

Abstract
Age-related macular degeneration (AMD), a progressive condition that is untreatable in up to 90% of patients, is a leading cause of blindness in the elderly worldwide. The two forms of AMD, wet and dry, are classified based on the presence or absence of blood vessels that have disruptively invaded the retina, respectively. A detailed understanding of the molecular mechanisms underlying wet AMD has led to several robust FDA-approved therapies. In contrast, there are not any approved treatments for dry AMD. In this review, we provide insight into the critical effector pathways that mediate each form of disease. The interplay of immune and vascular systems for wet AMD, and the proliferating interest in hunting for gene variants to explain AMD pathogenesis, are placed in the context of the latest clinical and experimental data. Emerging models of dry AMD pathogenesis are presented, with a focus on DICER1 deficit and the toxic accumulation of retinal debris. A recurring theme that spans most aspects of AMD pathogenesis is defective immune modulation in the classically immune-privileged ocular haven. Interestingly, the latest advances in AMD research highlight common molecular disease pathways with other common neurodegenerations. Finally, the therapeutic potential of intervening at known mechanisms of AMD pathogenesis is discussed.
I. Introduction
Age-related macular degeneration (AMD) is a principal cause of blindness in the United States and other industrialized nations. An estimated 10 million Americans are afflicted with AMD (Friedman et al., 2004), which is comparable in scope to the 12 million living with cancer (Hayat et al., 2007), or the 5 million with Alzheimer’s disease (Brookmeyer et al., 2007). The prevalence of AMD steadily increases with age, affecting 2% of the population at age 40, and one in four people by age 80 (Friedman et al., 2004). For reasons that are not fully understood, AMD is more common in lightly-pigmented and female populations (Friedman et al., 2004). Treatment of AMD is largely an unmet need: There are no FDA approved therapies except for a small percentage of individuals with end-stage disease.
There are two types of AMD, the “dry” and “wet” forms. Dry AMD is a chronic disease that usually causes some degree of visual impairment, and sometimes progresses to severe blindness. In contrast, wet AMD affects only 10–15% of AMD patients, emerges abruptly, and rapidly progresses to blindness if left untreated (Guyer et al., 1986; Wong et al., 2008). Since AMD patients typically develop the dry form first, wet AMD occurs on a background of dry AMD; as such, dry AMD can be considered a risk factor or even precursor state for wet AMD.
In the early stages of AMD, which is asymptomatic, insoluble extracellular aggregates called drusen accumulate in the retina (See Fig. 1 in (Bird, 2010)). The late stage of dry AMD, which is also known as geographic atrophy (GA), is characterized by scattered or confluent areas of degeneration of retinal pigment epithelium (RPE) cells and the overlying light-sensing retinal photoreceptors, which rely on the RPE for trophic support. The other late stage form of AMD, the wet form, is typified by choroidal neovascularization (CNV) wherein newly immature blood vessels grow towards the outer retina from the underlying choroid. These immature blood vessels leak fluid below or within the retina.
It is convenient to dichotomize the pathology of “wet” and “dry” forms of the disease based on the presence or absence, respectively, of CNV. However, as an understanding of AMD pathogenesis improves, emerging evidence indicates that significant overlap exists in the underlying mechanisms of these seemingly disparate clinical conditions. In spite of this apparent overlapping pathophysiology, the two forms of AMD are indeed somewhat clinically distinct: that is, effective treatment of wet AMD does not typically ameliorate the dry AMD component. Clearly, further clarification of the overlapping and unique processes that lead to wet and dry pathology will be essential for future advances in the prevention and treatment of AMD.
II. The Retina
In health
For a review of structural features in the healthy retina vs. the AMD-afflicted retina, the reader is referred to excellent reviews elsewhere (Bird, 2010; Rattner and Nathans, 2006). The features of a healthy ocular fundus is shown in Fig. 1A. Relative to the surrounding peripheral retina, the macular region has a high density of photoreceptors. As such, the macula subserves central vision and acuity that enables resolution of fine details, such as edges or borders. The retina consists of multiple cell layers that form an interdependent anatomical and metabolic network. Other notable features of the retina include: the selectively permeable blood-retinal barrier (Cunha-Vaz, 2004), the greatest oxygen consumption per weight of any organ in the body (Warburg, 1928), and immune privilege (Streilein, 2003).
Figure 1.
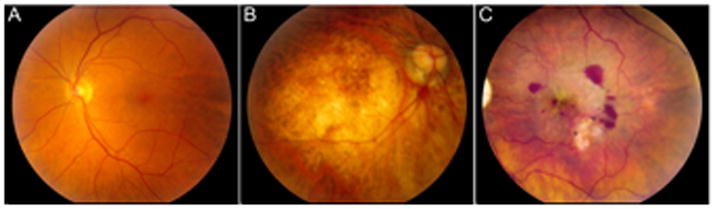
Fundus photographs in health and in AMD
In AMD
Geographic atrophy
A representative eye with GA is shown in Fig. 1B. AMD primarily affects the macular region of the retina, with relative sparing of the surrounding peripheral retina. AMD is defined by confluent regions of drusen, which are multi-component, heterogeneous aggregates that lie both external and internal to the RPE cells (Klein et al., 2008; Zweifel et al., 2010). The emergence and “growth” of drusen occurs slowly over years or decades. RPE cell death and synaptic dysfunction accompany underlying drusen (Johnson et al., 2005), although the cause-effect relationship of drusen and retinal degeneration (which may be reciprocal) is not fully understood.
Choroidal neovascularization
A representative eye with CNV is shown in Fig. 1C. CNV also primarily affects the macula. If left untreated, it can lead to severe blindness with scarring within several months. Assessment of CNV is typically made using fluorescein angiography or optical coherence tomography to measure characteristic lesions with leakage of blood or plasma proteins from immature choroidal blood vessels.
III. Introduction to the mechanisms of AMD pathogenesis
This Review is focused on the mechanistic underpinnings of AMD. Before delving into the molecular details of disease, we will provide a short account of the success of mechanism-based therapy in AMD.
From AMD disease mechanism to approved therapy
Understanding the molecular basis of disease empowers one to translate such knowledge into clinical use. A stellar example of mechanism-based therapy is anti-angiogenesis treatment for wet AMD. Ferrara’s cloning of vascular endothelial growth factor-A (VEGF-A) in 1989 (Leung et al., 1989), combined with knowledge derived from the pioneering work by Folkman and other investigators in the field of oncology (Folkman, 1995), revealed the centrality of VEGF in vascular biology. These seminal contributions enabled Ferrara et al. to develop the first anti-VEGF-A treatment, the monoclonal antibody Avastin (bevacizumab; Genentech), which received FDA approval for cancer treatment in 2004. Subsequently the importance of VEGF-A in ocular neovascularization was validated (Adamis et al., 1994; Aiello et al., 1994). Coupled with the identification of VEGF-A in surgically obtained CNV specimens from humans with AMD ((Frank et al., 1996; Kvanta et al., 1996; Lopez et al., 1996), the development of anti-VEGF-A agents to treat neovascular AMD quickly followed suit.
FDA-approved therapies for CNV emerged in 2004 (Macugen; pegaptanib sodium; Eyetech/Pfizer) (Gragoudas et al., 2004) and 2006 (Lucentis; ranibizumab; Genentech) (Brown et al., 2006; Rosenfeld et al., 2006). Although off-label bevacizumab is not FDA approved for use in neovascular AMD, it has assumed an equal footing with ranibizumab in clinical care because it has similar efficacy yet costs substantially less (Martin et al., 2012). Most recently, in 2011 Eylea (VEGF-TRAP-Eye; aflibercept; Regeneron)(Economides et al., 2003) received FDA approval for treatment of CNV.
Today, anti-VEGF-A therapy for CNV dramatically improves or stabilizes vision in the vast majority of patients (Martin et al., 2011). This phenomenal clinical success has set the stage for treatment of other ophthalmologic maladies (e.g. diabetic macular edema, retinal vein occlusion, iris or corneal neovascularization, uveitis) (Ciulla and Rosenfeld, 2009) and extraocular diseases (e.g. neoplasms, heart disease, neurodegeneration) (Carmeliet, 2005) that share a common VEGF-A-dependent pathway of angiogenesis. While anti-angiogenic therapy for wet AMD benefits many patients and typifies the success of mechanism-based translational medicine, there are no approved treatments for the more common dry form of AMD and progress towards the identification of molecular targets for this disease subset remains constrained.
The RPE: At the core of AMD pathogenesis
Whereas the step-wise development of certain maladies (e.g. cancers) is relatively well-defined (Hanahan and Weinberg, 2011), no such hallmarks of disease progression have been identified in AMD. Nonetheless, the literature abounds with various implicated causes of disease. For want of a comprehensive systems approach, one could navigate the numerous avenues and permutations by which an ever-growing list of mechanisms contribute to AMD pathology. Indeed, such an approach has been emphasized in previous reviews (Ambati et al., 2003a; Bird, 2010; Patel and Chan, 2008; Rattner and Nathans, 2006; Zarbin, 2004). Instead, since diverse etiologies may contribute to an AMD phenotype, we advance three models of disease mechanism that emphasize critical, non-redundant effector pathways.
In each of these models, the RPE is the fulcrum of AMD pathogenesis. In general, although inter-individual heterogeneity exists, RPE dysfunction and atrophy precedes the latter stages of AMD (GA or CNV). The RPE integrates numerous stimuli to define its own health, while also receiving and broadcasting signals to and from the retinal microenvironment. The capacity of the RPE to modulate diverse pathways of AMD pathogenesis can be gleaned from RNA transcriptome analyses of human AMD donor eyes (Booij et al., 2010; Newman et al., 2012) and in vitro RPE cells (Strunnikova et al., 2010). Importantly, human AMD samples display significant inter-individual variation in RPE transcript expression, which supports the concept that heterogenic stress responses underlie a categorical AMD phenotype. Genome-wide stress-response transcriptome and proteome assays have begun to catalog the effect of specific AMD-associated stresses (Kurji et al., 2010), and age-related changes in retinal molecular composition (Cai and Del Priore, 2006; Glenn et al., 2011) on whole-genome RPE gene expression. If these types of experimental approaches are applied to a multitude of AMD-associated stresses, the pooled results of these studies could reveal common protective and deleterious RPE gene responses, and would also help clarify the key molecular drivers of disease. Subsequently, the manipulation of critical pathways in stress-function assays and animal models of AMD could create new avenues of therapeutic strategy, and augment existing knowledge garnered from focused investigations of specific pathways or sets of genes.
An important route of communication and recurring theme in AMD pathology is the crosstalk of RPE with immune and vascular systems. This “immunovascular axis” drives CNV; however, whether this network modulates RPE cell viability is less clear. Although the vitality of the RPE cell is paramount to retinal health, it is also true that perturbations in other tissues, for example, the choroid, Bruch’s membrane and photoreceptors, are important burdens on the retinal microenvironment. Nevertheless, the critical event in AMD pathogenesis, from which there is no return, is RPE dysfunction and degeneration.
