

Abstract
Fibrillin microfibrils are extracellular matrix structures with essential functions in the development and the organization of tissues including blood vessels, bone, limbs and the eye. Fibrillin-1 and fibrillin-2 form the core of fibrillin microfibrils, to which multiple proteins associate to form a highly organized structure. Defining the components of this structure and their interactions is crucial to understand the pathobiology of microfibrillopathies associated with mutations in fibrillins and in microfibril-associated molecules. In this study, we have analyzed both in vitro and in vivo the role of fibrillin microfibrils in the matrix deposition of latent TGF-β binding protein 1 (LTBP-1), -3 and -4; the three LTBPs that form a complex with TGF-β. In Fbn1-/- ascending aortas and lungs, LTBP-3 and LTBP-4 are not incorporated into a matrix lacking fibrillin-1 microfibrils, whereas LTBP-1 is still deposited. In addition, in cultures of Fbn1-/- smooth muscle cells or lung fibroblasts, LTBP-3 and LTBP-4 are not incorporated into a matrix lacking fibrillin-1 microfibrils, whereas LTBP-1 is still deposited. Fibrillin-2 is not involved in the deposition of LTBP-1 in Fbn1-/- extracellular matrix as cells deficient for both fibrillin-1 and fibrillin-2 still incorporate LTBP-1 in their matrix. However, blocking the formation of the fibronectin network in Fbn1-/- cells abrogates the deposition of LTBP-1. Together, these data indicate that LTBP-3 and LTBP-4 association with the matrix depends on fibrillin-1 microfibrils, whereas LTBP-1 association depends on a fibronectin network.
Keywords: TGF-β, LTBP, fibrillin, fibronectin, Marfan syndrome, extracellular matrix
Introduction
The latent TGF-ß binding proteins (LTBP) comprise a family of four proteins structurally similar to fibrillins-1 (Fbn1), -2 and -3, the major constituents of microfibrils (Ramirez and Dietz, 2009; Todorovic and Rifkin, 2012). LTBPs are multi-domain proteins with molecular masses of 150-220 kDa and are composed primarily of repeating calcium-binding EGF-like domains and domains containing eight cysteine residues, named 8-Cysteine (8-Cys) or TGF-ß-binding protein-like (TB) domains (Gleizes et al., 1996; Handford et al., 2000; Saharinen et al., 1996). The 8-Cys domains are unique to the LTBP-fibrillin superfamily (Robertson et al., 2011). LTBPs-1, -3 and -4 covalently bind to the prodomain of TGF-β through the third 8-Cys domain, whereas LTBP-2 and fibrillins do not (Gleizes et al., 1996; Saharinen et al., 1996).
TGF-β is secreted from cells as a biologically inactive large latent complex (LLC), composed of LTBP-1, -3 or -4, the prodomain dimer of TGF-β, also referred to as the latency associated peptide (LAP), and the mature TGF-β dimer. LAP associates noncovalently with the mature TGF-β to form the small latent complex (SLC). The covalent binding of the SLC to an LTBP occurs intracellularly in the secretory pathway through the formation of two disulfide bonds between LAP and the third 8-Cys domain of LTBP-1, -3 or -4. For binding of TGF-β to its receptors, the interaction between LAP and TGF-β must be disrupted, which is known as latent TGF-β activation (Hyytiainen et al., 2004; Rifkin, 2005).
LTBPs regulate TGF-ß activity by facilitating its secretion, localizing the latent TGF-ß to specific sites in the extracellular matrix (ECM), and participating in latent TGF-β activation (Annes et al., 2004; Dallas et al., 1995; Flaumenhaft et al., 1993; Miyazono et al., 1991; Taipale et al., 1994). The importance of ECM sequestration of TGF-ß through its association with LTBPs is highlighted in Marfan syndrome (MFS), a condition caused by mutations in fibrillin-1. The defects in fibrillin-1 are accompanied by inappropriate activation of TGF-ß, perhaps due to faulty interaction of the LLC with the ECM (Neptune et al., 2003). In addition, a region of LTBP-1 (amino acids 403-449) must interact with fibronectin for effective activation of TGF-ß1 by the integrin αVß6 (Annes et al., 2004; Fontana et al., 2005).
Although LTBP-1, -2 and –4 bind to fibrillin-1 and –2 by non-covalent interactions through their C-terminus and LTBPs have been detected in microfibrils of multiple tissues, published results concerning LTBP assembly into a matrix devoid of fibrillin-1 are somewhat conflicting (Dallas et al., 2000; Isogai et al., 2003; Ono et al., 2009; Rifkin, 2005). Ono et al. proposed that the deposition of LTBP-1 in cell cultures is dependent on the assembly of fibrillin-1 microfibrils (Ono et al., 2009). However, others have not observed a defect in LTBP-1 matrix deposition in the absence of fibrillin-1 (Massam-Wu et al., 2010; Vehvilainen et al., 2009). In addition, the N-terminal regions of LTBP-1 and LTBP-4 interact with fibronectin, thus providing a second site for LTBPs to interact with the ECM (Dallas et al., 2005; Kantola et al., 2008). The significance of this interaction with fibronectin on LTBP-1 and -4 matrix deposition is difficult to assess, since fibrillin-1 assembly requires fibronectin too (Kinsey et al., 2008; Sabatier et al., 2009). Importantly, despite similar domain organization of all LTBPs, the C-terminal region of LTBP-3 reportedly does not interact with fibrillin-1 or -2 or with the ECM of fibroblasts, whereas the N-terminal region yields a localization pattern similar to that of the fibronectin network (Isogai et al., 2003; Koli et al., 2005). Because of the potential significance of LTBP assembly into the ECM in the pathobiology of microfibrillopathies, an understanding of the mechanism of LTBP deposition into the ECM is important. Therefore, we attempted to define the role of fibrillin-1, fibrillin-2 and fibronectin in matrix deposition of LTBPs. We provide in vitro and in vivo evidence that (1) fibrillin-1 is essential for the incorporation of LTBP-3 and LTBP-4, but not LTBP-1; (2) the presence of a fibronectin network is essential for the ECM association of LTBP-1; and (3) matrix from cells with a MFS mutation displays a perturbed LTBP-3 and -4 deposition, whereas LTBP-1 is incorporated.
Materials and Methods
Antibodies and reagents
Ab39, a rabbit antiserum against LTBP-1, was a gift from Dr. Carl-Henrik Heldin (Ludwig Institute for Cancer. Uppsala, Sweden) and Kohei Miyazono (Tokyo University). Ab951, a rabbit polyclonal antiserum directed against the C-terminal portion of mouse LTBP-3 was described previously (Chen et al., 2002). The specificity of the Ab951 against LTBP-3 was confirmed by immunofluorescence using wild type and Ltbp3-/- lung fibroblasts. Rabbit polyclonal antibodies against LTBP-4 (pAb2101), fibrillin-1 (pAb 9543), LTBP-1 (pAb8579) were previously described (Charbonneau et al., 2003; Ono et al., 2009). Goat polyclonal antibodies against LTBP-4 were purchased from R&D. Mouse monoclonal anti-β-actin (clone AC-15) and mouse monoclonal anti-fibronectin (clone FN-3E2) were purchased from Sigma. FUD and Del29 peptides were a gift from Dr. Deane Mosher (University of Wisconsin-Madison, Madison, USA).
Mice
Fbn1-/- and Fbn2-/- mice were a gift from Dr. Francesco Ramirez (Mount Sinai School of Medicine, New York, USA) and were previously described (Arteaga-Solis et al., 2001; Carta et al., 2006). For MEFs isolation, female and male mice were housed together overnight. Noon of the following day was considered E0.5 (Embryonic day 0.5). Mice were killed by CO2 asphyxiation. All procedures were conducted according to the regulations of the NYU Langone Medical Center IACUC.
