

Abstract
Prokineticin receptors (PROKR) are G protein-coupled receptors (GPCR) that regulate diverse biological processes, including olfactory bulb neurogenesis and GnRH neuronal migration. Mutations in PROKR2 have been described in patients with varying degrees of GnRH deficiency and are located in diverse functional domains of the receptor. Our goal was to determine whether variants in the first intracellular loop (ICL1) of PROKR2 (R80C, R85C, and R85H) identified in patients with hypogonadotropic hypogonadism interfere with receptor function and to elucidate the mechanisms of these effects. Because of structural homology among GPCR, clarification of the role of ICL1 in PROKR2 activity may contribute to a better understanding of this domain across other GPCR. The effects of the ICL1 PROKR2 mutations on activation of signal transduction pathways, ligand binding, and receptor expression were evaluated. Our results indicated that the R85C and R85H PROKR2 mutations interfere only modestly with receptor function, whereas the R80C PROKR2 mutation leads to a marked reduction in receptor activity. Cotransfection of wild-type (WT) and R80C PROKR2 showed that the R80C mutant could exert a dominant negative effect on WT PROKR2 in vitro by interfering with WT receptor expression. In summary, we have shown the importance of Arg80 in ICL1 for PROKR2 expression and demonstrate that R80C PROKR2 exerts a dominant negative effect on WT PROKR2.
Prokineticins (PROK) are bioactive peptides that bind to PROK receptors (PROKR), PROKR1 and PROKR2, to effect a wide spectrum of biological functions (1). PROKR1 and PROKR2 belong to the family A or rhodopsin family of G protein-coupled receptors (GPCR), the largest and most diverse of the GPCR families. Both PROKR were cloned and characterized simultaneously by three different groups (2–4). Human PROKR are approximately 80% identical to the mouse orphan receptor GPR73 and were initially known as orphan receptors GPR73a and GPR73b (2). PROKR1 and PROKR2 share 85% sequence identity and common signaling pathways. Activation of PROKR by PROK leads to mobilization of calcium, stimulation of phosphoinositide turnover, and activation of p42/p44 MAPK signaling pathways, presumably through a receptor/Gαq protein interaction (2–4). Some studies also suggest that PROKR are capable of coupling to Gs and Gi proteins (5, 6).
PROKR are distributed in a variety of human tissues (ovary, testis, adrenal gland, placenta, uterus, brain, intestinal tract, heart, bone marrow, and peripheral blood) and regulate diverse biological processes, including angiogenesis, neurogenesis, circadian rhythms, metabolism, pain perception, muscle contractility, hematopoiesis, immune response, and reproduction (1, 7–13). PROK have also been related to pathological processes such as tumorigenesis, participating as growth factors for cancer cells, as well as angiogenic and chemotactic factors for proinflammatory neutrophils (14–16).
The PROKR2 is highly expressed in the central nervous system and plays an important role in olfactory bulb neurogenesis and in GnRH neuronal migration (12, 17–19). Prokr2 knockout mice presented with malformation of olfactory bulbs and reproductive system hypoplasia due to abnormal GnRH neuronal migration (18). In recent years, mutations of PROKR2 have been described in patients with varying degrees of GnRH deficiency and are located in different functional domains of the receptor (20–28). We previously described a novel mutation in the first intracellular loop (ICL1) of PROKR2 in a patient with hypogonadotropic hypogonadism and anosmia (20). Additional mutations in ICL1 of PROKR2, R85C and R85H, have also been reported in patients with hypogonadotropic hypogonadism (22, 23, 27).
A considerable body of evidence has shown that ICL2, ICL3, and the carboxy-terminal domain of GPCR act in a cooperative fashion to dictate proper G protein recognition and efficient G protein activation (29, 30). The carboxy-terminal tail also plays an important role in receptor intracellular trafficking (31–33). However, the contribution of ICL1 to the function of GPCR is poorly understood. Moreover, the mechanism by which mutations in ICL1 affect receptor function is not clear. The length of the ICL1 loop is highly conserved among GPCR, indicating that it may play an important structural role (34). There are relatively few studies suggesting that the ICL1 loop is directly involved in G protein recognition (35). Functional analysis of a series of hybrid receptors demonstrated that ICL1 of the cholecystokinin-A receptor confers the ability to couple to Gs on the cholecystokinin-B receptor (36). Furthermore, mutational analysis showed that residues in the GnRH receptor ICL1 are required for selective coupling to adenylyl cyclase but are not essential for coupling to the phospholipase C pathway (37). ICL1 has also been shown to be important for export from the endoplasmic reticulum for α1B-, α2B-, and β2-adrenergic receptors as well as the angiotensin-1 receptor (38).
In an effort to understand the involvement of ICL1 in PROKR2 function and to determine whether ICL1 mutations may lead to GnRH deficiency in patients, we performed functional studies of the ICL1 mutations identified in patients with hypogonadotropic hypogonadism. Because of the structural homology among GPCR, the elucidation of the mechanisms by which ICL1 mutations of PROKR2 affect receptor function may contribute to a better understanding of this domain not only in PROKR2 but also across other GPCR. Here, we describe the importance of Arg80 in ICL1 to PROKR2 stability and proper localization in the plasma membrane and demonstrate that its substitution disturbs posttranslational processing and impairs receptor function. We also demonstrate that R80C PROKR2 exerts a dose-dependent dominant negative effect on wild-type (WT) PROKR2 function.
Materials and Methods
Reagents
Chemicals used were purchased from either Fisher Scientific (Pittsburgh, PA) or Sigma Chemical Co. (St. Louis, MO). Cell culture medium was from Invitrogen (Carlsbad, CA). Transfection reagents used were GenePORTER from Gene Therapy Systems (San Diego, CA) or FuGENE HD from Promega (Madison, WI). Antibody against V5 was obtained from Abcam (Cambridge, MA), Na/K ATPase antibody was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), and β-actin (AC-74) antibody was obtained from Sigma-Aldrich (St. Louis, MO). Radioisotopes, including 125I-mamba intestinal toxin-1 (MIT-1), a homologue of PROK1 (4), were from PerkinElmer (Waltham, MA). 125I-MIT-1-specific activity equals 2200 Ci (81.4 TBq)/mmol. PROK2 peptide was from Abcam.
Expression vectors
WT and mutant R85C PROKR2 cloned in pCDNA3.1/V5HisD TOPO expression vector (Invitrogen) were a kind gift from Nelly Pitteloud (Massachusetts General Hospital, Boston, MA). The V5-His tags are on the carboxy-terminal domain of PROKR2. These tags did not affect PROKR2 cell surface expression, ligand binding, or signaling properties (data not shown). Mutations (R80C and R85H) were introduced into the WT vector using the QuikChange Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's protocol. Oligonucleotides used for mutagenesis are described in Table 1. Plasmids were prepared using QIAGEN maxiprep kits (QIAGEN, Valencia, CA), and sequences were confirmed by bidirectional sequencing using the dideoxynucleotide chain-termination method on ABI 3730XL machines at the Dana-Farber/Harvard Cancer Center DNA Resource Core (Boston, MA).
Table 1.
Synthetic oligonucleotides used to perform site-directed mutagenesis
| Name | Sequence (5′–3′) | Size (bp) | Application |
|---|---|---|---|
| R80C | TAT CGC TGC CCT CAC CtG CTA TAA GAA GTT GCG | 33 | Mutagenesis |
| R85H | CCG CTA TAA GAA GTT GCa CAA CCT CAC CAA TCT GC | 35 | Mutagenesis |
All sequences are shown from 5′ to 3′ in the sense orientation. The bold nucleotides represent mutations: R80C c.238c>t; R85H c.254g>a.