IV. Molecular mechanisms of neovascular AMD: RPE stress and the immunovascular axis
Our first of three paradigms of AMD molecular pathogenesis is an integrated view of CNV that is supported by an abundance of successful therapeutic efforts in human and animal models. Figure 2 details the molecular mechanisms of CNV pathogenesis. As will be discussed, the RPE response to heterogeneous stressors is an integral process in CNV. RPE-independent mechanisms of CNV may also exist, though such pathways are not incongruent with the centrality of RPE immunovascular signaling in CNV.
Figure 2.

The Immunovascular axis of wet AMD
RPE Vascular Response
Blockade of VEGF-A, a potent pro-angiogenic messenger, is the basis of available therapies for neovascular AMD. The two major pathways by which the RPE produces and secretes VEGF-A are in response to complement (Nozaki, Raisler et al. 2006; Rohrer, Coughlin et al. 2011) (Fig 2, 1) and oxidative stress (Pons and Marin-Castano, 2011) (Fig 2, 2). Simply defined, oxidative stress is the oxidation of cellular macromolecules, and the complement system is a set of about 30 proteins that are an important component of the innate immune response to microbes (Bradley et al., 2011). If left unregulated, activation of complement proteins can directly damage host tissue and recruit immune cells to the vicinity of active complement activation. It is presumed that protection against complement is achieved through a variety of complement regulatory molecules that are expressed in and localize to the retina (Anderson et al., 2010).
These primary stresses may act independently to induce angiogenesis, but they also synergize. For example, oxidative stress potentiates complement-induced RPE secretion of VEGF-A (Thurman et al., 2009). Besides VEGF, other directly vasculogenic molecules (i.e. that act on endothelial cells, Fig. 2) are also secreted by the RPE in response to activated complement (Fukuoka et al., 2003) and oxidative stress (Higgins et al., 2003). Many such RPE-elaborated cytokines have been identified in human and experimental CNV specimens (Amin et al., 1994; Bhutto et al., 2006; Grossniklaus et al., 2002; Lopez et al., 1996). Analysis of human tissue is an important counterpart to information derived from experimental disease models, although it must be noted that these human data are somewhat limited by small sample sizes and also subject to variability introduced in part by technical and logistical challenges of post mortem tissue isolation.
Still, the RPE need not be the only source of pro-angiogenic factors, which could originate from various immune cells or other cell types (Fig. 2). Importantly, the focus of the present model is to display the multiple, redundant pathways via which CNV could be augmented. We emphasize the RPE as a central player in CNV in order to demonstrate two key mechanistic points: 1) The potential for multiple distinct stresses to converge to produce a common (pro-angiogenic) effect (Fig. 2); and 2) The diversity of response molecules produced by the RPE that could drive angiogenesis. Although VEGF-A blockade has dominated CNV treatment, it is reasonable to expect that future endeavors will lead to CNV therapeutics that block other angiogenesis-promoting molecules (Noel et al., 2007).
RPE Immune Response
A pro-inflammatory retinal milieu, which is promoted by RPE response to heterogeneous stresses, appears to be a key modulator of CNV development and progression. The RPE regulation of the complement system here too plays an important role: Besides inducing RPE secretion of pro-angiogenic VEGF, active complement factors C3a and C5a are potent chemotactic agents, and recruit leukocytes to the choroid (Nozaki et al., 2006) (Fig. 2). Furthermore, oxidative stress of the RPE by photooxidation products activates complement (Zhou et al., 2006), and an oxidative damaged-induced autoimmune reaction results in complement deposition in the retina (Hollyfield et al., 2008). Thus, just as the RPE secretes diverse direct effectors of angiogenesis in response to heterogeneous stressors, there are multiple pathways by which the RPE can regulate the retinal immune-landscape, which in turn can regulate neovascularization in AMD.
In particular, in CNV, the macrophage is the king of vascular-modifying immune cells that are attracted to the retina in disease; an increase in the number of retinal macrophages is a hallmark of CNV (Cherepanoff et al., 2010; Grossniklaus et al., 2000; Skeie and Mullins, 2009) (Fig. 2). However, whether macrophages are critical for CNV development or progression is not clear- their increase in CNV could either represent an exacerbation of disease, or a compensatory vascular-dampening response. In support of their pro-angiogenic properties, inhibition of monocyte migration to the retina reduced CNV in a laser-induced mouse model of disease (Espinosa-Heidmann et al., 2003; Sakurai et al., 2003). In contrast, in a non-injury mouse model of AMD, mice that are genetically deficient for either CCR2 or its cognate ligand (CCL2) - and consequently possess defects in macrophage mobilization - develop choroidal neovascularization (Ambati et al., 2003b), suggesting that macrophages somehow also protect against CNV (Ambati et al., 2003b; Molday et al., 2000). The reader is directed to an excellent review of the role of macrophages in CNV (Skeie and Mullins, 2009). Given the available evidence, the most likely role for macrophages in CNV is determined by local macrophage-polarizing factors (Kelly et al., 2007; Patel et al., 2008). Indeed, work in tumor biology has revealed complex local regulation of macrophage vascular-modifying activity. In light of current interest in immune-modulating interventions for CNV (Wang et al., 2011b), the particular microenvironmental influences governing macrophage activity in CNV remains an area of needed research.
The potential for immune contribution to CNV begs several salient questions about disease mechanism. For one, if certain pro-angiogenic factors are also pro-inflammatory, does anti-angiogenesis therapy achieve its clinical effect by reducing both direct vascular and indirect immune effects? Among the many factors that control macrophage chemotaxis, VEGF-A has a well-defined role in recruitment of pro-angiogenic macrophages (Cursiefen et al., 2004). Therefore, it is reasonable to expect that anti-VEGF therapy might reduce macrophage infiltration of the retina in CNV.
On the contrary, bevacizumab treatment of CNV dramatically increased the number of retinal macrophages within human neovascular membranes (Tatar et al., 2008). Subsequent work showed that blockade of VEGF-A increased leukocyte-endothelial adhesion, which could explain the increase in retinal macrophages following anti-VEGF-A therapy (Walshe et al., 2009). In addition, compensatory elevation in VEGF-A levels following anti-VEGF-A therapy (Willett et al., 2005) might promote inflammatory cell recruitment. These findings raise the following question, the answer to which has important therapeutic implications: What is the ratio of pro- and anti-angiogenic macrophages that accumulate after anti-VEGF-A treatment? If the proportion or relative activity of pro-angiogenic macrophages increased following anti-VEGF treatments, this finding could explain the tachyphylaxis (desensitization) that occurs with multiple anti-VEGF-A treatments (Forooghian et al., 2009). In that case, the selective inhibition of pro-angiogenic macrophages would be an appealing adjunct to anti-VEGF therapy. This is in contrast to the current, broad-spectrum use of immunosuppression in AMD, which does not target a certain immune cell or subtype.
Microglia are another immune cell type that might modulate human CNV pathogenesis. These resident retinal macrophages accumulate in the subretinal space in a CX3CR1-deficient mouse after light-induced and aging models of retinal degeneration; the accumulation of microglia in these mice appears to exacerbate laser-induced CNV (Combadiere et al., 2007). The CX3CR1−/−, CCL2−/−, CCR2−/− mouse models of AMD predominantly exhibit defects in microglia and macrophage function (Reviewed: (Raoul et al., 2010)). When compared to mouse models deficient in either one of these chemokine receptors, a double knockout mouse genetically deficient for both CX3CR1 and CCR2 produced a more penetrant, and earlier-onset, spontaneous phenotype of retinal lesions similar to those seen in dry AMD; also, a subset (15%) of mice spontaneously developed choroidal neovascularization (Tuo et al., 2007). However, while macrophages accumulate in human CNV membranes, it is not known whether microglia do, too. In the largest histopathologic characterization of microglia in AMD to date, which observed microglia at various stages of AMD pathology, there was a change in microglia morphology, but not number, in AMD compared to non-diseased retinas (Penfold et al., 1997). The precise functional ramifications of such altered microglia morphology remain to be elucidated. Very recently it has been claimed that many of the early-onset retinal degenerative phenotypes observed in the CX3CR1−/− mice are attributable to the fact that these mice were generated on the C57BL/6N background, which contains the rd8 retinal degeneration mutation of the Crb1 gene (Mattapallil et al., 2012). However, the CCL2−/− and CCR2−/− mice were generated on the C57BL/6J background, which does not contain the rd8 mutation, and develop retinal findings many months later than rd8 mice (Ambati et al., 2003b). Therefore, the retinal phenotype in CCL2−/− and CCR2−/− mice are attributable to microglia/macrophages and point to an important role for these myeloid-derived cells in maintaining retinal homeostasis.
Immune cells besides macrophages and microglia could also modify CNV (Fig. 2). The cellular infiltrate in experimental CNV is a motley band of circulating and resident, immune and non-immune cells. Interestingly, one-third of all infiltrating cells were not classified (Espinosa-Heidmann et al., 2005); future work could provide a comprehensive assessment of the composition of cellular infiltrate in human CNV specimens. Indeed, other myeloid-derived immune cells are increasingly implicated in vascular modification in other systems. For example, neutrophils and other non-macrophage immune cells increase in cancer tumors following anti-VEGF-A treatment (Ferrara, 2010). In fact, neutrophils contribute to CNV pathogenesis in experimental animal models (Sun and Nathans, 1997; Zhou et al., 2005); it would be interesting to learn whether neutrophils are present in human CNV specimens or the retinal immune infiltrate that follows anti-VEGF treatment. As is the case with macrophages, it is becoming increasingly clear that subsets of other immune cells can have dramatically different effect on vasculature (Sica et al., 2008). Thus, a full understanding of the immunopathology of CNV will require an assessment of all potential vascular-modifying immune cells, and their subsets, in health, disease, and following therapeutic intervention.
Looking forward, the mechanism of immune suppression in reducing CNV requires extensive clarification. While it is known that steroids reduce the net pro-angiogenic cytokine secretion by the RPE (Tong et al., 2006), the effect of immune suppressive agents on immune cell activity (Ehrchen et al., 2007) in CNV remains undefined. As such, targeted immune suppression or modulation of specific immune cells is one avenue of research that could yield valuable therapeutic advances in CNV.
Section V: Towards a Model of Dry AMD
In contrast to wet AMD, clinical success in treating dry AMD remains elusive. The molecular hallmarks of dry AMD are toxic accumulations, either within the RPE cell or at the RPE-BrM interface (Fig. 3). As such, dry AMD may be thought of as an insidious form of a metabolic storage disease. Two approaches to reducing these lingering burdens are: 1) Preventing their formation; or 2) Removing them after formation. Attempts to prevent RPE damage have been unsuccessful, although the removal of toxic accumulations as a therapeutic strategy remains largely unexplored. In search of the “holy grail” of AMD treatment, we will discuss two emerging conceptual frameworks that offer fresh research avenues and the promise to help fill the gaping therapeutic void in dry AMD.