Isolation of Vascular Smooth Muscle Cells, Lung Fibroblasts and Mouse Embryonic Fibroblasts
Vascular smooth muscle cells (VSMCs) were isolated from 12-day old Fbn1-/- ascending aortas and their respective wild-type littermates. The ascending aortas were digested with collagenase Type II (Worthington) in order to remove the adventitia and the endothelium was scraped-off by gently rubbing the intimal surface with a scalpel blade. The aortas were transferred to 35 mm culture dishes containing Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine and 100 units/ml penicillin/streptomycin and left overnight in a cell culture incubator. Tissues were subsequently digested with Elastase (Sigma) and Collagenase type II for 20 min. Enzyme digestion was stopped by adding DMEM supplemented with 20% FBS and the aorta fragments plated on 35 mm Primaria tissue culture dishes. VSMCs were expanded until passage 3 and frozen for subsequent experiments. Cells between passages 3 and 6 were employed in the experiments. Fbn1C1039G/+ ASMCs were a gift from Dr. Harry C. Dietz (Johns Hopkins School of Medicine, Baltimore, Maryland, USA).
Primary lung fibroblasts were isolated from 12-day-old Fbn1-/- mice and their respective wild-type littermates. In brief, the lungs were finely chopped and digested for 90 min with 2.4 U/ml Dispase II (Roche) and 0.1 % Collagenase A (Roche) in buffer I (2 mM CaCl2, 10 mM Hepes, 150 mM NaCl). The digested lungs were washed twice with PBS, 0.05 M EDTA and plated on 10-cm tissue culture dishes. Cells were allowed to adhere to the bottom of the culture plates and the remaining tissue pieces were removed. Cells were cultured until confluent in minimum essential medium (MEM) supplemented with 10% FBS, 2 mM L-glutamine and 100 units/ml penicillin/streptomycin before being frozen at passage 1 (P1). Lung fibroblasts between passages 1 and 3 were employed in all experiments.
Mouse embryonic fibroblasts (MEFs) were isolated from WT, Fbn1-/-/Fbn2+/+ and Fbn1-/-/Fbn2-/- E12.5 embryos. In brief, individual embryos were separated from their placentas and surrounding membranes and the head, internal organs, and limbs were removed. The embryos were minced in small pieces and digested for 10 min at 37 C with 0.125 % trypsin-EDTA. The enzymatic reaction was stopped by adding DMEM supplemented with 10 % FBS and the resulting cell suspension was plated in a 60 mm tissue culture dish. MEFs were frozen at passage 1 for subsequent experiments.
RNA isolation, cDNA Synthesis and Quantitative Real-Time PCR Analysis
Total RNA from cells, lungs and ascending aortas was extracted using the RNeasy Protect Mini kit (Qiagen) with DNase treatment included. Reverse transcription of 50 ng ascending aortic RNA was performed using the Sensiscript Reverse Transcriptase (Qiagen). Reverse transcription of 500 ng of lung or cellular RNAs was performed using the SuperScript III Reverse Transcriptase (Qiagen). The resulting cDNA was used for quantitative real-time PCR (qPCR) analysis. qPCRs were performed using specific primers and QuantiFast SYBR Green PCR Kit (Qiagen) on an iCycler Thermal Cycler (Bio-Rad, Hercules, CA). The amount of each gene was calculated using the Ct value and corresponding standard curve. Each target transcript expression was quantified relative to the Actb gene. Primers used: Actb sense AGCCTTCCTTCTTGGGTATGG, antisense GCCACCGATCCACACAGAGTA; Hprt sense CGCAGTCCCAGCGTCGTGAT, antisense CCCTTGAGCACACAGAGGGC; Ltbp-1 sense AGCACCATCACCTCTGCTCT, antisense CAGACACTGCTGTCCTCCAA; Ltbp-3 sense ACGGCCTCAGTTGCATAGAC, antisense AAAGAGCCTGGTGTGTTCGT; Ltbp-4 sense TGACCTCCGATACAACACCA, antisense AGGCAGAAAGCCTGTAGGTG; Fbn1 sense GATCAACGGCTACCCAAAAC, antisense GTTGGCTTCCATCTCAGACC; Fbn2 sense TGGCTGTGGGTGTGGACGGA, antisense CACCGCACCCGGGAATGGAC. Annealing temperature was 60°C.
Immunocytochemistry
ASMCs, lung fibroblasts and MEFs were plated on glass coverslips (Fishers Scientific) in either 6-well or 24-well plates and grown for up to 14 days post confluence. Postconfluent cultures were washed twice with PBS, fixed with 100% ethanol at room temperature for 10 min. Fixed cells were washed three times in PBS and were blocked by incubation with 2% bovine serum albumin in PBS for 1 h at room temperature. Cells were stained with primary antibodies against LTBP-1 (pAb39 or pAb8579), LTBP-4 (pAb2101), fibrillin-1 (pAb9543), LTBP-3 (pAb951) and fibronectin (mAbFN3E2). After three washes with PBS, the cells were incubated with secondary antibodies: anti-mouse Alexa Fluor 488 or anti-rabbit Alexa Fluor 594 (Invitrogen). Nuclei were stained with DAPI. The coverslips were mounted on glass slides with Prolong Gold anti-fading reagent (Invitrogen) and examined under the Axioskop 2 Mot Plus imaging microscope (Zeiss) with a 20x or 40x objective. Images were acquired with an AxioCam MRm camera (Zeiss) and AxioVision 3.1 software (Zeiss).
Conditioned media analysis
Cell cultures were washed with serum-free DMEM and incubated with fresh serum-free DMEM for 24 h. The conditioned media (CM) were collected, and protease inhibitors were added. The CM were clarified by centrifugation and concentrated using Amicon Ultracel 30K membrane (Millipore) and stored at −80 C.
Isolation and plasmin digestion of ECM-associated proteins
To isolate the insoluble ECM, lung and ascending aortic tissues were treated on ice with deoxycholate buffer (0.5% Na deoxycholate, 50 mM Tris-HCL (pH 8.0), 150 mM NaCl and 1% NP-40) and RIPA buffer, respectively. The insoluble material representing ECM was collected by centrifugation. After washing twice with ice-cold PBS, the ECM preparations were digested with 0.3 U/ml of plasmin (Sigma) in PBS containing 1 mM MgCl2, 1 mM CaCl2 and 0.1% n-octyl-b-D-glucopyranoside at 37°C for 1 h to release LTBPs from the ECM structures. Protease inhibitors were added; the supernatants clarified by centrifugation and concentrated using Amicon Ultracel 30 K membrane (Millipore).
Immunoblotting
Proteins were separated by SDS-PAGE using 4%–20% gradient polyacrylamide mini gels (Thermo scientific) in non-reducing condition and transferred to nitrocellulose membranes (Whatman) by electroblotting. Membranes were blocked in PBS/0.1% Tween-20 containing 5% nonfat milk for 1 h at room temperature and incubated with primary antibodies overnight at 4°C. The membranes were washed five times in PBS/0.1% Tween-20 and incubated with horseradish peroxidase-linked anti-Rabbit secondary antibody (GE Healthcare) for 1 h at room temperature. After washing, the immunostained bands were detected with an enhanced chemiluminescence (ECL) detection kit (Thermo Scientific).
Immunohistochemistry
Tissues were fixed in 4% paraformaldehyde for 12–16 h, dehydrated and embedded in paraffin. Sections (6–8 μm) were subjected to immunostaining. Antigen retrieval was performed by a short trypsin digestion of de-paraffinized tissue sections (0.1% trypsin in PBS, 1 min at 37°C). The protocol for immunofluorescent staining included a 30 min treatment with 100 mM CuSO4 to quench auto-fluorescence prior to a blocking step with 10% goat serum/1% BSA-PBS (Hu et al., 2010). The following primary antibodies were used for immunostainings: rabbit polyclonal to LTBP-1 (pAb8579), rabbit polyclonal to LTBP-3 (pAb951), rabbit polyclonal to LTBP-4 (pAb2101). The following secondary antibody was used: Alexa Fluor 594 goat anti-rabbit IgG (Invitrogen). Nuclei were stained with DAPI. The sections were mounted in Prolong Gold anti-fading reagent (Invitrogen) and examined under the Axioskop 2 Mot Plus imaging microscope (Zeiss) with a 20x or 40x objective. Images were acquired with an AxioCam MRm camera (Zeiss) and AxioVision 3.1 software (Zeiss).