Cell culture and transient and stable cell transfection
HEK293 and COS7 cells were grown and maintained in DMEM supplemented with 10% (vol/vol) fetal bovine serum (Omega, Tarzana, CA), 100 U/ml of penicillin, and 100 μg/ml of streptomycin sulfate (Invitrogen) in 5% CO2 humidified air at 37 C. For luciferase assays, HEK293 cells were transiently transfected in 24-well plates with 5 ng of WT or mutant PROKR2 plasmid, 5 ng of early growth response (Egr)1-luciferase (Luc) reporter, and 140 ng of empty vector. Because HEK293 cells detach easily from the cell culture plates, COS7 cells were used to perform inositol phosphate (IP) and binding assays. The COS7 cells were transiently transfected in six-well plates with 500 ng of WT or mutant PROKR2 and 500 ng of empty vector. For Western blot analyses, HEK293 cells stably transfected with WT or mutant PROKR2 were established by selecting cells transfected with 500 ng of PROKR2 plasmid in 10 ml/100-mm dishes in culture media containing 1000 μg/ml of geneticin. The cell lines were then maintained in the selective media for at least 2 wk before use in experiments.
To perform experiments to assess for dominant negative activity, COS7 cells were cotransfected with varying amounts of WT and R80C PROKR2 plasmids as indicated. In a given experiment, the total amount of plasmid transfected was kept constant by the addition of empty vector. In the Western blotting to assess R80C dominant negative effects, V5-His-tagged WT PROKR2 was transfected alone or with untagged WT or R80C PROKR2.
Luciferase assay
A luciferase reporter assay was performed using the murine Egr1 gene promoter, a target of the MAPK signaling cascade (kindly provided by Nelly Pitteloud) (39). The reporter construct used was generated by inserting the Egr1 promoter region (−1023 to −1) into the pGL3-basic luciferase vector (Egr1-Luc) (22). The luciferase assay was performed as previously described (22). Briefly, 24 h after transfection in 24-well plates, HEK293 cells were stimulated with increasing doses of PROK2 (10−10 to 10−7 m) in serum-free medium containing 0.1% BSA. After a 16-h incubation, the cells were lysed with 125 mm Tris-HCl (pH 7.6) and 0.5% Triton. Luciferase activity was measured in the cell lysates. Results were expressed as fold of untreated samples. All assay points were performed in triplicate, and each experiment was repeated at least three times.
IP assay
Total [3H]IP were measured as previously described (40) with slight modifications. Briefly, 48 h after transfection, COS7 cells were inositol starved for 2 h before the addition of 1 ml of the same medium containing 2 μCi of myo-(2-3H) inositol, followed by the addition of 10 mm LiCl 15 min later. After an additional 16-h incubation at 37 C, cells were stimulated with increasing concentrations of PROK2 (2 × 10−11 to 10−7 m) for 1 h. Cells were extracted twice for 30 min on ice with 20 mm formic acid, and lysates were neutralized to pH 7.5 with 7.5 mm HEPES and 150 mm KOH. After centrifuging for 2 min at 14,000 rpm, the supernatants were loaded onto Ag-X8 resin anion exchange columns (Bio-Rad Laboratories, Inc., Hercules, CA), previously equilibrated with 2 ml of 1 m NaOH, 2 ml of 1 m formic acid, and five times 5 ml of ddH2O. The columns were washed with 5 ml of double distilled water, and then 5 ml of 5 mm borax, 60 mm sodium formate, and [3H]IP were extracted with 3 ml of 0.9 m ammonium formate and 0.1 m formic acid. The incorporation of radioactivity in the eluates was measured in a scintillation counter. All assay points were performed in triplicate, and each experiment was repeated at least three times.
Receptor binding assay
COS7 cells were plated in six-well plates and transiently transfected with WT or mutant PROKR2 as indicated. After a 24-h incubation at 37 C, cells were washed with DMEM and 1% BSA and 5 mm HEPES and incubated for 15 min at room temperature with 100,000 cpm 125I-MIT-1 and with increasing concentrations (10−10 to 10−6 m) of unlabeled PROK2. Cells were washed five times with ice-cold PBS and BSA 0.5% and HEPES 10 mm; and lysed with 2 ml of 0.2 m NaOH. The radioactivity was measured in a γ-counter. Each experiment was repeated at least three times.
Western blot analysis and N-glycosylation experiments
HEK293 cells stably transfected with WT or mutant PROKR2 were grown in 100-mm dishes to confluence. Cell surface and total cellular proteins were extracted using the Pierce Cell Surface Protein Isolation kit (Thermo Scientific, Rockford, IL) according to the manufacturer's suggested protocol. Thirty micrograms of soluble proteins per lane were resolved by SDS-PAGE on 10% polyacrylamide gels under reducing conditions at 4 C and at a constant voltage (100 V). Proteins were electrotransferred overnight at 4 C, 30 V onto polyvinylidene fluoride membranes (Millipore Corp., Bedford, MA). Nonspecific binding was blocked with 5% nonfat dry milk in Tris-buffered saline with Tween 20 (TBST) for 1 h at room temperature with shaking. The blots were then incubated with anti-V5 antibody (1:1000) overnight at 4 C with gentle shaking. Blots were subsequently rinsed with TBST and incubated with antimouse secondary antibodies linked to horseradish peroxidase; after rinsing again with TBST, the antibody-antigen complexes were visualized using an enhanced chemiluminescence reagent (PerkinElmer). Molecular mass was approximated using molecular mass markers. Blots were stripped [100 mm β-MeOH, 2% sodium dodecyl sulfate, and 62.5 mm Tris (pH 6.8)] and reprobed with either β-actin antibody (1:10,000) or Na/K ATPase antibody (1:1000) for normalization purposes.
For experiments designed to remove N-glycosylation, protein extracted from HEK293 cells stably expressing WT or mutant PROKR2 were subject to endoglycosidase H (Endo H) digestion. This enzyme cleaves core N-glycans from proteins (41). Twenty micrograms of protein were digested with 2 U of Endo H for 2 h at 37 C.
Computational and statistical analysis
For IP and luciferase assays, the data for each set of experiments were subjected to nonlinear regression analysis, and the EC50 for each study was calculated using Prism 5.0 (GraphPad Software, Inc., San Diego, CA). For binding assays, the dissociation constant (Kd) and maximum binding (Bmax) were calculated based on nonlinear regression of homologous competition-binding analysis using Prism 5.0. Western blot quantification was done using ImageJ (Wayne Rasband; National Institute of Health, Bethesda, MD). Data were analyzed using one-way ANOVA (P < 0.05).
Results
The R80C PROKR2 mutation interferes with PROKR2 signaling
The ability of PROK2 to activate intracellular signaling cascades was evaluated by measuring [3H]IP accumulation and using an Egr1-Luc reporter assay to measure MAPK activation. WT PROKR2 and ICL1 PROKR2 mutants were stimulated with increasing concentrations of the ligand, PROK2. The stimulation of WT PROKR2 generated sigmoidal dose-response curves for both [3H]IP and Egr1-Luc induction (Fig. 1, A and B).
Fig. 1.

Effects of ICL1 PROKR2 mutations on ligand-mediated activation of signal transduction. PROKR2 signaling activity in response to ligand was evaluated by measuring IP accumulation and using an Egr1-Luc reporter assay to measure MAPK activation. The PROK2 dose-response curves of [3H]IP accumulation and Egr1-Luc activity were used to calculate the EC50 for each receptor. Results are expressed as percentage of the maximal stimulation of WT PROKR2 (mean ± se for three independent experiments, each performed in triplicate). A, Activation of Egr1-Luc by WT and ICL1 PROKR2 mutants. HEK293 cells were transiently transfected with WT PROKR2, R80C PROKR2, R85C PROKR2, or R85H PROKR2 together with the Egr1-Luc reporter and PGL3 empty vector. Twenty-four hours after transfection, cells were stimulated with increasing concentrations of PROK2 for 16 h, and cell extracts were assayed for luciferase activity. B, Stimulation of PROK2-mediated [3H]IP production by WT and ICL1 PROKR2 mutants. COS7 cells were transiently transfected with WT PROKR2, R80C PROKR2, R85C PROKR2, or R85H PROKR2. Forty-eight hours after transfection, cells were stimulated with increasing concentrations of PROK2 for 1 h, and [3H]IP were measured. *, P < 0.05 at that specific dose compared with WT.