Figure 3.

Toxic accumulations in dry AMD
The “Damaged Goods” Model
Simply put, AMD and other neurodegenerative disorders occur when a particular cell or group of cells dies. Although the distressed cells in each disease are often different, their pathologies share several themes. For one, some have recognized that common neurodegenerative diseases of aging, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and Huntington’s disease (HD), are similar in that they accumulate mitochondrial defects (Lin and Beal, 2006). We propose adding AMD to this list. The mitochondrial defects observed in the RPE of AMD eyes include DNA mutations, impaired structural integrity, and defective mitochondrial function (Fig. 3). The consequences of damaged mitochondria can be dire: in particular, diminished energy production and imbalance of pro- and anti-apoptotic signals lead to cell death (Lin and Beal, 2006). Mitochondrial damage also leads to increased ROS production, which, in turn, may tarnish other key cellular components.
Besides defective mitochondria, other toxins accumulate in AMD and other common neurodegenerative diseases. For example, an excessive amount of “lipofuscin”, which is non-degradable debris that accumulates in the RPE with age, is associated with AMD (Schmitz-Valckenberg et al., 2009). In the presence of light, lipofuscin forms ROS and is toxic to RPE cells (Winkler et al., 1999). Analogous lipofuscin-like substances that occur in other neurodgenerations include beta-amyloid or tau-protein inclusions in Alzheimer’s disease, huntingtin protein in Huntington’s disease, Lewy Bodies in Parkinson’s disease, and non-amyloid aggregates in amyotrophic lateral sclerosis. In general, mtDNA dysfunction precedes the accumulation of these substances (Lin and Beal, 2006).
The various forms of neurodegeneration each can be described in terms of such signature pathologies, which is likely the result of cell or tissue-specific stresses or response to stress. The diverse microenvironments and unique biological flux experienced in these heterogeneous cell types make it difficult to assign common inciting stressors or stress responses -which are likely to be many and overlapping in effect- of these diseases. Still, mitochondrial dysfunction appears to be a common co-pathology of neurodegeneration, and it is an appealing concept that the persistence of damaged mitochondria and other cellular detritus represents a common node at which point myriad stressors converge. In this respect, we view AMD as a disease that has many potential upstream causes or damage-inducing stimuli that funnel into downstream and less-redundant pathways. To date, nearly all attempts to revert AMD have focused on preventing toxin accumulation; yet if there are diverse causes of toxin formation, then it is worth defining the potential therapeutic role of filtering the cellular milieu at the confluence, rather than the source, of the disease pathogenesis watershed.
Autophagy
Cells are equipped with machinery to discard toxic accumulations. In a self-cleansing process called macroautophagy, the cell can be rid of large, damaged cellular contents such as organelles or proteins. Here forward we will use the term “autophagy” in place of “macroautophagy”, although it is important to recognize that other forms of autophagy also exist (Klionsky, 2007). In essence, autophagy of the mitochondria (a.k.a. “mitophagy”), and other cellular debris, could rejuvenate cells by disposing of defunct organelles, a concept which has been reviewed for AMD (Mitter et al., 2012) and other neurodegenerative disorders (Wong and Cuervo, 2010). Future work should address several basic questions about this cell survival mechanism in AMD, such as whether the various animal models of disease undergo autophagic changes, and if autophagy-modulating compounds can reverse experimental disease.
Damage control
Since ROS damage is a common feature of neurodegenerative diseases, anti-oxidant supplementation has been an area of intense therapeutic investigation. Unfortunately, this approach has failed to ameliorate manifest neurodegenerative disease (Boothby and Doering, 2005; Evans, 2008; Shen and Ji, 2010). Indeed, a cocktail of anti-oxidants has not shown benefit in progression to advanced dry AMD, although they were reported to have a small effect in reducing rate of progression to CNV (2001). Given the widespread shortcomings of anti-oxidant supplementation for dry AMD and other diseases in clinical trials, a new wave of neurodegeneration research focuses, appropriately, on combating lingering oxidative damage in an effort to renew cellular robustness.
Ironically, ROS damages the cellular components that are disposed by autophagy, yet ROS are also critical for induction of autophagy (Scherz-Shouval et al., 2007). The counterpoint is also true: It has been shown that anti-oxidants inhibit autophagy (Underwood et al., 2010). Thus, in theory, flooding the retina with anti-oxidants, which did not significantly prevent progression of or vision loss from AMD by main outcome measures (2001), could be counterproductive to removing biological garbage. The interplay of ROS and autophagy is expansive, and has been reviewed elsewhere (Szumiel, 2011). Autophagy may also regulate RPE health by reducing cytotoxicity that is secondary to a primary insult. For example, a mitochondrion that has been damaged by ROS overproduces even more ROS; therefore mitophagy would reduce both the root and downstream ROS burden (Zhou et al., 2011).
DICER1: The common thread?
Until now, we have discussed the mechanistic underpinnings for some of the many identified RPE stressors. Yet, because these injurious agents are so vast and heterogeneous, the “RPE stress” that they cause is necessarily a nebulous term. The cumulative burden on the RPE may, or may not, converge to a single pathway that determines RPE cell viability. Thus, in contrast to wet AMD – in which VEGF-A is the linchpin of blood vessel growth – the search for a single molecule or pathway that is critical in preventing RPE cell death in dry AMD remains elusive. Therapeutic attempts to promote the robustness of the RPE would be bolstered by the discovery of a common node that tied together at least some of the seemingly disparate stressors that are implicated in AMD pathogenesis. Is there an integrative hub of RPE viability that coordinates the effect of multiple, redundant stressors?
The activity of the enzyme DICER1 is sufficiently broad-reaching that it is an attractive candidate as a choreographer of retinal health and homeostasis (Fig. 4). Specifically, the literature supports an emerging role for DICER1 in governing RPE cell health and function via several mechanisms, including its influence on inflammation and global (coding and non-coding) RNA expression. DICER1, a ribonuclease, was specifically reduced in the RPE of GA patients (Kaneko et al., 2011); moreover, this pathological decrease in DICER1 was accompanied by the aberrant overabundance of the non-coding Alu RNA, which is toxic to RPE cells. In that study, Kaneko et al. also present a new disease model of GA: the genetic ablation or knockdown of DICER1 in the mouse RPE.
Figure 4.
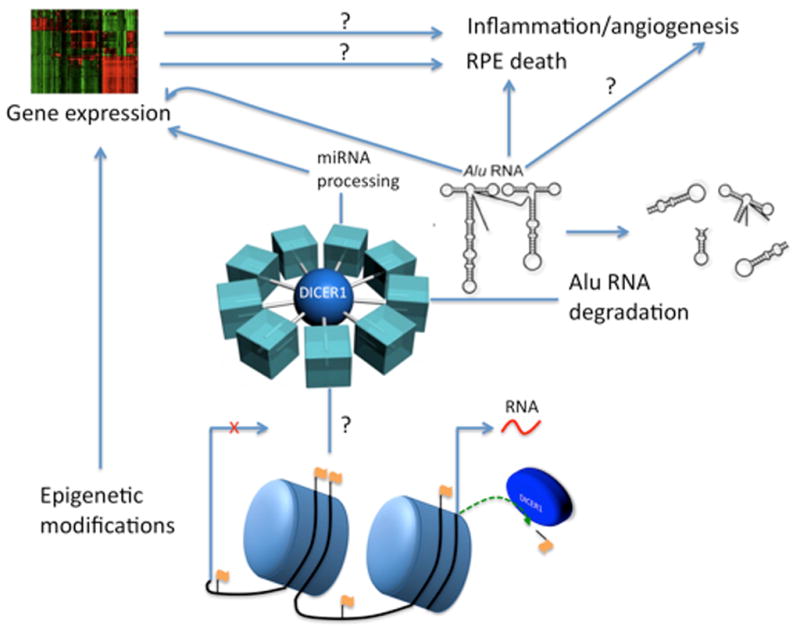
DICER1: Central hub of the RPE
The Alu RNAs that accumulate in DICER1 deficiency are transcribed from Alu DNA sequences in the nuclear genome. Sometimes described as “genomic parasites”, these ~300 nt DNA sequences constitute at least 11% of all genomic DNA (Batzer and Deininger, 2002). Alus are retrotransposons, meaning they “jump” around the genome by 1) Transcription; 2) Reverse transcription; and 3) Genomic integration at a new locus. The deleterious effect of Alu sequences is often ascribed to a single retrotransposition event; for example, an Alu sequence may insert into a critical gene, thereby disrupting gene function (Belancio et al., 2008). However, the mechanism of Alu RNA-induced toxicity in GA appears to occur by a novel pathway.
Recent work has identified an innate immune complex called the NLRP3 inflammasome as the response platform that mediates Alu RNA-induced RPE cell death (Tarallo et al., 2012). That study provided evidence of inflammasome activation in the RPE of human GA donor eyes, and showed that in experimental DICER1 deficit, activation of the NLRP3 inflammasome by Alu RNA leads to RPE IL-18 secretion, which induces MyD88-dependent RPE cell death. This finding solidifies the central role of the RPE in AMD pathogenesis. Interestingly, to date, NLRP3 inflammasome activation is almost exclusively confined to immune cells, thereby presenting an identity crisis for the RPE, which can now be re-defined, in part, in terms of its immune function.
As the mechanism of Alu RNA toxicity continues to be refined, one question remains unresolved: Why do Alu RNAs accumulate in the RPE of GA patients? Because DICER1 cleaves Alu RNA, it is reasonable to expect that DICER1 deficit precedes Alu RNA accumulation. Therefore, it is important to ask: why does DICER1 decrease in GA? Recent studies show that a variety of stresses can regulate DICER1 expression. It is possible that multiple RPE insults implicated in AMD pathogenesis could cause DICER1 to decrease (Wiesen and Tomasi, 2009). Interestingly, general stresses such as heat shock, viral infection, or translational inhibition also causes Alu to increase (Li and Schmid, 2001). Future intersecting projects could determine whether AMD-associated events (e.g. complement activation, mtDNA damage, oxidation of lipofuscin) lead to DICER1 deficit, Alu RNA accumulation and NLRP3 inflammasome activation. One recent example of such work was the finding that complement C1q, which is present in human AMD drusen, can activate the NLRP3 inflammasome (Doyle et al., 2012).
Do other neurodegenerative disorders also have a pathophysiologic decrease in DICER1? DICER1 is well known for its key role in the biogenesis of miRNAs, which facilitate the degradation or translational inhibition of most mRNAs (Friedman et al., 2009). Indeed, miRNA deficiency occurs in diseased but not age-matched controls in Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease (Christensen and Schratt, 2009; Eacker et al., 2009); whether DICER1 levels are similarly decreased in neurodegenerative diseases other than AMD remains to be seen. Intriguingly, microarray data reveal a reduction of DICER1 in the hippocampus of human Alzheimer’s disease donor tissue (Blalock et al., 2011).
