, and there is mounting evidence for a physiological role of arteriolar H2O2 in local coronary blood flow regulation (18, 29, 45). Our laboratory has recently reported that endogenous H2O2 can also contribute to the arteriolar dilation and hyperemia that accompanies skeletal muscle contraction (26)
Other studies from our laboratory have documented that postnatal growth of the microvasculature in skeletal muscle is accompanied by progressive changes in a number of factors that can influence arteriolar tone and blood flow (1, 23, 24, 32). For example, skeletal muscle arterioles from young rats exhibit endothelium-dependent dilation, but these responses are not mediated by either nitric oxide (NO) or cyclooxygenase metabolites, as they are in arterioles from mature rats (1). The specific endothelial factors that mediate this dilation in young arterioles remain obscure. Because H2O2 has been identified as an important endothelium-derived vasoactive factor in the cerebral microcirculation of newborn pigs (21), we undertook the current study to determine if H2O2 could also mediate endothelium-dependent dilation in skeletal muscle arterioles of young rats. A second aim of this study was to determine if there are any changes in the mechanism by which H2O2 can influence arteriolar tone during rapid juvenile growth.
Materials and Methods
Animals
All surgical and experimental procedures were approved by the West Virginia University Animal Care and Use Committee. Experiments were performed on gracilis muscle arterioles isolated from male Sprague-Dawley rats (Harlan Sprague Dawley, Indianapolis, IN) at 3-4 weeks of age (“weanlings”) or 6-7 weeks of age (“juveniles”).
Rats were anesthetized by intraperitoneal injection of sodium pentobarbital (50 mg/kg body weight), with supplemental anesthetic (10% of original dose) administered if needed. The right carotid artery was cannulated with polyethylene tubing (PE-10 for weanlings, PE-50 for juveniles) to measure mean arterial pressure before removal of the gracilis arteriole.
Preparation of Isolated Vessels
An arteriolar branch of the femoral artery supplying the gracilis muscle was removed, handling only the surrounding connective tissue to minimize vessel stretching or damage. The rat was sacrificed by intracardiac injection of sodium pentobarbital immediately after vessel removal. The vessel was placed in warmed physiological salt solution (PSS) equilibrated with 21% O2, 5% CO2, and 74% N2, and having the following composition (mM): 119 NaCl, 4.7 KCl, 1.17 MgSO4, 1.6 CaCl2, 1.18 NaH2PO4, 24 NaHCO3, 0.026 EDTA, and 5.5 glucose. After isolation, each vessel was prepared for in vitro video microscopy as previously described (12). Briefly, the vessel was mounted in a heated (37° C) chamber that allowed for its lumen and exterior surface to be perfused and superfused, respectively, with PSS from separate reservoirs. The vessel was cannulated at both ends with glass micropipettes (50- and 70-μm tip diameters for weanling and juvenile vessels, respectively) and secured to the inflow and outflow pipettes using 9-0 nylon suture. Any side branches were ligated with a single strand teased from 6-0 silk suture. The inflow pipette was connected to a reservoir perfusion system for control of intralumenal pressure and flow. The vessel was then extended to its in situ length and equilibrated at 80% of the animal's mean arterial pressure to approximate its normal in vivo perfusion pressure (10).
Vessel diameter was measured using an onscreen video micrometer. Any vessel that did not demonstrate endothelial viability, as judged by at least a 10% dilation in response to 10−7 M acetylcholine (ACh, Sigma Chemical, St. Louis, MO), was not used in the study. Using this criterion, approximately 1 in 10 vessels from each age group were discarded. In such cases, we then isolated and studied the anatomically similar arteriole from the animal's contralateral gracilis muscle, so that we were able to obtain a viable arteriole from each animal. Diameter measurements were made under static, zero-flow conditions after a 30-minute equilibration period with continuous perfusion. Resting vascular tone was calculated as (ΔD/Dmax) × 100, where ΔD is the diameter increase from rest in response to Ca2+-free PSS (30-40 minute equilibration with Ca2+-free bath solution and no luminal flow), and Dmax is the maximum diameter measured under these conditions.
Agonists
ACh, at bath concentrations of 10−5 or 10−7 M, was used to assess arteriolar capacity for Ca2+-dependent endothelial NO formation (42). Simvastatin (Merck Research Laboratories, Rathway, NJ) was used to assess arteriolar capacity for Ca2+-independent endothelial NO formation (32). Simvastatin stimulates endothelial NO release via activation of the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, which in turn leads to direct activation of eNOS through phosphorylation of its key serine residues (20). Simvastatin was activated by alkaline lysis (5.25 ml of 0.1 N NaOH per 140 mg, dissolved in 3.5 ml of ETOH) at 50°C for 2 h. The resulting solution was then diluted to a volume of 35 ml with PBS, and neutralized to pH 7.4 with HCl. One-ml aliquots of this solution were then serially diluted with phosphate-buffered saline (PBS), producing a final bath concentration of 10−5 or 10−7 M. The two concentrations of each agonist were applied in random order, separated by a washout period to allow the vessel to regain resting tone.
For exogenous H2O2 application, a 3% aqueous H2O2 solution was diluted with PSS to concentrations of 1×10−6, 5×10−6, 1×10−5, 5×10−5, and 1×10−4 M, and these increasing concentrations were sequentially added to the bath. After each application, the H2O2 was washed out of the chamber with PSS, and the vessel was allowed to return to its control diameter before the next H2O2 application.
