

Abstract
Lung cancer is the leading cause of cancer-related death. Despite a number of studies that have provided prognostic biomarkers for lung cancer, a paucity of reliable markers and therapeutic targets exist to diagnose and treat this aggressive disease. In this study we investigated the potential of nuclear receptors (NRs), many of which are well-established drug targets, as therapeutic markers in lung cancer. Using quantitative real-time PCR, we analyzed the expression of the 48 members of the NR superfamily in a human panel of 55 normal and lung cancer cell lines. Unsupervised cluster analysis of the NR expression profile segregated normal from tumor cell lines and grouped lung cancers according to type (i.e. small vs. non-small cell lung cancers). Moreover, we found that the NR signature was 79% accurate in diagnosing lung cancer incidence in smokers (n = 129). Finally, the evaluation of a subset of NRs (androgen receptor, estrogen receptor, vitamin D receptor, and peroxisome proliferator-activated receptor-γ) demonstrated the therapeutic potential of using NR expression to predict ligand-dependent growth responses in individual lung cancer cells. Preclinical evaluation of one of these receptors (peroxisome proliferator activated receptor-γ) in mouse xenografts confirmed that ligand-dependent inhibitory growth responses in lung cancer can be predicted based on a tumor's receptor expression status. Taken together, this study establishes NRs as theragnostic markers for predicting lung cancer incidence and further strengthens their potential as therapeutic targets for individualized treatment.
Lung cancer incidence has progressively increased to approximately 223,500 new cases/yr, with approximately 157,000 deaths/yr reported in the United States in 2010 (1). The disease is now the major cause of cancer-related death with the highest mortality rate (1). A hurdle to improving these statistics has been the lack of innovative and alternative approaches that can be used to diagnose various types of lung tumors and/or guide therapy. Histopathological review of biopsies has been the historical standard for diagnosing and treating lung cancers, which are classified into small-cell lung carcinoma (SCLC) of neuroendocrine cell origin, and non-small-cell lung carcinoma (NSCLC) of epithelial origin. NSCLC are further subcategorized into adenocarcinoma (ADK), squamous cell carcinoma (SCC), and large-cell carcinoma. A promising and objective, cost- and time-saving alternative to histopathological analyses (which often require additional invasive procedures) would be the use of molecular theragnostic biomarkers that have both therapeutic and diagnostic value. To that end, a number of studies have sought to identify unique gene-based biomarker sets derived from genome-wide microarray analyses (2–6). However, the clinical applicability of these markers remains uncertain as they await further validation. Furthermore, in addition to the conventional cytotoxic chemotherapies (e.g. paclitaxel, gemcitabine, cisplatinum) that are based on histological characterization of lung cancers, the two biomarker-based therapies that do exist (e.g. gefitinib and erlotinib) have been used in the clinic with significant efficacies among affected populations (7, 8). Nevertheless, the identification of new markers that would have both diagnostic and therapeutic value would be an important milestone in improving the clinical outcome of lung cancer.
The human nuclear receptor (NR) superfamily consists of 48 members of transcription factors, most of which are ligand activated and known for their crucial roles in diverse physiological processes including metabolism, reproduction, and development (9, 10). Functional alterations in several NRs also are associated with chronic diseases such as diabetes, atherosclerosis, metabolic syndrome, and cancer; and importantly, these NRs are well-documented targets of approved drugs that are used to these diseases (11–13). Several lines of evidence indicate the relevance of NRs to tumor pathogenesis, including estrogen receptor in breast cancer, androgen receptor (AR) in prostate cancer, retinoic acid receptor-β in lung cancer, and further the human NR superfamily in NCI-60 cancer cell panel (14–17). Recently Bouchardy et al. (18) reported that breast cancer patients with antiestrogen treatment showed lower lung cancer mortality. Clinical trials have also been initiated using a peroxisome proliferator-activated receptor (PPAR)-γ agonist or an antiestrogen in combination with drugs targeting mutant epidermal growth factor receptor (e.g. erlotinib) or vascular endothelial growth factor receptor (e.g. ZD6474 or bevacizumab) (19). Finally, we have shown recently that the mRNA expression profile of NRs can predict survival of early stage NSCLC patients, thereby demonstrating the potential of NRs as clinically useful biomarkers (20).
In this study, we assessed the potential of using mRNA expression of the entire NR superfamily as a tractable, diagnostic biomarker in lung cancer. Using data obtained from human lung cancers and mouse xenograft models, we show that NR profiling provides a promising new strategy for diagnosing and potentially treating lung cancer.
Results
Expression of NRs in lung cell lines
To explore the expression profile of NR in a variety of lung cancer types, quantitative, real-time PCR for the 48 members of the human NR superfamily was performed as previously described (9, 20, 21). A panel of 55 lung cell lines was surveyed, which included five normal human bronchial epithelial cells (HBEC), 10 immortalized cell lines, 13 SCLC, and 27 NSCLC. The immortalized cell lines included four HBEC lines immortalized with cyclin-dependent kinase 4 and the catalytic subunit of human telomerase reverse transcriptase (HBEC KT), two BEAS2B cell lines that express SV40 T antigen, three HBEC KT lines transformed with oncogenic K-rasV12 (HBEC KTRB), and one HBEC KT line expressing the human papilloma virus E6/E7 oncoproteins (HBEC KT+E6/E7). These immortalized HBEC lines have previously been shown to represent various stages in oncogenic progression toward lung cancer (22, 23). The SCLC and NSCLC lines were derived from human primary tumors as described (24). The expression profiles of the 48 NRs for each of the 55 cell lines are shown in Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org, and the results are summarized according to the relative expression levels of each NR in Supplementary Table 1.
To determine the potential diagnostic utility of the NR expression profile, we first investigated whether it could be used to distinguish tumor types. Unsupervised cluster analysis of the quantitative PCR (QPCR) data set differentiated normal and immortalized HBEC from cancer cells and NSCLC from SCLC (Fig. 1A). We also evaluated whether the progression of a normal HBEC into a cancer cell correlated with NR expression. The relative levels of NR expression in the lung cancer cell lines were plotted on a color-coded heat map after normalizing the expression value for each receptor to the five normal HBEC lines (Fig. 1B). Interestingly, the HBEC KT and BEAS2B cell lines (which have been immortalized but are not neoplastic) exhibited altered expression for relatively few receptors when compared with the normal HBEC. In contrast, all of the cancer cells showed large changes in receptor expression levels with up-regulation of expression being the predominant change observed. These data suggest that events other than the introduction of an oncogene are required for the deregulation of NR expression seen with disease progression. Unsupervised cluster analysis of NR expression from these data revealed two major NR groups, one which encompassed a subset of NRs overexpressed in cancer (cluster I), and a second group NR that were notably down-regulated in cancer (cluster II). Intriguingly, these two NR groups are similar, albeit not identical, to the two clusters previously identified from anatomical profiling of NR expression to govern developmental (cluster I) and metabolic (cluster II) pathways (9). Taken together, these data indicate that NR expression reflects the pathogenic state of lung cancer cells and suggests that NR profiling might be used to predict pathogenic progression.