Interestingly, in contrast to the proposed role of DICER1 deficit in other disorders, the phenotypic outcome of DICER1 deficiency in the experimental model of AMD was independent of miRNA perturbation. Instead, the accumulation of Alu RNA was the major driver of RPE toxicity. Based on this finding, it will be interesting to see if Alu RNA plays a role in the expanding compendium of diseases that are defined by DICER1 deficit.
Even though perturbation of miRNA maturation appeared to be dispensable for RPE cell health in the DICER1 deficit-induced animal model of AMD, miRNAs might still play a key role in determining the cell viability of RPE. Importantly, in the Kaneko et al. study, the mice were not exposed to the various stressors implicated in AMD – perhaps miRNA perturbation in AMD serves a key role only when coupled with some other RPE insult. Notably, miRNA expression regulates AMD-associated events, including inflammation (O’Neill et al., 2011) and angiogenesis (Sen et al., 2009) (Fig. 4).
Finally, DICER1 regulation of gene expression might also be achieved by miRNA-independent mechanisms, such as Dicer-dependent chromatin modifications (Woolcock et al., 2011); also, the Alu RNAs that accumulate in DICER1 deficiency may modulate translation (Hasler and Strub, 2006), or repress gene and miRNA transcription (Yakovchuk et al., 2009). In conclusion, there is great potential for DICER1 to mediate the intersection of multiple AMD-associated mechanisms of disease (Fig. 4). To be sure, there is no shortage of future research directions that revolve around the broad-reaching functions of DICER1. As the mechanistic role of DICER1 and Alu RNA in AMD are deciphered, neutralization of Alu RNA (e.g. antisense oligonucleotides), restoration of DICER1 levels (e.g. gene replacement therapy), or pharmacological targeting of the downstream MyD88 effector (e.g. small molecule or siRNA) are possible strategies to address this imbalance in AMD pathophysiology.
Environmental risk factors
Smoking is the most consistently documented modifiable risk factor for developing AMD. Smoking also confers the greatest numerical risk for AMD: smokers are 2–3 times as likely than non-smokers to develop AMD (Chen et al., 2011), and smoking cessation reduces the risk of developing AMD (Thornton et al., 2005). Several nutritional deficiencies are associated with AMD risk. Low dietary intake of anti-oxidants is associated with increased AMD risk, and a large clinical trial reported that high-dose anti-oxidant supplementation modestly reduced AMD progression (Group, 2001). However, even these benefits are restricted to progression to CNV and do not alter the risk of developing GA. In a recent epidemiologic study, omega-3 fatty acid (FA) intake was associated with a lower risk of AMD (Christen et al., 2011). The protective effect of statins on AMD is not well established, and would require long-term prospective interventional studies to confirm its relevance to AMD pathogenesis. Lifetime exposure to sunlight is not consistently associated with AMD. Ongoing clinical trials will assess the potential benefit of various nutritional supplements for treatment of AMD.
Genetics
The last 15 years of gene hunting have provided a foundation for population-based studies in dry AMD. However, the lack of breakthroughs in diagnostic or therapeutic strategies, or even in fundamentally advancing pathogenetic insights, has been disappointing. In contrast, over the same period of time, five different therapies were developed and are now in use for neovascular AMD.
Genome-wide association studies (GWAS) represent one prevailing approach in AMD research that has been used in attempt to predict risk of disease, understand pathogenesis, and identify potential therapeutic targets. GWAS ascribe specific gene variations to a group of people that have a common disease phenotype (e.g. those with or without AMD). GWAS have indeed identified several genetic loci, which harbor genetic variants known as single nucleotide polymorphisms (SNPs) that associate with an increased risk of AMD. An extensive review of genetic variation in AMD has been published elsewhere (Patel et al., 2008).
Risk assessment
In contrast to most diseases in which common risk variants do not explain the majority of genetic heritability ((Goldstein, 2009; Manolio et al., 2009; McClellan and King, 2010; Paynter et al., 2010), aggregate gene variation accounts for a bulk of the statistical risk of AMD (Edwards et al., 2005; Klein et al., 2010; Scholl et al., 2009) or CNV (Hageman et al., 2005). In addition, other models may use genetic information to predict progression from early (asymptomatic, no vision loss) to advanced (symptomatic, vision loss) AMD (Reviewed in:(Charbel Issa et al., 2011)), although they do not predict disease progression once advanced AMD has developed (Klein et al., 2010; Scholl et al., 2009). Also, since there are no approved treatments for GA, any analysis of factors that increase GA risk or progression, including information gleaned from AMD biomarkers (Gu et al., 2009; Guymer et al., 2011), provides therapeutically inactionable information at present.
Variation in multiple complement system genes (Bird, 2010; Bradley et al., 2011) is one of the most consistent statistical associations with AMD risk. It bears noting that the discovery of complement dysregulation in AMD based on biochemical approaches (Baudouin et al., 1992; Hageman et al., 2001; Johnson et al., 2001) predated the identification of sequence variations in complement genes (Edwards et al., 2005; Hageman et al., 2005).
The complement gene variant conferring the greatest quantitative statistical AMD risk is factor H (CFH). CFH inhibits a key activation step in complement activation, thereby reducing complement-induced host cell damage and inflammation. Still, there exists a very low sensitivity and specificity in terms of using genetic variation in CFH alone to determine AMD risk. In fact, when taking into consideration disease prevalence, the positive predictive value of genetic variation to assess AMD risk is anemic, even when multiple genetic loci are considered (Jakobsdottir et al., 2009).
Nevertheless, the predictive power of AMD risk assessment can be augmented greatly by considering genetic information from multiple loci in combination with epidemiologic and environmental risk factors. Indeed, many non-genetic, environmental risk factors for AMD have been identified, and for further discussion on this topic the reader is directed to excellent and comprehensive reviews (Ambati et al., 2003a; Chakravarthy et al., 2010; Krishnadev et al., 2010). In addition, onset and affection status of the fellow eye can be strongly influenced by an aggregated genetic risk (Chen et al., 2011). Next generation sequencing technologies combined with rigorous biological definition of mechanistic implications of the identified variants are likely to yield more valuable insights both into disease pathogenesis and rational development of novel diagnostics and therapeutics in the coming decade.
Current therapeutic prospects
Here, we will review the frontline experimental approaches and potential future directions for the treatment of AMD. We will also discuss the mechanistic justification of these interventions.
Targeting complement in GA
Complement inhibition as a therapeutic strategy represents the culmination of numerous research articles that focus on the role of complement in AMD pathogenesis. Although complement inhibition suppresses CNV in animal models of wet AMD, it has not been shown to ameliorate dry AMD in vivo. Indeed, there are robust human data that lead one to question the value of complement inhibition for dry AMD. First, SNPs in complement genes do not predict progression of dry AMD (Klein et al., 2010; Scholl et al., 2009). Second, complement deposition is not prominent in GA eyes (Ambati, unpublished data; Hageman, personal communication). Finally, RPE cells are extremely resistant to complement-induced cell death (Ambati, unpublished data; Dean Bok, personal communication) except when their rich cache of negative complement regulators is simultaneously antagonized or depleted (Lueck et al., 2011). However, such strategies may not be representative of the disease state as there is no apparent reduction in expression of these negative regulators with aging or in AMD (Lincoln Johnson, personal communication). Indeed, in a recent clinical trial, there was no benefit of an anti-C5 antibody in reducing drusen or expansion of GA (Filho, 2012). The rationale for ongoing clinical trials investigating complement inhibition appears to rest primarily with genetic association; robust pre-clinical experimentation is still required to resolve the ostensibly therapeutic effect of complement inhibition for dry AMD.
Targeting complement in CNV treatment
With respect to complement inhibition for the treatment of CNV, this strategy may have a dual mechanism of action: Reduction in secretion of VEGF-A by RPE, or inhibiting the retinal infiltration of pro-angiogenic leukocytes (Nozaki et al., 2006). Several studies show that a variety of anti-complement agents reduce CNV in animal models of disease (Bora et al., 2007; Nozaki et al., 2006; Rohrer et al., 2009). There are plans to test the safety of one complement inhibitor (POT-4) in a phase I clinical trial in patients with CNV (NCT 00473928). In summary, complement inhibitors suppress CNV in animal models of disease, thus supporting clinical investigation of their use in humans.
TLR3 in GA and CNV: A precarious target
A SNP in the gene coding for the dsRNA sensor toll-like Receptor 3 (TLR3) was initially reported to be associated with protection against developing GA (Yang et al., 2008). However, this association was not confirmed in other studies. Genetic association or not, TLR3 knockout mice are protected against RPE degeneration caused by exogenous dsRNA (Kleinman et al., 2012) or by accumulation of all-trans retinaldehyde (Shiose et al., 2011). Certain viruses contain dsRNA genomes, while other viruses may elaborate dsRNA intermediates during their replication cycle. Therefore, it is tempting to speculate that there might be a viral etiology of GA- an under-investigated area of research in AMD. Another potential source of TLR3 activation in GA could be endogenous mRNA (Kariko et al., 2004). On the other hand, it is important to recognize that TLR3 stimulation causes CNV suppression (Kleinman et al., 2008); therefore, although modulation of TLR3 activity shows promise in treating either dry or wet AMD, it also risks potential exacerbation of the other form.
Autophagy
Autophagy-promoting molecules may also provide a beneficial therapeutic effect for dry and wet AMD. One experimental model showed that the classic autophagy inducer rapamycin inhibits angiogenesis sprouting and VEGF-A production by RPE cells (Stahl et al., 2008). Also, in a small pilot study, systemic rapamycin reduced the number of anti-VEGF-A injections required to treat CNV; although the authors attributed this effect to immune suppression, it is possible that rapamycin also directly inhibited endothelial cell proliferation and also modulated RPE secretion of VEGF-A (Nussenblatt et al., 2010). Rapamycin was used in the EMERALD Clinical Trial (Phase II, NCT 00766337), which included of ranibizumab plus rapamycin for CNV. However, this study was terminated and we are not aware of any published results.
In theory, targeting autophagy appears to be a promising avenue for future endeavors in AMD research. However, there are several stipulations to this strategy. First, induction of autophagy would require careful dosing and timing. Under some circumstances, especially in feeble or dying cells, autophagy can cause cell death (Kourtis and Tavernarakis, 2009). Furthermore, since there is some crosstalk between autophagic and apoptotic machinery, healthier cells may also undergo apoptosis if they register a strong enough pro-autophagic signal (Maiuri et al., 2007). In light of these considerations, one might expect autophagy induction to be a reasonable treatment for early macular degeneration, when signs of RPE damage are just beginning. On the other hand, if the RPE is damaged past a critical point, such as in the later stages of AMD, autophagy might cause cell death and thereby exacerbate the disease. Indeed, this concept has been demonstrated in an animal model of AD (Majumder et al., 2011); in the case of autophagy, timing is of the essence. The global immune-modulatory effect of mTOR inhibition on retinal health would also be important to discern before its clinical investigation.