Assessment of Arteriolar H2O2 Production
2′,7′ dichlorodihydrofluorescein diacetate (DCFH-DA) was used to detect intracellular H2O2 production (29, 33) in arterioles at rest or during exposure to 10−5 M ACh, either with or without simultaneous exposure to catalase (Sigma, from bovine liver, 80 U/ml bath concentration). To ensure maximal scavenging of H2O2, this concentration of catalase was higher than the concentration we have previously found to scavenge all H2O2 formed during stimulation of arteriolar superoxide (O2 −) generation in vivo (22).
Non-fluorescent DCFH-DA freely enters the cell, where it is de-acetated to DCFH and then oxidized by H2O2 to form fluorescent 2′,7′ dichlorofluorescein (DCF) (37). Under some conditions, DCFH can also be oxidized in vivo by cellular peroxidases, peroxynitrite, or even hypochlorous acid (33, 37). Therefore, we considered only the portion of DCF fluorescence that could be reduced by catalase as an indicator of H2O2 activity.
After the arteriole was isolated, cannulated and allowed to develop spontaneous tone at 80% of mean arterial pressure, 5×10−5 M DCFH-DA was added to the vessel bath under one of the conditions described above. After 20 minutes of exposure to DCFH-DA, the arteriole was quickly removed from the glass micropipettes and transferred to the stage of a fluorescence microscope coupled to a high-sensitivity black and white CCD video camera (Dage-MTI, Michigan City, IN), and then epi-illuminated for 1 second using a mercury lamp and the appropriate excitation and emission filters for detection of DCF fluorescence (460-490 nm excitation, 515-550 nm barrier). In all cases, an instantaneous image of the vessel was obtained immediately upon opening of the illumination diaphragm, with identical illumination settings and optical parameters used for each vessel. Metamorph software (v. 6.01, Universal Imaging, Downingtown PA) was used to acquire, digitize and store images for subsequent analysis (46). Average fluorescence intensity was determined for the entire vessel wall (V) as well as for the background (B) of each vessel. Fluorescence intensity, based on the standard 0-255 gray scale (17), was quantified using the following equation:
where %IMAX is percent of maximum fluorescence intensity, V is the intensity of the vessel wall, and B is the background.
Scavengers and Inhibitors
To assess the role of reactive oxygen species in arteriolar dilation, vessels were pretreated with catalase (see above) or the membrane-permeable superoxide dismutase (SOD) mimetic 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl (tempol) (39) (Sigma, 10−4 M), alone or in combination. These agents were added to the bath for 30 minutes before vessels were challenged with agonists. As a control for active catalase, arteriolar responses to ACh and H2O2 were also assessed after treatment with denatured catalase. For denaturization, catalase was boiled for 10 minutes. To determine the contribution of endothelial NO production to vessel responses, the NOS inhibitor Nω-nitro-L-arginine methyl ester (L-NAME) was added to the bath at a concentration of 10−4 M for 20 minutes before agonist challenge (13).
Since many endothelium-derived relaxing factors reduce arteriolar tone at least in part through activation of smooth muscle K+ channels (6, 41, 43), the contribution of these channels to arteriolar dilation was assessed by using tetraethylammonium (TEA, Sigma) and the antidiabetic sulphonylurea, glibenclamide (Sigma), in both intact and endothelium-denuded vessels (see below). We used a 10−3 M bath concentration of TEA to selectively block Ca2+-activated K+ (KCa) channels (31), and 10−6 M glibenclamide to selectively block KATP channels (36).
Endothelial Denudation
To determine the role of the endothelium in mediating arteriolar responses to exogenous H2O2, the endothelium was removed from some vessels by mechanical abrasion (40). The pipette tip at each end of the vessel was gently advanced through the vessel lumen at least three times to ensure elimination of the endothelium. We have previously verified that this method successfully denudes the endothelium of gracilis muscle arterioles without affecting the underlying smooth muscle, as judged by the preservation of normal constrictor responses to phenylephrine and normal endothelium-independent dilator responses to sodium nitroprusside (1).
All data are presented as mean ± SE. For all analyses, a probability value of p < 0.05 was considered to be statistically significant. Dilation in response to Ca2+-free PSS is expressed as percent increase from control diameter. Differences between the means of individual experimental groups were determined by ANOVA/Newman-Keuls test, or by an unpaired Student's t-test when two independent means were compared.
Results
General characteristics of all rats from which vessels were removed for functional studies are reported in Table 1. Age, body weight, and mean arterial pressure were significantly greater in juvenile rats than in weanling rats. Table 1 also summarizes the general characteristics of all arterioles utilized for functional studies. Resting and passive diameters of arterioles from juvenile rats were significantly greater than those of arterioles from weanling rats, whereas there was no difference between groups in the level of spontaneous tone (resting vascular tone) that developed with vessel pressurization.
Table 1.
| Animal Characteristics | Weanlings | Juveniles |
|---|---|---|
| N | 42 | 39 |
| Age (days) | 25 ± 1 | 46 ± 1 * |
| Body Weight (g) | 58 ± 1 | 170 ± 3 * |
| MAP (mmHg) | 80 ± 2 | 96 ± 2 * |
| Vessel Characteristics | Weanlings | Juveniles |
| n | 42 | 39 |
| Resting Diameter (μm) | 39 ± 1 | 56 ± 2 * |
| Passive Diameter (μm) | 58 ± 2 | 83 ± 2 * |
| Resting Vascular Tone (%) | 32 ± 2 | 32 ± 2 |
General characteristics of all rats and vessels used for functional studies. MAP = mean arterial pressure. Values are given as means ± SE. N = number of animals; n = number of vessels.
p<0.05 vs. weanling group.
