

Abstract
The CD40-CD154 system controls various aspects of the host inflammatory response in models of cellular and humoral immunity. Recently, we described a role for CD40 in the innate immune response in polymicrobial sepsis. However, recent data suggests that CD40 maybe activated by CD154 or directly via bacterial heat shock protein (HSP) 70. Therefore, we decided to test the mechanism of CD40 activation in murine polymicrobial sepsis. Wild-type (WT), CD40–/–, and CD154–/– underwent cecal ligation and puncture (CLP). Compared with WT mice, CD40–/– had improved survival in association with attenuated production of IL-12, TNF-α, and IL-6. In contrast, CD154–/– mice behaved similar to WT mice with regard to mortality and cytokine production. The differential response of CD40–/– and CD154–/– mice to CLP was not due to a general attenuated response to inflammatory stimuli, as all three strains had similar survival after LPS administration, and CD40–/– macrophages had normal production of IL-12 in response to lipopolysaccharide. In contrast, CD40–/– macrophages had attenuated IL-12 production in response to Escherichia coli HSP70 (DnaK). Furthermore, intraperitoneal administration of DnaK resulted in a 4-fold increase in IL-12 in WT mice, which was absent in CD40–/– mice. This data demonstrates CD154-independent CD40 activation in polymicrobial sepsis and suggests that bacterial HSP70 is capable of stimulating CD40 in vitro and in vivo.
Keywords: DnaK, cecal ligation and puncture, IL-12, LPS, macrophage
INTRODUCTION
Sepsis and septic shock affects nearly 500,000 people a year with an approximate 35% to 45% mortality (1). Mortality in sepsis is frequently the result of the development of multisystem organ failure (MSOF) (1, 2). The development of MSOF is associated with increased production of proinflammatory cytokines, including interleukin (IL)-6, IL-1β, and tumor necrosis factor-α (TNF-α) (3, 4). In addition, recent data suggests an important role for TH-1 cytokines, including interferon (IFN)-γ and IL-12 (5–7).
Control of cytokine production during the initial stages of infection is partly controlled by the innate immune response. Before the development of cellular or humoral immunity, mediators in the innate immune response such as Toll-like receptors (TLR) are capable of binding bacterial products including lipopolysaccharide (LPS), lipoteichoic acid (LTA), and CpG DNA, resulting in inflammatory cytokine production (1, 8, 9). However, TLR4 knockout mice, although protected from endotoxemia, have no survival advantage in murine models of polymicrobial sepsis, suggesting an important role for additional receptors in the innate immune response during sepsis (10).
One potential candidate to mediate the innate immune response to polymicrobial sepsis is CD40. CD40 (TNF receptor superfamily member 5) is a 48-kDa protein expressed primarily on B cells, macrophages, dendritic cells, vascular endothelial cells, and fibroblasts (11). CD40 expression is regulated at the transcriptional level with signal transducer and activator of transcription-1 (STAT-1) and NF-κB (12, 13). CD40 is classically activated by CD154, soluble or expressed on platelets and activated T Cells (11). Activation is a potent inducer of NF-κB and proinflammatory cytokine production, specifically TH-1 cytokines such as IL-12 (14, 15). The ability of CD40 to strongly regulate TH-1 cytokines gives it a prominent role in control of TH-1-mediated diseases such as Crohn’s disease and tuberculosis (16, 17).
Recently, we described a role for CD40 in the innate immune response. CD40–/– mice are protected from the lethality of polymicrobial sepsis, with a marked attenuation in IL-12 production and NF-κB DNA binding (18). In addition, recent data describes upregulation of CD40 in human sepsis and its ligand, CD154, in mice after CLP (19, 20). However, the mechanism of CD40 activation in polymicrobial sepsis remains unclear. Recent data suggests bacterial heat shock protein (HSP) 70 kDa (HSP70) binds to and activates CD40 directly (21). Mycobacterium tuberculosis HSP70 and Escherichia coli HSP70 (DnaK) increased β-chemokine production from macrophages in a CD40-dependent manner (21). Furthermore, recent in vivo data described increased mortality in CD40–/– mice but not CD154–/– mice in response to M. tuberculosis. The differential response was attributed to M. tuberculosis HSP70 binding to CD40 on dendritic cells (22). Combined, these data suggest CD40 may participate in the innate immune response by binding directly to bacterial products.