In luciferase assays measuring induction of Egr1 gene promoter activity, stimulation of R85C and R85H PROKR2 by PROK2 resulted in dose-response curves with a modest but significant 30% reduction in maximal MAPK activation but no significant change in the EC50 for the response to PROK2 (EC50: WT PROKR2, 3.3 × 10−9 m; R85C PROKR2, 3.5 × 10−9 m; R85H PROKR2, 3.0 × 10−9 m; and R80C PROKR2, 1.0 × 10−9 m). R80C PROKR2 showed a marked and significant 70% reduction in activation of the MAPK signaling pathway (Fig. 1A).
In IP assays, R85C and R85H PROKR2 showed similar dose-response curves to WT PROKR2 (Fig. 1B), with EC50 values not statistically different from that of WT (EC50: WT PROKR2, 5.0 × 10−0 m; R85C PROKR2, 1.0 × 10−9 m; R85H PROKR2, 2.3 × 10−9 m; and R80C PROKR2, 1.4 × 10−9 m). Maximal [3H]IP accumulation by PROK2 for R85C and R85H PROKR2 were not different from WT PROKR2. In contrast, stimulation of R80C PROKR2 by PROK2 showed a marked reduction in the maximal [3H]IP accumulation, with an EC50 value not statistically different from that of WT (Fig. 1B). Taken together, the impaired Egr1-Luc and [3H]IP responses to PROK2 for the R80C PROKR2 mutant indicate that this mutation results in severe impairment of PROKR2 signaling activation.
The R80C PROKR2 mutation interferes with PROK2 ligand binding
The effects of the ICL1 PROKR2 mutations on ligand binding were studied using 125I-MIT-1, together with increasing concentrations of unlabeled PROK2 to produce competition binding curves. The displacement curves for R85C and R85H PROKR2 showed a 15% decrease in Bmax compared with WT PROKR2, but this reduction was not statistically significant (Table 2 and Fig. 2). The R80C PROKR2 mutant had an approximately 64% reduction in Bmax compared with WT PROKR2. The R80C, R85C, and R85H PROKR2 mutants had Kd values not statistically different from WT PROKR2 (Fig. 2 and Table 1). These results suggest that R80C PROKR2 cell surface expression is markedly reduced. R85C and R85H PROKR2 have similar ligand affinities as WT PROKR2.
Table 2.
Binding parameters for WT PROKR2 and ICL1 mutants
| PROKR2 | Bmax (cpm) | Log Kd (mol) |
|---|---|---|
| WT | 11,911 ± 1014 | −7.4 ± 0.03 |
| R80C | 4461 ± 463a | −7.3 ± 0.83 |
| R85C | 9898 ± 315 | −7.6 ± 0.41 |
| R85H | 10,454 ± 2113 | −7.4 ± 0.34 |
The amount of cell surface receptor is expressed as Bmax and receptor affinity for the radioligand as Kd. Results are the mean for three independent experiments.
P < 0.05 compared with WT.
Fig. 2.

Displacement binding assay of WT and ICL1 PROKR2 mutants. To measure PROK2 binding by displacement analysis, COS7 cells transfected with either WT, R80C, R85C, or R85H PROKR2 were incubated with 125I-MIT-1 in the presence of increasing concentrations (10−10 to 10−6 m) of unlabeled PROK2 for 15 min, after which cells were washed and radioactivity assayed in cell lysates. The PROK2 displacement curves were used to calculate Bmax and Kd for each PROKR2 receptor. Results are expressed as percentage of the WT PROKR2 maximal binding. Each point is the mean ± se of three independent experiments.
The role of Arg80 in PROKR2 expression and glycosylation
To further examine PROKR2 expression, whole-cell and plasma membrane protein fractions were isolated and analyzed by Western blot analysis, using stably transfected HEK293 cells expressing V5-tagged WT PROKR2 or ICL1 mutants (Fig. 3, A and B). Western blot analysis showed a marked reduction of R80C PROKR2 expression in whole-cell protein preparations, to 25% of WT PROKR2 expression levels, with a modest but significant reduction of R85H PROKR2 expression of approximately 30% and no difference in expression of R85C PROKR2 compared with the WT receptor (Fig. 3, upper panels). In agreement with the results of the binding assays, Western blot analysis confirmed a reduction of R80C PROKR2 expression of approximately 60% in membrane preparations, whereas R85H and R85C PROKR2 were expressed at levels similar to WT PROKR2 in membrane fractions (Fig. 3, lower panels).
Fig. 3.

Analysis of WT and ICL1 PROKR2 mutant protein expression. A, Representative Western blot analysis showing total cellular (upper panel) and membrane (lower panel) expression of WT and ICL1 mutant V5-tagged PROKR2 in stable transfectants of HEK293 cells. B, Quantification of the relative total cellular (upper panel) and membrane (lower panel) expression of the PROKR2 mutants compared with WT PROKR2, using data pooled from at least three independent experiments. The intensity of the PROKR2 band (detected using anti-V5 antibody) was normalized to β-actin in whole-cell lysates and to Na+/K+ATPase in the membrane fraction. Both the 45- and 30-kDa bands were included in the quantification. HEK, Untransfected HEK293 cells. Bars represent mean ± sem. *, P < 0.05 compared with WT.
WT PROKR2 was visualized by Western blot analysis as a 45-kDa band, consistent with Marsango et al. (42). A similar band was observed for all of the ICL1 mutants studied. Interestingly, an additional lower molecular mass band was observed for R80C PROKR2, at 30 kDa. This 30-kDa band was barely visualized in the other lanes, corresponding to WT PROKR2, R85C PROKR2, and R85H PROKR2. We hypothesized that this lower molecular mass band observed for R80C PROKR2 might represent immature, incompletely glycosylated receptor. The absence of the 30-kDa band in membrane fraction proteins supports this hypothesis and suggests that only mature glycosylated receptor is expressed on the cell surface (Fig. 3A). To test this hypothesis further, proteins extracted from stably transfected HEK293 cells expressing V5-tagged WT or R80C PROKR2 were subjected to enzymatic digestion using Endo H to remove N-linked glycans (Fig. 4). In lanes containing untreated proteins, WT PROKR2 was resolved almost exclusively as a single band at 45 kDa, with an additional faint band seen at 30 kDa. After treatment of protein extracts from cells expressing WT PROKR2 with Endo H, the 30-kDa band increased in intensity, whereas the 45-kDa band decreased in intensity (Fig. 4, lanes 2 and 3). These results indicate that the 45-kDa band represents mature, N-glycosylated PROKR2. The presence of a greater proportion of the immature, deglycosylated 30-kDa band in the protein extracts from cells expressing the R80C PROKR2 mutant indicates that substitution of the Arg80 in ICL1 results in a decrease in receptor glycosylation (Fig. 4, lanes 4 and 5).
Fig. 4.
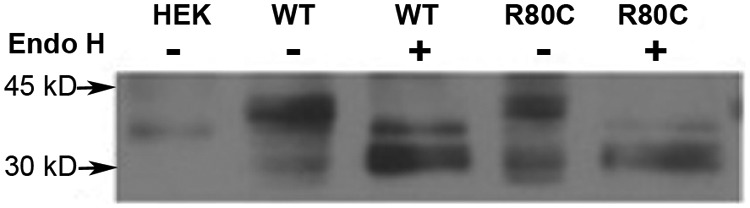
Evaluation of PROKR2 glycosylation. Evaluation of N-glycosylation of WT and R80C PROKR2 by means of Endo H treatment. HEK293 cells stably expressing WT and R80C V5-tagged PROKR2 were used to prepare whole-cell protein extracts. These samples were either left untreated (−) or treated with Endo H (+) and then analyzed by reducing SDS-PAGE and Western blot analysis using an anti-V5 antibody; 45-kDa (glycosylated) and 30-kDa (deglycosylated) bands were visualized as marked. HEK, Untransfected HEK293 cells.