Statistical Analysis
Data are expressed as mean ±SD. The significance of differences between groups was determined by unpaired student’s t test. Values of P ≤ 0.05 were considered significant.
Fibrillin-1 expression is essential for the incorporation of LTBP-3 and -4 but not for LTBP-1 in aorta and lung tissues
Relatively little information is available concerning the mechanisms and protein interactions involved in LTBP incorporation into the ECM of aorta and lungs, two tissues that show defects in both humans and mice with Fbn1 mutations. To determine if fibrillin-1 is involved in LTBP matrix incorporation in vivo, we initially investigated the localization of LTBP-1, -3 and -4 in the ascending aortas of 10-day-old WT and Fbn1-/- mice by immunohistology. In WT aortas, LTBP-1, -3 and -4 were detected throughout the media with LTBP-3 and LTBP-4 staining most intensely in the elastic lamellae within the media (Fig. 1A). LTBP-3 and -4 staining signal was brightest in the internal elastic lamina (IEL) and LTBP-3 staining was also intense in the external elastic lamina (EEL). In Fbn1 null aortas, LTBP-1 staining was equivalent or slightly less than WT tissues, whereas LTBP-3 and -4 staining were greatly decreased compared to WT (Fig. 1A). We performed qPCR analyses to assess the relative levels of LTBP mRNA in WT and Fbn1-/- ascending aortas. No significant differences in the expression levels of Ltbp-1, -3 and -4 were observed between WT and Fbn1-/-, indicating that decreased matrix incorporation of LTBP-3 and -4 in Fbn1-/- ascending aortas is not a result of a reduction in the expression of Ltbp-3 and -4 (Fig. 1B).
Figure 1.
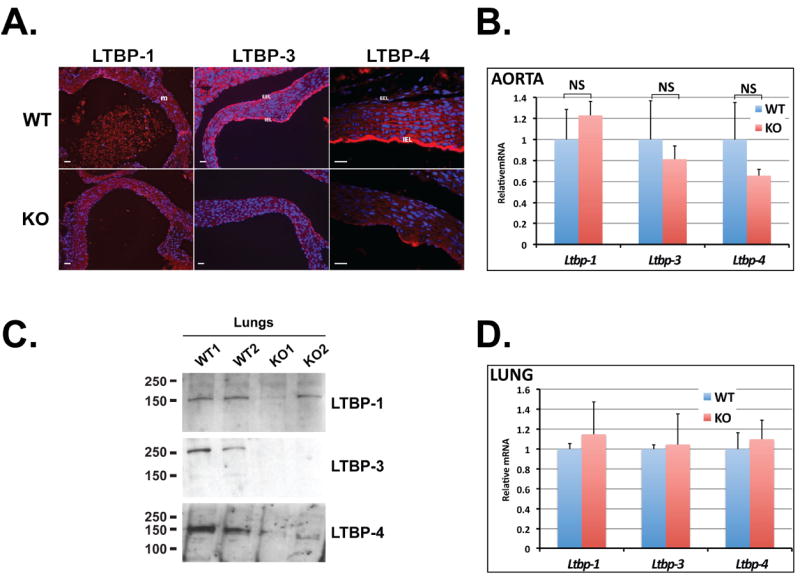
Fibrillin expression is essential for matrix incorporation of LTBP-3 and -4. (A) Immunofluorescence using antibodies against LTBP-1, -3 and 4 in WT and Fbn1-/- aortas from 10-day-old mice. WT and Fbn1-/- tissues showed equivalent amounts of LTBP-1 deposition in the matrix. In contrast, both LTBP-3 and -4 staining were reduced in Fbn1-/- aortas compared with WT. (m) media; (IEL) internal elastic lamina; (EEL) external elastic lamina. Scale bars, 20 μm. (B) Quantitative PCR analysis from WT and Fbn1-/- aortas. Total cellular RNA was isolated from WT and Fbn1-/- 10-day-old aortas, and the expression of Ltbp-1, -3 and -4 was analyzed. mRNA expression levels were normalized to the expression of the Hprt gene and the results are expressed as fold change over the wild type set to 1. Data are presented as the mean ± SD of four wild type and four Fbn1-/- samples, each performed in duplicate. NS, not significant, unpaired t test. (C) Analysis of sodium deoxycholate insoluble lung matrix after digestion with plasmin to solubilize the LTBPs. Plasmin digests of lungs from two lines each of wild type and Fbn1 null mice were analyzed by Western blotting using antibodies specific for each LTBP isoform. One representative immunoblot of two independent experiments is shown. The migration of the molecular mass markers (kDa) is indicated on the left. (D) Quantitative PCR analysis from wild type and Fbn1-/- lungs. Total cellular RNA was isolated from wild type and Fbn1-/- lungs, and the expression of Ltbp-1, -3 and -4 was analyzed. The mRNA expression levels were normalized to the expression of the β-actin gene and the results expressed as fold change over the wild type set to 1. Data are presented as the mean ± SD of five wild types and five Fbn1-/-, each performed in duplicate.
To confirm the previous results, we examined by Western blotting the amounts of LTBPs in sodium deoxycholate insoluble matrices of WT and Fbn1 null aortic tissues after digestion with plasmin to solubilize matrix-associated LTBPs. Our results indicate that LTBP-1 was present and LTBP-4 absent in Fbn1-/- tissues but despite several attempts after plasmin digestion we were unable to detect LTBP-3 in WT and mutant aortic tissues by immunoblotting (Fig. S1). Since LTBP-1, -3 and -4 are expressed in the lung and mice devoid of fibrillin-1 expression display developmental impairment of distal alveolar septation, we measured the amount of LTBPs associated with the ECM of lung tissue. (Dabovic et al., 2010; Neptune et al., 2003). LTBP-1-specific antibodies revealed one fragment of ~150 kDa in both WT and Fbn1-/- tissues, representing the uncomplexed form of LTBP-1. However, we observed some variability in the intensity of the LTBP-1 band among the fibrillin-1 null lung samples (Fig. 1C). The reason for this variability is unclear. In WT lungs, immunoblotting with an antibody specific for LTBP-3 detected a fragment of ~250 kDa, corresponding to the proteolytically processed complex composed of LTBP-3 and LAP (Fig. 1C). In WT samples, anti-LTBP-4 antibody revealed one fragment of ~150 kDa, probably representing the uncomplexed form of LTBP-4 (Fig. 1C). In Fbn1 null lungs, LTBP-3 was almost undetectable and LTBP-4 was substantially reduced in the insoluble ECM (Fig. 1C). We performed qPCR analysis to ensure that differences in LTBP deposition between WT and Fbn1-/- samples were not caused by decreased LTBP mRNA expression levels. We observed similar levels of Ltbp-1, -3 and -4 transcripts in both WT and Fbn1-/- lungs, ruling out the possibility that differences in LTBP ECM incorporation between WT and Fbn1-/- lungs were due to changed Ltbp expression levels (Fig. 1D).
In summary, in vivo data suggest that matrix association of LTBP-3 and -4 depends on fibrillin-1 microfibrils, whereas LTBP-1 is incorporated into the matrix even in the absence of fibrillin-1, perhaps through its interaction with other matrix proteins.