In this study, we demonstrate that in contrast to CD40–/– mice, CD154–/– mice are not protected from the lethality of polymicrobial sepsis and do not have attenuated cytokine production. Furthermore, although CD40–/– mice have a similar response to wild-type (WT) mice during endotoxemia, CD40–/– mice fail to induce IL-12 in response to DnaK in vitro and in vivo. Together, these data suggest that CD40 activation in sepsis may be in part mediated via DnaK.
MATERIALS AND METHODS
Mice
C57BL/6 WT, CD40–/–, and CD154–/– female mice (all on the same genetic background), 5 to 6 weeks old at the time of delivery, were obtained from Jackson Laboratories (Bar Harbor, ME) and were allowed to acclimatize for 1 week before use. All were provided with free access to food and water and 12-h light and dark cycles in accordance with animal care guidelines. The animal use committee of New York University approved all studies. The experiments were performed in adherence to the National Institutes of Health Guidelines on the Use of Laboratory Animals.
Cecal ligation and puncture (CLP)
CLP was done using a modification of the procedure as previously described (18). Briefly, mice were anesthetized using 2% isofluorane anesthesia with supplemental oxygen. A 1- to 2-cm midline abdominal incision was made and the cecum was ligated below the ileocecal valve and was punctured once through with a 19-gauge needle. The incision was then sutured with 3.0 silk. All mice were given 1 mL of normal saline subcutaneously postoperatively. They were then allowed free access to food and water. Plasma and bronchoalveolar lavage fluid (BALF) were collected at 18 h as previously described (18). Survival analysis for CD40–/– mice was in part published previously (18). For endotoxemia experiments, mice were injected with 500 μg of LPS (Sigma, St. Louis, MO) and were monitored for 7 days for survival.
In vitro experiments
Murine peritoneal macrophages were obtained as previously described (18). Briefly, mice were injected with 2 mL of 3% brewer’s thioglycollate intraperitoneally (Sigma) 3 to 5 days before use and were subsequently euthanized as previously described. This procedure resulted in >95% macrophages. Cells were washed and resuspended in RPMI media 1640 (Gibco, Gaithersburg, MD) with 10% fetal calf serum (HyClone Laboratories, Logan, UT), penicillin-streptomycin (Gibco), and fungizone-amphotericin B (Gibco) at 4 × 105/mL. Cells were treated with 100 U/mL IFN-γ (R&D Systems, Minneapolis, MN) or 10 ng of LPS for 24 h followed by treatment with 10 μg of DnaK (StressGen Biotechnologies, Victoria, British Columbia, Canada) or saline. Supernatants were collected at 24 h and were stored at −40°C for cytokine analysis. Conditions were repeated in duplicate.
In vivo experiments
WT or CD40–/– mice were treated with saline or 100 μg of LPS i.p. for 24 h. This was followed by the administration of 100 μg of DnaK or saline control for 4 h at which point mice were harvested and plasma was collected.
Cytokine analysis
IL-6, TNF-α, and IL-12 (p40) levels were determined using commercially available enzyme-linked immunosorbent assay (ELISA) kits purchased from R&D Systems. All samples were prepared and analyzed as per the protocols provided.
Statistical analysis
All numerical data were expressed as means ± SEM. P values were derived from a two-tailed Mann-Whitney test or a log-rank test for median survival. For comparisons among multiple groups, a one-way analysis of variance (ANOVA) with Bonferroni post hoc analysis was performed for all groups. All statistical analysis and graphing were determined using the GraphPad Prism statistical software (version IV; GraphPad, San Diego, CA).
RESULTS
CD40–/– and CD154–/– mice have differential response to CLP
To investigate the role of CD154 in activation of CD40 during polymicrobial sepsis, we performed CLP as previously described (18). Consistent with data from our laboratory and others, WT mice had a median survival of 25 h after CLP, which was nearly 2-fold higher in CD40–/– mice (48 vs. 25 h; P = 0.001), although there was no difference in overall survival (0% vs. 10%) (18, 23, 24). In contrast, CD154–/– mice had a similar median survival compared with WT mice (26 vs. 25 h; P = NS), which was also significantly reduced compared with CD40–/– mice (26 vs. 48 hrs; P = 0.001; Fig. 1).