R80C PROKR2 exerts a dominant negative effect on WT PROKR2
The R80C PROKR2 mutation was identified previously in the heterozygous state in a female Brazilian patient with Kallmann syndrome (20). The great majority of PROKR2 mutations identified in patients with gonadotropin deficiency are heterozygous mutations (27). It has been reported previously that heterozygous PROKR2 mutations do not exert dominant negative effects on the WT receptor (24). However, on the other hand, it has been demonstrated previously that PROKR2 can form dimers (42). We hypothesized that the R80C mutant PROKR2 might interfere with the function or processing of the WT PROKR2, contributing to the pathogenesis of the disease phenotype in the patient with the heterozygous mutation. To determine whether the R80C PROKR2 mutant can exert a dominant negative effect on WT PROKR2 signaling, we performed transient transfections of COS7 cells with both WT and R80C PROKR2 plasmids in varying amounts and ratios, followed by stimulation of the transfected cells with 10−8 m PROK2 and measurement of [3H]IP production (Fig. 5A). Cotransfection of equal amounts of 10, 25, or 100 ng of WT and R80C PROKR2 plasmids in combination significantly reduced the PROK2-stimulated [3H]IP response by 35 ± 6, 33 ± 4, and 59 ± 3%, respectively, compared with that of WT PROKR2 alone (Fig. 5A). When the cotransfection ratio of R80C:WT PROKR2 plasmids was 4:1 (WT 25 ng/R80C 100 ng), the PROK2-stimulated [3H]IP response was even further reduced by 70 ± 3% when compared with that of 25 ng of WT PROKR2 alone (Fig. 5A).
Fig. 5.

Evaluation for dominant negative effects of R80C PROKR2 on the WT receptor. A, Measurement of [3H]IP accumulation in COS7 cells after cotransfection with varying amounts of WT and R80C PROKR2 plasmids as indicated and stimulation with 10−8 m PROK2. Results are expressed as percentage of the maximal stimulation of WT PROKR2 (100 ng) alone. Bars represent mean ± sem for three independent experiments, each performed in triplicate. B, Displacement binding assay in COS7 cells transfected with either WT PROKR2 alone or cotransfected with WT and R80C PROKR2 in varying amounts as indicated. Cells were incubated with 125I-MIT-1 with or without 10−7 m of unlabeled PROK2 for 15 min, after which cells were washed and radioactivity assayed in cell lysates. Specific binding activity was calculated and expressed as percentage of binding to WT PROKR2 (500 ng) alone. Bars represent mean ± sem for three independent experiments, each performed in triplicate. *, P < 0.05 compared with the same amount of WT PROKR2 alone; (−) untreated and (+) treated with 10−8 m PROK2; 10, 25, 100, 400, and 500 ng represent the amount of each PROKR2 plasmid transfected.
To begin to elucidate the mechanism by which the dominant negative effect of R80C on WT PROKR2 occurs, we next performed ligand binding studies, cotransfecting both WT and R80C PROKR2 plasmids in varying amounts and ratios. Cotransfection of 500 ng of both WT and R80C PROKR2 plasmids reduced specific binding activity by 34% when compared with 500 ng of WT PROKR2 alone, a statistically significant difference. Cotransfection of 100 ng of both WT and R80C PROKR2 plasmids decreased specific binding activity by approximately 20% compared with 100 ng of WT PROKR2 plasmid alone, which did not reach statistical significance. However, when the cotransfection ratio of R80C:WT plasmids was 4:1 (WT 100 ng/R80C 400 ng), the specific binding activity was significantly reduced, by 36% (Fig. 5B).
To determine whether the reduced ligand binding of WT PROKR2 in the presence of cotransfected R80C mutant PROKR2 was the result of inhibitory effects of the mutant receptor on WT PROKR2 expression, we performed Western blot analysis on whole-cell lysate and membrane proteins of cells cotransfected with plasmids encoding WT and mutant receptor (Fig. 6). The WT receptor carried a C-terminal V5-tag, whereas the R80C mutant did not, so that Western blot analysis using an anti-V5 antibody would detect only WT PROKR2. Cotransfection of 100 ng of R80C PROKR2 plasmid with 100 ng of WT-V5 PROKR2 plasmid reduced WT PROKR2 protein levels in whole-cell lysates and in plasma membrane as determined by Western blotting by 32 and 55%, respectively, compared with transfection with 100 ng of WT PROKR2 plasmid alone. When the cotransfection ratio of R80C:WT PROKR2 was 4:1 (WT-V5 100 ng/R80C 400 ng), the WT PROKR2 expression was further reduced, to 20% of WT alone in both whole-cell and membrane lysate preparations (Fig. 6, A and B). To ensure that the decrease in WT PROKR2 expression was not a nonspecific effect of the cotransfected plasmid, we also performed Western blot analysis on whole-cell protein lysate of HEK293 cells cotransfected with plasmid encoding 100 ng of WT PROKR2 tagged with V5 (WT-V5 100 ng) and untagged WT PROKR2 plasmid (WT 100 ng or WT 400 ng). No reduction in WT-V5 PROKR2 expression was visualized, even when 400 ng of untagged WT receptor plasmid was cotransfected (Fig. 6C).
Fig. 6.
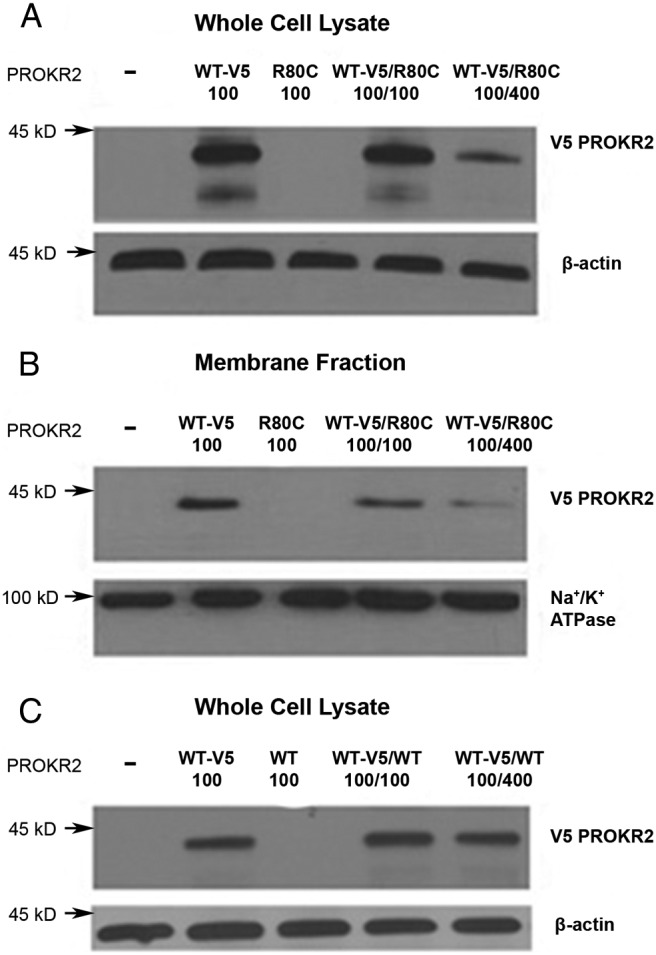
Evaluation for potential dominant negative effects of R80C PROKR2 on WT PROKR2 expression. Representative Western blot analysis of whole-cell lysates (A) and membrane fraction (B) from HEK 293 cells transiently transfected with either 100 ng of V5-tagged WT PROKR2 plasmid alone (WT-V5 100), 100 ng of untagged R80C PROKR2 plasmid alone (R80C 100), or 100 ng of V5-tagged WT PROKR2 cotransfected with 100 ng (WT-V5/R80C 100/100) or 400 ng (WT-V5/R80C 100/400) of untagged R80C PROKR2. C, Representative Western blot analysis of whole-cell lysates from HEK 293 cells transiently transfected with either 100 ng of V5-tagged WT PROKR2 plasmid alone (WT-V5 100), 100 ng of untagged WT PROKR2 plasmid alone (WT 100), or 100 ng of V5-tagged WT PROKR2 cotransfected with 100 ng (WT-V5/WT 100/100) or 400 ng (WT-V5/WT 100/400) of untagged WT PROKR2. The intensity of the PROKR2 band (detected using anti-V5 antibody) was normalized to β-actin in whole-cell lysates and to Na+/K+ATPase in the membrane fractions. −, Untransfected HEK 293 cells.