Fibrillin-1 is necessary for in vitro incorporation of LTBP-3 & -4 but not LTBP-1 in ASMC and lung fibroblast matrix
We next determined whether fibrillin-1 is dispensable for matrix targeting of LTBP-1, -3 and -4 in cell cultures. We established an in vitro model using WT and Fbn1-/- ASMCs isolated from the ascending aorta and analyzed the ECM for the presence of LTBP-1, -3 and -4 by immunofluorescence. After 14 days in culture, strong immunoreactivity was detected in WT cultures with antibodies against LTBP-1, -3 and -4 as well as fibrillin-1 (Fig. 2A). In Fbn1-/- cultures there were no detectable fibrillin-1 fibers and this absence correlated with the total ablation of the incorporation of LTBP-3 and -4 into the ECM. However, LTBP-1 fibers were observed in cultures of Fbn1-/- ASMCs, concordant with the results observed in vivo with aorta and lung tissues (Fig. 2A). In addition, qPCR analysis indicated that WT and Fbn1-/- cells express Ltbp-1, -3 and -4 transcripts at similar levels (Fig. 2B). Immunoblotting of 24 h conditioned media from ASMCs showed that the amount of secreted LTBP-1 was slightly lower in Fbn1 null compared to WT cell conditioned media, but similar band intensities were detected between WT and Fbn1-/- for LTBP-3 and -4 (Fig. 2C). Our results suggest that in ASMC cultures, LTBP-3 and -4 deposition into the ECM is dependent on fibrillin-1, whereas LTBP-1 incorporation is independent of fibrillin-1.
Figure 2.
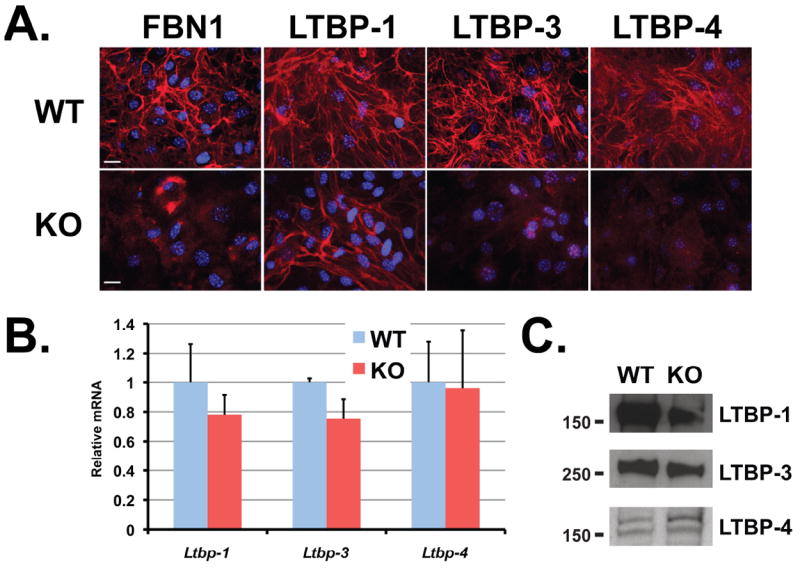
Incorporation of LTBP-1, -3 and 4 into WT and Fbn1-/- ASMC matrices. (A) Assembly of LTBP-1, -3 and -4 in WT and Fbn1-/- ASMCs. Cells were cultured for 14 days on glass coverslips, fixed with ethanol and stained with specific antibodies against LTBPs (red). Nuclei are stained with DAPI (blue). Scale bars, 20 μm all panels. (B) Quantitative PCR analysis from WT and Fbn1-/- ASMCs. Total cellular RNA was isolated from WT and Fbn1 null cells, and the expression of Ltbp-1, -3 and -4 was analyzed. The mRNA expression levels were normalized to the expression of the β-actin gene and the results expressed as fold change over the wild type set to 1. Data are presented as the mean ± SD of three independent experiments, each performed in duplicate. (C) Analyses of 24 h conditioned media from WT and Fbn1 null ASMCs. Conditioned media were collected and concentrated ten fold and analyzed by Western blotting using antibodies specific for each LTBP isoform.
We next examined whether the patterns of LTBP incorporation in cultured ASMCs could be reproduced in other cell types. We used primary lung fibroblasts isolated from 12-day-old WT and Fbn1-/- mice. The pattern of LTBP assembly in WT and Fbn1-/- lung fibroblasts resembled that obtained by immunofluorescence in ASMCs. The extracellular matrix of WT lung fibroblasts incorporated LTBP-1, -3 and -4, whereas Fbn1-/- lung fibroblasts incorporated LTBP-1 but not LTBP-3 and -4 (Fig. 3A). Quantitative PCR analyses were performed to assess whether the differential ECM incorporation of LTBPs in Fbn1-/- lung fibroblasts compared to WT was due to differences in mRNA expression levels. Ltbp-1, -3 and -4 transcript levels in WT and Fbn1-/- lung fibroblasts were similar (Fig. 3B). In addition, we performed immunoblotting of the secreted proteins from serum-free conditioned media (Fig. 3C). LTBP-1, -3 and -4 were present in the media of both WT and Fbn1-/- lung fibroblasts. Thus, in cultures of both lung fibroblasts and ASMCs, LTBP-3 and -4 deposition, but not their secretion, is governed by the presence of fibrillin-1, whereas LTBP-1 is still incorporates in a matrix devoid of fibrillin-1. This suggests that the role of fibrillin-1 in LTBP deposition into the matrix is LTBP isoform specific.
Figure 3.
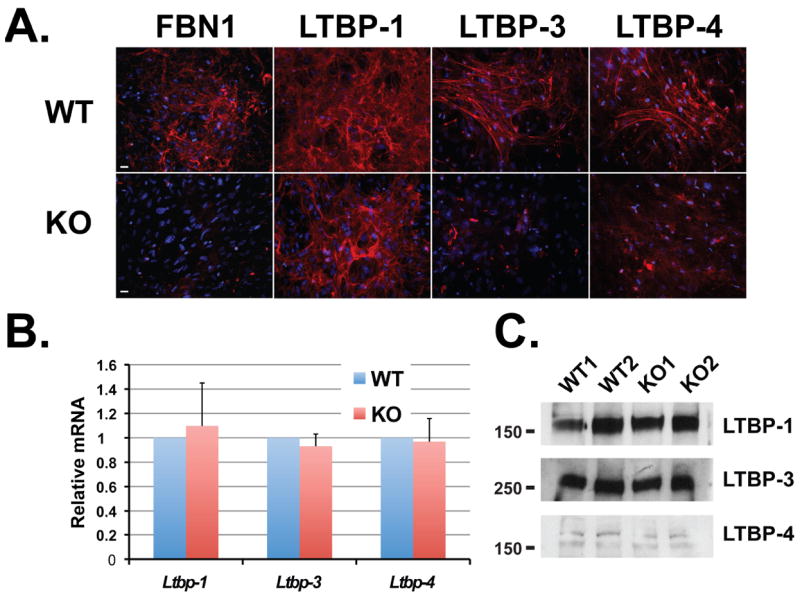
Incorporation of LTBP-1, -3 and 4 into WT and Fbn1 null lung fibroblast matrices. (A) Assembly of LTBP-1, -3 and -4 in WT and Fbn1 null lung fibroblasts. Cells were cultured for 14 days on glass coverslips, fixed with ethanol and stained with specific antibodies against LTBPs (red). Nuclei are stained with DAPI (blue). Scale bars, 20 μm all panels. (B) Quantitative PCR analysis from WT and Fbn1 null lung fibroblasts. Total cellular RNA was isolated from WT and Fbn1 null cells, and the expression of Ltbp-1, -3 and -4 was analyzed. The mRNA expression levels were normalized to the expression of the β-actin gene and the results expressed as fold change over the wild type set to 1. Data are presented as the mean ± SD of three independent experiments, each performed in duplicate. (C) Analyses of 24 h conditioned media from two WT and two Fbn1-/- lung fibroblasts. Conditioned media were collected and concentrated ten fold and analyzed by Western blotting using antibodies specific for each LTBP isoform.