FIG. 1. CD40–/– and CD154–/– mice have differential response to CLP.
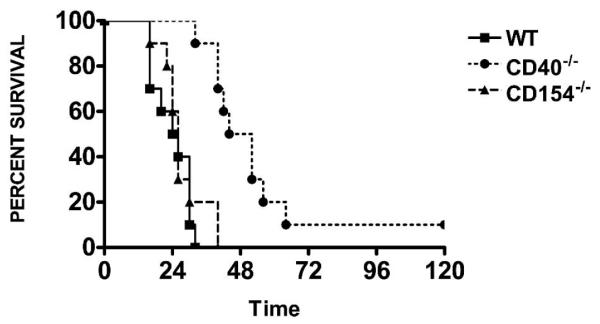
WT, CD40–/–, and CD154–/– underwent CLP for survival. Time 0 represents time of surgery. P < 0.0001 for CD40–/– compared with WT or CD154–/– mice. Twelve to 15 mice per group.
We next investigated whether the alterations in survival correlated to changes in circulating cytokines. Similar to previous data, CD40–/– mice subjected to CLP had attenuated plasma levels of IL-12 when compared with WT mice (1297 ± 184 pg/mL vs. 159 ± 42 pg/mL; P < 0.001; Fig. 2A) and levels that were not significantly different from unoperated controls (159 ± 42 pg/mL vs. 109.1 ± 12 pg/mL; P = NS). In contrast, CD154–/– mice had reduced levels of IL-12 compared with WT mice but had significantly greater IL-12 levels than those observed in CD40–/– mice (Fig. 2A). CD40–/– mice also had attenuated circulating levels of IL-6 and TNF-α compared with WT mice 18 h after CLP (Fig. 2, B and C). In contrast, CD154–/– mice had circulating levels of IL-6 and TNF-α that were indistinguishable from WT mice (Fig. 2, B and C). Similar results were obtained for IL-6 and IL-12 in BALF (Table 1). Together, these data demonstrate a CD154-independent mechanism of CD40 activation in polymicrobial sepsis, which is present in multiple tissue compartments.
FIG. 2. CD40–/– and CD154–/– have differential cytokine production after CLP.

WT, CD40–/–, and CD154–/– underwent CLP and plasma was collected at 18 h. (A) IL-12; (B) IL-6; (C) TNF-α. P < 0.001 by ANOVA for all three cytokines. *P < 0.05 for WT versus CD40–/– on Bonferroni post hoc analysis. Five to seven mice per group.
Table 1.
CD40–/– but not CD154–/– mice have attenuated BALF IL-6 and Il-12 production after CLP
| WT | CD40–/– | CD154–/– | |
|---|---|---|---|
| IL-12 (pg/mL) | 9.5 ± 2.6 | 0.98 ± 0.5* | 2.7 ± 2.2 |
| IL-6 (pg/mL) | 1133 ± 211 | 135 ± 33.3* | 738 ± 295.7 |
BALF collected 18 h after CLP. P < 0.01 for IL-6 and IL-12 via ANOVA.
P < 0.05 compared with WT via Bonferroni post hoc analysis. Data represents 8–10 mice per group.
One potential explanation for the altered phenotype is that CD40 is required for the general response to multiple inflammatory stimuli. To test if a global reduction in the responsiveness accounted for the blunted inflammation during CLP, WT, CD154–/–, and CD40–/– mice were treated with LPS, a stimulus transduced by the CD14-TLR4 system. In contrast to what was observed after CLP, WT, CD154–/–, and CD40–/– mice all had similar clinical appearance and survival to LPS challenge (Fig. 3). This suggests that the changes observed in CD40–/– and CD154–/– mice after CLP are not due to altered responsiveness to LPS and that additional bacterial products maybe further activating CD40.
FIG. 3. CD40–/– and CD154–/– mice have similar response to endotoxemia.