Fig. 1.

NR gene expression signature in lung cancer cell lines. A, Unsupervised cluster analysis for 55 lung cell lines using the NR QPCR mRNA expression data. Colors depict immortalized and normal HBEC (green), SCLC (red), and NSCLC (blue) cell lines. B, Heat map of relative NR expression in lung cancer cell lines. Expression values for each NR were normalized to the average receptor value (arbitrarily set to 1) obtained from the five normal HBEC cell lines. Red and green colors represent values that were higher or lower, respectively, than the average value observed in the normal HBEC. The horizontal axis shows unsupervised clustering of 47 NRs (including the alternative splice variants for PPARγ and PPARδ), and the vertical axis displays the 55 lung cell lines ordered based on their pathological classification. ME, Mesothelioma; BA, bronchioalveolar. Constitutive androstane receptor, farnesoid X receptor, and HNF4α were not included due to their low expression (Ct > 34). The scale represents log 2 ratio of the normalized NR expression values.
NRs as diagnostic biomarkers of lung cancer subtypes
Nuclear receptor expression is known to vary significantly between normal lung epithelial cells as well as different types of lung tumors (25–27). For example, higher expression of estrogen receptor (ER)-α protein has been shown to be associated with both adenocarcinoma and squamous cell carcinoma compared with normal bronchial epithelium (26). In addition, PPARγ expression was reported to be higher in primary NSCLC tumors compared with normal surrounding tissue (27). These observations taken together from the data shown above prompted us to evaluate whether expression of specific NRs might have utility in distinguishing the major histological subtypes of lung cancer. Analysis of the NR mRNA profiling revealed that specific types of lung cancer express discrete subsets of NRs (Fig. 2). For example, mineralocorticoid receptor and PPARγ expression were associated with NSCLC (Fig. 2A). Estrogen-related receptor (ERR)-β, ERRγ, nerve growth factor-induced gene B3 (NGFIB3), progesterone receptor, and retinoic acid-related orphan receptor-α showed significantly higher expression in SCLC compared with NSCLC. Receptors primarily expressed in SCLC included retinoid X receptor-γ, steroidogenic factor 1, and small heterodimeric partner (SHP). In contrast, retinoic acid receptor-γ, thyroid hormone receptor-β and vitamin D receptor (VDR) showed moderately or dramatically reduced expression in SCLC compared with the NSCLC or HBEC (Fig. 2A). In addition, we analyzed differences in expression in SCC vs. ADK and found that a group of receptors, e.g. ERα, hepatocyte nuclear factor 4 (HNF4)-γ, liver receptor homolog-1, mineralocorticoid receptor, PPARγ2, PPARγ, and retinoic acid-related orphan receptor-γ showed significantly increased expression in the ADK vs. SCC (Fig. 2B). We also analyzed tissue microarray data from 309 NSCLC including 196 adenocarcinomas (AC) and 113 SCC for several expression of PPARγ protein. PPARγ protein was expressed in both AC and SCC. Interestingly, although SCC showed a significantly higher expression of cytoplasmic PPARγ (mean score of AC, 4.3; SCC, 33.3; P < 0.001), AC had a significantly higher expression of nuclear PPARγ (mean score of AC, 35.7; SCC, 16.7; P < 0.001). In this analysis, protein expression was quantified as a semiquantitative score (ranging from 0 to 300) using intensity and extension of the immunostaining results (Supplemental Table 2). Collectively these data define discrete subgroups of NRs that distinguish, and therefore may be involved in, specific types of lung tumors.
Fig. 2.

Classification of tumor types based on NR expression. A, Distribution of NRs in normal and immortalized, SCLC, and NSCLC. The subsets of NRs were grouped in the Venn diagram based on their expression pattern using the statistical U test. The panels display representative NR expression profiles for each class identified in the Venn diagram. Constitutive androstane receptor, farnesoid X receptor, and HNF4α were not included due to their low expression (Ct > 34). B, Distribution of NRs in adenocarcinoma and squamous cell carcinoma cells are depicted in the Venn diagram using the statistical U test. The panels display representative NRs expression profiles that distinguish adenocarcinoma cells. Constitutive androstane receptor, ERRβ, ERRγ, farnesoid X receptor, HNF4α, pregnane X receptor, retinoid X receptor-γ, steroidogenic factor 1, SHP, and TLX were excluded due to their low expression (Ct > 34) in these two groups of cell lines.
NRs as diagnostic biomarkers of lung cancer incidence
We next sought to investigate whether the NR gene signature could be used as a diagnostic biomarker for lung cancer incidence in smokers who are at risk. To that end, we excerpted the 110 NR probe sets from 129 Affymetrix HG-U133A microarray samples (Santa Clara, CA) that describe gene expression in the lungs of patients with a history of smoking (5). Using the excerpted NR gene expression profile from a training set of 77 samples, a prediction model was developed to distinguish tumor and nontumor samples using a penalized logistic regression approach. The model was then validated in an independent testing set (n = 52) (5). Figure 3A shows the receiving operation curve of the NR prediction model in the testing data set. The area under the curve (AUC) is 0.84 (95% confidence interval 0.72–0.96), which indicates that the NR signature has significant prediction power for lung cancer diagnosis. With a 73% positive predictive value and an 81% negative predictive value, the NR biomarker had a diagnostic sensitivity of 70% in patients who had lung cancer and a diagnostic specificity of 84% in diagnosing patients without cancer (Fig. 3B). The NR signature had an overall predictive accuracy of 79% (41 of 52) for lung cancer incidence in the testing set, which was comparable with the 80-gene signature previously reported from the genome-wide study using the same sample sets (5). These data implicate the NR gene signature as a diagnostic biomarker set that may be used both to classify types of lung cancer and predict lung cancer incidence in smokers.
Fig. 3.
