

Abstract
Endometriosis results from ectopic invasion of endometrial tissue within the peritoneal cavity. Aberrant levels of the estrogen receptor (ER), ERα and ERβ, and higher incidence of autoimmune disorders are observed in women with endometriosis. An immunocompetent mouse model of endometriosis was used in which minced uterine tissue from a donor was dispersed into the peritoneal cavity of a recipient. Wild-type (WT), ERα-knockout (αERKO), and βERKO mice were donors or recipients to investigate the roles of ERα, ERβ, and estradiol-mediated signaling on endometriosis-like disease. Mice were treated with vehicle or estradiol, and resulting location, number, and size of endometriosis-like lesions were assessed. In comparison with WT lesions in WT hosts, αERKO lesions in WT hosts were smaller and fewer in number. The effect of ER status and estradiol treatment on nuclear receptor status, proliferation, organization, and inflammation within lesions were examined. αERKO lesions in WT hosts did not form distal to the incision site, respond to estradiol, or proliferate but did have increased inflammation. WT lesions in αERKO hosts did respond to estradiol, proliferate, and show decreased inflammation with treatment, but surprisingly, progesterone receptor expression and localization remained unchanged. Only minor differences were observed between WT lesions in βERKO hosts and βERKO lesions in WT hosts, demonstrating the estradiol-mediated signaling responses are predominately through ERα. In sum, these results suggest ER in both endometriosis-like lesions and their environment influence lesion characteristics, and understanding these interactions may play a critical role in elucidating this enigmatic disease.
Endometriosis is a gynecological disease affecting 10–14% of reproductive-aged women with symptoms such as dysmenorrhea, dyspareunia, and infertility. This number increases to 35–50% in women with pelvic pain, infertility, or both (1, 2). Endometriosis is most commonly believed to occur via retrograde menstruation, also known as the Sampson hypothesis (3), in which viable endometrial tissue flows retrograde through the fallopian tube and into the peritoneal cavity, where it attaches and can invade tissues and organs within the cavity. Because at least 90% of women experience retrograde menstruation, the lower incidence (10–14%) of endometriosis suggests additional elements impact its etiology (4, 5). Current hypotheses regarding disease pathogenesis and pathophysiology suggest inherent defects in the immune system, the uterus, the endometrium, or even the peritoneal environment of women with endometriosis (6–12).
The endometrium, whether it is within the uterine cavity (eutopic) or found in endometriotic lesions (ectopic) is highly regulated by hormone action (13). Estrogen mediates its endometrial effects through activity of the estrogen receptors (ERα and ERβ), whereas progesterone regulation occurs by way of the progesterone receptors (PRA, PRB, and PRC) (13–16). Gene-targeted knockout animal models of these receptors exhibit distinct phenotypes and allow for examination of receptor function in normal physiology and disease. Mice lacking ERα [ERα-knockout (αERKO)] have dysregulation of the hypothalamic-pituitary-gonad axis and hypoplastic uteri resulting in infertility (17, 18), whereas mice lacking ERβ (βERKO) primarily exhibit ovarian defects that lead to compromised fertility (19, 20). Both ER are implicated in endometriosis; peritoneal macrophages from women with endometriosis overexpress ERα and ERβ, endometriosis lesions have altered methylation patterns of ERβ, and the ER may mediate regulation of one another (21–23). The variants of PR exhibit distinct phenotypes. PRA-knockout mice are infertile due to ovarian and uterine abnormalities, PRB-knockout mice have altered progesterone responses in the mammary glands, and the PRC isoform is N-terminally truncated, able to bind ligand, but does not bind DNA (16, 24). PRA and PRB are expressed in the endometrium, but endometriosis lesions can lack PRA, whereas PRC expression is observed to increase in endometriosis (8, 25). Estrogen is mitogenic and can be both proinflammatory and antiinflammatory depending on the cell type, but progesterone inhibits the mitogenic action of estrogen and promotes endometrial cell differentiation (26). Therefore, current therapies for endometriosis aim to decrease ovarian estrogen production and or counteract estrogen effects with the use of GnRH agonists, progestins (including oral contraceptives), and androgens, but side effects limit their long-term use (5).