Immunovascular modification
Whereas anti-VEGF-A treatment is directly anti-angiogenic to the CNV vasculature, the mechanisms of immune cell contribution to CNV are less clear. Addressing the functional effect of anti-VEGF-A therapy on specific immune cell types will be essential in understanding the proposed inflammatory link to CNV. The reader is directed to further discussion of the need for strategies to target both vascular and extravascular components in treatment of CNV (Spaide, 2006).
If CNV is immune driven, then another pertinent question is: Does dampening the immune response suppress CNV? Although anti-VEGF therapy is the current standard of care for CNV, the use of steroids to inhibit the immune system was once a front-line clinical option. Triamcinolone is one example of a steroid that was once widely used for treatment of CNV, but does not provide long-term improvement in vision (Reviewed in (Becerra et al., 2011)). Nevertheless, triamcinolone and other steroids are still sometimes used as adjunct therapies because they can reduce the frequency of other treatments in tandem. There are some case reports and small-scale studies showing that other immunosuppressants (e.g. methotrexate, non-steroidal anti-inflammatory drugs) might somewhat ameliorate CNV (Reviewed here: (Wang et al., 2011b)), although larger studies are required to validate these findings.
Treatment of CNV: Beyond VEGF-A
The immune and vascular systems that feed CNV are intertwined, and modulation of either shows clinical benefit for CNV. Anti-VEGF-A therapy is currently the most effective single agent for the majority of CNV patients. A better understanding of specific immune effectors will be important in designing improved immune-modifying CNV therapy. Future experimentation in humans is required to confirm the potential of complement inhibition or anti-oxidants in treating CNV. All of these aforementioned interventions hold a common link in that they somehow dampen the immunovascular axis of disease. But might there be an intervention that affects the CNV vasculature with minimal effect on the immune component? In light of the potential efficacy-reducing immune modulation resulting from anti-VEGF-A therapy, a specific vascular-acting molecule would be a novel therapeutic target in CNV.
In fact, it seems that such a target exists. The eotaxin family of chemokines and their receptor CCR3 are found in human CNV specimens but not in the undiseased choroid (Takeda et al., 2009). Despite the known role of eotaxins in eosinophil and mast cell chemotaxis, these eotaxins did not promote immune cell migration to the retina in this system; instead, they acted on the endothelial receptor CCR3, which in turn stimulated angiogenesis (Takeda et al., 2009). CCR3 inhibition was slightly more effective than anti-VEGF-A in suppressing CNV in a mouse model of disease. Furthermore, CNV suppression occurred without altering levels of VEGF-A, although more subtle interactions between these pathways have been identified (Wang et al., 2011a). Thus, unlike anti-VEGF-A or anti-inflammatory treatment, blocking the eotaxin-CCR3 axis in CNV might avoid major modulation of immune and inflammatory elements. These findings are buttressed by other studies validating the efficacy of CCR3 targeting in laser-induced CNV (Mizutani et al., 2011), the overexpression of CCR3 and its ligand in a spontaneous mouse model of CNV (Nagai et al., 2011), and by studies showing increased circulating eotaxins in AMD patients (Mo et al., 2010). Looking forward to chemokine-targeting therapy, bertilimumab, a monoclonal antibody targeting eotaxin-1, is slated for clinical trials in CNV.
It is also important to investigate disease mechanisms other than VEGF-A in CNV, especially in light of mechanistic findings that VEGF-A has a physiologic cytoprotective role in the retina, and that quenching or altering VEGF expression can be toxic to multiple cell types in rodent retina (Ford et al., 2011; Murakami et al., 2010; Nishijima et al., 2007; Saint-Geniez et al., 2008; Takeda et al., 2009). Recently, evidence has accumulated to support the concept that anti-VEGF-A therapy can contribute to physiological alterations in the retinal vasculature in the short-term (Papadopoulou et al., 2009; Sacu et al., 2011) and frank RPE toxicity in the long-term (Bhisitkul, 2011, 2012).
Retinal Replacement
An alternative approach to restoring the health of moribund RPE cells is to replace them. Indeed, RPE cell transplant with fetal neural tissue or human embryonic stem cell (hESC)-derived RPE-like cells is the focus of current clinical trials (NCT00346060; NCT01344993). Following implantation of hESC-derived RPE cells into mice, increased eye movement to light stimulus was reported (Lu et al., 2009). In contrast to the foreseeable complications of autophagy-induction in late AMD, replacing the dying RPE with sprightly implanted cells might be preferable in such cases of advanced disease. RPE cell replacement may even complement efforts to perform retinal transplant, which is the transfer of the neural retina from areas of dying RPE cells to healthier regions (Sheridan et al., 2009; Zarbin, 2008). An alternative approach to retinal replacement is the Argus II “bionic eye”, recently approved for use in Europe and awaiting US FDA approval. Argus II is a microchip implanted in the retina that transmits environmental light stimuli to the brain via the optic nerve, thus providing some visual, albeit artificial, relief to a small fraction of treated patients (Humayun et al., 2012). If the pharmacologic rescue of near-deteriorated cells remains exceedingly inactionable, then perhaps cell replacement or microchip implants will arrive at the forefront of therapeutic hope.
Conclusions and outlook
Ostensibly, the numerous influences that underlie AMD provide multiple potential targets for disease treatment (Ambati et al., 2003a; Bird, 2010). However, despite their apparent heterogeneity, these pathways are highly redundant and can produce similar pathologic effects. Therefore, any therapeutic intervention that addresses an isolated injurious mechanism is unlikely to counter the convergence of parallel conduits.
Looking towards the future of AMD therapy, an emerging paradigm diverges from the conventional approach of preventing retinal dysfunction and death. Instead, empowering retinal health in spite of injury, rather than attempting to eliminate numerous overlapping insults, deserves appreciable investigation in AMD prevention and treatment. Modulating or enhancing specific RPE coping mechanisms, rather than attempting to remove modify heterogeneous barrage of insults, is an intriguing conceptual scaffold on which to base future therapeutic developments.
Acknowledgments
We thank D.H. Fowler, A.M. Rao, G.S. Rao and K. Ambati for discussions., and T. Dolan and M. Hazzard for figure assistance. J.A. was supported by National Eye Institute (NEI)/National Institutes of Health (NIH) grants R01EY018350, R01EY018836, R01EY020672, R01EY022238, R21EY019778, RC1EY020442, Doris Duke Distinguished Clinical Scientist Award, Burroughs Wellcome Fund Clinical Scientist Award in Translational Research, Dr. E. Vernon Smith and Eloise C. Smith Macular Degeneration Endowed Chair, and B.J.F. by NIH T32HL091812 and UL1RR033173.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamis AP, Miller JW, Bernal MT, D’Amico DJ, Folkman J, Yeo TK, Yeo KT. Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. American journal of ophthalmology. 1994;118:445–450. doi: 10.1016/s0002-9394(14)75794-0. [DOI] [PubMed] [Google Scholar]
- Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, Pasquale LR, Thieme H, Iwamoto MA, Park JE, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. The New England journal of medicine. 1994;331:1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- Ambati J, Ambati BK, Yoo SH, Ianchulev S, Adamis AP. Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol. 2003a;48:257–293. doi: 10.1016/s0039-6257(03)00030-4. [DOI] [PubMed] [Google Scholar]
- Ambati J, Anand A, Fernandez S, Sakurai E, Lynn BC, Kuziel WA, Rollins BJ, Ambati BK. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med. 2003b;9:1390–1397. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- Amin R, Puklin JE, Frank RN. Growth factor localization in choroidal neovascular membranes of age-related macular degeneration. Investigative ophthalmology & visual science. 1994;35:3178–3188. [PubMed] [Google Scholar]
- Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Progress in retinal and eye research. 2010;29:95–112. doi: 10.1016/j.preteyeres.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- Baudouin C, Peyman GA, Fredj-Reygrobellet D, Gordon WC, Lapalus P, Gastaud P, Bazan NG. Immunohistological study of subretinal membranes in age-related macular degeneration. Jpn J Ophthalmol. 1992;36:443–451. [PubMed] [Google Scholar]
- Becerra EM, Morescalchi F, Gandolfo F, Danzi P, Nascimbeni G, Arcidiacono B, Semeraro F. Clinical evidence of intravitreal triamcinolone acetonide in the management of age-related macular degeneration. Curr Drug Targets. 2011;12:149–172. doi: 10.2174/138945011794182746. [DOI] [PubMed] [Google Scholar]
- Belancio VP, Hedges DJ, Deininger P. Mammalian non-LTR retrotransposons: for better or worse, in sickness and in health. Genome Res. 2008;18:343–358. doi: 10.1101/gr.5558208. [DOI] [PubMed] [Google Scholar]
- Bhisitkul RB. In 2011 UCSF Ophthalmology Update. San Francisco: 2011. 7 Year Update on the ANCHOR/MARINA Ranibizumab Cohort: The SEVEN UP Study. [Google Scholar]
- Bhisitkul RB, Rofagha S, Boyer DS, Sadda S, Zhang K. Anchor/marina: A Multicenter, Prospective Cohort Study. Association for Research in Vision and Ophthalmology; Ft. Lauderdale, Florida: 2012. Year 7 Outcomes For Ranibizumab-treated Subjects. [Google Scholar]
- Bhutto IA, McLeod DS, Hasegawa T, Kim SY, Merges C, Tong P, Lutty GA. Pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in aged human choroid and eyes with age-related macular degeneration. Experimental eye research. 2006;82:99–110. doi: 10.1016/j.exer.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird AC. Therapeutic targets in age-related macular disease. J Clin Invest. 2010;120:3033–3041. doi: 10.1172/JCI42437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Buechel HM, Popovic J, Geddes JW, Landfield PW. Microarray analyses of laser-captured hippocampus reveal distinct gray and white matter signatures associated with incipient Alzheimer’s disease. J Chem Neuroanat. 2011;42:118–126. doi: 10.1016/j.jchemneu.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booij JC, ten Brink JB, Swagemakers SM, Verkerk AJ, Essing AH, van der Spek PJ, Bergen AA. A new strategy to identify and annotate human RPE-specific gene expression. PLoS One. 2010;5:e9341. doi: 10.1371/journal.pone.0009341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boothby LA, Doering PL. Vitamin C and vitamin E for Alzheimer’s disease. Ann Pharmacother. 2005;39:2073–2080. doi: 10.1345/aph.1E495. [DOI] [PubMed] [Google Scholar]