Taken together, these results indicate that the ICL1 mutant R80C PROKR2 interferes with ligand binding and ligand-activated signal transduction by the WT receptor through inhibitory effects of the R80C mutant on WT PROKR2 expression in vitro. These dominant negative effects were most evident when the mutant receptor was transfected in excess of the WT PROKR2.
Discussion
In recent years, several PROKR2 mutations have been described in patients with GnRH deficiency (28, 43). More recently, variants in PROKR2 have also been identified in patients with Hirschsprung's disease, suggesting the association of PROKR2 with the proliferation and differentiation of the enteric nervous system in humans (44). PROKR2 variants in patients with combined pituitary hormone deficiency and septo-optic dysplasia were also recently described, but the exact role of the PROK system in these diseases is still to be elucidated (45). In vitro studies have demonstrated that different mutations affect PROKR2 function to varying degrees (22, 24). It has been shown that mutations in extracellular loops abolish ligand binding, that mutations in transmembrane domains potentially affect cell surface receptor expression, and that mutations in ICL2 or ICL3 affect G protein interaction and/or activation (22, 24, 46).
There is very little known about the role of ICL1 in GPCR, much less in PROKR2 (35). The present study was undertaken to determine whether mutations in the ICL1 of PROKR2 interfere with the function of the receptor, thereby causing the phenotype of hypogonadotropic hypogonadism. Furthermore, clarification of the mechanisms by which these mutations affect receptor function will lead to a better understanding of the role of ICL1 in PROKR2 biology, and perhaps in GPCR in general. We studied three mutations identified in ICL1 of PROKR2: the R80C mutation, which we previously reported, and the R85C and R85H mutations, which had been reported by other groups when we first began this study (20, 22, 24). Subsequent to our studies, two additional mutations were reported in residue 85 in the ICL1 of PROKR2, R85G and R85L, both of which were identified in patients with hypogonadotropic hypogonadism (27). Data from a recent study suggest that the mutant R85G PROKR2 has a defect in protein folding and expression (45). The mutant R85L PROKR2 has not yet been tested functionally. The alignment of the ICL1 of PROKR2 across species reveals that the ICL1 is highly conserved, including the arginines at positions 80 and 85, among primates, rodents, fish, birds, and other eukaryotes, suggesting an important role for ICL1 in general and for these arginines in particular in PROKR2 function (Fig. 7). The data we have presented here indicate the importance of Arg80 for PROKR2 expression and consequently its intracellular trafficking to plasma membrane.
Fig. 7.
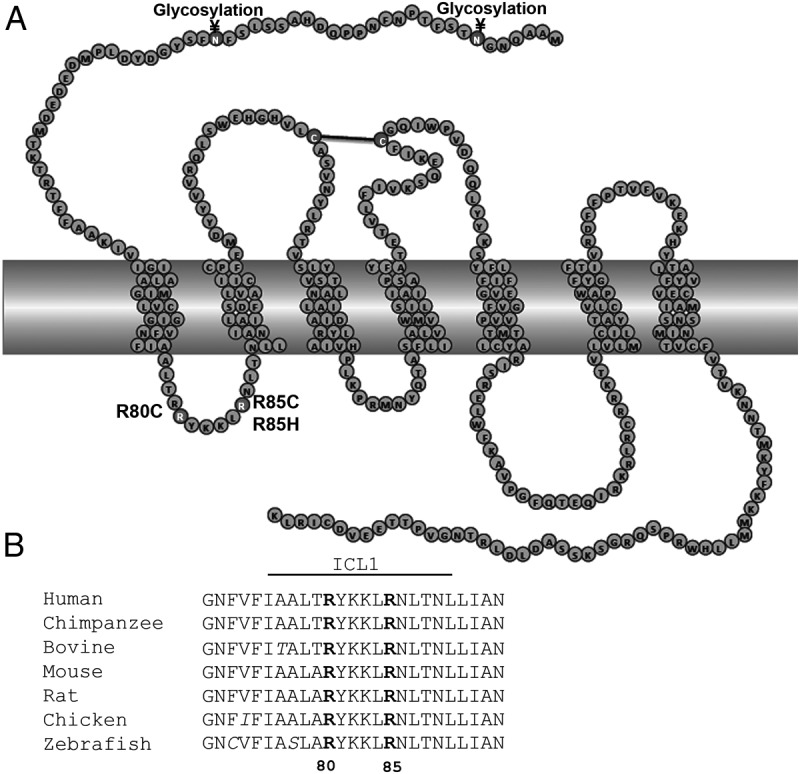
PROKR2 ICL1 amino acid sequence alignment among species. A, Schematic representation of mutations in the ICL1 of PROKR2. Residues in dark gray represent the ICL1 mutations. Residues involved in disulfide bond formation (black line) and in glycosylation are represented in gray. B, Amino acid sequence alignment among species, showing that arginines in position 80 and 85 are highly conserved among different species. Sequences were taken from the GPCR database (www.gpcr.org), and the alignment was produced in ClustalW.
The R80C PROKR2 mutation resulted in a marked decrease in activation of intracellular signal transduction in response to PROK2 as well as a marked decrease in PROK2 binding capacity; the dose-response and binding displacement curves suggested a reduction in the number of receptors in the plasma membrane. Western blot analysis confirmed a decrease in R80C PROKR2 expression in the membrane fraction. Moreover, expression of the mutant receptor was reduced in whole-cell lysates as well. The presence of a lower molecular mass band (∼30 kDa) in protein lysates from cells transfected with R80C PROKR2 was particularly notable (Fig. 3A). This band was nearly absent in protein extracts from cells expressing WT, R85C or R85H PROKR2. It has been demonstrated previously that immature GPCR are manifest as lower molecular mass bands in Western blot analysis, representing nonglycosylated receptors (47, 48). Indeed, Endo H digestion of the protein extracts confirmed that the 30-kDa band in the lane containing R80C PROKR2 represents nonglycosylated receptors (Fig. 4A).
N-glycosylation is the most common posttranslational modification of GPCR and is necessary for functions ranging from agonist binding to GPCR folding, maturation, stability, and internalization. N-glycosylation of the human thromboxane A2 receptor has been reported to affect intracellular trafficking (49). However, N-glycosylation of the porcine muscarinic acetylcholine receptor subtype 2 is not required for cell surface expression (50). Melanocortin 2 receptor N-glycosylation is important for intracellular trafficking only in the absence of melanocortin 2 receptor accessory protein (48). We observed a decrease in R80C PROKR2 expression in membrane preparations and a similar decrease in receptor expression in whole-cell lysates. Thus, there is no evidence of a specific defect in targeting of the receptor to the cell surface. Substitution of the Arg80 in PROKR2 ICL1 may lead to a conformational change that impairs PROKR2 stability and glycosylation resulting in reduced whole-cell expression levels. The loss of the basic arginine residue may also impair insertion of the mature receptor into the plasma membrane.
Most GPCR possess one or more consensus sequences (Asn-X-Ser/Thr) for N-glycosylation in their extracellular domains (51). The N-terminal domain of PROKR2 contains two potential glycosylation sites, Asn7 and Asn21 (www.uniprot.org) (Fig. 7A). In a recent study, it was demonstrated that PROKR2 lacking the N-terminal domain was expressed in membranes from yeast cells (42). However, the N-linked glycans from yeast differ significantly from those of mammalian cells, and this may explain the differences observed (52). Mammalian cells and yeast share the initial biosynthetic pathway for the synthesis of N-glycans, but the pathways diverge significantly as the proteins reach the Golgi apparatus (52).