The etiology of endometriosis remains elusive with current studies focusing on heredity, immune regulation, angiogenesis, or environmental toxicant exposure. Consistent with an immune dysfunction, women with endometriosis also have associated conditions with higher incidences of systemic lupus erythematosus, Sjogren's syndrome, rheumatoid arthritis, multiple sclerosis, and allergies (27). Cytokine regulation within the endometrium plays a normal role in menstruation, implantation, and the defense mechanisms of the mucosal epithelium (28). However, women with endometriosis have increased activation of peritoneal macrophages and elevated levels of inflammatory cytokines (29–31). Illustrating another potential defect underlying this disease and suggesting misregulation of immune surveillance may contribute to the attachment of ectopic endometrium within the peritoneal cavity (30).
Limited studies exist regarding endometrial-peritoneal attachment and invasion in the development of endometriosis. We hypothesized that the ER, dependent on estradiol-mediated activity, would affect endometriosis-like lesion growth and development. To test our hypothesis, we used syngeneic immunocompetent mice to address the role of ERα and ERβ in the evolution of endometriosis-like disease from a host- and donor-specific manner. The αERKO and βERKO mice were used as recipients and as donors for the development of endometriosis-like disease. In this immune-competent mouse model of endometriosis, our data provide evidence for the crucial importance of ERα activity in both the development of blood vessels that support endometriosis-like lesion growth and on donor tissue responsiveness.
Materials and Methods
Animal care and treatment
All animal studies were conducted in accordance with the National Institutes of Health Guidelines for Humane Use and Care of Animals and with approved National Institute of Environmental Health Sciences (NIEHS) animal protocol. Esr1 (ERα) with exon 3 deleted (αERKO) was generated as described previously (17). Esr2 (ERβ) with exon 3 flanked by loxP sites was generated by Xenogen (Caliper Life Sciences, Cranbury, NJ) using a strategy similar to that described by Dupont et al. (32). The phenotype of the exon 3 deleted βERKO animals are comparable to previously published βERKO phenotypes (in submission). Adult female C57/BL6J mice were purchased from Charles River Laboratories (Raleigh, NC). ER-knockout mice were generated from the NIEHS αERKO or βERKO colonies at Charles River Laboratories (Wilmington, MA) or were generated by in-house breeding at NIEHS. Mice were in a controlled temperature range (72–74 F) on a 12-h light, 12-h dark cycle. Mice were given food and water ad libitum.
Recipient mice were ovariectomized through two 0.5-cm dorsolateral skin incisions and were then divided into two treatment groups, estradiol valerate (2.5 μg/mouse·wk; Sigma-Aldrich, St. Louis, MO) in corn oil or corn oil vehicle (n = 6–12 mice per group). Mice were dosed sc once per week for 2 wk before experimental endometriosis induction. Donor mice were primed 41 h before uterus removal with pregnant mare serum gonadotropin (10 IU ip). The donor uterus was removed en bloc after euthanasia, cleaned of excess tissue, and washed thrice in sterile PBS. The uterus was slit with a linear incision longitudinally and minced (≤1.5 mm). Recipient mice were anesthetized using isoflurane/oxygen and given buprenorphine (0.1 mg/kg) for pain management. A 0.5-cm right dorsolateral incision was made, minced donor tissue in 500 μl PBS was injected into the peritoneal cavity of the recipient, and a gentle massage was given to disperse the tissue. An equivalent amount (∼100 mg) of minced tissue was transferred into all recipients [wild-type (WT) and βERKO donors were used at a 1:1 donor uterus to host ratio, whereas the αERKO donors, with hypoplastic uteri, were used at a 5:1 donor uterus to host ratio]. Mice were treated for 3 additional weeks with estradiol valerate or vehicle (Fig. 1). Groups were designated in the following manner (donor to host): WT to WT, WT to αERKO, WT to βERKO, αERKO to WT, and βERKO to WT.
Fig. 1.

Endometriosis experimental protocol. WT, αERKO, and βERKO host mice were ovariectomized (OVEX) on d −14 and were given a sc injection of corn oil or estradiol valerate (2.5 μg/mouse·wk) once a week for the duration of the study beginning at OVEX (*). WT, αERKO, and βERKO donor mice were primed with pregnant mare serum gonadotropin (PMSG) (10 IU) 41 h before removal of uterus for uterine synchronization. Donor uteri were cleanly removed, washed thrice in PBS, slit longitudinally, and finely minced. Minced uterine tissue was injected into the peritoneal cavity of a host mouse (n = 6–12 per group). Endometriosis-like lesions were removed 21 d after minced uterine tissue injection.