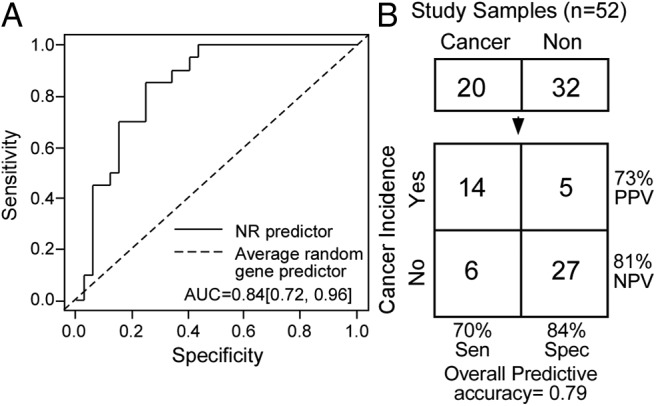
The NR gene signature as a biomarker for lung cancer incidence in smokers. A, Receiving operation curve analysis using the NR gene expression signature from 129 smokers. NR lung expression data from the microarray data sets (accession no. GSE 4115) for these patients were trained in one set (n = 77) and validated in an independent testing set (n = 52). A, The 95% confidence interval for the area under the curve (AUC) is 0.84 (0.72, 0.96). B, Diagnostic utility of the NR gene signature. The NR gene signature showed a diagnostic sensitivity of 70% (14 of 20 cancer cases) with only a 16% false-positive rate (five of 32 noncancer cases). This analysis revealed a prediction accuracy of 79% for lung cancer incidence in tested smokers. Cancer and Non indicate patients with and without cancer, respectively. Cancer incidence indicates the predicted cancer diagnosis based on the NR signature. Sens, Sensitivity; Spec, specificity; PPV, positive predictive value; NPV, negative predictive value.
Prediction of NR ligand responsiveness in lung cancer
Having established that the NR mRNA expression profile has diagnostic value, we asked whether this signature might have therapeutic potential as a predictor of NR ligand responsiveness in lung cancer. AR, ERα, VDR, and PPARγ were selected for further analysis because they have been extensively studied in several types of cancer such as breast, prostate, and colon cancers (17, 27–32). Moreover, ligands for these receptors are available and have been used clinically. Receptor-positive and -negative lung cancer cell lines (shown in Supplemental Fig. 1) were evaluated for cognate ligand responsiveness as measured by cell growth. To rule out potential effects due to differing culture conditions, the cell lines were selected randomly from those cell lines that grow well under the same culture conditions. Figure 4A shows the results from evaluating a subset of AR-positive [cycle time (Ct) < 28] and AR-negative (Ct > 35) cells in response to the agonist dihydrotestosterone (DHT). Note that Ct indicates the threshold cycle correlated to initial copy number for the gene of interest in QPCR reaction. LnCaP, an AR-positive prostate cancer cell line, was used as a positive control and showed consistent growth stimulation with DHT treatment at a physiological concentrations (<1 nm) as previously described (33). Two AR-negative lung cancer cell lines, H1299 and H2009, showed no significant response to DHT treatment. H1184 cells, a SCLC line that has the highest mRNA expression of AR, showed significant growth stimulation in the physiological subnanomolar to nanomolar concentration ranges of DHT but was growth inhibited at supraphysiological concentrations (1 μm). Interestingly, H2122 (a NSCLC line) showed a strong DHT dose-dependent growth stimulation, whereas H1993 (a NSCLC line) showed a growth-inhibitory response to DHT. For ERα, two positive expressing NSCLC lines, H2052 and HCC78 (Ct < 27), were treated either with a receptor agonist (17β-estradiol) or antagonist (ICI 182,780) and monitored for growth (Fig. 5A). The breast cancer cell line MCF-7 was used as a positive control, and its growth was stimulated or repressed by agonist or antagonist as expected. Agonist and antagonist treatment of the H2052 cells also caused a marked dose-dependent stimulation or inhibition, respectively, in cell growth. Interestingly, HCC78 cells were growth inhibited by 17β-estradiol as well as the ICI 182,780 antagonist. Two ERα-negative lines, H1299 and H2009 (Ct > 34), were unresponsive to either ligand. Note that ERβ was not expressed in any of the tested cell lines.
Fig. 4.

Evaluation of AR in lung cancer cells. A, Lung cancer cell growth response to DHT treatment. Relative growth responses were assessed as described in Materials and Methods. Asterisks show the statistically significant points evaluated by ANOVA. B, Hierarchical clustering of AR-dependent gene signatures and lung cancer cells. A genetic signature of 380 genes showing more than a 2-fold difference between AR-positive and AR-negative lung cancer cells was used for the cluster analysis using Matrix 1.29.
Fig. 5.

Evaluation of ERα in lung cancer cells. A, Lung cancer cell growth response to ERα ligands. Relative growth responses were assessed as described in Materials and Methods. Asterisks show the statistically significant points evaluated by ANOVA. B, Hierarchical clustering of ERα-dependent gene signatures and lung cancer cells. A genetic signature of 540 genes showing more than a 2-fold difference between ERα-positive and ERα-negative lung cancer cells was performed as in Fig. 4.
In a similar fashion, we evaluated the response of various cells to 1α, 25-(OH)-dihydroxyvitamin D3, the physiological ligand of VDR. Because most of the lung cancer cells express quantitatively detectable levels of VDR, the SCLC line H841 was the only low-abundance VDR cell line (Ct > 32) that could be tested along with three high VDR-expressing lines (Ct < 26): H2122, H1993, and H2347 (Fig. 6A). As predicted by their VDR mRNA expression status, the VDR-deficient cell did not respond, whereas the three VDR-positive cell lines responded to vitamin D and were growth inhibited in a dose-dependent fashion. Likewise, PPARγ ligand-dependent growth was evaluated in six different cell lines (Fig. 7A). The PPARγ-specific agonist troglitazone showed significant growth inhibition in a dose-dependent manner in three high PPARγ-expressing lines (H2347, H1993, Calu-1) (Ct < 24) but no significant responses in three low abundance-expressing lines (H1770, HCC1195, H1299) (Ct > 30). Overall, the analysis of these receptors in the surveyed cell lines suggests that NR mRNA expression profile can be used to predict a cell's ability to respond to a given receptor's ligand.
Fig. 6.