- Bora NS, Kaliappan S, Jha P, Xu Q, Sivasankar B, Harris CL, Morgan BP, Bora PS. CD59, a complement regulatory protein, controls choroidal neovascularization in a mouse model of wet-type age-related macular degeneration. Journal of immunology. 2007;178:1783–1790. doi: 10.4049/jimmunol.178.3.1783. [DOI] [PubMed] [Google Scholar]
- Bradley DT, Zipfel PF, Hughes AE. Complement in age-related macular degeneration: a focus on function. Eye (Lond) 2011 doi: 10.1038/eye.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007;3:186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- Brown DM, Kaiser PK, Michels M, Soubrane G, Heier JS, Kim RY, Sy JP, Schneider S. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. The New England journal of medicine. 2006;355:1432–1444. doi: 10.1056/NEJMoa062655. [DOI] [PubMed] [Google Scholar]
- Cai H, Del Priore LV. Bruch membrane aging alters the gene expression profile of human retinal pigment epithelium. Curr Eye Res. 2006;31:181–189. doi: 10.1080/02713680500514628. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- Chakravarthy U, Wong TY, Fletcher A, Piault E, Evans C, Zlateva G, Buggage R, Pleil A, Mitchell P. Clinical risk factors for age-related macular degeneration: a systematic review and meta-analysis. BMC Ophthalmol. 2010;10:31. doi: 10.1186/1471-2415-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbel Issa P, Chong NV, Scholl HP. The significance of the complement system for the pathogenesis of age-related macular degeneration - current evidence and translation into clinical application. Graefes Arch Clin Exp Ophthalmol. 2011;249:163–174. doi: 10.1007/s00417-010-1568-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zeng J, Zhao C, Wang K, Trood E, Buehler J, Weed M, Kasuga D, Bernstein PS, Hughes G, et al. Assessing susceptibility to age-related macular degeneration with genetic markers and environmental factors. Archives of ophthalmology. 2011;129:344–351. doi: 10.1001/archophthalmol.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanoff S, McMenamin P, Gillies MC, Kettle E, Sarks SH. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. The British journal of ophthalmology. 2010;94:918–925. doi: 10.1136/bjo.2009.165563. [DOI] [PubMed] [Google Scholar]
- Christen WG, Schaumberg DA, Glynn RJ, Buring JE. Dietary {omega}-3 Fatty Acid and Fish Intake and Incident Age-Related Macular Degeneration in Women. Arch Ophthalmol. 2011 doi: 10.1001/archophthalmol.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen M, Schratt GM. microRNA involvement in developmental and functional aspects of the nervous system and in neurological diseases. Neurosci Lett. 2009;466:55–62. doi: 10.1016/j.neulet.2009.04.043. [DOI] [PubMed] [Google Scholar]
- Ciulla TA, Rosenfeld PJ. Anti-vascular endothelial growth factor therapy for neovascular ocular diseases other than age-related macular degeneration. Curr Opin Ophthalmol. 2009;20:166–174. doi: 10.1097/ICU.0b013e328329d173. [DOI] [PubMed] [Google Scholar]
- Combadiere C, Feumi C, Raoul W, Keller N, Rodero M, Pezard A, Lavalette S, Houssier M, Jonet L, Picard E, et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest. 2007;117:2920–2928. doi: 10.1172/JCI31692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha-Vaz JG. The blood-retinal barriers system. Basic concepts and clinical evaluation. Exp Eye Res. 2004;78:715–721. doi: 10.1016/s0014-4835(03)00213-6. [DOI] [PubMed] [Google Scholar]
- Cursiefen C, Chen L, Borges LP, Jackson D, Cao J, Radziejewski C, D’Amore PA, Dana MR, Wiegand SJ, Streilein JW. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. The Journal of clinical investigation. 2004;113:1040–1050. doi: 10.1172/JCI20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, Farrar GJ, Kiang AS, Humphries MM, Lavelle EC, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nature medicine. 2012;18:791–798. doi: 10.1038/nm.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eacker SM, Dawson TM, Dawson VL. Understanding microRNAs in neurodegeneration. Nature reviews Neuroscience. 2009;10:837–841. doi: 10.1038/nrn2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economides AN, Carpenter LR, Rudge JS, Wong V, Koehler-Stec EM, Hartnett C, Pyles EA, Xu X, Daly TJ, Young MR, et al. Cytokine traps: multi-component, high-affinity blockers of cytokine action. Nature medicine. 2003;9:47–52. doi: 10.1038/nm811. [DOI] [PubMed] [Google Scholar]
- Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- Ehrchen J, Steinmuller L, Barczyk K, Tenbrock K, Nacken W, Eisenacher M, Nordhues U, Sorg C, Sunderkotter C, Roth J. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood. 2007;109:1265–1274. doi: 10.1182/blood-2006-02-001115. [DOI] [PubMed] [Google Scholar]
- Espinosa-Heidmann DG, Reinoso MA, Pina Y, Csaky KG, Caicedo A, Cousins SW. Quantitative enumeration of vascular smooth muscle cells and endothelial cells derived from bone marrow precursors in experimental choroidal neovascularization. Experimental eye research. 2005;80:369–378. doi: 10.1016/j.exer.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Espinosa-Heidmann DG, Suner IJ, Hernandez EP, Monroy D, Csaky KG, Cousins SW. Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularization. Investigative ophthalmology & visual science. 2003;44:3586–3592. doi: 10.1167/iovs.03-0038. [DOI] [PubMed] [Google Scholar]
- Evans J. Antioxidant supplements to prevent or slow down the progression of AMD: a systematic review and meta-analysis. Eye. 2008;22:751–760. doi: 10.1038/eye.2008.100. [DOI] [PubMed] [Google Scholar]
- Ferrara N. Role of myeloid cells in vascular endothelial growth factor-independent tumor angiogenesis. Curr Opin Hematol. 2010;17:219–224. doi: 10.1097/MOH.0b013e3283386660. [DOI] [PubMed] [Google Scholar]
- Filho CAAG, Zohar Y, Gregori G, Li Y, Feuer W, Penha FM, Sadda SR, Zhang L, Zhang K, Rosenfeld PJ. AMD Patients With Drusen: The COMPLETE Study. Association for Research in Vision and Ophthalmology; Ft. Lauderdale, FL, IOVS: 2012. Efficacy Of The Systemic Complement Inhibitor Eculizumab. [Google Scholar]
- Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nature medicine. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- Ford KM, Saint-Geniez M, Walshe T, Zahr A, D’Amore PA. Expression and role of VEGF in the adult retinal pigment epithelium. Investigative ophthalmology & visual science. 2011;52:9478–9487. doi: 10.1167/iovs.11-8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forooghian F, Cukras C, Meyerle CB, Chew EY, Wong WT. Tachyphylaxis after intravitreal bevacizumab for exudative age-related macular degeneration. Retina. 2009;29:723–731. doi: 10.1097/IAE.0b013e3181a2c1c3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank RN, Amin RH, Eliott D, Puklin JE, Abrams GW. Basic fibroblast growth factor and vascular endothelial growth factor are present in epiretinal and choroidal neovascular membranes. American journal of ophthalmology. 1996;122:393–403. doi: 10.1016/s0002-9394(14)72066-5. [DOI] [PubMed] [Google Scholar]
- Friedman DS, O’Colmain BJ, Munoz B, Tomany SC, McCarty C, de Jong PT, Nemesure B, Mitchell P, Kempen J. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–572. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoka Y, Strainic M, Medof ME. Differential cytokine expression of human retinal pigment epithelial cells in response to stimulation by C5a. Clin Exp Immunol. 2003;131:248–253. doi: 10.1046/j.1365-2249.2003.02087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn JV, Mahaffy H, Dasari S, Oliver M, Chen M, Boulton ME, Xu H, Curry WJ, Stitt AW. Proteomic profiling of human retinal pigment epithelium exposed to an advanced glycation-modified substrate. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle. Ophthalmologie. 2011 doi: 10.1007/s00417-011-1856-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- Gragoudas ES, Adamis AP, Cunningham ET, Jr, Feinsod M, Guyer DR. Pegaptanib for neovascular age-related macular degeneration. The New England journal of medicine. 2004;351:2805–2816. doi: 10.1056/NEJMoa042760. [DOI] [PubMed] [Google Scholar]
- Grossniklaus HE, Cingle KA, Yoon YD, Ketkar N, L’Hernault N, Brown S. Correlation of histologic 2-dimensional reconstruction and confocal scanning laser microscopic imaging of choroidal neovascularization in eyes with age-related maculopathy. Arch Ophthalmol. 2000;118:625–629. doi: 10.1001/archopht.118.5.625. [DOI] [PubMed] [Google Scholar]
- Grossniklaus HE, Ling JX, Wallace TM, Dithmar S, Lawson DH, Cohen C, Elner VM, Elner SG, Sternberg P., Jr Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Molecular vision. 2002;8:119–126. [PubMed] [Google Scholar]
- Group AREDSR. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119:1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Pauer GJ, Yue X, Narendra U, Sturgill GM, Bena J, Gu X, Peachey NS, Salomon RG, Hagstrom SA, et al. Assessing susceptibility to age-related macular degeneration with proteomic and genomic biomarkers. Mol Cell Proteomics. 2009;8:1338–1349. doi: 10.1074/mcp.M800453-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyer DR, Fine SL, Maguire MG, Hawkins BS, Owens SL, Murphy RP. Subfoveal choroidal neovascular membranes in age-related macular degeneration. Visual prognosis in eyes with relatively good initial visual acuity. Archives of ophthalmology. 1986;104:702–705. doi: 10.1001/archopht.1986.01050170092029. [DOI] [PubMed] [Google Scholar]
- Guymer RH, Tao LW, Goh JK, Liew D, Ischenko O, Robman LD, Aung K, Cipriani T, Cain M, Richardson AJ, et al. Identification of Urinary Biomarkers for Age-related Macular Degeneration. Invest Ophthalmol Vis Sci. 2011 doi: 10.1167/iovs.10-7120. [DOI] [PubMed] [Google Scholar]
- Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–732. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hasler J, Strub K. Alu RNP and Alu RNA regulate translation initiation in vitro. Nucleic Acids Res. 2006;34:2374–2385. doi: 10.1093/nar/gkl246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayat MJ, Howlader N, Reichman ME, Edwards BK. Cancer statistics, trends, and multiple primary cancer analyses from the Surveillance, Epidemiology, and End Results (SEER) Program. Oncologist. 2007;12:20–37. doi: 10.1634/theoncologist.12-1-20. [DOI] [PubMed] [Google Scholar]
- Higgins GT, Wang JH, Dockery P, Cleary PE, Redmond HP. Induction of angiogenic cytokine expression in cultured RPE by ingestion of oxidized photoreceptor outer segments. Investigative ophthalmology & visual science. 2003;44:1775–1782. doi: 10.1167/iovs.02-0742. [DOI] [PubMed] [Google Scholar]
- Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L, Ufret RL, Salomon RG, Perez VL. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008;14:194–198. doi: 10.1038/nm1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humayun MS, Dorn JD, da Cruz L, Dagnelie G, Sahel JA, Stanga PE, Cideciyan AV, Duncan JL, Eliott D, Filley E, et al. Interim Results from the International Trial of Second Sight’s Visual Prosthesis. Ophthalmology. 2012;119:779–788. doi: 10.1016/j.ophtha.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsdottir J, Gorin MB, Conley YP, Ferrell RE, Weeks DE. Interpretation of genetic association studies: markers with replicated highly significant odds ratios may be poor classifiers. PLoS Genet. 2009;5:e1000337. doi: 10.1371/journal.pgen.1000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001;73:887–896. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
- Johnson PT, Brown MN, Pulliam BC, Anderson DH, Johnson LV. Synaptic pathology, altered gene expression, and degeneration in photoreceptors impacted by drusen. Investigative ophthalmology & visual science. 2005;46:4788–4795. doi: 10.1167/iovs.05-0767. [DOI] [PubMed] [Google Scholar]
- Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, Kleinman ME, Ponicsan SL, Hauswirth WW, Chiodo VA, et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011 doi: 10.1038/nature09830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004;279:12542–12550. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- Kelly J, Ali Khan A, Yin J, Ferguson TA, Apte RS. Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. The Journal of clinical investigation. 2007;117:3421–3426. doi: 10.1172/JCI32430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein ML, Ferris FL, 3rd, Francis PJ, Lindblad AS, Chew EY, Hamon SC, Ott J. Progression of geographic atrophy and genotype in age-related macular degeneration. Ophthalmology. 2010;117:1554–1559. e1551. doi: 10.1016/j.ophtha.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Meuer SM, Knudtson MD, Iyengar SK, Klein BE. The epidemiology of retinal reticular drusen. Am J Ophthalmol. 2008;145:317–326. doi: 10.1016/j.ajo.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinman ME, Kaneko H, Cho WG, Dridi S, Fowler BJ, Blandford AD, Albuquerque RJ, Hirano Y, Terasaki H, Kondo M, et al. Short-interfering RNAs Induce Retinal Degeneration via TLR3 and IRF3. Mol Ther. 2012;20:101–108. doi: 10.1038/mt.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, Albuquerque RJ, Yamasaki S, Itaya M, Pan Y, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591–597. doi: 10.1038/nature06765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- Kourtis N, Tavernarakis N. Autophagy and cell death in model organisms. Cell death and differentiation. 2009;16:21–30. doi: 10.1038/cdd.2008.120. [DOI] [PubMed] [Google Scholar]
- Krishnadev N, Meleth AD, Chew EY. Nutritional supplements for age-related macular degeneration. Curr Opin Ophthalmol. 2010;21:184–189. doi: 10.1097/ICU.0b013e32833866ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurji KH, Cui JZ, Lin T, Harriman D, Prasad SS, Kojic L, Matsubara JA. Microarray analysis identifies changes in inflammatory gene expression in response to amyloid-beta stimulation of cultured human retinal pigment epithelial cells. Investigative ophthalmology & visual science. 2010;51:1151–1163. doi: 10.1167/iovs.09-3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvanta A, Algvere PV, Berglin L, Seregard S. Subfoveal fibrovascular membranes in age-related macular degeneration express vascular endothelial growth factor. Investigative ophthalmology & visual science. 1996;37:1929–1934. [PubMed] [Google Scholar]
- Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- Li TH, Schmid CW. Differential stress induction of individual Alu loci: implications for transcription and retrotransposition. Gene. 2001;276:135–141. doi: 10.1016/s0378-1119(01)00637-0. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Lopez PF, Sippy BD, Lambert HM, Thach AB, Hinton DR. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Investigative ophthalmology & visual science. 1996;37:855–868. [PubMed] [Google Scholar]
- Lu B, Malcuit C, Wang S, Girman S, Francis P, Lemieux L, Lanza R, Lund R. Long-term safety and function of RPE from human embryonic stem cells in preclinical models of macular degeneration. Stem Cells. 2009;27:2126–2135. doi: 10.1002/stem.149. [DOI] [PubMed] [Google Scholar]
- Lueck K, Wasmuth S, Williams J, Hughes TR, Morgan BP, Lommatzsch A, Greenwood J, Moss SE, Pauleikhoff D. Sub-lytic C5b-9 induces functional changes in retinal pigment epithelial cells consistent with age-related macular degeneration. Eye (Lond) 2011;25:1074–1082. doi: 10.1038/eye.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One. 2011;6:e25416. doi: 10.1371/journal.pone.0025416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DF, Maguire MG, Fine SL, Ying GS, Jaffe GJ, Grunwald JE, Toth C, Redford M, Ferris FL., 3rd Ranibizumab and Bevacizumab for Treatment of Neovascular Age-Related Macular Degeneration: Two-Year Results. Ophthalmology. 2012 doi: 10.1016/j.ophtha.2020.01.029. [DOI] [PubMed] [Google Scholar]
- Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL, Jaffe GJ. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. The New England journal of medicine. 2011;364:1897–1908. doi: 10.1056/NEJMoa1102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattapallil MJ, Wawrousek EF, Chan CC, Zhao H, Roychoudhury J, Ferguson TA, Caspi RR. The rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Investigative ophthalmology & visual science. 2012 doi: 10.1167/iovs.12-9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- Mitter SK, Rao HV, Qi X, Cai J, Sugrue A, Dunn WA, Jr, Grant MB, Boulton ME. Autophagy in the retina: a potential role in age-related macular degeneration. Adv Exp Med Biol. 2012;723:83–90. doi: 10.1007/978-1-4614-0631-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani T, Ashikari M, Tokoro M, Nozaki M, Ogura Y. CCR3 Antagonist Suppress Laser-induced Choroidal Neovascularization In Mice. Paper presented at: ARVO; Ft. Lauderdale, FL. 2011. [Google Scholar]
- Mo FM, Proia AD, Johnson WH, Cyr D, Lashkari K. Interferon gamma-inducible protein-10 (IP-10) and eotaxin as biomarkers in age-related macular degeneration. Investigative ophthalmology & visual science. 2010;51:4226–4236. doi: 10.1167/iovs.09-3910. [DOI] [PubMed] [Google Scholar]
- Molday LL, Rabin AR, Molday RS. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat Genet. 2000;25:257–258. doi: 10.1038/77004. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Ikeda Y, Yonemitsu Y, Miyazaki M, Inoue M, Hasegawa M, Sueishi K, Ishibashi T. Inhibition of choroidal neovascularization via brief subretinal exposure to a newly developed lentiviral vector pseudotyped with Sendai viral envelope proteins. Hum Gene Ther. 2010;21:199–209. doi: 10.1089/hum.2009.102. [DOI] [PubMed] [Google Scholar]
- Nagai N, Izumi-Nagai K, Robbie S, Bainbridge J, Chang B, Hurd R, Ng Y-S, Shima D. Spontaneous Cnv In A Novel Mutant Mouse Is Associated With Early Chorio-retinal Para-inflammation And Vegf Driven Angiogenesis. Paper presented at: ARVO ; Ft. Lauderdale, FL. 2011. [Google Scholar]
- Newman AM, Gallo NB, Hancox LS, Miller NJ, Radeke CM, Maloney MA, Cooper JB, Hageman GS, Anderson DH, Johnson LV, et al. Systems-level analysis of age-related macular degeneration reveals global biomarkers and phenotype-specific functional networks. Genome Med. 2012;4:16. doi: 10.1186/gm315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijima K, Ng YS, Zhong L, Bradley J, Schubert W, Jo N, Akita J, Samuelsson SJ, Robinson GS, Adamis AP, et al. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171:53–67. doi: 10.2353/ajpath.2007.061237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel A, Jost M, Lambert V, Lecomte J, Rakic JM. Anti-angiogenic therapy of exudative age-related macular degeneration: current progress and emerging concepts. Trends Mol Med. 2007;13:345–352. doi: 10.1016/j.molmed.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, Chen Y, Zhang K, Ambati BK, Baffi JZ, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2328–2333. doi: 10.1073/pnas.0408835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenblatt RB, Byrnes G, Sen HN, Yeh S, Faia L, Meyerle C, Wroblewski K, Li Z, Liu B, Chew E, et al. A randomized pilot study of systemic immunosuppression in the treatment of age-related macular degeneration with choroidal neovascularization. Retina. 2010;30:1579–1587. doi: 10.1097/IAE.0b013e3181e7978e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol. 2011;11:163–175. doi: 10.1038/nri2957. [DOI] [PubMed] [Google Scholar]
- Papadopoulou DN, Mendrinos E, Mangioris G, Donati G, Pournaras CJ. Intravitreal ranibizumab may induce retinal arteriolar vasoconstriction in patients with neovascular age-related macular degeneration. Ophthalmology. 2009;116:1755–1761. doi: 10.1016/j.ophtha.2009.03.017. [DOI] [PubMed] [Google Scholar]
- Patel M, Chan CC. Immunopathological aspects of age-related macular degeneration. Semin Immunopathol. 2008;30:97–110. doi: 10.1007/s00281-008-0112-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N, Adewoyin T, Chong NV. Age-related macular degeneration: a perspective on genetic studies. Eye (Lond) 2008;22:768–776. doi: 10.1038/sj.eye.6702844. [DOI] [PubMed] [Google Scholar]
- Paynter NP, Chasman DI, Pare G, Buring JE, Cook NR, Miletich JP, Ridker PM. Association between a literature-based genetic risk score and cardiovascular events in women. JAMA. 2010;303:631–637. doi: 10.1001/jama.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penfold PL, Liew SC, Madigan MC, Provis JM. Modulation of major histocompatibility complex class II expression in retinas with age-related macular degeneration. Invest Ophthalmol Vis Sci. 1997;38:2125–2133. [PubMed] [Google Scholar]
- Pons M, Marin-Castano ME. Cigarette smoke-related hydroquinone dysregulates MCP-1, VEGF and PEDF expression in retinal pigment epithelium in vitro and in vivo. PLoS One. 2011;6:e16722. doi: 10.1371/journal.pone.0016722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul W, Auvynet C, Camelo S, Guillonneau X, Feumi C, Combadiere C, Sennlaub F. CCL2/CCR2 and CX3CL1/CX3CR1 chemokine axes and their possible involvement in age-related macular degeneration. J Neuroinflammation. 2010;7:87. doi: 10.1186/1742-2094-7-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattner A, Nathans J. Macular degeneration: recent advances and therapeutic opportunities. Nat Rev Neurosci. 2006;7:860–872. doi: 10.1038/nrn2007. [DOI] [PubMed] [Google Scholar]