There were no differences between age groups in overall arteriolar responsiveness to ACh or simvastatin (Figure 1). Treatment with catalase, tempol, or catalase + tempol had no effect on the resting diameter of either weanling or juvenile arterioles (Table 2). However, catalase dramatically reduced responses to ACh and simvastatin in juvenile arterioles, but not in weanling arterioles (Figure 2, left panels). Denatured catalase had no effect on responses to ACh in either group (Table 3), verifying that the effects of catalase on juvenile arterioles were specifically due to the enzymatic activity of the molecule. Tempol by itself was without effect on responses to ACh or simvastatin in either group. In the juvenile arterioles, treatment with catalase + tempol reduced ACh and simvastatin responses to levels that were not different from those with catalase treatment alone (top right panel). In weanling arterioles, catalase + tempol had no effect on responses to either agonist (bottom right panel).
Figure 1.
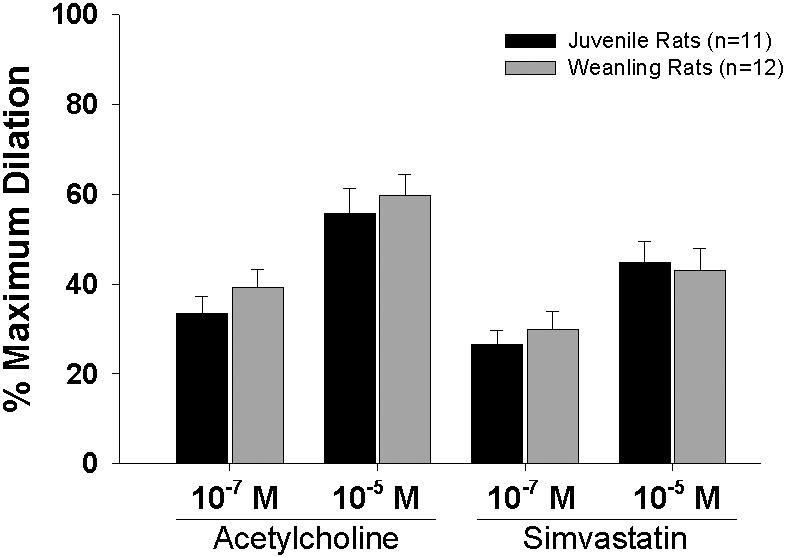
Responses of gracilis muscle arterioles from juvenile and weanling rats to acetylcholine and simvastatin. n = number of vessels. Values are given as means ± SE.
Table 2. Arteriolar Diameter (μm).
| Control | Catalase | Control | Tempol | Control | Catalase + Tempol | |
|---|---|---|---|---|---|---|
| Weanlings | 45 ± 4 | 43 ± 4 | 45 ± 4 | 43 ± 5 | 43 ± 3 | 43 ± 4 |
| Juveniles | 64 ± 4 | 62 ± 5 | 64 ± 4 | 58 ± 4 | 64 ± 4 | 60 ± 6 |
Summary of resting vessel diameters under control conditions and after treatment with 80 U/ml catalase and 10−4 M tempol, separately or in combination.Values are given as means ± SE.
Figure 2.

Effect of catalase (80 U/ml) and tempol (10−4 M), alone and in combination, on responses of juvenile and weanling arterioles to acetylcholine and simvastatin. n = number of vessels. Values are given as means ± SE. * p<0.05 vs. Control.
Table 3.
| Weanlings (n=3) | Juveniles (n=3) | |||
|---|---|---|---|---|
| Control | Inactivated catalase | Control | Inactivated catalase | |
| 10−5M ACh | 32 ± 4 | 41 ± 8 | 33 ± 3 | 40 ± 8 |
| 10−6 M H2O2 | -22 ± 10 | -22 ± 9 | -13 ± 3 | -12 ± 5 |
| 10−5 M H2O2 | -22 ± 9 | -27 ± 11 | -1 ± 4 | -2 ± 1 |
| 10−4 M H2O2 | -7 ± 4 | -3 ± 3 | 17 ± 4 | 18 ± 8 |
Magnitude of arteriolar diameter changes in response to ACh and H2O2 before and after treatment with denatured catalase (80 U/ml). Values are given as means (% maximum dilation) ± SE. n = number of vessels.
A total of 26 juvenile arterioles and 22 weanling arterioles were examined after DCFH-DA exposure for determination of relative H2O2 levels. Compared to their respective control levels, 10−5 M ACh significantly increased DCF fluorescence in juvenile arterioles (from 29 ± 4 to 44 ± 4% of maximum intensity), but not in weanling arterioles (Figure 3). Catalase reduced DCF fluorescence under control conditions as well as during ACh exposure in juvenile arterioles, but had no significant effect in weanling arterioles.
Figure 3.