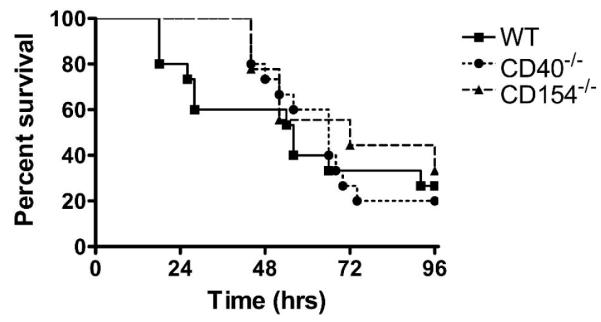
WT, CD40–/–, and CD154–/– mice received 500 μg of LPS intraperitoneally at time 0 and survival was assessed over 5 days. Data represents 10 mice per group.
DnaK ( E. coli HSP70) induces IL-12 in vitro and in vivo via CD40
Bacterial HSP70 has been shown to bind and activate CD40 in vitro, resulting in β-chemokine production (21). However, murine peritoneal macrophages from WT and CD40–/– mice showed similar induction IL-12 after stimulation with DnaK (Fig. 4A). Because CD40 expression is strongly regulated by LPS (12), and peritoneal macrophages from WT and CD40–/– mice exhibited similar IL-12 production to LPS ex vivo (Fig. 4B), we used a strategy of LPS priming to test the effect of DnaK. When WT peritoneal macrophages were pretreated with LPS, DnaK increased IL-12 levels by 150% compared with pretreatment controls. In contrast, peritoneal macrophages from CD40–/– mice showed no additional IL-12 production (Fig. 4C). This was specific for DnaK, as IFN-γ stimulation (23 ± 14 pg/mL vs. 29 ± 6.2 pg/mL; P = NS), and repeat LPS administration to LPS primed cells resulted in a similar induction of IL-12 in WT compared with CD40–/– macrophages (2.8 vs. 9.8; P = 0.15). Similar results were obtained with IL-6 (18.3% vs. 1.3%; P = 0.01). Again, this was specific for DanK, as repetitive LPS stimulation was similar in both groups (53% vs. 34%; P = 0.53).
FIG. 4. CD40–/– macrophages have a differential IL-12 response to LPS and DnaK.

Thioglycollate elicited peritoneal macrophages from WT and CD40–/– mice. (A) DnaK macrophages were stimulated with 10 μg of DnaK for 24 h. (B) LPS macrophages were stimulated with 10 ng/mL LPS for 24 h and IL-12 was quantified in supernatants by ELISA. (C) LPS-DnaK macrophages were pretreated with LPS and were subsequently stimulated with 10 μg of DnaK for 24 h. IL-12 levels presented as the percentage of change from non-DnaK-treated cells. *P = 0.02. Data represents cells from six different mice.
We next wished to test whether DnaK could further augment IL-12 in vivo via CD40. WT mice pretreated with LPS demonstrated a 4-fold increase in IL-12 4 h after administration of DnaK. In contrast, CD40–/– mice had only a minimal increase in IL-12 after DnaK, and absolute levels were 2-fold less than WT mice (178 ± 15 vs. 351 ± 53; P < 0.02; Fig. 5). There was no difference in systemic IL-12 levels in response to LPS by itself between WT and CD40–/– mice (100.5 ± 12.9 pg/mL vs. 95.5 ± 8.6 pg/mL; P = NS). Together, these data suggest that CD40 is required for DnaK-mediated IL-12 production.
FIG. 5. CD40–/– mice have impaired IL-12 production in response to DnaK in vivo.
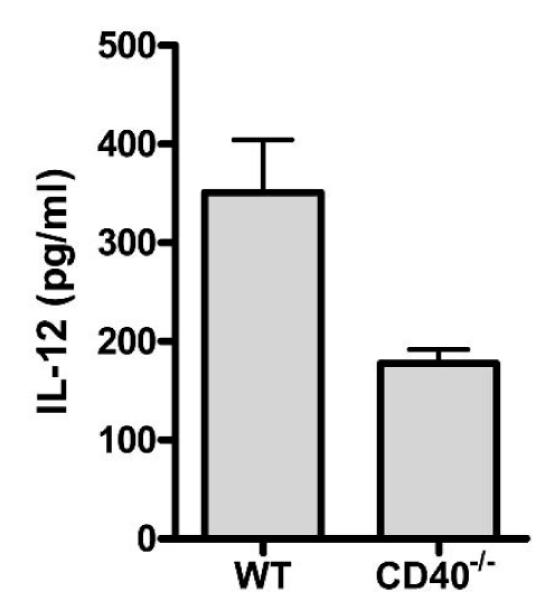
WT and CD40–/– mice pretreated with LPS were given 100 μg of DnaK intraperitoneally and plasma was harvested at 4 h. IL-12 was measured by ELISA. P < 0.02. Four to five mice per group.