After 3 wk, mice were euthanized with CO2, the peritoneal cavity was opened, and endometriosis-like lesions were removed. To assess the effects of genotype on ectopic uterine tissue, ectopic lesions were photographed to document in situ images of endometriosis-like lesions (Leica dissecting microscope MZ16FA and Leica camera DFC490; Leica, Wetzlar, Germany). Endometriosis-like lesions were visualized, dissected, measured, and weighed. Endometriosis-like lesions were removed and either fixed in 10% formalin or snap-frozen on dry ice and stored at −80 C until use.
To evaluate the effects of ERα and estradiol on the regulation of angiogenic factors, ovariectomized WT and αERKO mice (n = 5 per group) were injected ip with 100 μl 0.85% saline/0.25% ethanol vehicle or with 2.5 μg/ml estradiol (10 μg/kg body weight). Uterine tissue was collected after 2 h of saline vehicle or estradiol and snap-frozen on dry ice and stored at −80 C until use.
Immunohistochemical analysis
The fixed tissues were routinely processed for paraffin embedding. Five-micrometer sections were cut, and the slides were used for hematoxylin and eosin (H&E) staining and for immunohistochemistry (IHC). For IHC, all the slides were deparaffinized and hydrated through descending grades of alcohol followed by Tris-buffered saline. Antigen retrieval was performed using citrate buffer in a decloaking chamber (Biocare, Walnut Creek, CA) for 3 min. Endogenous peroxidase was blocked by incubating the slides with 3% hydrogen peroxide for 10 min. For all IHC, sections were incubated with blocking reagents, antibodies, and detection reagents at room temperature (Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org). All slides were counterstained in hematoxylin (Sigma, St. Louis, MO), dehydrated, and coverslipped. For PR and ERα, the sections were incubated with the blocking reagent from the VECTOR MOM kit (Vector Laboratories, Inc., Burlingame, CA). Then sections were incubated with anti-PR (Beckman Coulter, Marseille, France; no. 1546) or anti-ERα (Beckman Coulter; PN IM1545) primary antibodies, followed by biotinylated antimouse IgG (Vector). ExtrAvidin peroxidase (Sigma), and then liquid DAB+ substrate chromogen system (Dako North America, Inc., Carpinteria, CA) was used to develop the signal.
For Ki67, sections were blocked with 5% normal rabbit serum (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Sections were incubated with anti-Ki67 (TEC 3; DakoCytomation, Carpinteria, CA) antibody, followed by biotinylated rabbit antirat IgG (Vector). For CD3, sections were blocked with 5% normal goat serum (Jackson Immunoresearch Laboratories, West Grove, PA). An avidin-biotin complex (Vector) was applied for 15 min. Sections were incubated with anti-CD3 (Abcam, Cambridge, MA; no. ab5690) antibody, followed by goat antirabbit biotin-conjugated secondary antibody (Vector). The Vectastain R.T.U. Elite label was applied and incubated for 30 min. For F4/80, sections were blocked with 10% normal rabbit serum (Vector). An avidin-biotin complex was applied for 15 min. Sections were incubated with anti-F4/80 (BioLegend, San Diego, CA; no. 123102) antibody, followed by biotinylated rabbit antirat IgG (Vector). The Vectastain R.T.U. Elite label was applied for 30 min.
For smooth muscle actin (SMA), sections were blocked with 5% normal goat serum (Santa Cruz). Sections were incubated with polyclonal SMA antibody (Abcam; no. ab295238), followed by goat antirabbit biotin-conjugated secondary antibody (VectorRabbit Elite kit). An avidin-biotin complex was applied for 30 min. For von Willebrand factor (vWF)/factor VII, antigen retrieval was performed using Biocare Pepsin Carezyme II for 3 min at 37 C. Sections were washed in Tris/HCl and blocked with 5% normal goat serum. An avidin-biotin complex was applied for 15 min. Sections were incubated with anti-vWF (Biocare Medical; no. CP039A) antibody, followed by biotinylated goat antirabbit conjugated secondary (VectorRabbit Elite kit). An avidin-biotin complex was applied for 30 min.
Slides stained with Masson's Tricrhome were stained following the Leica ST5020 Slide Stainer manufacturer's protocol (Leica, Germany).