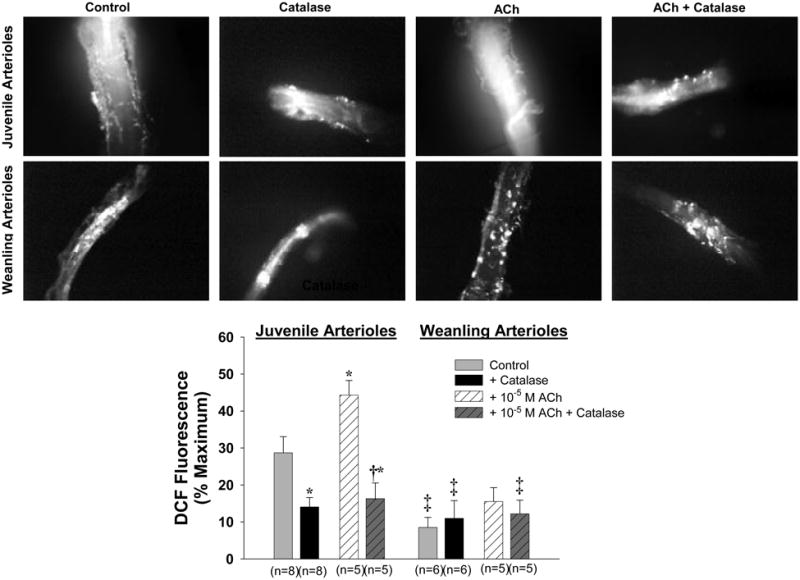
Representative microscopic images showing DCF fluorescence in arterioles exposed to DCFH-DA. Upper row: juvenile vessels. Lower row: weanling vessels. Bottom panel: Effect of 10−5 M ACh, alone or in the presence of catalase, on mean DCF fluorescence intensity in weanling and juvenile arterioles. * p<0.05 vs. Control in same age group. † p<0.05 vs. ACh in same age group. ‡ p<0.05 vs. Control in juvenile arterioles.
Application of exogenous H2O2 at bath concentrations ranging from 1×10−6 M to 1×10−4 M elicited concentration-dependent responses from juvenile arterioles, with the lower concentrations causing constriction and the higher concentrations causing dilation (Figure 4). Weanling arterioles also constricted in response to H2O2 at the lower concentrations, and these constrictions were significantly greater than those of the juvenile arterioles. However, whereas juvenile arterioles clearly dilated in response to higher concentrations of H2O2, weanling vessels did not.
Figure 4.

Responses of juvenile and weanling arterioles to exogenous hydrogen peroxide (H2O2). n = number of vessels. Values are given as means ± SE. * p<0.05 vs. juvenile arterioles.
To gain further insight into the contribution of H2O2 to ACh- and simvastatin-induced dilation of juvenile arterioles, we explored the mechanism(s) by which exogenous H2O2 induces the dilation of these vessels. Treatment with L-NAME had no significant effect on the resting diameters of either juvenile arterioles (51 ± 4 μm before vs. 48 ± 6 μm after L-NAME) or weanling arterioles (42 ± 3 μm vs. 39 ± 3 μm). However, L-NAME significantly reduced the H2O2induced dilation of juvenile arterioles, with subsequent addition of catalase completely abolishing the L-NAME-insensitive component of this dilation (Figure 5, top panel). Weanling arterioles remained unresponsive to these higher concentrations of H2O2 in the presence of L-NAME or L-NAME + catalase (bottom panel).
Figure 5.

Responses of juvenile and weanling arterioles to exogenous hydrogen peroxide (H2O2), before and after inhibition of nitric oxide synthase with Nω-nitro-L-arginine methyl ester (L-NAME), alone and in combination with catalase. n = number of vessels. Values are given as means ± SE. * p<0.05 vs. Control.
Treatment with TEA + glibenclamide significantly reduced the resting diameters of juvenile arterioles (51 ± 5 μm before vs. 36 ± 3 μm after treatment) and weanling arterioles (36 ± 3 μm vs. 30 ± 2 μm). When expressed as percent reduction from control diameter, the magnitude of this effect was similar in the two groups (25-29%). TEA + glibenclamide also converted the H2O2 -induced dilation of juvenile arterioles into constrictor responses (Figure 6, top panel), and unmasked a constrictor response of weanling arterioles to these higher concentrations of H2O2 (bottom panel).
Figure 6.
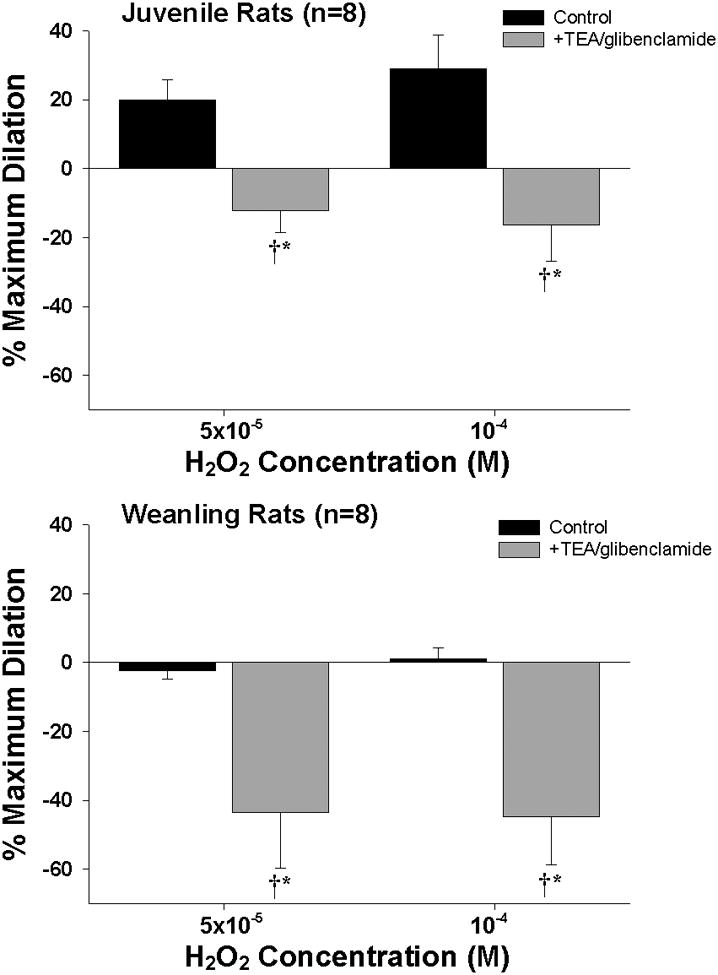
Responses of juvenile and weanling arterioles to exogenous hydrogen peroxide (H2O2) before and after inhibition of KCa channels with TEA (10−3 M) and KATP channels with glibenclamide (10−6 M). n = number of vessels. Values are given as means ± SE. * p<0.05 vs. Control. † indicates significant difference from zero (p<0.05).