Substitution of Arg85 with either cysteine or histidine resulted in mutant receptors with a 30% reduction of maximal Egr1-Luc activation, reflecting activation of the MAPK pathway, whereas induction of IP accumulation was similar to WT PROKR2. These discordant effects on various intracellular signaling pathways have been demonstrated previously for other PROKR2 mutants, such as R248Q, V331M, and R357W, demonstrating complexity in the activation of PROKR2 signaling pathways (22). The possibility that the longer treatment duration used in the luciferase assay (16 h) compared with the IP assay (1 h) may have contributed to the discordant effects observed also needs to be considered; the Arg85 mutations may have caused changes in PROKR2 trafficking that were manifest only after longer incubation periods. Bands corresponding to immature nonglycosylated receptors were not detected by Western blot analysis for R85C and R85H PROKR2. Previous observations strongly suggest that the accessibility of the Asn-X-Ser/Thr sequence to oligosaccharide transferase is the predominant structural feature of a protein that determines whether or not it is glycosylated (53). Our results suggest that the substitution of the basic Arg80 to cysteine leads to a conformational change that impairs PROKR2 glycosylation and its cell surface expression. The substitution of the arginine at position 85 to cysteine did not result in immature bands nor in a reduction of PROKR2 plasma membrane expression, whereas the substitution at the same position to a histidine resulted in a small reduction in whole-cell and membrane expression (Fig. 3), and to a glycine also reduced PROKR2 at the cell surface (45), indicating that not only the amino acid position is important in determining the effects on conformational change but also the nature of the amino acid substitution.
Previous studies of Arg85 PROKR2 mutants showed similar results in signal transduction experiments (22, 24, 54). However, in Western blot analysis, the results are conflicting for R85H PROKR2: one study did not show a difference in R85H PROKR2 expression compared with WT expression, whereas another study showed a decrease by approximately 60%, and our study showed a 30% reduction in R85H expression (Fig. 3) (24, 54). The use of different detergents to extract proteins from the plasma membrane may explain the differences observed in these studies. Additionally, differences were observed in the molecular mass of PROKR2 in Western blot analyses among the different studies. In the study that showed similar levels of expression of R85H PROKR2 to WT receptor (24), the PROKR2 molecular mass was approximately 85 kDa, which may correspond to PROKR2 dimers. In the study that found a 60% reduction in R85H expression, the molecular mass was not shown (54).
Marsango et al. (42) showed that PROKR2 can form dimers. The ability of PROKR2 to form dimers suggests that dimerization of a mutant PROKR2 receptor with the WT receptor might interfere with the function of WT PROKR2, contributing to the pathogenesis of the disease phenotype in patients with heterozygous mutations through dominant negative effects. Hence, we tested the loss-of-function PROKR2 mutation, R80C, to assess whether this mutant receptor exerts a dominant negative effect on WT PROKR2. Our functional cotransfection studies of WT and R80C PROKR2 indicated that the R80C mutant exerts a dominant negative effect on the WT receptor (Figs. 5 and 6). The decreased expression levels of WT PROKR2 in cotransfected cells suggest that the R80C PROKR2 mutant may interact with the WT receptor, potentially through dimerization, thereby targeting the dimers to degradation (Fig. 6). These findings are different from those of a previous study, in which PROKR2 mutants did not impair WT PROKR2 function (24). The differences between these studies may be due to diverse effects of different PROKR2 mutants, or alternatively may reflect differences in PROKR2 expression levels between the studies. High levels of expression of WT PROKR2 can overcome and obscure dominant negative effects of mutant PROKR2. In our study, transfection of reduced amounts of the PROKR2 expression vectors, such that the receptor was not overexpressed, was required to demonstrate a dominant negative effect. The results of our in vitro studies suggest that the presence of R80C PROKR2 in the heterozygous state may contribute to hypogonadotropic hypogonadism through a dominant negative mechanism. However, the identification of asymptomatic first-degree relatives carrying the R80C PROKR2 mutation argues against this supposition. The extent of the dominant negative effect was dependent on the relative proportion of WT to R80C PROKR2 in vitro; the presence or extent of the dominant negative effect in vivo may likewise depend on the relative amounts of WT and R80C mutant receptors. The R80C mutation leads to a conformational change in PROKR2 that targets the receptor to degradation. The manifestation of the hypogonadotropic hypogonadism phenotype in individuals harboring the R80C PROKR2 mutation may thus depend on additional factors, such as the activity of PROKR2 degradation pathways, differences in the relative expression of the two copies of the PROKR2 gene, and polymorphisms in regulatory genes. Although differential allelic expression of PROKR2 in humans is not well known, especially in the central nervous system, it is a prevalent phenomenon affecting the expression of 20% of human genes (55) and may play an important role in the hypogonadotropic hypogonadism phenotype. The same argument can be applied to R85 mutations; these mutations do not seem to cause the disease alone, but they may contribute to the disease phenotype in association with other factors, such as polymorphisms, unidentified mutations in additional genes (i.e. digenic or multigenic disorders) (26, 56), or epigenetic and environmental effects. Nevertheless, the precise role of heterozygous PROKR2 mutations in hypogonadotropic hypogonadism remains to be elucidated (57).
In conclusion, we have shown that Arg80 in the ICL1 of PROKR2 is important for receptor expression and exerts a dominant negative effect on the WT receptor.
Acknowledgments
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development/National Institutes of Health through Cooperative Agreement U54 HD28138, as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research (to U.B.K.), and Grant F05 HD072773 (to A.P.A.); Fundação de Amparo à Pesquisa do Estado de São Paulo Grants 06/56753-3R (to A.P.A.) and 05/04726-0 (to A.P.A. and A.C.L.); and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior Grant 3806/11-1 (to A.P.A.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Bmax
- Maximum binding
- Egr
- early growth response
- Endo H
- endoglycosidase H
- GPCR
- G protein-coupled receptor
- ICL1
- first intracellular loop
- IP
- inositol phosphate
- Kd
- dissociation constant
- Luc
- luciferase
- MIT-1
- mamba intestinal toxin-1
- PROK
- prokineticin
- PROKR
- prokineticin receptor
- TBST
- Tris-buffered saline with Tween 20
- WT
- wild type.