Reduced deposition of LTBP-3 and -4 in Fbn1C1039G/+ ASMCs
We next investigated if a matrix with a fibrillin-1 mutation similar to that found in MFS has an effect on LTBP assembly. Therefore, we analyzed LTBP incorporation into the matrix from Fbn1C1039G/+ ASMCs. This mutation is representative of the most common class of mutations causing human MFS; that is, mutations of cysteine residues in the EGF-like domains of fibrillin-1 (Boileau et al., 2005; Robinson et al., 2006). Matrix from cells heterozygous for the C1039G Fbn1 mutation displayed decreased amount of fibrillin-1 microfibrils compared to matrix from WT cells (Fig. 4A). Like the Fbn1-/- cells, matrix from Fbn1C1039G/+ cell cultures also displayed a lack of LTBP-3 and -4 deposition compared to WT matrix, whereas LTBP-1 was still deposited into both WT and mutant matrices (Fig. 4A). We performed immunoblotting of the secreted proteins from serum-free conditioned media of WT and Fbn1C1039G/+ ASMCs. Similar amounts of LTBP-1, -3 and -4 were present in the medium of both cell types (Fig. 4B). Therefore the C1039G point mutation reduced the amount of fibrillin-1 microfibrils and consequently the incorporation of LTBP-3 and -4 into the ECM.
Figure 4.
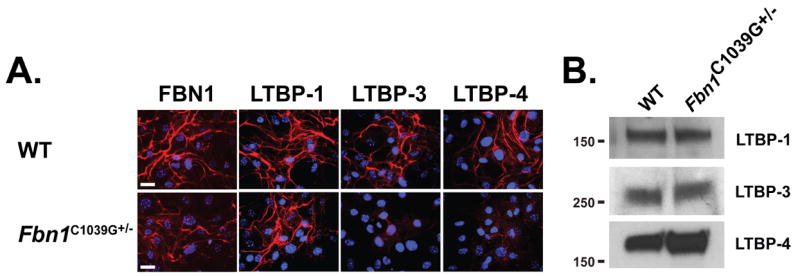
Incorporation of LTBP-1, -3 and 4 into WT and Fbn1C1039G/+ ASMC matrices. (A) Analyses by immunofluorescence of the deposition of LTBP-1, -3 and -4 into the ECM of WT and Fbn1C1039G/+ ASMCs. Cells were cultured for 7 days on glass coverslips, fixed with ethanol and stained with specific antibodies against LTBPs (red). Nuclei are stained with DAPI (blue). Scale bars, 20 μm all panels. (B) Analyses of 24 h conditioned media from WT and Fbn1C1039G/+ ASMCs. Conditioned media were collected and concentrated ten fold and analyzed by Western blotting using antibodies specific for each LTBP isoform.
Matrix devoid of fibrillin-1 and -2 incorporates LTBP-1
Because the LTBP-1 C-terminus can interact with both fibrillin-1 and fibrillin-2, we reasoned that the deposition of LTBP-1 into a matrix devoid of fibrillin-1 might be due to the presence of fibrillin-2 microfibrils (Isogai et al., 2003). To test this hypothesis, we isolated E12.5 WT, Fbn1-/- and Fbn1-/-/Fbn2-/- mouse embryonic fibroblasts (MEFs) and monitored the incorporation of LTBP-1 into the matrix (Fig. 5A). In Fbn1-/- ECM, we observed fibrillin-2 fibrils and LTBP-1 was incorporated into the ECM with an immunostaining pattern similar to that of WT MEFs. More importantly, a matrix devoid of fibrillin-1 and fibrillin-2 still incorporated LTBP-1 indicating a fibrillin-independent mechanism for LTBP-1 deposition into the matrix (Fig. 5A). Cells were also monitored for the expression of Fbn1, Fbn2 and Ltbp-1 mRNAs. Quantitative PCR confirmed the absence of Fbn1 and Fbn2 mRNA in Fbn1-/-/Fbn2-/- MEFs and Fbn1 mRNA in Fbn1-/- MEFs (Fig. 5B). In addition, a substantial amount of Ltbp-1 mRNA was expressed in WT, Fbn1-/- and Fbn1-/-/Fbn2-/- MEFs. Thus in the absence of fibrillin-1 and -2, the two main constituents of microfibrils, LTBP-1 is incorporated, probably through its interaction with another protein of the ECM. Therefore, whereas fibrillin-1 and -2 can interact with LTBP-1, they are not essential for its deposition.
Figure 5.

LTBP-1 incorporation into a matrix devoid of fibrillin-1 and -2. (A) Analyses by immunofluorescence of the deposition of LTBP-1, fibrillin-1 and fibrillin-2 into the ECM of WT, Fbn1-/-Fbn2+/+ and Fbn1-/-Fbn2-/- mouse embryonic fibroblasts. Cells were cultured for 7 days on glass coverslips, fixed with ethanol and stained with specific primary antibodies (red). Nuclei are stained with DAPI (blue). Scale bars, 20 μm all panels. (B) Quantitative PCR analysis from WT, Fbn1-/-Fbn2+/+ and Fbn1-/-Fbn2-/- mouse embryonic fibroblasts. Total cellular RNA was isolated and the expression levels of Fbn1, Fbn2 and LTBP-1 were analyzed. The mRNA expression levels were normalized to the expression of the β-actin gene and the results expressed as fold change over the wild type set to 1. Data are presented as the mean ± SD of two independent experiments, each performed in duplicate.
LTBP-1 matrix incorporation is fibronectin dependent
Because the LTBP-1 N-terminal region interacts with fibronectin, we examined whether fibronectin is required for LTBP-1 ECM deposition (Fontana et al., 2005). We first assessed the matrix deposition of LTBP-1 and fibronectin in WT and Fbn1-/- ASMCs. We observed that LTBP-1 is incorporated into the ECM of WT and Fbn1-/- ASMCs with a pattern of deposition similar to fibronectin fibers (Fig. 6A). These data indicate that within the ECM, independent of fibrillin-1 microfibrils, LTBP-1 and fibronectin are in close physical proximity. Interestingly, we also observed similar pattern of localization comparing LTBP-3 or LTBP-4 with fibronectin in WT ASMCs (Fig. S2). In order to assess the importance of fibronectin in the assembly of LTBP-1, we treated Fbn1-/- ASMCs with FUD peptide, which blocks fibronectin fiber assembly (Sabatier et al., 2009; Tomasini-Johansson et al., 2001). Blocking the assembly of fibronectin with 500 nM of FUD peptide in cultures of Fbn1-/- ASMCs resulted in the absence of LTBP-1 fibers compared to Fbn1-/- cells treated with the inactive mutant of FUD, Del29 (Fig. 6B). WT ASMCs treated with FUD peptides resulted in the disruption of fibronectin assembly and the absence of fibrillin-1 microfibrils and LTBP-1 incorporation (Fig. S3). We obtained similar results with Fbn1-/-/Fbn2-/- MEFs treated with FUD peptides (Fig. S4). Immunoblotting of 24 hr conditioned media of cell cultures treated with FUD peptides showed that the secretion of fibronectin and LTBP-1 was not affected by the FUD peptides compared to Del29 treated cells (Fig. 6C). Taken together, our results indicate that the formation of a fibronectin network but not fibrillin-1 microfibrils is essential for the extracellular deposition of LTBP-1.
Figure 6.
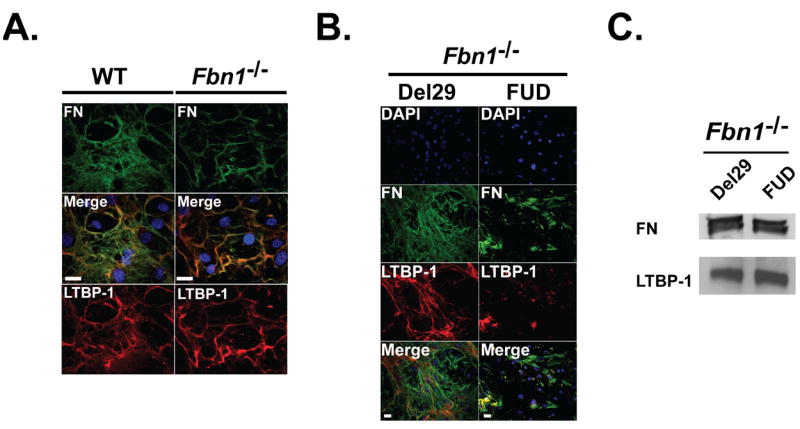
Deposition of LTBP-1 depends on the formation of a fibronectin network. (A) LTBP-1 localization with fibronectin. WT ASMCs were stained with specific primary antibodies against LTBP-1 (red) and fibronectin (green). The nuclei were stained with DAPI (blue). Merging of LTBP-1 and fibronectin images show similar localization. Scale bars, 20 mm all panels. (B) Fbn1-/- ASMCs were treated with either 500 nM FUD peptide or 500 nM control Del29 peptide, the inactive mutant of FUD for 7 days before immunofluorescence analyses. Cells were stained with specific primary antibodies against LTBP-1 (red), fibronectin (green) and the nuclei were stained with DAPI (blue). Merging of LTBP-1 and fibronectin staining with DAPI nuclear staining show similar localization of LTBP-1 and some fibronectin fibrils in cells treated with control Del29 peptide. Scale bars, 20 μm all panels (C) Analyses of 24 h conditioned medium from Fbn1-/- ASMCs treated with either 500 nM FUD peptide or 500 nM Del29 peptide. Conditioned media were collected and concentrated ten fold and analyzed by Western blotting using antibodies specific for LTBP-1 and fibronectin. The migration of the molecular mass markers (kDa) is indicated on the left.