Endothelial removal tended to reduce the resting diameters of juvenile arterioles (54 ± 2 μm before vs. 47 ± 4 μm after de-endothelialization) and weanling arterioles (36 ± 3 μm vs. 29 ± 2 μm), but these reductions did not reach statistical significance. However, removal of the endothelium significantly reduced the H2O2 -induced dilation of juvenile arterioles, with subsequent exposure to TEA + glibenclamide completely abolishing the remaining endothelium-independent component of these responses (Figure 7, top panel). Weanling arterioles remained unresponsive to higher concentrations of H2O2 after endothelial removal, and in contrast to our findings in intact weanling arterioles, TEA + glibenclamide did not unmask a significant constrictor response to higher concentrations of H2O2 in these vessels after endothelial removal (bottom panel).
Figure 7.

Effect of endothelial removal on responses of juvenile and weanling arterioles to exogenous hydrogen peroxide (H2O2) before and after combined treatment with TEA and glibenclamide. n = number of vessels. Values are given as means ± SE. * p<0.05 vs. Control.
Discussion
Growth-related changes in microvascular endothelial function have been reported by our laboratory and others (1, 24, 32, 44). In a recent study (1), we found that skeletal muscle arterioles from mature rats exhibit endothelium-dependent dilations to ACh and simvastatin that are partially due to NO release, with vasodilator prostanoids also contributing to these responses. However, in that study, we also found that the endothelium-dependent dilation of arterioles from younger (weanling) rats is not mediated by any of these factors. Since H2O2 has been found to mediate endothelium-dependent dilation of the cerebral vasculature in newborn pigs (21), we undertook the current study in part to determine if H2O2 could be playing an analogous role in the skeletal muscle circulation of weanling rats. However, we found that catalase had no effect on the responses of weanling arterioles to ACh or simvastatin (Figure 2, bottom panel), or on DCF fluorescence in these vessels either at rest or during ACh exposure (Figure 3, bottom panel). These findings suggest that H2O2 does not contribute to the endothelium-dependent dilation of skeletal muscle arterioles in young rats. In further support of this conclusion, exogenous H2O2, at bath concentrations of up to 100 μM, did not elicit a dilation of weanling arterioles (Figure 4). Due to the activity of catalase, glutathione peroxidase and other H2O2 -scavenging molecules in both smooth muscle and endothelial cells (5, 8), the extent to which vascular wall H2O2 actually increased during these exogenous applications is unknown. However, the concentrations that were achieved are most likely above normal, steady-state in vivo levels, which have been estimated to range from 1-100 nM within cells (14), and have been measured extracellularly at 1-10 nM within the vascular wall (7). During severe inflammation, tissue H2O2 levels can increase to as high as 100 μM (15). In the presence of K+ channel inhibitors, weanling arterioles did respond (i.e., constricted) to higher concentrations of exogenous H2O2 (Figure 6, bottom panel). This suggests that the signaling pathway for constriction that is activated by H2O2 at lower concentrations is overridden at higher concentrations of H2O2 by a dilator mechanism involving vascular smooth muscle hyperpolarization.
The current findings, in conjunction with those of our earlier study (1), rule out the contributions of NO, cyclooxygenase products and H2O2 to the endothelium-dependent dilation of skeletal muscle arterioles in rats between 3 and 4 weeks of age. Further studies will be necessary to identify the factor(s) that do mediate such responses at this early stage of life. It should also be noted that we did not find a significant difference between age groups in overall arteriolar responsiveness to ACh (Figure 1), which contrasts with our earlier findings that weanling arterioles exhibited a slightly greater responsiveness to ACh (1). Because this was found to be a relatively modest difference in the previous study, we may simply have been unable to statistically resolve that difference in the current study due to the much smaller number of vessels studied here (“n” values of 11-12 in the current study vs. 37-52 in the earlier study).
Contribution of H2O2 to endothelium-dependent responses of juvenile arterioles
In contrast to our findings in weanling arterioles, exogenous catalase greatly reduced the dilation of juvenile arterioles to both ACh and simvastatin (Figure 2). Furthermore, we found a large, catalase-sensitive component of DCF fluorescence in these vessels at rest (approximately 50% of total fluorescence) that was further increased (to approximately 63% of total fluorescence) when the vessels were challenged with ACh (Figure 3, bottom panel). These findings indicate that endogenous H2O2 does contribute to the dilation of these vessels to ACh and simvastatin, and in light of our findings in weanling arterioles, suggest that this mechanism becomes operative sometime between 4 and 6 weeks of age.