Evaluation of VDR in lung cancer cells. A, Lung cancer cell growth response to 1α, 25-dihydroxyvitamin D3. Relative growth responses were assessed as described in Materials and Methods. Asterisks show the statistically significant points evaluated by ANOVA. B, Hierarchical clustering of VDR-dependent gene signatures and lung cancer cells. A genetic signature of 717 genes showing more than an 8-fold difference between VDR-positive and VDR-negative lung cancer cells was performed as in Fig. 4.
Fig. 7.

Evaluation of PPARγ in lung cancer cells and tumors. A, Lung cancer cell growth response to troglitazone. Relative growth responses were assessed as described in Materials and Methods. Asterisks show the statistically significant points evaluated by ANOVA. B, Hierarchical clustering of PPARγ-dependent gene signatures and lung cancer cells. A genetic signature of 1010 genes showing more than a 2-fold difference between PPARγ-positive and PPARγ-negative lung cancer cells was performed as in Fig. 4. C, Response of PPARγ-positive and PPARγ-negative xenograft lung cancer tumors to pioglitazone. H1299 and H2347 cells were sc injected into athymic nude mice and allowed to form tumors. Mice were treated with 25 mg/kg pioglitazone or vehicle control four times a week (indicated on the x-axis). Tumor volumes were measured twice a week. Tumor volume represents the tumor size (n = 4 per group) ± sem and statistical analysis determined using a Student t test.
Genome-wide mRNA expression correlates with NR expression
As a first attempt to characterize the gene expression networks that might be regulated by specific NRs, we tested the correlation of global gene expression with individual NR expression in the lung cancer cell lines. We performed an Affymetrix U133AB microarray in the lung cancer cell lines that had been evaluated for AR, ERα, VDR, and PPARγ activity. Biostatistic analyses showed 210 genes differentially overexpressed and 170 genes differentially underexpressed (>2-fold) in the selected AR-positive cells vs. negative cells (Fig. 4B); and 295 genes overexpressed and 245 genes underexpressed (>2-fold) in the ERα-positive vs. negative cells (Fig. 5B). Consistent with the QPCR data, this analysis confirmed the higher expression of AR (>2.6-fold) in the AR-positive cells (P < 0.05) and ERα (>3.3-fold) in the ERα-positive cells (P < 0.05). In a similar analysis with VDR, 3277 genes were differentially up-regulated and 5178 genes were differentially down-regulated greater than 2-fold in high vs. low-expressing cells (Fig. 6B). Note that the average expression of VDR was 26-fold higher in the high-expressing group compared with the low-expressing group. The microarray data for the PPARγ correlation study identified 633 genes overexpressed vs. 377 genes underexpressed greater than 2-fold in the PPARγ high- vs. low-expressing cell lines (Fig. 7B). Consistently, PPARγ expression was greater than 27-fold in the high expression group compared with low expression group. Notably, for all these correlations, hierarchical clustering permitted segregation of the cell lines based on expression of each NR.
The hierarchical clustering analyses described above for AR, ERα, VDR, and PPARγ were analyzed further to determine the number of statistically significant (P < 0.05) genes that correlated with NR expression. The statistically significant, correlated gene sets for these NRs consisted of 195 genes (51 positively and 144 negatively correlated) for AR, 145 genes (75 positively and 70 negatively correlated) for ERα, 155 genes (144 positively and 11 negatively correlated) for VDR, and 895 genes (568 positively and 327 negatively correlated) for PPARγ. These statistically filtered genes are listed in the Supplemental Data and provide a worthwhile resource for identifying common transcriptional regulatory networks or direct target genes of these NRs.
PPARγ agonist-dependent inhibition of tumor growth
We next evaluated the possibility of guiding lung cancer therapy by targeting specific NRs expressed in individual cancers. Several lines of evidence have suggested the use for PPARγ agonists as therapeutics for treating cancer (13, 32, 34). Thus, as a proof of principle, we validated the potential of using PPARγ expression as a predictor of thiazolidinedione responsiveness in mouse xenograft tumors established by sc injection of PPARγ-positive (H2347) or -negative (H1299) cells into the flank of immune-compromised nude mice. Although the data shown above (Figs. 4A, 5A, 6A, and 7A) indicated that mRNA expression alone might predict a cell line's response to a given receptor's ligand, we also confirmed the expression of PPARγ protein levels by immunoblot analysis in the PPARγ-positive and -negative cell lines (Supplemental Fig. 2A) and in the xenograft tumors (Supplemental Fig. 2B). A dose of 25 mg/kg of pioglitazone, a PPARγ agonist that is used to treat type 2 diabetes, was ip administered four times a week for 5 wk, starting 5 d after cancer cell injection into mice. Pioglitazone treatment had no effect on H1299 (PPARγ negative) tumor growth (Fig. 7C). In contrast, H2347 (PPARγ positive) cells showed a marked, statistically significant inhibition of tumor growth with pioglitazone treatment compared with the vehicle (Fig. 7C). These data provide further evidence that PPARγ may be a relevant therapeutic target for lung cancer treatment and moreover suggest that NR expression may be a viable predictor of a tumor's response to therapy. This result provides a strong rationale for using systematic profiling of NR mRNA expression as a potential therapeutic biomarker for individual cancers.
Discussion
We previously demonstrated that the NR superfamily is a prognostic biomarker set for survival and relapse of lung cancer patients and further provided SHP as the first single-gene prognostic biomarker for early stage (stage I) of lung cancer patients (20, 26, 27). These findings prompted us to develop a rationale of investigating the diagnostic potential of the NR superfamily. In the present study, we investigated the potential use of the NR superfamily gene signature for the diagnosis and treatment of lung cancer. Several independent preclinical and clinical approaches were used to demonstrate that NR profiling of lung cancer can be used to do the following: 1) distinguish between normal bronchial epithelial cells and lung cancer; 2) classify different types of lung cancer (e.g. NSCLC vs. SCLC); 3) diagnose lung cancer incidence in smokers at risk; and 4) predict NR ligand responsiveness and provide tumor-specific therapeutic targets. Previous work has suggested that a key advantage to developing a biomarker that can diagnose lung cancer in smokers at risk is that it might eliminate the need for other costly tests and even provide a rationale for treating healthy smokers prophylactically (5). The expression profiling of the NR superfamily takes this strategy one step further by providing a diagnostic biomarker that can detect both the presence and type of lung cancer and that might be used to discover novel therapeutic targets for individual patients.