- Rohrer B, Long Q, Coughlin B, Wilson RB, Huang Y, Qiao F, Tang PH, Kunchithapautham K, Gilkeson GS, Tomlinson S. A targeted inhibitor of the alternative complement pathway reduces angiogenesis in a mouse model of age-related macular degeneration. Investigative ophthalmology & visual science. 2009;50:3056–3064. doi: 10.1167/iovs.08-2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, Kim RY. Ranibizumab for neovascular age-related macular degeneration. The New England journal of medicine. 2006;355:1419–1431. doi: 10.1056/NEJMoa054481. [DOI] [PubMed] [Google Scholar]
- Sacu S, Pemp B, Weigert G, Matt G, Garhofer G, Pruente C, Schmetterer L, Schmidt-Erfurth U. Response of retinal vessels and retrobulbar hemodynamics to intravitreal anti-VEGF treatment in eyes with branch retinal vein occlusion. Investigative ophthalmology & visual science. 2011;52:3046–3050. doi: 10.1167/iovs.10-5842. [DOI] [PubMed] [Google Scholar]
- Saint-Geniez M, Maharaj AS, Walshe TE, Tucker BA, Sekiyama E, Kurihara T, Darland DC, Young MJ, D’Amore PA. Endogenous VEGF is required for visual function: evidence for a survival role on muller cells and photoreceptors. PLoS One. 2008;3:e3554. doi: 10.1371/journal.pone.0003554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai E, Anand A, Ambati BK, van Rooijen N, Ambati J. Macrophage depletion inhibits experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3578–3585. doi: 10.1167/iovs.03-0097. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Embo J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz-Valckenberg S, Fleckenstein M, Scholl HP, Holz FG. Fundus autofluorescence and progression of age-related macular degeneration. Surv Ophthalmol. 2009;54:96–117. doi: 10.1016/j.survophthal.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Scholl HP, Fleckenstein M, Fritsche LG, Schmitz-Valckenberg S, Gobel A, Adrion C, Herold C, Keilhauer CN, Mackensen F, Mossner A, et al. CFH, C3 and ARMS2 are significant risk loci for susceptibility but not for disease progression of geographic atrophy due to AMD. PLoS One. 2009;4:e7418. doi: 10.1371/journal.pone.0007418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen CK, Gordillo GM, Khanna S, Roy S. Micromanaging vascular biology: tiny microRNAs play big band. J Vasc Res. 2009;46:527–540. doi: 10.1159/000226221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Ji HF. Insights into the disappointing clinical trials of antioxidants in neurodegenerative diseases. Journal of Alzheimer’s disease: JAD. 2010;19:1141–1142. doi: 10.3233/JAD-2010-1307. [DOI] [PubMed] [Google Scholar]
- Sheridan CM, Mason S, Pattwell DM, Kent D, Grierson I, Williams R. Replacement of the RPE monolayer. Eye. 2009;23:1910–1915. doi: 10.1038/eye.2008.420. [DOI] [PubMed] [Google Scholar]
- Shiose S, Chen Y, Okano K, Roy S, Kohno H, Tang J, Pearlman E, Maeda T, Palczewski K, Maeda A. Toll-like receptor 3 is required for development of retinopathy caused by impaired all-trans-retinal clearance in mice. The Journal of biological chemistry. 2011;286:15543–15555. doi: 10.1074/jbc.M111.228551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18:349–355. doi: 10.1016/j.semcancer.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Skeie JM, Mullins RF. Macrophages in neovascular age-related macular degeneration: friends or foes? Eye. 2009;23:747–755. doi: 10.1038/eye.2008.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaide RF. Rationale for combination therapies for choroidal neovascularization. American journal of ophthalmology. 2006;141:149–156. doi: 10.1016/j.ajo.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Stahl A, Paschek L, Martin G, Gross NJ, Feltgen N, Hansen LL, Agostini HT. Rapamycin reduces VEGF expression in retinal pigment epithelium (RPE) and inhibits RPE-induced sprouting angiogenesis in vitro. FEBS Lett. 2008;582:3097–3102. doi: 10.1016/j.febslet.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Streilein JW. Ocular immune privilege: therapeutic opportunities from an experiment of nature. Nat Rev Immunol. 2003;3:879–889. doi: 10.1038/nri1224. [DOI] [PubMed] [Google Scholar]
- Strunnikova NV, Maminishkis A, Barb JJ, Wang F, Zhi C, Sergeev Y, Chen W, Edwards AO, Stambolian D, Abecasis G, et al. Transcriptome analysis and molecular signature of human retinal pigment epithelium. Hum Mol Genet. 2010;19:2468–2486. doi: 10.1093/hmg/ddq129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Nathans J. Stargardt’s ABCR is localized to the disc membrane of retinal rod outer segments. Nat Genet. 1997;17:15–16. doi: 10.1038/ng0997-15. [DOI] [PubMed] [Google Scholar]
- Szumiel I. Autophagy, reactive oxygen species and the fate of mammalian cells. Free radical research. 2011;45:253–265. doi: 10.3109/10715762.2010.525233. [DOI] [PubMed] [Google Scholar]
- Takeda A, Baffi JZ, Kleinman ME, Cho WG, Nozaki M, Yamada K, Kaneko H, Albuquerque RJ, Dridi S, Saito K, et al. CCR3 is a target for age-related macular degeneration diagnosis and therapy. Nature. 2009;460:225–230. doi: 10.1038/nature08151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, Cho WG, Kaneko H, Fowler BJ, Bogdanovich S, et al. DICER1 Loss and Alu RNA Induce Age-Related Macular Degeneration via the NLRP3 Inflammasome and MyD88. Cell. 2012;149:847–859. doi: 10.1016/j.cell.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatar O, Yoeruek E, Szurman P, Bartz-Schmidt KU, Adam A, Shinoda K, Eckardt C, Boeyden V, Claes C, Pertile G, et al. Effect of bevacizumab on inflammation and proliferation in human choroidal neovascularization. Archives of ophthalmology. 2008;126:782–790. doi: 10.1001/archopht.126.6.782. [DOI] [PubMed] [Google Scholar]
- Thornton J, Edwards R, Mitchell P, Harrison RA, Buchan I, Kelly SP. Smoking and age-related macular degeneration: a review of association. Eye (Lond) 2005;19:935–944. doi: 10.1038/sj.eye.6701978. [DOI] [PubMed] [Google Scholar]
- Thurman JM, Renner B, Kunchithapautham K, Ferreira VP, Pangburn MK, Ablonczy Z, Tomlinson S, Holers VM, Rohrer B. Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J Biol Chem. 2009;284:16939–16947. doi: 10.1074/jbc.M808166200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong JP, Lam DS, Chan WM, Choy KW, Chan KP, Pang CP. Effects of triamcinolone on the expression of VEGF and PEDF in human retinal pigment epithelial and human umbilical vein endothelial cells. Molecular vision. 2006;12:1490–1495. [PubMed] [Google Scholar]
- Tuo J, Bojanowski CM, Zhou M, Shen D, Ross RJ, Rosenberg KI, Cameron DJ, Yin C, Kowalak JA, Zhuang Z, et al. Murine ccl2/cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Investigative ophthalmology & visual science. 2007;48:3827–3836. doi: 10.1167/iovs.07-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood BR, Imarisio S, Fleming A, Rose C, Krishna G, Heard P, Quick M, Korolchuk VI, Renna M, Sarkar S, et al. Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease. Human molecular genetics. 2010;19:3413–3429. doi: 10.1093/hmg/ddq253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walshe TE, Dole VS, Maharaj AS, Patten IS, Wagner DD, D’Amore PA. Inhibition of VEGF or TGF-{beta} signaling activates endothelium and increases leukocyte rolling. Arterioscler Thromb Vasc Biol. 2009;29:1185–1192. doi: 10.1161/ATVBAHA.109.186742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wittchen ES, Jiang Y, Ambati B, Grossniklaus HE, Hartnett ME. Upregulation of CCR3 by age-related stresses promotes choroidal endothelial cell migration via VEGF-dependent and independent signaling. Invest Ophthalmol Vis Sci. 2011a doi: 10.1167/iovs.11-8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wang VM, Chan CC. The role of anti-inflammatory agents in age-related macular degeneration (AMD) treatment. Eye (Lond) 2011b;25:127–139. doi: 10.1038/eye.2010.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. Uber die klassifizierung tierischer gewebe nach ihrem stoffwechsel. Biochem Z. 1928;184:484–488. [Google Scholar]
- Wiesen JL, Tomasi TB. Dicer is regulated by cellular stresses and interferons. Mol Immunol. 2009;46:1222–1228. doi: 10.1016/j.molimm.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett CG, Boucher Y, Duda DG, di Tomaso E, Munn LL, Tong RT, Kozin SV, Petit L, Jain RK, Chung DC, et al. Surrogate markers for antiangiogenic therapy and dose-limiting toxicities for bevacizumab with radiation and chemotherapy: continued experience of a phase I trial in rectal cancer patients. J Clin Oncol. 2005;23:8136–8139. doi: 10.1200/JCO.2005.02.5635. [DOI] [PubMed] [Google Scholar]
- Winkler BS, Boulton ME, Gottsch JD, Sternberg P. Oxidative damage and age-related macular degeneration. Mol Vis. 1999;5:32. [PMC free article] [PubMed] [Google Scholar]
- Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:805–811. doi: 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong TY, Chakravarthy U, Klein R, Mitchell P, Zlateva G, Buggage R, Fahrbach K, Probst C, Sledge I. The natural history and prognosis of neovascular age-related macular degeneration: a systematic review of the literature and meta-analysis. Ophthalmology. 2008;115:116–126. doi: 10.1016/j.ophtha.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Woolcock KJ, Gaidatzis D, Punga T, Buhler M. Dicer associates with chromatin to repress genome activity in Schizosaccharomyces pombe. Nat Struct Mol Biol. 2011;18:94–99. doi: 10.1038/nsmb.1935. [DOI] [PubMed] [Google Scholar]
- Yakovchuk P, Goodrich JA, Kugel JF. B2 RNA and Alu RNA repress transcription by disrupting contacts between RNA polymerase II and promoter DNA within assembled complexes. Proc Natl Acad Sci U S A. 2009;106:5569–5574. doi: 10.1073/pnas.0810738106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Stratton C, Francis PJ, Kleinman ME, Tan PL, Gibbs D, Tong Z, Chen H, Constantine R, Yang X, et al. Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N Engl J Med. 2008;359:1456–1463. doi: 10.1056/NEJMoa0802437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004;122:598–614. doi: 10.1001/archopht.122.4.598. [DOI] [PubMed] [Google Scholar]
- Zarbin MA. Retinal pigment epithelium-retina transplantation for retinal degenerative disease. Am J Ophthalmol. 2008;146:151–153. doi: 10.1016/j.ajo.2008.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2006;103:16182–16187. doi: 10.1073/pnas.0604255103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Pham L, Zhang N, He S, Gamulescu MA, Spee C, Ryan SJ, Hinton DR. Neutrophils promote experimental choroidal neovascularization. Molecular vision. 2005;11:414–424. [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- Zweifel SA, Imamura Y, Spaide TC, Fujiwara T, Spaide RF. Prevalence and significance of subretinal drusenoid deposits (reticular pseudodrusen) in age-related macular degeneration. Ophthalmology. 2010;117:1775–1781. doi: 10.1016/j.ophtha.2010.01.027. [DOI] [PubMed] [Google Scholar]