Our recent finding that endothelial removal completely abolishes the ACh- and simvastatin-induced dilation of juvenile arterioles (1) verifies that this H2O2 is endothelial in origin. The O2− from which H2O2 is formed could be generated during mitochondrial electron transport, or from the intracellular activity of membrane-bound NADPH oxidase, xanthine oxidase, cyclooxygenase, cytochrome P450 monooxygenase or nitric oxide synthase (3). Because any agonist-induced increase in H2O2 production must first trigger increased O2− formation, we initially expected that in the presence of the SOD mimetic tempol, ACh or simvastatin would lead to greater H2O2 production and therefore a larger dilation. The fact that this did not occur (Figure 2, top panel) suggests that the intrinsic SOD capacity of these vessels may be high enough to dismutate all of the additional O2− that is formed in response to these agonists, such that treatment with tempol would have no additional effect.
The responses of juvenile arterioles to exogenously applied H2O2 were complex, with lower concentrations of H2O2 eliciting constriction and higher concentrations of H2O2 eliciting dilation (Figure 4). This is consistent with a report by Cseko et al (9), who found that constriction of rat gracilis muscle arterioles to lower concentrations of H2O2 is completely abolished by prostaglandin H2 /thromboxane A2 (PGH2/TxA2) receptor blockade, but only reduced (not abolished) by endothelial removal. These authors concluded that at low concentrations, H2O2 increases arteriolar tone via the formation of constrictor prostanoids in both endothelial and vascular smooth muscle cells.
The dilation of juvenile arterioles to higher concentrations of H2O2 was reduced following endothelial removal (Figure 7), implying an important role for endothelium-derived mediators in these responses as well. Endothelial removal has also been shown to reduce H2O2 -induced dilation in mouse coronary arterioles (38), but not in human coronary arterioles, as reported by Miura and colleagues (29). Although this may reflect species-specific differences, it could also reflect the fact that the arterioles studied by Miura et al. were obtained from patients with various diseases (e.g., coronary artery disease, diabetes mellitus, hypertension), and therefore could have displayed some degree of endothelial cell dysfunction (34).
As shown in the top panel of Figure 5, inhibition of NOS in intact juvenile arterioles reduced the magnitude of H2O2- induced dilation to a level similar to that achieved by de-endothelialization, which implicates endothelium-derived NO as a major contributor to this response. We also found that blockade of KCa and KATP channels completely abolished the residual endothelium-independent portion of this dilation (Figure 7, top panel), suggesting a direct relaxing effect of H2O2 on the vascular smooth muscle due to K+ channel activation. This is consistent with previous findings that H2O2 can induce smooth muscle hyperpolarization and vasodilation by opening K+ channels, although the main contributing channel type (i.e., KCa vs. KATP channels) can vary with the species or vascular bed studied (4, 11, 16, 28, 30).
In conclusion, our results suggest that sometime between 4 and 6 weeks of age, H2O2 assumes an important role in the endothelium-dependent control of arteriolar tone through both an increase in the capacity of these vessels to produce H2O2 (Figure 3) and the onset of a vascular smooth muscle dilator response to H2O2 (Figure 4). The mechanisms that underlie these complex changes are unknown and will require further study. Our current findings seem to indicate that most or all of the elements of the cell signaling pathways that mediate H2O2 –induced dilation at 6 weeks of age are at least capable of being activated at 4 weeks of age. For example, because H2O2 and simvastatin both activate eNOS through AKT-dependent phosphorylation of its serine residues (2, 20), our finding that weanling and juvenile arterioles are both responsive to simvastatin (Figure 1) suggests that the signaling pathway through which H2O2 can induce endothelium-dependent smooth muscle relaxation is present in the younger animals. Furthermore, H2O2 can also activate smooth muscle potassium channels in weanling arterioles (Figure 6). However, the lack of any net dilator response to H2O2 in these vessels suggests that other unknown factors are involved.
Acknowledgments
This work was funded by the American Heart Association (0330194N to JCF and 0150199N to MAB) and the National Institutes of Health (R01 DK64668 to JCF NIH and RO1 HL44012 to MAB).
References
- 1.Balch Samora J, Frisbee JC, Boegehold MA. Growth-dependent changes in endothelial factors regulating arteriolar tone. Am J Physiol Heart Circ Physiol. 2007;292:H207–H214. doi: 10.1152/ajpheart.00677.2006. [DOI] [PubMed] [Google Scholar]
- 2.Cai H. NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ Res. 2005;96:818–822. doi: 10.1161/01.RES.0000163631.07205.fb. [DOI] [PubMed] [Google Scholar]