RNA isolation real-time PCR
Frozen endometriosis-like lesions were pulverized under liquid nitrogen, and RNA was isolated using TRIzol as per manufacturer's instructions (Invitrogen, Carlsbad, CA). Using a previously described method, cDNA was synthesized and analyzed by real-time PCR using Fast SYBR (33). Relative transcript levels were quantified in comparison with the WT to WT vehicle group and normalized to Rpl7 using the model described by Pfaffl (34). Primer sequences (Supplemental Table 2) purchased from Sigma were selected using Primer Express (Applied Biosystems, Foster City, CA) or Harvard Primer Bank (Harvard University, Boston, MA).
Grading of inflammation
Endometriosis like-lesions fixed for histology and stained by H&E and immunohistochemically for CD3 (T-cell marker) and F4/80 (macrophage marker) were used to assess degree of inflammation and cellular infiltrates. Lesions were blinded and randomized for independent scoring. For the grading of inflammation, when the inflammatory cells involved less than 10, 11–30, 31–60, and more than 60% of the tissue section, the inflammation was graded as minimal (1), mild (2), moderate (3), and severe (4), respectively.
Statistical analysis
One-way ANOVA with Tukey's posttest, two-way ANOVA with Bonferroni posttest, unpaired one-tailed t tests, and one-way ANOVA with Bonferroni's multiple-comparison posttest P < 0.05 were performed using GraphPad Prism version 5.00 for Windows, GraphPad Software, San Diego, CA.
Results
A syngeneic mouse model of endometriosis was used to assess endometriosis-like lesion formation. At necropsy, attached lesions per animal were counted. Figure 2A shows that there was both a host and a treatment effect on the numbers of endometriosis-like lesions per mouse. All groups, except the αERKO tissue into a WT host (α to WT), demonstrated a significant increase in lesion weight with estradiol treatment compared with vehicle (Fig. 2B). In the βERKO to WT (β to WT) estradiol-treated group, the weight increase was slightly attenuated compared with the weight increase of the WT to WT estradiol-treated group but remained statistically increased. When an αERKO was used as either the host or the donor uterine tissue, without treatment, only one attached lesion was found at the injection site (WT to α; α to WT). However, estrogen treatment increased the numbers of lesions, especially in the WT to αERKO group and modestly increased lesion numbers in all other groups but not in αERKO to WT groups. After estrogen treatment, the endometriosis-like lesions increased in weight (Fig. 2B) and were fluid filled and distended (Fig. 2C, 19–24). These data demonstrate that ERα in the lesion was critical for a weight increase in response to estradiol treatment, consistent with findings indicating the role of ERα in uterine growth (35).
Fig. 2.

Evaluation of endometriosis-like lesion formation using WT, αERKO, and βERKO immunocompetent mice as donors or recipients. A, Estradiol signaling mediated through ERα confers a maximal number of endometriosis-like lesions. Adherent lesions were removed from WT to WT, WT to αERKO, WT to βERKO, αERKO to WT, and βERKO to WT lesions from mice treated with vehicle or estradiol (E2) (2.5 μg/mouse·wk) for 3 wk and counted. Letters indicate P < 0.05 by one-way ANOVA with Tukey's multiple-comparison posttest. B, Endometriosis-like lesions require ERα to increase in weight with estradiol treatment. *, P < 0.05, unpaired one-tailed t test. C, Representative macroscopic and microscopic photomicrographs of endometriosis-like lesions. Macroscopic images are at ×7.5 or ×14.5 magnification, and microscopic images stained with H&E are ×100 or ×400 magnification. Yellow arrows indicate endometriosis-like lesions; white arrows in 1 and 19 indicate uterus; white arrows in 24 indicate intestine.