DISCUSSION
In this study, we document that in contrast to CD40–/– mice, CD154–/– mice are not protected from the lethality of polymicrobial sepsis. This is associated with little to no attenuation in IL-6, TNF-α, or IL-12 production, suggesting a CD154-independent mechanism of CD40 activation in sepsis. These differences were not due to differences in endotoxin sensitivity, as all three strains had similar survival to LPS administration. However, CD40–/– macrophages, while exhibiting no alterations in cytokine production after LPS administration, exhibited significant attenuation in IL-6 and IL-12 production after DnaK compared with control macrophages. This was recapitulated in vivo, where CD40–/– mice had attenuated IL-12 production in response to DnaK compared with WT mice, suggesting that DnaK is capable of directly stimulating CD40 in vivo and in vitro.
The finding of a differential response between CD40–/– and CD154–/– mice to polymicrobial sepsis is surprising. This is relatively specific for polymicrobial sepsis, as all three strains of mice had a similar response to endotoxemia. In addition, the severity of the sepsis in our model with near 100% mortality at 48 h, suggests that these pathways are relevant in fulminant sepsis/septic shock. Few studies have directly compared CD40–/– and CD154–/– mice in the same study. In those that have, most failed to show significant differences. Specifically, CD40–/– and CD154–/– mice behave similarly in models of hyperoxic lung injury and malaria (25, 26). However, our data is consistent with those of Lazarevic et al. (22). In that study, CD40–/– but not CD154–/– were susceptible to aerosolized M. tuberculosis with increased mortality and bacterial growth only in CD40–/– mice. The differential phenotype of CD40–/– mice in tuberculosis and sepsis is further evidence the effect of CD40 activation is in part dependent upon the necessity of CD40 activation to control bacterial growth versus pathologically augmenting inflammation. In addition, the strength and nature of CD40 stimulation may also be important and regulates which intracellular signaling cascades are activated (27).
There are many possible explanations for the observed differences between CD40–/– and CD154–/– mice in our study. One possibility is that congenital absence of CD40 may render these mice globally unable to respond to numerous inflammatory stimuli. This is unlikely as there was no observed difference between WT, CD40–/–, and CD154–/– in response to endotoxemia. Furthermore, macrophages from WT and CD40–/– mice exhibited similar production of IL-12 in response to IFN-γ and LPS stimulation in vitro, suggesting that the IFN-γ receptor and the TLR4-CD14 system are unaffected by congenital absence of CD40.
Another potential explanation is the presence of a CD154-independent mechanism of CD40 activation during polymicrobial sepsis. The differential cytokine production in serum and BALF in CD40–/– and CD154–/– mice suggests this effect is not localized to a specific tissue compartment. Bacterial HSP70s are a recently described stimulator of inflammatory cytokines from human macrophages in vitro (21, 28). In our study, we demonstrate that DnaK (E. coli HSP70) is capable of eliciting IL-6 and IL-12 production from primary murine macrophages but not those from CD40–/– mice. This is consistent with other in vitro studies documenting that M. tuberculosis HSP70 and DnaK are capable of inducing β-chemokines, IL-6, and TNF-α from human macrophages and murine dendritic cells in a CD40-dependent manner (21, 22, 28). However, our study now extends these in vitro observations and is the first to demonstrate that DnaK can stimulate inflammatory cytokine production in vivo, suggesting that resident macrophages are also capable of responding to DnaK. The lack of IL-12 induction by DnaK in CD40–/– mice suggests that this phenomenon is also CD40 dependent. Together, this implicates CD40 as a potentially important member of the innate immune response.