References
- 1. Ngan ES, Tam PK. 2008. Prokineticin-signaling pathway. Int J Biochem Cell Biol 40:1679–1684 [DOI] [PubMed] [Google Scholar]
- 2. Lin DC, Bullock CM, Ehlert FJ, Chen JL, Tian H, Zhou QY. 2002. Identification and molecular characterization of two closely related G protein-coupled receptors activated by prokineticins/endocrine gland vascular endothelial growth factor. J Biol Chem 277:19276–19280 [DOI] [PubMed] [Google Scholar]
- 3. Soga T, Matsumoto S, Oda T, Saito T, Hiyama H, Takasaki J, Kamohara M, Ohishi T, Matsushime H, Furuichi K. 2002. Molecular cloning and characterization of prokineticin receptors. Biochim Biophys Acta 1579:173–179 [DOI] [PubMed] [Google Scholar]
- 4. Masuda Y, Takatsu Y, Terao Y, Kumano S, Ishibashi Y, Suenaga M, Abe M, Fukusumi S, Watanabe T, Shintani Y, Yamada T, Hinuma S, Inatomi N, Ohtaki T, Onda H, Fujino M. 2002. Isolation and identification of EG-VEGF/prokineticins as cognate ligands for two orphan G-protein-coupled receptors. Biochem Biophys Res Commun 293:396–402 [DOI] [PubMed] [Google Scholar]
- 5. Chen J, Kuei C, Sutton S, Wilson S, Yu J, Kamme F, Mazur C, Lovenberg T, Liu C. 2005. Identification and pharmacological characterization of prokineticin 2β as a selective ligand for prokineticin receptor 1. Mol Pharmacol 67:2070–2076 [DOI] [PubMed] [Google Scholar]
- 6. Lin R, LeCouter J, Kowalski J, Ferrara N. 2002. Characterization of endocrine gland-derived vascular endothelial growth factor signaling in adrenal cortex capillary endothelial cells. J Biol Chem 277:8724–8729 [DOI] [PubMed] [Google Scholar]
- 7. Cheng MY, Bullock CM, Li C, Lee AG, Bermak JC, Belluzzi J, Weaver DR, Leslie FM, Zhou QY. 2002. Prokineticin 2 transmits the behavioural circadian rhythm of the suprachiasmatic nucleus. Nature 417:405–410 [DOI] [PubMed] [Google Scholar]
- 8. Hu WP, Li JD, Zhang C, Boehmer L, Siegel JM, Zhou QY. 2007. Altered circadian and homeostatic sleep regulation in prokineticin 2-deficient mice. Sleep 30:247–256 [PMC free article] [PubMed] [Google Scholar]
- 9. LeCouter J, Lin R, Tejada M, Frantz G, Peale F, Hillan KJ, Ferrara N. 2003. The endocrine-gland-derived VEGF homologue Bv8 promotes angiogenesis in the testis: localization of Bv8 receptors to endothelial cells. Proc Natl Acad Sci USA 100:2685–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. LeCouter J, Zlot C, Tejada M, Peale F, Ferrara N. 2004. Bv8 and endocrine gland-derived vascular endothelial growth factor stimulate hematopoiesis and hematopoietic cell mobilization. Proc Natl Acad Sci USA 101:16813–16818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li JD, Hu WP, Boehmer L, Cheng MY, Lee AG, Jilek A, Siegel JM, Zhou QY. 2006. Attenuated circadian rhythms in mice lacking the prokineticin 2 gene. J Neurosci 26:11615–11623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ng KL, Li JD, Cheng MY, Leslie FM, Lee AG, Zhou QY. 2005. Dependence of olfactory bulb neurogenesis on prokineticin 2 signaling. Science 308:1923–1927 [DOI] [PubMed] [Google Scholar]
- 13. Prosser HM, Bradley A, Chesham JE, Ebling FJ, Hastings MH, Maywood ES. 2007. Prokineticin receptor 2 (Prokr2) is essential for the regulation of circadian behavior by the suprachiasmatic nuclei. Proc Natl Acad Sci USA 104:648–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goi T, Fujioka M, Satoh Y, Tabata S, Koneri K, Nagano H, Hirono Y, Katayama K, Hirose K, Yamaguchi A. 2004. Angiogenesis and tumor proliferation/metastasis of human colorectal cancer cell line SW620 transfected with endocrine glands-derived-vascular endothelial growth factor, as a new angiogenic factor. Cancer Res 64:1906–1910 [DOI] [PubMed] [Google Scholar]
- 15. Monnier J, Piquet-Pellorce C, Feige JJ, Musso O, Clement B, Turlin B, Theret N, Samson M. 2008. Prokineticin 2/Bv8 is expressed in Kupffer cells in liver and is down regulated in human hepatocellular carcinoma. World J Gastroenterol 14:1182–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, Blanchard D, Bais C, Peale FV, van Bruggen N, Ho C, Ross J, Tan M, Carano RA, Meng YG, Ferrara N. 2007. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 450:825–831 [DOI] [PubMed] [Google Scholar]
- 17. Cheng MY, Leslie FM, Zhou QY. 2006. Expression of prokineticins and their receptors in the adult mouse brain. J Comp Neurol 498:796–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matsumoto S, Yamazaki C, Masumoto KH, Nagano M, Naito M, Soga T, Hiyama H, Matsumoto M, Takasaki J, Kamohara M, Matsuo A, Ishii H, Kobori M, Katoh M, Matsushime H, Furuichi K, Shigeyoshi Y. 2006. Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc Natl Acad Sci USA 103:4140–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prosser HM, Bradley A, Caldwell MA. 2007. Olfactory bulb hypoplasia in Prokr2 null mice stems from defective neuronal progenitor migration and differentiation. Eur J Neurosci 26:3339–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abreu AP, Trarbach EB, de Castro M, Frade Costa EM, Versiani B, Matias Baptista MT, Garmes HM, Mendonca BB, Latronico AC. 2008. Loss-of-function mutations in the genes encoding prokineticin-2 or prokineticin receptor-2 cause autosomal recessive Kallmann syndrome. J Clin Endocrinol Metab 93:4113–4118 [DOI] [PubMed] [Google Scholar]
- 21. Canto P, Munguía P, Söderlund D, Castro JJ, Méndez JP. 2009. Genetic analysis in patients with Kallmann syndrome: coexistence of mutations in prokineticin receptor 2 and KAL1. J Androl 30:41–45 [DOI] [PubMed] [Google Scholar]
- 22. Cole LW, Sidis Y, Zhang C, Quinton R, Plummer L, Pignatelli D, Hughes VA, Dwyer AA, Raivio T, Hayes FJ, Seminara SB, Huot C, Alos N, Speiser P, Takeshita A, Van Vliet G, Pearce S, Crowley WF, Jr, Zhou QY, Pitteloud N. 2008. Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin-releasing hormone deficiency: molecular genetics and clinical spectrum. J Clin Endocrinol Metab 93:3551–3559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dodé C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, Kottler ML, Lespinasse J, Lienhardt-Roussie A, Mathieu M, Moerman A, Morgan G, Murat A, Toublanc JE, Wolczynski S, Delpech M, Petit C, Young J, Hardelin JP. 2006. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet 2:e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Monnier C, Dodé C, Fabre L, Teixeira L, Labesse G, Pin JP, Hardelin JP, Rondard P. 2009. PROKR2 missense mutations associated with Kallmann syndrome impair receptor signalling activity. Hum Mol Genet 18:75–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sinisi AA, Asci R, Bellastella G, Maione L, Esposito D, Elefante A, De Bellis A, Bellastella A, Iolascon A. 2008. Homozygous mutation in the prokineticin-receptor2 gene (Val274Asp) presenting as reversible Kallmann syndrome and persistent oligozoospermia: case report. Hum Reprod 23:2380–2384 [DOI] [PubMed] [Google Scholar]
- 26. Sykiotis GP, Plummer L, Hughes VA, Au M, Durrani S, Nayak-Young S, Dwyer AA, Quinton R, Hall JE, Gusella JF, Seminara SB, Crowley WF, Jr, Pitteloud N. 2010. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci USA 107:15140–15144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sarfati J, Guiochon-Mantel A, Rondard P, Arnulf I, Garcia-Piñero A, Wolczynski S, Brailly-Tabard S, Bidet M, Ramos-Arroyo M, Mathieu M, Lienhardt-Roussie A, Morgan G, Turki Z, Bremont C, Lespinasse J, Du Boullay H, Chabbert-Buffet N, Jacquemont S, Reach G, De Talence N, Tonella P, Conrad B, Despert F, Delobel B, Brue T, Bouvattier C, Cabrol S, Pugeat M, Murat A, Bouchard P, Hardelin JP, Dodé C, Young J. 2010. A comparative phenotypic study of Kallmann syndrome patients carrying monoallelic and biallelic mutations in the prokineticin 2 or prokineticin receptor 2 genes. J Clin Endocrinol Metab 95:659–669 [DOI] [PubMed] [Google Scholar]