Discussion
The purpose of the present study was to elucidate the role of fibrillin-1 and fibronectin in the modulation of the deposition of LTBPs into the ECM. We focused on LTBP-1, -3 and -4 because these proteins interact with TGF-β to form the large latent complex. Targeting LLC to the ECM is a major step in the process of activation of TGF-β. Default of specific localization of this potent growth factor can alter TGF-β activity. For example, overexpression of a mutated form of LTBP-1 that binds TGF-β but does not interact with the ECM results in an increase of TGF-β activity (Mazzieri et al., 2005). In addition, the cleavage of LTBP-1 by a bone morphogenetic protein 1 (BMP1)-like metalloproteinase, thereby liberating LLC from the ECM, leads to consequent activation of TGF-β1 by MMP2 (Ge and Greenspan, 2006).
LTBP-1 and -4 interact with several matrix molecules, including fibronectin, fibrillin-1 and -2 (Dallas et al., 2005; Fontana et al., 2005; Isogai et al., 2003; Kantola et al., 2008). Ablation of fibrillin-1 in mice is sufficient to raise TGF-β activity in aorta and lung tissues (Neptune et al., 2003). This lack of fibrillin-1 microfibrils is believed to disrupt LTBP-1 and -4 incorporation into the matrix and may result in the activation of TGF-β. Our results both in vitro and in vivo indicate that the lack of fibrillin-1 microfibrils significantly decreases the deposition of not only LTBP-4 but also LTBP-3 into the matrix, whereas there is no significant effect on LTBP-1 incorporation. The same pattern of altered LTBP incorporation in the absence of fibrillin-1 was observed in vivo in lung and aorta tissues and in vitro in both smooth muscle cells and lung fibroblasts. Two groups have reported the importance of fibrillin-1 in targeting LTBP-1 to the ECM in cell culture. Ono and colleagues showed that Fbn1-/- neonatal dermal fibroblasts secrete but do not deposit LTBP-1 into the ECM (Ono et al., 2009). Similarly, Massam-Wu et al. demonstrated that fibrillin-1 knockdown in retinal epithelial cells significantly decreases LTBP-1 ECM incorporation but not secretion (Massam-Wu et al., 2010). However, the same authors claimed that fibrillin-1 knockdown in human dermal fibroblasts does not impair LTBP-1 extracellular matrix deposition significantly (Massam-Wu et al., 2010). Furthermore, in UMR-106 rat osteosarcoma cultures, which do not express endogenous fibrillin-1, or in human embryonic lung fibroblasts stably transfected with fibrillin-1 shRNA, LTBP-1 incorporates into the ECM (Dallas et al., 2005; Vehvilainen et al., 2009). It is possible that the apparent discrepancies between those results are due to variable compensation of fibrillin-1 by fibrillin-2 depending upon the model system used. Both fibrillin-1 and -2 interact with the C-terminus of LTBP-1 with comparable affinities (Ono et al., 2009). A report suggested a model of microfibril structure in which fibrillin-2 forms a core within the microfibrils. The core is subsequently overlaid by fibrillin-1. In the absence of fibrillin-1, the core becomes accessible and might interact with LTBP-1 (Charbonneau et al., 2010). However, we show that LTBP-1 fibrils were still present in the matrix of mouse embryonic fibroblasts that lack both fibrillin-1 and fibrillin-2, indicating that fibrillin-2 is not required for LTBP-1 deposition into the ECM in the absence of fibrillin-1.
Another candidate for targeting LTBP-1 in a matrix lacking fibrillin-1 is fibronectin (Dallas et al., 2005). A sequence of 24 amino acid (aa 414-437) in the hinge region of LTBP-1 binds to fibronectin and FN-/- mouse embryonic fibroblasts do not deposit LTBP-1 in the ECM (Fontana et al., 2005). However, it is not clear if this absence is due to the lack of fibronectin, fibrillin-1 or both since fibronectin is also important for fibrillin-1 microfibril assembly (Dallas et al., 2005; Sabatier et al., 2009). We show that FUD peptides, which abrogate fibronectin assembly in Fbn1-/- ASMCs, prevent the assembly of LTBP-1, indicating that fibronectin provides docking for LTBP-1.
LTBP-1 and -4 molecules are structurally similar and both can interact with fibrillin-1 and fibronectin (Dallas et al., 2005; Fontana et al., 2005; Kantola et al., 2008). However, our data indicate that in a matrix devoid of fibrillin-1 microfibrils, only LTBP-1 is deposited in the ECM. Our in vivo results showed that in wild type lungs and aortas, LTBP-1 and -4 are both covalently linked to the ECM and can be released through plasmin digestion of insoluble ECM-associated proteins. Nunes et al. showed that transglutaminase crosslinks LTBP-1 to the matrix and that the N-terminal residues 294–441 are critical to the transglutaminase reactivity of LTBP-1 (Nunes et al., 1997). Interestingly, those residues contain the domain that interacts with fibronectin (Fontana et al., 2005). To date, it is not known to which proteins of the ECM LTBP-4 is covalently linked and if transglutaminase is involved in that process. It is interesting to speculate that, whereas LTBP-1 is covalently bound to fibronectin in the ECM, LTBP-4 may covalently bind fibrillin-1. Thus in the absence of fibrillin-1, LTBP-4 is not incorporated into the ECM. Further studies need to be done in order to understand which domains of LTBP-1 and -4 determine their specificity of deposition into the ECM.
Our data show that fibrillin-1 is required for the incorporation of LTBP-3 into the matrix. Absence of fibrillin-1 microfibrils both in vitro and in vivo prevents the association of LTBP-3 to the ECM. A previous study reported no interaction between a C-terminal fragment of LTBP-3 and the N-terminal region of fibrillin-1 in ELISA binding assays (Isogai et al., 2003). The reason for this discrepancy is unclear, but it is possible that domains of LTBP-3 other than its C-terminus mediate its interaction with fibrillin-1 or that regions of fibrillin-1 other than its N-terminal region are involved. We also cannot rule out an indirect interaction between LTBP-3 and fibrillin-1 through their association with another protein in the ECM. Consistent with this hypothesis, LTBP-1 and -2 interact with fibulin-4 and -5 respectively, and fibulin-2, -4 and -5 can associate with fibrillin-1 (El-Hallous et al., 2007; Massam-Wu et al., 2010). In addition, interactions between LTBP-1 and ADAMTSL-2 and -3 have been reported (Sengle et al., 2012). Further studies will be required to explore the nature of the interaction between LTBP-3 and fibrillin-1.
Although informative regarding the ability of fibrillin-1 to affect the incorporation of LTBPs into the ECM, studies with cells that do not express fibrillin-1 do not provide an accurate model with which to study the defective matrix found in MFS, since most patients with MFS harbor heterozygous missense mutations (Judge and Dietz, 2005; Robinson et al., 2006). To our knowledge, there is no report showing an effect of a Marfan-like matrix on LTBP deposition. Mice heterozygous for the C1039G mutation (Fbn1C1039G/+) display MFS-like phenotypic manifestations in the pulmonary, cardiovascular and skeletal systems (Judge et al., 2004). In Fbn1C1039G/+ ASMCs, we observed an important decrease in LTBP-3 and -4 ECM incorporation, whereas LTBP-1 was still present in both mutant and WT cells, thus providing results showing defects in LTBP deposition in a Marfan-like ECM.