Our study also provides valuable information regarding the difference between murine models of sepsis. Currently, there are many murine models of human sepsis, the most common of which are endotoxemia and polymicrobial sepsis (CLP). In studies comparing the two models, CLP more closely resembles human sepsis in regards to mitochondrial and cytokine gene regulation and in response to immunomodulatory therapy and regulation (24, 29, 30). Furthermore, mice devoid of endotoxin signaling (TLR4–/–) have similar outcomes compared with WT mice after CLP, suggesting the role for additional stimuli and receptors in the innate immune response (10). Our data now suggests that bacterial HSP70s can further stimulate inflammation and augment LPS-induced cytokine production via CD40. Although our study only investigated DnaK, it is likely other bacterial HSP70 have a similar effect as already described with Salmonella typheri and Mycobacterium tuberculosis HSP70 in vitro (22, 31). Furthermore, HSP70 has been found in certain disease states. Patients with streptococcal disease or rheumatic fever have circulating bacterial HSP70 in their sera (32). In addition, recent studies also demonstrate that large amounts of E. coli HSP70 are released from damaged cell envelopes in the absence of heat shock (33). Consequently, it is likely that multiple bacterial HSP70s are released and are capable of activating CD40 at sites of active bacterial clearance.
Finally, the presence of CD154-independent means of CD40 activation has significant implications for any future therapeutic interventions. Currently, targeting of the CD40-CD154 systems has been focused almost exclusively on CD154 monoclonal antibodies. Our data suggests that in certain disease states, this may result in incomplete blockade of CD40 signaling. One such disease is rheumatoid arthritis. Patients with rheumatoid arthritis have a higher incidence of HLA-DR with higher affinity for DnaK (34). Furthermore, patients with systemic lupus erythematosis have antibodies to M. tuberculosis HSP70 (35). The significance of this will not be fully known until better blocking agents of CD40 become available.
In conclusion, our data further supports an essential role for CD40 but not CD154 in the innate immune response during polymicrobial sepsis. E. coli HSP70 is capable of augmenting inflammatory cytokine production in vitro and in vivo via CD40 and likely explains this differential phenotype. These findings have significant implications for any future therapeutic trials aimed at targeting the CD40 system.
Acknowledgments
This work was supported by the National Institutes of Health (grants KO8 HL070710, MO1 RR0096, and RO1 HL57879).
REFERENCES
- 1.Wheeler AP, Bernard GR. Treating patients with severe sepsis. N Engl J Med. 1999;340:207–214. doi: 10.1056/NEJM199901213400307. [DOI] [PubMed] [Google Scholar]
- 2.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 3.Meduri GU, Headley S, Kohler G, Stentz F, Tolley E, Umberger R, Leeper K. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1β and IL-6 levels are consistent and efficient predictors of outcome over time. Chest. 1995;107:1062–1073. doi: 10.1378/chest.107.4.1062. [DOI] [PubMed] [Google Scholar]
- 4.Meduri GU, Kohler G, Headley S, Tolley E, Stentz F, Postlethwaite A. Inflammatory cytokines in the BAL of patients with ARDS. Persistent elevation over time predicts poor outcome. Chest. 1995;108:1303–1314. doi: 10.1378/chest.108.5.1303. [DOI] [PubMed] [Google Scholar]
- 5.Matsukawa A, Hogaboam CM, Lukacs NW, Lincoln PM, Evanoff HL, Kunkel SL. Pivotal role of the CC chemokine, macrophage-derived chemokine, in the innate immune response. J Immunol. 2000;164:5362–5368. doi: 10.4049/jimmunol.164.10.5362. [DOI] [PubMed] [Google Scholar]
- 6.Silva AT, Cohen J. Role of interferon-γ in experimental gram-negative sepsis. J Infect Dis. 1992;166:331–335. doi: 10.1093/infdis/166.2.331. [DOI] [PubMed] [Google Scholar]