One of the more intriguing findings from this work was that the NR-specific pattern of mRNA expression in an individual tumor might be used to predict that tumor's response to a given receptor's ligand. As a proof of principle, we tested randomly chosen subsets of NR-positive and -negative cell lines (for AR, ERα, VDR, and PPARγ) and found a remarkable concordance between the presence of a given receptor's mRNA and the cell's growth response to that receptor's ligand. Although the functional data support the notion that mRNA expression may be used in some cases to indicate the presence or absence of functional receptors, mRNA expression may not always be reliable (25). At present, the paucity of adequate antibodies for most NRs prevents a robust atlas of protein expression. Knowing a given tumor's NRs profile might then be used to guide therapeutic options. As a case in point, we used the cancer cell-specific pattern of PPARγ expression to evaluate growth responses to a PPARγ agonist. The potential benefits of targeting PPARγ in cancer (including NSCLC) have been suggested previously (13, 32, 34). In this study, we showed that the PPARγ mRNA profile in tumor cells predicted both the presence of PPARγ protein (as measured by immunoblot) and cell growth inhibition in response to thiazolidinedione. The ability of thiazolidinedione treatment to inhibit PPARγ-positive xenograft tumors provides a promising rationale for the theragnostic use of NR profiling. It is worth noting that treatment and prevention of a number of diseases have been successful by specifically targeting NRs (35–40); thus, this approach is standard practice for guiding antihormonal therapy of breast and prostate cancer (41–43). Nevertheless, to develop NRs as therapeutic targets for lung cancer treatment, profiling of NR protein expression in pair-matched patient samples from normal lung vs. corresponding tumors might be advantageous because different NRs are known to be expressed in various cell types in normal lung tissues (25).
In addition to the diagnostic and therapeutic potential of the NR profiling in lung cancer, this work also reveals a strategy to study the pathogenesis and progression of the disease. To that end, we note that there was a substantial change in the NR expression profile when comparing normal HBECs and nontumorigenic, immortalized cell lines harboring oncogenic alterations (e.g. by expressing K-rasV12, E6/E7, CDK4, and hTERT) to the fully tumorigenic clones (Fig. 1B). The NR expression changes marking the progression of normal cells into cancer cells imply NR regulation of transcriptional networks plays a role in tumorigenesis. Furthermore, the unsupervised clustering of NR expression in the various cancer cell lines revealed the presence of two major classes of NRs that are remarkably similar to the two clusters found previously by analyzing whole-body anatomical expression of NRs in normal tissues (9). These two NR clusters represent a hierarchical network that governs two distinct physiological processes, development, and metabolism. The finding that the developmental and metabolic NR-dependent transcriptional networks are dysregulated in lung cancer suggests that NRs play an important role in the homeostatic balance that maintains normal differentiated lung tissue. Closer inspection of the NR expression signature revealed specific NRs appear to be associated with specific types of lung cancer and their transcriptional programs. Notable examples included the expression of SHP in SCLC and also PPARγ in NSCLC. SHP is an atypical nuclear receptor that is known for its role in enterohepatic lipid metabolism in which it interacts and negatively regulates the activity of other transcription factors, including many NRs (44, 45). The expression of a potent transcriptional repressor like SHP in a cell type in which it is not normally expressed may be expected to have significant pathological effects on cell function, and this finding provides a rationale for further mechanistic studies. The role of PPARγ in multiple cancer types has implicated this receptor in the progression of the disease as a therapeutic target (13, 32, 34), which we have validated further in this study. Finally, by evaluating global changes in gene expression that were associated with specific NRs, we found significant, NR-specific changes in expression of discrete subsets of genes (Figs. 4–7). These findings imply that this type of analysis might be used to find the downstream target genes that may also be important in lung cancer pathogenesis.
Collectively, our studies support the idea of targeting individual receptors, particularly those that are already well-documented targets of Food and Drug Administration-approved drugs for treating lung cancer. By profiling all 48 NR family members and validating the preclinical implications of the selected NRs as diagnostic biomarkers and therapeutic targets, our work provides a new theragnostic approach that could simultaneously decide diagnosis as well as guide individual-specific therapeutic treatment schemes in the future.
Materials and Methods
Cell culture
NHBEC, HBEC UI, and BEAS2B cells (American Type Culture Collection, Manassas, VA) were cultured in BGEM media (Cambrex Bio Science, Walkersville, MD) supplemented with the supplied nutrients according to the manufacturer. Patient-derived HBEC were immortalized by introducing CDK4 and hTERT as previously described (23) and are referred to as HBEC2-KT, HBEC3-KT, HBEC4-KT, and HBEC5-KT. Genetic manipulations such as introducing K-rasV12, or human papilloma vial oncoproteins E6/E7 into the immortalized HBEC were performed to investigate the effect of such oncogenic alterations (23). This series of HBEC were grown in keratinocyte-serum-free medium (Invitrogen Corp., Grand Island, NY) supplemented with epithelial growth factor and pituitary gland extract (22, 23). Lung cancer cells derived from primary tumors (24) were maintained in RPMI 1640 media (Invitrogen) with 5 or 10% heat-inactivated fetal bovine serum (ΔFBS; Gemini BioProducts, Woodland, CA). Genomic DNA fingerprinting was used to confirm the identity of the cell lines used in this study (46).
Immunoblot analysis
Following a standard protocol for immunoblot assays, 50 μg of whole-cell lysates was assayed with a mouse monoclonal anti-PPARγ antibody (47) or antiactin antibody (Sigma, St. Louis, MO) to confirm PPARγ protein expression.
QPCR assay and data analysis
Total RNA were purified using QIAGEN RNeasy minikit (QIAGEN Sciences, Gaithersburg, MD) following the manufacturer's instructions. The quality and concentration of each RNA was assessed using either ethidium bromide-stained agarose gel or Agilent 2100 bioanalyzer (Quantum Analytics Inc., Foster City, CA). Efficiency-corrected standard curve-based QPCR assays were optimized for multiple plate comparisons and were performed in the 55 lung cell panel using primer and probe sets as described previously (48). Raw data from this analysis has been deposited on the NURSA web site (www.NURSA.org) and is also available on request.