Based upon our findings, we propose a model where in a wild type matrix, LTBP-3 and -4 are incorporated into the ECM mainly through their interaction with fibrillin-1, whereas LTBP-1 interacts with both fibronectin and probably fibrillin-1 (Fig. 7, left panel). In a MFS-like matrix, the deposition of LTBP-3 and -4 is abrogated, whereas LTBP-1 is still deposited into the matrix through its interaction with fibronectin (Fig 7, right panel). Our data suggest that in MFS, TGF-β in LTBP-3 or -4 containing LLCs might be activated excessively because of its defective sequestration into the ECM. On the other hand, although LTBP-1 is still incorporated into a MFS-matrix, the quality of the anchoring might be altered, and this might lead to improper activation of TGF-β. It remains unknown which LTBP isoforms are involved in different manifestations observed in MFS patients. It might be possible to dissect out the importance of each LTBP in MFS by generating mice with mutations in both Fbn1 and the genes for the different TGF-β binding LTBPs (Ltbp-1, Ltbp-3, and Ltbp-4). Experiments are in progress to elucidate the implications of LTBP-fibrillin-1 interaction in the pathophysiological mechanisms of MFS.
Figure 7.
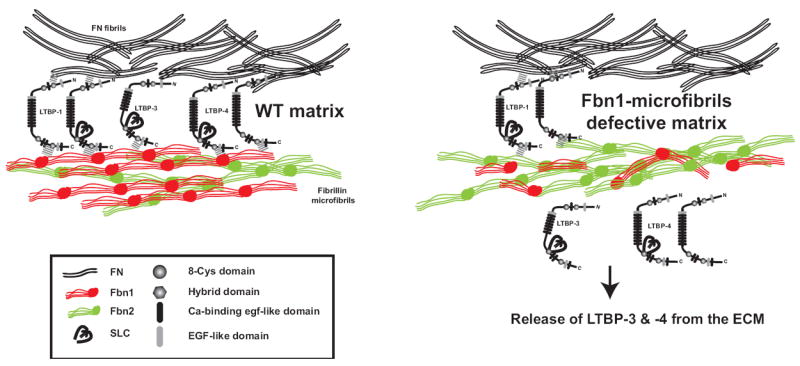
A model for LTBP assembly into WT and fibrillin-1-microfibril defective matrix. In the absence of fibrillin-1 microfibrils, the deposition of LTBP3-TGF-β and LTBP4-TGF-β complexes into the ECM is abrogated and might lead to improper TGF-β activation. On the other hand, LTBP1-TGF-b is assembled into the ECM but only through its interaction with fibronectin. Although LTBP-1 is still incorporated into a MFS-like matrix, the quality of the anchoring is altered and might lead to improper activation of TGF-β.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank Melinda Vassallo for technical support, Susan Oliveri for manuscript proofreading, Drs Harry C Dietz, Francesco Ramirez and Deane F Mosher for reagents.
Contract grant sponsor: NIH; Contract grant numbers: CA034282, AR49698 to DBR.
Footnotes
The authors have no conflict of interest to declare.
References
- Annes JP, Chen Y, Munger JS, Rifkin DB. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J Cell Biol. 2004;165(5):723–734. doi: 10.1083/jcb.200312172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteaga-Solis E, Gayraud B, Lee SY, Shum L, Sakai L, Ramirez F. Regulation of limb patterning by extracellular microfibrils. The Journal of cell biology. 2001;154(2):275–281. doi: 10.1083/jcb.200105046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau C, Jondeau G, Mizuguchi T, Matsumoto N. Molecular genetics of Marfan syndrome. Curr Opin Cardiol. 2005;20(3):194–200. doi: 10.1097/01.hco.0000162398.21972.cd. [DOI] [PubMed] [Google Scholar]
- Carta L, Pereira L, Arteaga-Solis E, Lee-Arteaga SY, Lenart B, Starcher B, Merkel CA, Sukoyan M, Kerkis A, Hazeki N, Keene DR, Sakai LY, Ramirez F. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. The Journal of biological chemistry. 2006;281(12):8016–8023. doi: 10.1074/jbc.M511599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbonneau NL, Carlson EJ, Tufa S, Sengle G, Manalo EC, Carlberg VM, Ramirez F, Keene DR, Sakai LY. In vivo studies of mutant fibrillin-1 microfibrils. J Biol Chem. 2010 doi: 10.1074/jbc.M110.130021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbonneau NL, Dzamba BJ, Ono RN, Keene DR, Corson GM, Reinhardt DP, Sakai L. Fibrillins can co-assemble in fibrils, but fibrillin fibril composition displays cell-specific differences. J Biological Chemistry. 2003;278(4):2740–2749. doi: 10.1074/jbc.M209201200. [DOI] [PubMed] [Google Scholar]
- Chen Y, Dabovic B, Annes JP, Rifkin DB. Latent TGF-beta binding protein-3 (LTBP-3) requires binding to TGF-beta for secretion. FEBS Lett. 2002;517(1-3):277–280. doi: 10.1016/s0014-5793(02)02648-0. [DOI] [PubMed] [Google Scholar]
- Dabovic B, Chen Y, Choi J, Davis EC, Sakai LY, Todorovic V, Vassallo M, Zilberberg L, Singh A, Rifkin DB. Control of lung development by latent TGF-ss binding proteins. J Cell Physiol. 2010 doi: 10.1002/jcp.22479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallas SL, Keene DR, Bruder SP, Saharinen J, Sakai LY, Mundy GR, Bonewald LF. Role of the latent transforming growth factor beta binding protein 1 in fibrillin-containing microfibrils in bone cells in vitro and in vivo. J Bone Miner Res. 2000;15(1):68–81. doi: 10.1359/jbmr.2000.15.1.68. [DOI] [PubMed] [Google Scholar]
- Dallas SL, Miyazono K, Skerry TM, Mundy GR. Dual role for the latent transforming growth factor-ß binding protein in storage of latent TGF-ß in the extracellular matrix and as a structural matrix protein. J Cell Biol. 1995;131(2):539–549. doi: 10.1083/jcb.131.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallas SL, Sivakumar P, Jones CJ, Chen Q, Peters DM, Mosher DF, Humphries MJ, Kielty CM. Fibronectin Regulates Latent Transforming Growth Factor-ß (TGF-ß) by Controlling Matrix Assembly of Latent TGF-ß-binding Protein-1. J Biol Chem. 2005;280(19):18871–18880. doi: 10.1074/jbc.M410762200. [DOI] [PubMed] [Google Scholar]
- El-Hallous E, Sasaki T, Hubmacher D, Getie M, Tiedemann K, Brinckmann J, Batge B, Davis EC, Reinhardt DP. Fibrillin-1 interactions with fibulins depend on the first hybrid domain and provide an adaptor function to tropoelastin. J Biol Chem. 2007;282(12):8935–8946. doi: 10.1074/jbc.M608204200. [DOI] [PubMed] [Google Scholar]
- Flaumenhaft R, Abe M, Sato Y, Miyazono K, Harpel J, Heldin CH, Rifkin DB. Role of the latent TGF-beta binding protein in the activation of latent TGF-beta by co-cultures of endothelial and smooth muscle cells. J Cell Biology. 1993;120(4):995–1002. doi: 10.1083/jcb.120.4.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Chen Y, Prijatelj P, Sakai T, Fassler R, Sakai LY, Rifkin DB. Fibronectin is required for integrin alphavbeta6-mediated activation of latent TGF-beta complexes containing LTBP-1. FASEB J. 2005;19(13):1798–1808. doi: 10.1096/fj.05-4134com. [DOI] [PubMed] [Google Scholar]
- Ge G, Greenspan DS. BMP1 controls TGFbeta1 activation via cleavage of latent TGFbeta-binding protein. J Cell Biol. 2006;175(1):111–120. doi: 10.1083/jcb.200606058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleizes PE, Beavis RC, Mazzieri R, Shen B, Rifkin DB. Identification and characterization of an eight-cysteine repeat of the latent transforming growth factor-beta binding protein-1 that mediates bonding to the latent transforming growth factor-beta 1. J Biol Chem. 1996;271:29891–29896. doi: 10.1074/jbc.271.47.29891. [DOI] [PubMed] [Google Scholar]