Representative macroscopic images of endometriosis-like lesions are shown (Fig. 2C, 1–6 and 19–24; yellow arrows). The majority of the lesions found in the vehicle group were at the site of incision used for injection of uterine tissue, were smaller in size, were not fluid filled, and appeared visually to exhibit less vascularization (Fig. 2C, 1–6). The lesions removed in the estradiol-treated groups were found at the injection site as well as within the intestinal mesentery, the cul-de-sac area (the area anterior and posterior to the base of the uterus), and around the uterus, spleen, and associated fat pads (Fig. 2C, 19–24). The lesions from animals receiving estrogen, with the exception of the αERKO to WT group, were filled with fluid and were visibly vascularized. The αERKO to WT groups, both vehicle- and estradiol-treated animals, had one dense, encapsulated attached lesion at the injection site that was not fluid filled. The attached lesions from the αERKO to WT groups had to be dissected and removed for proper macroscopic imaging (Fig. 2C, 4 and 27; ×14.5). Interestingly, unadhered and unattached pieces of uterine tissue floating in the peritoneal cavity were observed, but only in the αERKO to WT groups. Examples of the floaters are shown macroscopically (Fig. 2C, 5 and 23). This tissue took on a different, nonvascularized, coloration (white) than all the other lesions where no blood supply or attachment to tissues within the peritoneal cavity was ever apparent. Endometriosis is histologically defined as the presence of endometrial-like glands and/or stroma outside the uterus (36). Representative microscopic images (×100 and ×400) of H&E-stained lesions demonstrate the endometriosis-like appearance of the lesions, including glandular epithelial and stromal areas (Fig. 2C, 7–18 and 25–36). With the focus to identify glandular structures, we were able to find glandular structures in all lesions but the floaters, albeit this was much more difficult in the WT to βERKO and βERKO to WT vehicle groups. As expected, with estradiol treatment, the epithelial cell height increased in all groups except the αERKO to WT group (Fig. 2C, 31–36).
In normal uterine tissue, SMA is detected only in the myometrium; however, SMA is a frequent component of human endometriotic lesions, with fibrosis and smooth muscle metaplasia occurring outside the endometriotic foci in 88.2% of patients (36–38). Organization of smooth muscle in the endometriosis-like lesions was evaluated using SMA IHC and Masson's trichrome stain (Supplemental Table 3). The smooth muscle was scored as organized when the cells were arranged in bundles surrounding the glandular structures and as not organized when the cells were randomly present in the sections of endometriosis-like lesions. WT to WT lesions showed strong endometriosis-like tissue organization as well as the presence of glandular structures with polarized epithelial cell-lined lumens, regardless of treatment (eight of eight evaluated lesions in vehicle-treated animals showed organization; 11 of 11 evaluated lesions in estrogen-treated animals showed organization). WT to αERKO lesions without estrogen treatment lacked proper arrangement of smooth muscle cells (five of 10) but exhibited organization in all but one lesion with estrogen treatment (14 of 15). Lesions of WT to βERKO (zero of five) and βERKO to WT (zero of 11) lacked organization with vehicle but did demonstrate an increase in organization after estrogen treatment (six of eight from WT to βERKO and four of nine from βERKO to WT). The αERKO to WT groups were very different from the other experimental groups in that only one attached lesion was found per animal, and this lesion was only ever at the tissue injection site (zero of nine vehicle-treated and five of 10 estradiol-treated animals), suggesting a role for ERα and ERβ in the organization response. In the floaters, no organization or SMA staining was observed in either treatment group (zero of six vehicle-treated and zero of six estradiol-treated animals). vWF/factor VIII IHC was used to highlight blood vessels, and no vWF/factor VIII staining was observed in the floaters, but blood vessels were seen in all other lesions (data not shown). Proliferative activity in the lesions was assessed by Ki67 IHC. The lesions from estradiol-treated WT to WT mice exhibit increased proliferation in the epithelial layer of the lesions (Fig. 3). Ki67 staining was observed in the WT to WT vehicle group, but this was less evident in the other groups receiving vehicle treatment. The least Ki67 staining after estradiol treatment was seen in the αERKO to WT and βERKO to WT groups, with no Ki67 staining seen in any of the floaters from the αERKO to WT groups. The decreased Ki67 staining in the βERKO to WT group correlates with the somewhat attenuated endometriosis-like lesion growth. These data suggest that the presence of estrogen signaling though ERα and ERβ is involved during lesion development in proper organization of glandular/luminal epithelial and stromal cell areas. In addition, both ERα and ERβ responsiveness is needed in the lesions for them to reach full proliferative capacity.
Fig. 3.

An increase in the proliferative marker Ki67 is seen after estradiol (E2) treatment in endometriosis-like lesions. Representative photomicrographs demonstrating Ki67 expression in WT to WT, WT to αERKO, WT to βERKO, αERKO to WT, and βERKO to WT lesions from mice treated with vehicle or estradiol (2.5 μg/mouse·wk) for 3 wk. Magnification, ×400.