Cell line microarray data analysis
Microarray assays were performed using Affymetrix U133A and -B chips in the 48 human lung cell lines. The GSE accession of the microarray dataset is GSE4824 in GEOdatasets. Expression data were analyzed using unsupervised cluster analysis. Included in the analysis of the selected receptor correlation were the following lung cancer lines: four AR-positive cell lines (H2122, H460, H1993, and H1184) and eight AR-negative cell lines (H2009, HCC827, HCC1195, H1607, H1299, H2882, HCC366, and H289); five ERα-positive cell lines (H1607, H1993, HCC78, H2052, and HCC1195) and 23 ERα-negative cell lines (H1299, H157, H2882, H1819, H2087, H358, HCC44, H2887, HCC15, HCC366, HCC461, H526, H1672, H2107, H889, H289, H60, H82, H1963, H1184, H2171, HCC970, and H841); six high VDR-expressing cell lines (H1993, H2122, H2347, H460, H157, and H1299) and five low VDR-expressing cell lines (H82, H889, H2107, H1963, and H841); five high PPARγ-expressing cell lines (H1993, H2347, Calu-1, H2887, and H2882) and five low PPARγ-expressing cell lines (HCC1195, H1770, H841, H460, and H1299). Matrix 1.29 software (developed by Luc Girard) was used for the hierarchical cluster analysis of the QPCR data of the NR expression and the Affymetrix microarray data. Pearson correlation of each gene to each receptor expression was calculated and a significance test for the correlation was calculated with the formula: P = TDIST[ABS(r/SQRT[(1-r^2)/(n-2)]),n-2,2] where r is the Pearson correlation value and n is the number of samples tested. Genes successfully filtered to a significance of P < 0.05 are found in the Supplemental Data set.
Classification analysis of NR expression in smokers with cancer
Training and testing data sets for analyzing NR expression as a diagnostic marker in lung cancer were downloaded from a previous study (GSE4115 in Gene Expression Omnibus database) (5). These samples were collected originally from smokers and former smokers as part of a diagnostic study for suspicion of lung cancer (5). Postbiopsy analysis revealed a population of subjects with and without lung cancer. Sample information was kindly provided by Dr. Avrum Spira (Boston University, Boston, MA). Microarray data were processed by Robust Multi-array Average methods (49), and all gene expression values were log transformed (base 2 scale). One hundred ten transcripts (i.e. probes from the HG-U133A Affymetrix arrays) corresponding to 48 human NR genes were used to build a classifier to predict lung cancer diagnosis using penalized logistic regression model (50). All of the parameters used in building the model were default values in R version 2.92 and package step Plr version 0.91 (R Foundation for Statistical Computing, Vienna, Austria, http://www.R-project.org); the linear combination of all covariables are in Supplemental Table 3. The model accuracy was evaluated when using a probability cutoff value of P = 0.5.
Cell growth assays
For evaluation of AR activity, cells were maintained in phenol-red free RPMI 1640 media containing 5% heat-inactivated, charcoal-stripped ΔFBS. For the assay, 105 cells were split into six-well plates and grown in RPMI 1640 media containing 5% charcoal-stripped, heat-inactivated ΔFBS. DHT (Sigma) was added every day for 3 d in triplicate wells. Relative percentage cell growth response was calculated by counting Trypan blue-excluding cells (Sigma). For ERα evaluation, assays were performed in the same way as for AR except cells were maintained in phenol red-free containing media. Agonist 17β-estradiol (Sigma) or antagonist ICI 182, 780 (Tocris, Ellisville, MO) was added as indicated. VDR- and PPARγ-dependent cell growth was monitored using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt and 3-(4,5-dimethylthiazole-2-yl)-2,5 diphenyltetrazolium bromide (MTT) assays (Sigma), respectively. Cells were split into 96-well plates containing phenol red-free RPMI 1640 media and 5% charcoal-stripped ΔFBS in final volume of 100 μl/well and treated as indicated with 1α,25-dihydroxyvitamin D3 (Tocris) or troglitazone (Cayman Chemicals Co., Ann Arbor, MI). The MTT assays were performed by measuring absorbance of MTT metabolites at 560 nm wavelength.
Xenograft experiments
The care and treatment of experimental animals were performed with the approval of the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center (Dallas, TX). Athymic nude mice (5 to 6 wk old females) were purchased from Charles River Laboratories (Wilmington, MA) and maintained in specific pathogen-free conditions. H1299 (a PPARγ negative cell line, Ct > 30) and H2347 (a PPARγ positive cell line, Ct = 20) cells (2 × 106) were sc injected into the right flank areas of mice on d 0 (n = 8/group). The mice were randomly divided into two groups (n = 4) and treated with ip administration of 25 mg/kg pioglitazone (Actos; Takeda Pharmaceutical Co., Osaka, Japan) dissolved in 10% dimethylsulfoxide or the same volume of dimethylsulfoxide vehicle control four times a week. Tumor volumes were directly measured twice a week from d 5 with calipers and calculated by the formula π/6 × (large diameter) × (small diameter)2. Mice treated with vehicle were killed at d 37 before tumor necrosis occurred. The difference in tumor volume between treatment groups was statistically analyzed with a Student t test using SPSS 11.5 software (SPSS Inc., Chicago, IL).
Supplementary Material
Acknowledgments
We thank Avrum Spira for kindly providing clinical information for the microarray dataset (GSE4115) and Michael Peyton for discussions and comments.
This work was supported by the Howard Hughes Medical Institute (to D.J.M.), the Robert A. Welch Foundation (Grant I-1275 to D.J.M.), the National Institutes of Health (Grant U19 DK62434 to D.J.M.; Grant P50 CA70907 to J.D.M.; Grants UL1 RR024982 and Grant CA152301 to Y.X.; and Grant T32 GM007062 to A.L.B.), the Cancer Prevention Research Institute of Texas (Grant RP101251 to Y.X., D.J.M., and J.D.M.), a research grant from Yonsei University Wonju College of Medicine (Grant YUWCM-2011-18 to Y.J.), and the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science, and Technology (Grant 2011-0005682 to Y.J.). The funders had no role in the study design, the data collection and analysis, the decision to publish, or the preparation of the manuscript.
Disclosure Summary: The authors have no conflicts of interests to declare.
Footnotes
- AC
- Adenocarcinoma
- ADK
- adenocarcinoma
- AR
- androgen receptor
- Ct
- cycle time
- DHT
- dihydrotestosterone
- ER
- estrogen receptor
- ERR
- estrogen-related receptor
- ΔFBS
- fetal bovine serum
- HBEC
- human bronchial epithelial cell
- HBEC KT
- HBECs immortalized with cyclin dependent kinase 4 and the catalytic subunit of human telomerase reverse transcriptase
- HNF4
- hepatocyte nuclear factor 4
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NR
- nuclear receptor
- NSCLC
- non-small-cell lung carcinoma
- PPAR
- peroxisome proliferator-activated receptor
- QPCR
- quantitative PCR
- SCC
- squamous cell carcinoma
- SCLC
- small-cell lung carcinoma
- SHP
- small heterodimeric partner
- VDR
- vitamin D receptor.