- 3.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 4.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- 5.Chen K, Thomas SR, Keaney JF., Jr Beyond LDL oxidation: ROS in vascular signal transduction. Free Radic Biol Med. 2003;35:117–132. doi: 10.1016/s0891-5849(03)00239-9. [DOI] [PubMed] [Google Scholar]
- 6.Coleman HA, Tare M, Parkington HC. EDHF is not K+ but may be due to spread of current from the endothelium in guinea pig arterioles. Am J Physiol Heart Circ Physiol. 2001;280:H2478–2483. doi: 10.1152/ajpheart.2001.280.6.H2478. [DOI] [PubMed] [Google Scholar]
- 7.Cosentino F, Patton S, d'Uscio LV, Werner ER, Werner-Felmayer G, Moreau P, Malinski T, Luscher TF. Tetrahydrobiopterin alters superoxide and nitric oxide release in prehypertensive rats. J Clin Invest. 1998;101:1530–1537. doi: 10.1172/JCI650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coyle CH, Martinez LJ, Coleman MC, Spitz DR, Weintraub NL, Kader KN. Mechanisms of H2O2-induced oxidative stress in endothelial cells. Free Radic Biol Med. 2006;40:2206–2213. doi: 10.1016/j.freeradbiomed.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 9.Cseko C, Bagi Z, Koller A. Biphasic effect of hydrogen peroxide on skeletal muscle arteriolar tone via activation of endothelial and smooth muscle signaling pathways. J Appl Physiol. 2004;97:1130–1137. doi: 10.1152/japplphysiol.00106.2004. [DOI] [PubMed] [Google Scholar]
- 10.DeLano FA, Schmid-Schonbein GW, Skalak TC, Zweifach BW. Penetration of the systemic blood pressure into the microvasculature of rat skeletal muscle. Microvasc Res. 1991;41:92–110. doi: 10.1016/0026-2862(91)90011-y. [DOI] [PubMed] [Google Scholar]
- 11.Filipovic DM, Reeves WB. Hydrogen peroxide activates glibenclamide-sensitive K+ channels in LLC-PK1 cells. Am J Physiol. 1997;272:C737–743. doi: 10.1152/ajpcell.1997.272.2.C737. [DOI] [PubMed] [Google Scholar]
- 12.Fredricks KT, Liu Y, Lombard JH. Response of extraparenchymal resistance arteries of rat skeletal muscle to reduced PO2. Am J Physiol. 1994;267:H706–715. doi: 10.1152/ajpheart.1994.267.2.H706. [DOI] [PubMed] [Google Scholar]
- 13.Frisbee JC, Roman RJ, Murali Krishna U, Falck JR, Lombard JH. Altered mechanisms underlying hypoxic dilation of skeletal muscle resistance arteries of hypertensive versus normotensive Dahl rats. Microcirculation. 2001;8:115–127. [PubMed] [Google Scholar]
- 14.Giulivi C, Boveris A, Cadenas E. The steady-state concentrations of oxygen radicals in mitochondria. In: Gilbert DL, Colton CA, editors. Reactive oxygen species in biological systems. New York: Kluwer Academic/Plenum Publishers; 1999. [Google Scholar]
- 15.Halliwell B, Clement MV, Long LH. Hydrogen peroxide in the human body. FEBS Lett. 2000;486:10–13. doi: 10.1016/s0014-5793(00)02197-9. [DOI] [PubMed] [Google Scholar]
- 16.Iida Y, Katusic ZS. Mechanisms of cerebral arterial relaxations to hydrogen peroxide. Stroke. 2000;31:2224–2230. doi: 10.1161/01.str.31.9.2224. [DOI] [PubMed] [Google Scholar]
- 17.Klabunde RE, Anderson DE. Role of nitric oxide and reactive oxygen species in platelet-activating factor-induced microvascular leakage. J Vasc Res. 2002;39:238–245. doi: 10.1159/000063689. [DOI] [PubMed] [Google Scholar]
- 18.Koller A, Bagi Z. Nitric oxide and H2O2 contribute to reactive dilation of isolated coronary arterioles. Am J Physiol Heart Circ Physiol. 2004;287:H2461–2467. doi: 10.1152/ajpheart.00295.2004. [DOI] [PubMed] [Google Scholar]
- 19.Kunsch C, Medford RM. Oxidative stress as a regulator of gene expression in the vasculature. Circ Res. 1999;85:753–766. doi: 10.1161/01.res.85.8.753. [DOI] [PubMed] [Google Scholar]
- 20.Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lacza Z, Puskar M, Kis B, Perciaccante JV, Miller AW, Busija DW. Hydrogen peroxide acts as an EDHF in the piglet pial vasculature in response to bradykinin. Am J Physiol Heart Circ Physiol. 2002;283:H406–411. doi: 10.1152/ajpheart.00007.2002. [DOI] [PubMed] [Google Scholar]
- 22.Lenda DM, Sauls BA, Boegehold MA. Reactive oxygen species may contribute to reduced endothelium-dependent dilation in rats fed high salt. Am J Physiol Heart Circ Physiol. 2000;279:H7–H14. doi: 10.1152/ajpheart.2000.279.1.H7. [DOI] [PubMed] [Google Scholar]
- 23.Linderman JR, Boegehold MA. Arteriolar network growth in rat striated muscle during juvenile maturation. Int J Microcirc Clin Exp. 1996;16:232–239. doi: 10.1159/000179179. [DOI] [PubMed] [Google Scholar]
- 24.Linderman JR, Boegehold MA. Growth-related changes in the influence of nitric oxide on arteriolar tone. Am J Physiol. 1999;277:H1570–1578. doi: 10.1152/ajpheart.1999.277.4.H1570. [DOI] [PubMed] [Google Scholar]
- 25.Lum H, Roebuck KA. Oxidant stress and endothelial cell dysfunction. Am J Physiol Cell Physiol. 2001;280:C719–741. doi: 10.1152/ajpcell.2001.280.4.C719. [DOI] [PubMed] [Google Scholar]
- 26.Marvar PJ, Boegehold MA. Dietary salt reduces hydrogen peroxide-dependent dilation in contracting muscle. FASEB J. 2006;20:A268. doi: 10.1080/10739680701444057. [DOI] [PubMed] [Google Scholar]