- Handford PA, Downing AK, Reinhardt DP, Sakai LY. Fibrillin: from domain structure to supramolecular assembly. Matrix Biology. 2000;19(6):457–470. doi: 10.1016/s0945-053x(00)00100-1. [DOI] [PubMed] [Google Scholar]
- Hu Q, Shifren A, Sens C, Choi J, Szabo Z, Starcher BC, Knutsen RH, Shipley JM, Davis EC, Mecham RP, Urban Z. Mechanisms of emphysema in autosomal dominant cutis laxa. Matrix Biol. 2010;29(7):621–628. doi: 10.1016/j.matbio.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyytiainen M, Penttinen C, Keski-Oja J. Latent TGF-beta binding proteins: extracellular matrix association and roles in TGF-beta activation. Crit Rev Clin Lab Sci. 2004;41(3):233–264. doi: 10.1080/10408360490460933. [DOI] [PubMed] [Google Scholar]
- Isogai Z, Ono RN, Ushiro S, Keene DR, Chen Y, Mazzieri R, Charbonneau NL, Reinhardt DP, Rifkin DB, Sakai LY. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol Chem. 2003;278(4):2750–2757. doi: 10.1074/jbc.M209256200. [DOI] [PubMed] [Google Scholar]
- Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, Sakai LY, Dietz HC. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114(2):172–181. doi: 10.1172/JCI20641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge DP, Dietz HC. Marfan’s syndrome. Lancet. 2005;366(9501):1965–1976. doi: 10.1016/S0140-6736(05)67789-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantola AK, Keski-Oja J, Koli K. Fibronectin and heparin binding domains of latent TGF-beta binding protein (LTBP)-4 mediate matrix targeting and cell adhesion. Exp Cell Res. 2008;314(13):2488–2500. doi: 10.1016/j.yexcr.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Kinsey R, Williamson MR, Chaudhry S, Mellody KT, McGovern A, Takahashi S, Shuttleworth CA, Kielty CM. Fibrillin-1 microfibril deposition is dependent on fibronectin assembly. J Cell Sci. 2008;121(Pt 16):2696–2704. doi: 10.1242/jcs.029819. [DOI] [PubMed] [Google Scholar]
- Koli K, Hyytiainen M, Ryynanen MJ, Keski-Oja J. Sequential deposition of latent TGF-beta binding proteins (LTBPs) during formation of the extracellular matrix in human lung fibroblasts. Exp Cell Res. 2005;310(2):370–382. doi: 10.1016/j.yexcr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Massam-Wu T, Chiu M, Choudhury R, Chaudhry SS, Baldwin AK, McGovern A, Baldock C, Shuttleworth CA, Kielty CM. Assembly of fibrillin microfibrils governs extracellular deposition of latent TGF beta. J Cell Sci. 2010;123(Pt 17):3006–3018. doi: 10.1242/jcs.073437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzieri R, Jurukovski V, Obata H, Sung J, Platt A, Annes E, Karaman-Jurukovska N, Gleizes PE, Rifkin DB. Expression of truncated latent TGF-beta-binding protein modulates TGF-beta signaling. Journal of cell science. 2005;118(Pt 10):2177–2187. doi: 10.1242/jcs.02352. [DOI] [PubMed] [Google Scholar]
- Miyazono K, Olofsson A, Colosetti P, Heldin CH. A role of the latent TGF-beta 1-binding protein in the assembly and secretion of TGF-beta 1. EMBO J. 1991;10(5):1091–1101. doi: 10.1002/j.1460-2075.1991.tb08049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33(3):407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- Nunes I, Gleizes PE, Metz CN, Rifkin DB. Latent transforming growth factor-beta binding protein domains involved in activation and transglutaminase-dependent cross-linking of latent transforming growth factor-beta. The Journal of cell biology. 1997;136(5):1151–1163. doi: 10.1083/jcb.136.5.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono RN, Sengle G, Charbonneau NL, Carlberg V, Bachinger HP, Sasaki T, Lee-Arteaga S, Zilberberg L, Rifkin DB, Ramirez F, Chu ML, Sakai LY. Latent transforming growth factor beta-binding proteins and fibulins compete for fibrillin-1 and exhibit exquisite specificities in binding sites. J Biol Chem. 2009;284(25):16872–16881. doi: 10.1074/jbc.M809348200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Dietz HC. Extracellular microfibrils in vertebrate development and disease processes. J Biol Chem. 2009 doi: 10.1074/jbc.R900004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifkin DB. Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability. J Biol Chem. 2005;280(9):7409–7412. doi: 10.1074/jbc.R400029200. [DOI] [PubMed] [Google Scholar]
- Robertson I, Jensen S, Handford P. TB domain proteins: evolutionary insights into the multifaceted roles of fibrillins and LTBPs. Biochem J. 2011;433(2):263–276. doi: 10.1042/BJ20101320. [DOI] [PubMed] [Google Scholar]
- Robinson PN, Arteaga-Solis E, Baldock C, Collod-Beroud G, Booms P, De Paepe A, Dietz HC, Guo G, Handford PA, Judge DP, Kielty CM, Loeys B, Milewicz DM, Ney A, Ramirez F, Reinhardt DP, Tiedemann K, Whiteman P, Godfrey M. The molecular genetics of Marfan syndrome and related disorders. J Med Genet. 2006;43(10):769–787. doi: 10.1136/jmg.2005.039669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatier L, Chen D, Fagotto-Kaufmann C, Hubmacher D, McKee MD, Annis DS, Mosher DF, Reinhardt DP. Fibrillin assembly requires fibronectin. Mol Biol Cell. 2009;20(3):846–858. doi: 10.1091/mbc.E08-08-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saharinen J, Taipale J, Keski-Oja J. Association of the small latent transforming growth factor-beta with an eight cysteine repeat of its binding protein LTBP-1. EMBO J. 1996;15(2):245–253. [PMC free article] [PubMed] [Google Scholar]
- Sengle G, Tsutsui K, Keene DR, Tufa SF, Carlson EJ, Charbonneau NL, Ono RN, Sasaki T, Wirtz MK, Samples JR, Fessler LI, Fessler JH, Sekiguchi K, Hayflick SJ, Sakai LY. Microenvironmental regulation by fibrillin-1. PLoS Genet. 2012;8(1):e1002425. doi: 10.1371/journal.pgen.1002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J, Miyazono K, Heldin C-H, Keski-Oja J. Latent transforming growth factor-ß1 associates to fibroblast extracellular matrix via latent TGF-ß binding protein. J Cell Biol. 1994;124(1 & 2):171–181. doi: 10.1083/jcb.124.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic V, Rifkin DB. LTBPs, more than just an escort service. J Cell Biochem. 2012;113(2):410–418. doi: 10.1002/jcb.23385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasini-Johansson BR, Kaufman NR, Ensenberger MG, Ozeri V, Hanski E, Mosher DF. A 49-residue peptide from adhesin F1 of Streptococcus pyogenes inhibits fibronectin matrix assembly. J Biol Chem. 2001;276(26):23430–23439. doi: 10.1074/jbc.M103467200. [DOI] [PubMed] [Google Scholar]
- Vehvilainen P, Hyytiainen M, Keski-Oja J. Matrix association of latent TGF-beta binding protein-2 (LTBP-2) is dependent on fibrillin-1. J Cell Physiol. 2009;221(3):586–593. doi: 10.1002/jcp.21888. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.