References
- 1. Jemal A, Siegel R, Xu J, Ward E. 2010. Cancer statistics, 2010. CA Cancer J Clin 60:277–300 [DOI] [PubMed] [Google Scholar]
- 2. Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, Loda M, Weber G, Mark EJ, Lander ES, Wong W, Johnson BE, Golub TR, Sugarbaker DJ, Meyerson M. 2001. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci USA 98:13790–13795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Molina R, Augé JM, Bosch X, Escudero JM, Viñolas N, Marrades R, Ramírez J, Carcereny E, Filella X. 2009. Usefulness of serum tumor markers, including progastrin-releasing peptide, in patients with lung cancer: correlation with histology. Tumour Biol 30:121–129 [DOI] [PubMed] [Google Scholar]
- 4. Raponi M, Zhang Y, Yu J, Chen G, Lee G, Taylor JM, Macdonald J, Thomas D, Moskaluk C, Wang Y, Beer DG. 2006. Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res 66:7466–7472 [DOI] [PubMed] [Google Scholar]
- 5. Spira A, Beane JE, Shah V, Steiling K, Liu G, Schembri F, Gilman S, Dumas YM, Calner P, Sebastiani P, Sridhar S, Beamis J, Lamb C, Anderson T, Gerry N, Keane J, Lenburg ME, Brody JS. 2007. Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat Med 13:361–366 [DOI] [PubMed] [Google Scholar]
- 6. Wójcik E, Kulpa JK, Sas-Korczyska B, Korzeniowski S, Jakubowicz J. 2008. ProGRP and NSE in therapy monitoring in patients with small cell lung cancer. Anticancer Res 28:3027–3033 [PubMed] [Google Scholar]
- 7. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. 2004. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350:2129–2139 [DOI] [PubMed] [Google Scholar]
- 8. Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. 2004. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304:1497–1500 [DOI] [PubMed] [Google Scholar]
- 9. Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. 2006. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 126:789–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. 2001. Nuclear receptors and lipid physiology: opening the X-files. Science 294:1866–1870 [DOI] [PubMed] [Google Scholar]
- 11. Barish GD, Evans RM. 2004. PPARs and LXRs: atherosclerosis goes nuclear. Trends Endocrinol Metab 15:158–165 [DOI] [PubMed] [Google Scholar]
- 12. Evans RM, Barish GD, Wang YX. 2004. PPARs and the complex journey to obesity. Nat Med 10:355–361 [DOI] [PubMed] [Google Scholar]
- 13. Mansure JJ, Nassim R, Kassouf W. 2009. Peroxisome proliferator-activated receptor γ in bladder cancer: a promising therapeutic target. Cancer Biol Ther 8:6–15 [DOI] [PubMed] [Google Scholar]
- 14. Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY, Zhu Y, Skaar TC, Gomez B, O'Brien K, Wang Y, Hilakivi-Clarke LA. 2003. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene 22:7316–7339 [DOI] [PubMed] [Google Scholar]
- 15. Miyamoto H, Messing EM, Chang C. 2004. Androgen deprivation therapy for prostate cancer: current status and future prospects. Prostate 61:332–353 [DOI] [PubMed] [Google Scholar]
- 16. Virmani AK, Rathi A, Zöchbauer-Müller S, Sacchi N, Fukuyama Y, Bryant D, Maitra A, Heda S, Fong KM, Thunnissen F, Minna JD, Gazdar AF. 2000. Promoter methylation and silencing of the retinoic acid receptor-β gene in lung carcinomas. J Natl Cancer Inst 92:1303–1307 [DOI] [PubMed] [Google Scholar]
- 17. Holbeck S, Chang J, Best AM, Bookout AL, Mangelsdorf DJ, Martinez ED. 2010. Expression profiling of nuclear receptors in the NCI60 cancer cell panel reveals receptor-drug and receptor-gene interactions. Mol Endocrinol 24:1287–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bouchardy C, Benhamou S, Schaffar R, Verkooijen HM, Fioretta G, Schubert H, Vinh-Hung V, Soria JC, Vlastos G, Rapiti E. 2011. Lung cancer mortality risk among breast cancer patients treated with anti-estrogens. Cancer 117:1288–1295 [DOI] [PubMed] [Google Scholar]
- 19. www.clinicaltrials.gov.