- 27.Matoba T, Shimokawa H, Morikawa K, Kubota H, Kunihiro I, Urakami-Harasawa L, Mukai Y, Hirakawa Y, Akaike T, Takeshita A. Electron spin resonance detection of hydrogen peroxide as an endothelium-derived hyperpolarizing factor in porcine coronary microvessels. Arterioscler Thromb Vasc Biol. 2003;23:1224–1230. doi: 10.1161/01.ATV.0000078601.79536.6C. [DOI] [PubMed] [Google Scholar]
- 28.Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, Kanaide H, Takeshita A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. 2000;106:1521–1530. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res. 2003;92:e31–40. doi: 10.1161/01.res.0000054200.44505.ab. [DOI] [PubMed] [Google Scholar]
- 30.Miura H, Liu Y, Gutterman DD. Human coronary arteriolar dilation to bradykinin depends on membrane hyperpolarization: contribution of nitric oxide and Ca2+-activated K+ channels. Circulation. 1999;99:3132–3138. doi: 10.1161/01.cir.99.24.3132. [DOI] [PubMed] [Google Scholar]
- 31.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268:C799–822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- 32.Nurkiewicz TR, Boegehold MA. Calcium-independent release of endothelial nitric oxide in the arteriolar network: onset during rapid juvenile growth. Microcirculation. 2004;11:453–462. doi: 10.1080/10739680490475999. [DOI] [PubMed] [Google Scholar]
- 33.Silveira LR, Pereira-Da-Silva L, Juel C, Hellsten Y. Formation of hydrogen peroxide and nitric oxide in rat skeletal muscle cells during contractions. Free Radic Biol Med. 2003;35:455–464. doi: 10.1016/s0891-5849(03)00271-5. [DOI] [PubMed] [Google Scholar]
- 34.Simionescu M. Implications of early structural-functional changes in the endothelium for vascular disease. Arterioscler Thromb Vasc Biol. 2007;27:266–274. doi: 10.1161/01.ATV.0000253884.13901.e4. [DOI] [PubMed] [Google Scholar]
- 35.Sobey CG, Heistad DD, Faraci FM. Mechanisms of bradykinin-induced cerebral vasodilatation in rats. Evidence that reactive oxygen species activate K+ channels. Stroke. 1997;28:2290–2294. doi: 10.1161/01.str.28.11.2290. discussion 2295. [DOI] [PubMed] [Google Scholar]
- 36.Standen NB, Quayle JM, Davies NW, Brayden JE, Huang Y, Nelson MT. Hyperpolarizing vasodilators activate ATP-sensitive K+ channels in arterial smooth muscle. Science. 1989;245:177–180. doi: 10.1126/science.2501869. [DOI] [PubMed] [Google Scholar]
- 37.Tarpey MM, Fridovich I. Methods of detection of vascular reactive species: nitric oxide, superoxide, hydrogen peroxide, and peroxynitrite. Circ Res. 2001;89:224–236. doi: 10.1161/hh1501.094365. [DOI] [PubMed] [Google Scholar]
- 38.Thengchaisri N, Kuo L. Hydrogen peroxide induces endothelium-dependent and -independent coronary arteriolar dilation: role of cyclooxygenase and potassium channels. Am J Physiol Heart Circ Physiol. 2003;285:H2255–2263. doi: 10.1152/ajpheart.00487.2003. [DOI] [PubMed] [Google Scholar]
- 39.Thiemermann C. Membrane-permeable radical scavengers (tempol) for shock, ischemia-reperfusion injury, and inflammation. Crit Care Med. 2003;31:S76–84. doi: 10.1097/00003246-200301001-00011. [DOI] [PubMed] [Google Scholar]
- 40.Uluoglu C, Zengil H. Comparison of different de-endothelization procedures in the isolated rat thoracic aorta: a short communication. Res Commun Mol Pathol Pharmacol. 2003:113–114. 289–297. [PubMed] [Google Scholar]
- 41.Ungvari Z, Csiszar A, Koller A. Increases in endothelial Ca2+ activate KCa channels and elicit EDHF-type arteriolar dilation via gap junctions. Am J Physiol Heart Circ Physiol. 2002;282:H1760–1767. doi: 10.1152/ajpheart.00676.2001. [DOI] [PubMed] [Google Scholar]
- 42.Ungvari Z, Sun D, Huang A, Kaley G, Koller A. Role of endothelial [Ca2+]i in activation of eNOS in pressurized arterioles by agonists and wall shear stress. Am J Physiol Heart Circ Physiol. 2001;281:H606–612. doi: 10.1152/ajpheart.2001.281.2.H606. [DOI] [PubMed] [Google Scholar]
- 43.Welsh DG, Segal SS. Role of EDHF in conduction of vasodilation along hamster cheek pouch arterioles in vivo. Am J Physiol Heart Circ Physiol. 2000;278:H1832–1839. doi: 10.1152/ajpheart.2000.278.6.H1832. [DOI] [PubMed] [Google Scholar]
- 44.Willis AP, Leffler CW. Endothelial NO and prostanoid involvement in newborn and juvenile pig pial arteriolar vasomotor responses. Am J Physiol Heart Circ Physiol. 2001;281:H2366–2377. doi: 10.1152/ajpheart.2001.281.6.H2366. [DOI] [PubMed] [Google Scholar]
- 45.Yada T, Shimokawa H, Hiramatsu O, Kajita T, Shigeto F, Goto M, Ogasawara Y, Kajiya F. Hydrogen peroxide, an endogenous endothelium-derived hyperpolarizing factor, plays an important role in coronary autoregulation in vivo. Circulation. 2003;107:1040–1045. doi: 10.1161/01.cir.0000050145.25589.65. [DOI] [PubMed] [Google Scholar]
- 46.Zhu J, Mori T, Huang T, Lombard JH. Effect of high-salt diet on NO release and superoxide production in rat aorta. Am J Physiol Heart Circ Physiol. 2004;286:H575–583. doi: 10.1152/ajpheart.00331.2003. [DOI] [PubMed] [Google Scholar]