- 7.Bjerre A, Brusletto B, Hoiby EA, Kierulf P, Brandtzaeg P. Plasma interferon-γ and interleukin-10 concentrations in systemic meningococcal disease compared with severe systemic gram-positive septic shock. Crit Care Med. 2004;32:433–438. doi: 10.1097/01.CCM.0000104950.52577.97. [DOI] [PubMed] [Google Scholar]
- 8.Krug A, Towarowski A, Britsch S, Rothenfusser S, Hornung V, Bals R, Giese T, Engelmann H, Endres S, Krieg AM, Hartmann G. Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL-12. Eur J Immunol. 2001;31:3026–3037. doi: 10.1002/1521-4141(2001010)31:10<3026::aid-immu3026>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 9.Re F, Strominger JL. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J Biol Chem. 2001;276:37692–37699. doi: 10.1074/jbc.M105927200. [DOI] [PubMed] [Google Scholar]
- 10.Echtenacher B, Freudenberg MA, Jack RS, Mannel DN. Differences in innate defense mechanisms in endotoxemia and polymicrobial septic peritonitis. Infect Immun. 2001;69:7271–7276. doi: 10.1128/IAI.69.12.7271-7276.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Kooten C, Banchereau J. CD40-CD40 ligand. J Leukocyte Biol. 2000;67:2–17. doi: 10.1002/jlb.67.1.2. [DOI] [PubMed] [Google Scholar]
- 12.Tone M, Tone Y, Babik JM, Lin CY, Waldmann H. The Role of Sp1 and NF-κB in regulating CD40 gene expression. J Biol Chem. 2002;277:8890–8897. doi: 10.1074/jbc.M109889200. [DOI] [PubMed] [Google Scholar]
- 13.Nguyen VT, Benveniste EN. Involvement of STAT-1 and ets family members in interferon-γ induction of CD40 transcription in microglia/macrophages. J Biol Chem. 2000;275:23674–23684. doi: 10.1074/jbc.M002482200. [DOI] [PubMed] [Google Scholar]
- 14.Jyothi MD, Khar A. Regulation of CD40L expression on natural killer cells by interleukin-12 and interferon-γ: its role in the elicitation of an effective anti-tumor immune response. Cancer Immunol Immunother. 2000;49:563–572. doi: 10.1007/s002620000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McDyer JF, Goletz TJ, Thomas E, June CH, Seder RA. CD40 ligand/CD40 stimulation regulates the production of IFN-γ from human peripheral blood mononuclear cells in an IL-12- and/or CD28-dependent manner. J Immunol. 1998;160:1701–1707. [PubMed] [Google Scholar]
- 16.Henderson RA, Watkins SC, Flynn JL. Activation of human dendritic cells following infection with Mycobacterium tuberculosis. J Immunol. 1997;159:635–643. [PubMed] [Google Scholar]
- 17.De Jong YP, Comiskey M, Kalled SL, Mizoguchi E, Flavell RA, Bhan AK, Terhorst C. Chronic murine colitis is dependent on the CD154/CD40 pathway and can be attenuated by anti-CD154 administration. Gastroenterology. 2000;119:715–723. doi: 10.1053/gast.2000.16485. [DOI] [PubMed] [Google Scholar]
- 18.Gold JA, Parsey M, Hoshino Y, Hoshino S, Nolan A, Yee H, Tse DB, Weiden MD. CD40 contributes to lethality in acute sepsis: an in vivo role for CD40 in innate immunity. Infect Immun. 2003;71:3521–3528. doi: 10.1128/IAI.71.6.3521-3528.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding Y, Chung CS, Newton S, Chen Y, Carlton S, Albina JE, Ayala A. Polymicrobial sepsis induces divergent effects on splenic and peritoneal dendritic cell function in mice. Shock. 2004;22:137–144. doi: 10.1097/01.shk.0000131194.80038.3f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sugimoto K, Galle C, Preiser JC, Creteur J, Vincent JL, Pradier O. Monocyte CD40 expression in severe sepsis. Shock. 2003;19:24–27. doi: 10.1097/00024382-200301000-00005. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Kelly CG, Karttunen JT, Whittall T, Lehner PJ, Duncan L, MacAry P, Younson JS, Singh M, Oehlmann W, Cheng G, Bergmeier L, Lehner T. CD40 is a cellular receptor mediating mycobacterial heat shock protein 70 stimulation of CC-chemokines. Immunity. 2001;15:971–983. doi: 10.1016/s1074-7613(01)00242-4. [DOI] [PubMed] [Google Scholar]