- 20. Jeong Y, Xie Y, Xiao G, Behrens C, Girard L, Wistuba II, Minna JD, Mangelsdorf DJ. 2010. Nuclear receptor expression defines a set of prognostic biomarkers for lung cancer. PLoS Med 7:e1000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xie CQ, Jeong Y, Fu M, Bookout AL, Garcia-Barrio MT, Sun T, Kim BH, Xie Y, Root S, Zhang J, Xu RH, Chen YE, Mangelsdorf DJ. 2009. Expression profiling of nuclear receptors in human and mouse embryonic stem cells. Mol Endocrinol 23:724–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sato M, Vaughan MB, Girard L, Peyton M, Lee W, Shames DS, Ramirez RD, Sunaga N, Gazdar AF, Shay JW, Minna JD. 2006. Multiple oncogenic changes [K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase] are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res 66:2116–2128 [DOI] [PubMed] [Google Scholar]
- 23. Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, Peyton M, Zou Y, Kurie JM, Dimaio JM, Milchgrub S, Smith AL, Souza RF, Gilbey L, Zhang X, Gandia K, Vaughan MB, Wright WE, Gazdar AF, Shay JW, Minna JD. 2004. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res 64:9027–9034 [DOI] [PubMed] [Google Scholar]
- 24. Phelps RM, Johnson BE, Ihde DC, Gazdar AF, Carbone DP, McClintock PR, Linnoila RI, Matthews MJ, Bunn PA, Jr, Carney D, Minna JD, Mulshine JL. 1996. NCI-navy medical oncology branch cell line data base. J Cell Biochem Suppl 24:32–91 [DOI] [PubMed] [Google Scholar]
- 25. Townsend EA, Miller VM, Prakash YS. 2012. Sex differences and sex steroids in lung health and disease. Endocr Rev 33:1–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Raso MG, Behrens C, Herynk MH, Liu S, Prudkin L, Ozburn NC, Woods DM, Tang X, Mehran RJ, Moran C, Lee JJ, Wistuba II. 2009. Immunohistochemical expression of estrogen and progesterone receptors identifies a subset of NSCLCs and correlates with EGFR mutation. Clin Cancer Res 15:5359–5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Keshamouni VG, Reddy RC, Arenberg DA, Joel B, Thannickal VJ, Kalemkerian GP, Standiford TJ. 2004. Peroxisome proliferator-activated receptor-γ activation inhibits tumor progression in non-small-cell lung cancer. Oncogene 23:100–108 [DOI] [PubMed] [Google Scholar]
- 28. Liu H, Lee ES, Gajdos C, Pearce ST, Chen B, Osipo C, Loweth J, McKian K, De Los Reyes A, Wing L, Jordan VC. 2003. Apoptotic action of 17β-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J Natl Cancer Inst 95:1586–1597 [DOI] [PubMed] [Google Scholar]
- 29. Osipo C, Gajdos C, Liu H, Chen B, Jordan VC. 2003. Paradoxical action of fulvestrant in estradiol-induced regression of tamoxifen-stimulated breast cancer. J Natl Cancer Inst 95:1597–1608 [DOI] [PubMed] [Google Scholar]
- 30. Joly-Pharaboz MO, Ruffion A, Roch A, Michel-Calemard L, André J, Chantepie J, Nicolas B, Panaye G. 2000. Inhibition of growth and induction of apoptosis by androgens of a variant of LNCaP cell line. J Steroid Biochem Mol Biol 73:237–249 [DOI] [PubMed] [Google Scholar]
- 31. Umekita Y, Hiipakka RA, Kokontis JM, Liao S. 1996. Human prostate tumor growth in athymic mice: inhibition by androgens and stimulation by finasteride. Proc Natl Acad Sci USA 93:11802–11807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Allred CD, Kilgore MW. 2005. Selective activation of PPARγ in breast, colon, and lung cancer cell lines. Mol Cell Endocrinol 235:21–29 [DOI] [PubMed] [Google Scholar]
- 33. Okamoto M, Lee C, Oyasu R. 1997. Autocrine effect of androgen on proliferation of an androgen responsive prostatic carcinoma cell line, LNCAP: role of interleukin-6. Endocrinology 138:5071–5074 [DOI] [PubMed] [Google Scholar]
- 34. Girnun GD, Naseri E, Vafai SB, Qu L, Szwaya JD, Bronson R, Alberta JA, Spiegelman BM. 2007. Synergy between PPARγ ligands and platinum-based drugs in cancer. Cancer Cell 11:395–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wada T, Gao J, Xie W. 2009. PXR and CAR in energy metabolism. Trends Endocrinol Metab 20:273–279 [DOI] [PubMed] [Google Scholar]
- 36. Fruchart JC. 2009. Peroxisome proliferator-activated receptor-α (PPARα): at the crossroads of obesity, diabetes and cardiovascular disease. Atherosclerosis 205:1–8 [DOI] [PubMed] [Google Scholar]
- 37. Takahashi N, Goto T, Hirai S, Uemura T, Kawada T. 2009. Genome science of lipid metabolism and obesity. Forum Nutr 61:25–38 [DOI] [PubMed] [Google Scholar]
- 38. Ondrey F. 2009. Peroxisome proliferator-activated receptor γ pathway targeting in carcinogenesis: implications for chemoprevention. Clin Cancer Res 15:2–8 [DOI] [PubMed] [Google Scholar]
- 39. Shimizu M, Takai K, Moriwaki H. 2009. Strategy and mechanism for the prevention of hepatocellular carcinoma: phosphorylated retinoid X receptor α is a critical target for hepatocellular carcinoma chemoprevention. Cancer Sci 100:369–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jordan VC. 2008. Tamoxifen: catalyst for the change to targeted therapy. Eur J Cancer 44:30–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aquilina JW, Lipsky JJ, Bostwick DG. 1997. Androgen deprivation as a strategy for prostate cancer chemoprevention. J Natl Cancer Inst 89:689–696 [DOI] [PubMed] [Google Scholar]
- 42. Jordan VC. 2009. A century of deciphering the control mechanisms of sex steroid action in breast and prostate cancer: the origins of targeted therapy and chemoprevention. Cancer Res 69:1243–1254 [DOI] [PubMed] [Google Scholar]
- 43. Labrie F, Belanger A, Simard J, Labrie C, Dupont A. 1993. Combination therapy for prostate cancer. Endocrine and biologic basis of its choice as new standard first-line therapy. Cancer 71:1059–1067 [DOI] [PubMed] [Google Scholar]
- 44. Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ. 2000. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 6:507–515 [DOI] [PubMed] [Google Scholar]
- 45. Seol W, Hanstein B, Brown M, Moore DD. 1998. Inhibition of estrogen receptor action by the orphan receptor SHP (short heterodimer partner). Mol Endocrinol 12:1551–1557 [DOI] [PubMed] [Google Scholar]
- 46. Shames DS, Girard L, Gao B, Sato M, Lewis CM, Shivapurkar N, Jiang A, Perou CM, Kim YH, Pollack JR, Fong KM, Lam CL, Wong M, Shyr Y, Nanda R, Olopade OI, Gerald W, Euhus DM, Shay JW, Gazdar AF, Minna JD. 2006. A genome-wide screen for promoter methylation in lung cancer identifies novel methylation markers for multiple malignancies. PLoS Med 3:e486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tanaka T, Takeno T, Watanabe Y, Uchiyama Y, Murakami T, Yamashita H, Suzuki A, Aoi R, Iwanari H, Jiang SY, Naito M, Tachibana K, Doi T, Shulman AI, Mangelsdorf DJ, Reiter R, Auwerx J, Hamakubo T, Kodama T. 2002. The generation of monoclonal antibodies against human peroxisome proliferator-activated receptors (PPARs). J Atheroscler Thromb 9:233–242 [DOI] [PubMed] [Google Scholar]
- 48. Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. 2006. High-throughput real-time quantitative reverse transcription PCR. Curr Protoc Mol Biol Chapter 15:Unit 15:18. [DOI] [PubMed] [Google Scholar]
- 49. Bolstad BM, Irizarry RA, Astrand M, Speed TP. 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19:185–193 [DOI] [PubMed] [Google Scholar]
- 50. Zhu J, Hastie T. 2004. Classification of gene microarrays by penalized logistic regression. Biostatistics 5:427–443 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.