- 28. Abreu AP, Kaiser UB, Latronico AC. 2010. The role of prokineticins in the pathogenesis of hypogonadotropic hypogonadism. Neuroendocrinology 91:283–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oldham WM, Hamm HE. 2008. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol 9:60–71 [DOI] [PubMed] [Google Scholar]
- 30. Wess J. 1997. G-protein-coupled receptors: molecular mechanisms involved in receptor activation and selectivity of G-protein recognition. FASEB J 11:346–354 [PubMed] [Google Scholar]
- 31. Conn PM, Janovick JA. 2009. Trafficking and quality control of the gonadotropin releasing hormone receptor in health and disease. Mol Cell Endocrinol 299:137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Conn PM, Ulloa-Aguirre A, Ito J, Janovick JA. 2007. G protein-coupled receptor trafficking in health and disease: lessons learned to prepare for therapeutic mutant rescue in vivo. Pharmacol Rev 59:225–250 [DOI] [PubMed] [Google Scholar]
- 33. Duvernay MT, Dong C, Zhang X, Zhou F, Nichols CD, Wu G. 2009. Anterograde trafficking of G protein-coupled receptors: function of the C-terminal F(X)6LL motif in export from the endoplasmic reticulum. Mol Pharmacol 75:751–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wess J. 1998. Molecular basis of receptor/G-protein-coupling selectivity. Pharmacol Ther 80:231–264 [DOI] [PubMed] [Google Scholar]
- 35. Kleinau G, Jaeschke H, Worth CL, Mueller S, Gonzalez J, Paschke R, Krause G. 2010. Principles and determinants of G-protein coupling by the rhodopsin-like thyrotropin receptor. PLoS One 5:e9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu V, Yang M, McRoberts JA, Ren J, Seensalu R, Zeng N, Dagrag M, Birnbaumer M, Walsh JH. 1997. First intracellular loop of the human cholecystokinin-A receptor is essential for cyclic AMP signaling in transfected HEK-293 cells. J Biol Chem 272:9037–9042 [DOI] [PubMed] [Google Scholar]
- 37. Arora KK, Krsmanovic LZ, Mores N, O'Farrell H, Catt KJ. 1998. Mediation of cyclic AMP signaling by the first intracellular loop of the gonadotropin-releasing hormone receptor. J Biol Chem 273:25581–25586 [DOI] [PubMed] [Google Scholar]
- 38. Duvernay MT, Dong C, Zhang X, Robitaille M, Hébert TE, Wu G. 2009. A single conserved leucine residue on the first intracellular loop regulates ER export of G protein-coupled receptors. Traffic 10:552–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schwachtgen JL, Houston P, Campbell C, Sukhatme V, Braddock M. 1998. Fluid shear stress activation of egr-1 transcription in cultured human endothelial and epithelial cells is mediated via the extracellular signal-related kinase 1/2 mitogen-activated protein kinase pathway. J Clin Invest 101:2540–2549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bedecarrats GY, Linher KD, Kaiser UB. 2003. Two common naturally occurring mutations in the human gonadotropin-releasing hormone (GnRH) receptor have differential effects on gonadotropin gene expression and on GnRH-mediated signal transduction. J Clin Endocrinol Metab 88:834–843 [DOI] [PubMed] [Google Scholar]
- 41. Maley F, Trimble RB, Tarentino AL, Plummer TH., Jr 1989. Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal Biochem 180:195–204 [DOI] [PubMed] [Google Scholar]
- 42. Marsango S, Bonaccorsi di Patti MC, Barra D, Miele R. 2011. Evidence that prokineticin receptor 2 exists as a dimer in vivo. Cell Mol Life Sci 68:2919–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martin C, Balasubramanian R, Dwyer AA, Au MG, Sidis Y, Kaiser UB, Seminara SB, Pitteloud N, Zhou QY, Crowley WF., Jr 2011. The role of the Prokineticin 2 pathway in human reproduction: evidence from the study of human and murine gene mutations. Endocr Rev 32:225–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ruiz-Ferrer M, Torroglosa A, Núñez-Torres R, de Agustín JC, Antiñolo G, Borrego S. 2011. Expression of PROKR1 and PROKR2 in human enteric neural precursor cells and identification of sequence variants suggest a role in HSCR. PLoS One 6:e23475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Raivio T, Avbelj M, McCabe MJ, Romero CJ, Dwyer AA, Tommiska J, Sykiotis GP, Gregory LC, Diaczok D, Tziaferi V, Elting MW, Padidela R, Plummer L, Martin C, Feng B, Zhang C, Zhou QY, Chen H, Mohammadi M, Quinton R, Sidis Y, Radovick S, Dattani MT, Pitteloud N. 2012. Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab 97:E694–E699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Peng Z, Tang Y, Luo H, Jiang F, Yang J, Sun L, Li JD. 2011. Disease-causing mutation in PKR2 reveals a critical role of positive charges in the second intracellular loop for G-protein coupling and receptor trafficking. J Biol Chem 286:16615–16622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Clouser CL, Menon KM. 2005. N-linked glycosylation facilitates processing and cell surface expression of rat luteinizing hormone receptor. Mol Cell Endocrinol 235:11–19 [DOI] [PubMed] [Google Scholar]
- 48. Roy S, Perron B, Gallo-Payet N. 2010. Role of asparagine-linked glycosylation in cell surface expression and function of the human adrenocorticotropin receptor (melanocortin 2 receptor) in 293/FRT cells. Endocrinology 151:660–670 [DOI] [PubMed] [Google Scholar]
- 49. Kelley LP, Kinsella BT. 2003. The role of N-linked glycosylation in determining the surface expression, G protein interaction and effector coupling of the alpha (α) isoform of the human thromboxane A(2) receptor. Biochim Biophys Acta 1621:192–203 [DOI] [PubMed] [Google Scholar]
- 50. van Koppen CJ, Nathanson NM. 1990. Site-directed mutagenesis of the m2 muscarinic acetylcholine receptor. Analysis of the role of N-glycosylation in receptor expression and function. J Biol Chem 265:20887–20892 [PubMed] [Google Scholar]
- 51. Davidson JS, Flanagan CA, Zhou W, Becker II, Elario R, Emeran W, Sealfon SC, Millar RP. 1995. Identification of N-glycosylation sites in the gonadotropin-releasing hormone receptor: role in receptor expression but not ligand binding. Mol Cell Endocrinol 107:241–245 [DOI] [PubMed] [Google Scholar]
- 52. Hamilton SR, Gerngross TU. 2007. Glycosylation engineering in yeast: the advent of fully humanized yeast. Curr Opin Biotechnol 18:387–392 [DOI] [PubMed] [Google Scholar]
- 53. Hart GW, Brew K, Grant GA, Bradshaw RA, Lennarz WJ. 1979. Primary structural requirements for the enzymatic formation of the N-glycosidic bond in glycoproteins. Studies with natural and synthetic peptides. J Biol Chem 254:9747–9753 [PubMed] [Google Scholar]
- 54. Caronia LM, Martin C, Welt CK, Sykiotis GP, Quinton R, Thambundit A, Avbelj M, Dhruvakumar S, Plummer L, Hughes VA, Seminara SB, Boepple PA, Sidis Y, Crowley WF, Jr, Martin KA, Hall JE, Pitteloud N. 2011. A genetic basis for functional hypothalamic amenorrhea. N Engl J Med 364:215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Serre D, Gurd S, Ge B, Sladek R, Sinnett D, Harmsen E, Bibikova M, Chudin E, Barker DL, Dickinson T, Fan JB, Hudson TJ. 2008. Differential allelic expression in the human genome: a robust approach to identify genetic and epigenetic cis-acting mechanisms regulating gene expression. PLoS Genet 4:e1000006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pitteloud N, Quinton R, Pearce S, Raivio T, Acierno J, Dwyer A, Plummer L, Hughes V, Seminara S, Cheng YZ, Li WP, Maccoll G, Eliseenkova AV, Olsen SK, Ibrahimi OA, Hayes FJ, Boepple P, Hall JE, Bouloux P, Mohammadi M, Crowley W. 2007. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest 117:457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Balasubramanian R, Plummer L, Sidis Y, Pitteloud N, Martin C, Zhou QY, Crowley WF., Jr 2011. The puzzles of the prokineticin 2 pathway in human reproduction. Mol Cell Endocrinol 346:44–50 [DOI] [PMC free article] [PubMed] [Google Scholar]