- 22.Lazarevic V, Myers AJ, Scanga CA, Flynn JL. CD40, but not CD40L, is required for the optimal priming of T cells and control of aerosol M. tuberculosis infection. Immunity. 2003;19:823–835. doi: 10.1016/s1074-7613(03)00324-8. [DOI] [PubMed] [Google Scholar]
- 23.Soriano FG, Liaudet L, Szabo E, Virag L, Mabley JG, Pacher P, Szabo C. Resistance to acute septic peritonitis in poly(ADP-ribose) polymerase-1-deficient mice. Shock. 2002;17:286–292. doi: 10.1097/00024382-200204000-00008. [DOI] [PubMed] [Google Scholar]
- 24.Sachdeva K, Yan B, Chichester CO. Lipopolysaccharide and cecal ligation/ puncture differentially affect the subcellular distribution of the pregnane X receptor but consistently cause suppression of its target genes CYP3A. Shock. 2003;19:469–474. doi: 10.1097/01.shk.0000048903.46342.ec. [DOI] [PubMed] [Google Scholar]
- 25.Piguet PF, Da Kan C, Vesin C, Rochat A, Donati Y, Barazzone C. Role of CD40-CD40L in mouse severe malaria. Am J Pathol. 2001;159:733–742. doi: 10.1016/s0002-9440(10)61744-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adawi A, Zhang Y, Baggs R, Finkelstein J, Phipps RP. Disruption of the CD40-CD40 ligand system prevents an oxygen-induced respiratory distress syndrome. Am J Pathol. 1998;152:651–657. [PMC free article] [PubMed] [Google Scholar]
- 27.Mathur RK, Awasthi A, Wadhone P, Ramanamurthy B, Saha B. Reciprocal CD40 signals through p38MAPK and ERK-1/2 induce counteracting immune responses. Nat Med. 2004;10:540–544. doi: 10.1038/nm1045. [DOI] [PubMed] [Google Scholar]
- 28.Galdiero M, de l’Ero GC, Marcatili A. Cytokine and adhesion molecule expression in human monocytes and endothelial cells stimulated with bacterial heat shock proteins. Infect Immun. 1997;65:699–707. doi: 10.1128/iai.65.2.699-707.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Remick DG, Newcomb DE, Bolgos GL, Call DR. Comparison of the mortality and inflammatory response of two models of sepsis: lipopolysaccharide versus cecal ligation and puncture. Shock. 2000;13:110–116. doi: 10.1097/00024382-200013020-00004. [DOI] [PubMed] [Google Scholar]
- 30.Remick D, Manohar P, Bolgos G, Rodriguez J, Moldawer L, Wollenberg G. Blockade of tumor necrosis factor reduces lipopolysaccharide lethality, but not the lethality of cecal ligation and puncture. Shock. 1995;4:89–95. doi: 10.1097/00024382-199508000-00002. [DOI] [PubMed] [Google Scholar]
- 31.Takaya A, Tomoyasu T, Matsui H, Yamamoto T. The DnaK/DnaJ chaperone machinery of Salmonella enterica serovar Typhimurium is essential for invasion of epithelial cells and survival within macrophages, leading to systemic infection. Infect Immun. 2004;72:1364–1373. doi: 10.1128/IAI.72.3.1364-1373.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lemos JA, Giambiagi-Demarval M, Castro AC. Expression of heat-shock proteins in Streptococcus pyogenes and their immunoreactivity with sera from patients with streptococcal diseases. J Med Microbiol. 1998;47:711–715. doi: 10.1099/00222615-47-8-711. [DOI] [PubMed] [Google Scholar]
- 33.Vazquez-Laslop N, Lee H, Hu R, Neyfakh AA. Molecular sieve mechanism of selective release of cytoplasmic proteins by osmotically shocked Escherichia coli. J Bacteriol. 2001;183:2399–2404. doi: 10.1128/JB.183.8.2399-2404.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roth S, Willcox N, Rzepka R, Mayer MP, Melchers I. Major differences in antigen-processing correlate with a single Arg71↔Lys substitution in HLA-DR molecules predisposing to rheumatoid arthritis and with their selective interactions with 70-kDa heat shock protein chaperones. J Immunol. 2002;169:3015–3020. doi: 10.4049/jimmunol.169.6.3015. [DOI] [PubMed] [Google Scholar]
- 35.Tasneem S, Islam N, Ali R. Crossreactivity of SLE autoantibodies with 70-kDa heat shock proteins of Mycobacterium tuberculosis. Microbiol Immunol. 2001;45:841–846. doi: 10.1111/j.1348-0421.2001.tb01323.x. [DOI] [PubMed] [Google Scholar]