Because altered PR levels and progesterone sensitivity in endometriosis have been reported, it is important to examine the localization of PR expression in the endometriosis-like lesions (Fig. 4). As expected, PR localizes predominately to the glandular and luminal epithelial cells in all vehicle-treated groups. With estradiol treatment, the predominant localization of PR expression relocalizes to the stroma (14, 39), and this occurs in the WT to WT, WT to βERKO, and βERKO to WT groups. PR expression in αERKO uteri does not relocate from the epithelium to the stroma upon estrogen treatment due to the lack of ERα (13), and the same phenomenon was observed in the αERKO to WT lesions (Fig. 4). Surprisingly, PR expression was still seen, but the complete localization of PR to the stroma does not occur in the WT to αERKO lesions after estrogen treatment. PR was detected in both the epithelial and stromal areas of the lesions after estradiol treatment. ERα protein was also visualized by IHC in all compartments and in all lesions, except the αERKO to WT lesions, as expected (Supplemental Fig. 1).
Fig. 4.

PR localization does not redistribute from the epithelial to the stromal compartment with estradiol (E2) treatment in the WT to αERKO group. Representative photomicrograph demonstrating PR expression in WT to WT, WT to αERKO, WT to βERKO, αERKO to WT, and βERKO to WT lesions from mice treated with vehicle or estradiol (2.5 μg/mouse·wk) for 3 wk. Magnification, ×200.
To further examine the steroid receptor status, ERα and PR transcript levels were evaluated (Fig. 5A). In comparison with the ERα transcript levels in WT to WT vehicle-treated animals, a significant decrease in ERα was seen in the WT to βERKO and βERKO to WT vehicle-treated groups. When analyzing treatment effects on ERα expression, only WT to αERKO vehicle-treated vs. WT to αERKO estradiol-treated animals demonstrated a significant decrease in ERα levels. Upon transcript analysis of ERα and PR, the αERKO to WT lesions fell into two groups: the lesions that formed at the injection site and the floaters. A minimal amount of ERα could be detected in the injection site lesions most likely due to the presence of surrounding tissue that could not be fully dissected away from the lesions. PR transcript increased with estradiol treatment, and this increase was also seen when comparing WT to WT vehicle vs. WT to WT estradiol, WT to βERKO vehicle vs. WT to βERKO estradiol, and βERKO to WT vehicle vs. βERKO to WT estradiol. Surprisingly, no increase in PR expression with estradiol was seen when comparing WT to αERKO vehicle vs. WT to αERKO estradiol, suggesting a host-mediated effect, consistent with the lack of PR switch. The expression levels of PR in the vehicle group significantly decreased in WT to βERKO, αERKO to WT, and βERKO to WT compared with WT to WT, suggesting a donor genotype effect on basal PR levels. As expected without the ERα, no changes in PR expression levels were seen in the WT to αERKO lesions upon estradiol treatment. Collectively, these data suggest both a host and treatment effect on the ERα and PR expression levels and suggest the involvement of paracrine factors in lesion maintenance.
Fig. 5.

PR gene expression does not increase with estradiol treatment in WT to αERKO lesions, and all lesions except the αERKO to WT lesions are responsive to estradiol (E2) treatment when examining estradiol-responsive genes Ltf and Muc4. A, ERα and PR gene expression. B, Ltf, Muc4, and Krt18 gene expression. Total RNA was isolated from WT to WT, WT to αERKO, WT to βERKO, αERKO to WT, and βERKO to WT endometriosis-like lesions. Mice were treated with vehicle (corn oil) or estradiol (2.5 μg/mouse·wk) for 3 wk. Transcripts were quantified by real-time PCR as described under Materials and Methods and were normalized relative to RpL7. Letters indicate P < 0.05 by one-way ANOVA with Bonferroni's posttest; *, P < 0.05, unpaired one-tailed t test.
To examine the functionality of the ERα-mediated signaling in the lesions in response to estrogen treatment, two estrogen-mediated ERα target genes were examined (33, 40). Lactoferrin (Ltf) was significantly increased in all lesions except for the αERKO to WT lesions, whereas mucin 4 (Muc4) trended to increase in the WT to WT estradiol-treated lesions and was significantly increased in WT to αERKO, WT to βERKO, and βERKO to WT (Fig. 5B). These increases demonstrate that the responses were mediated by the ERα expression in the lesions, because lesions without ERα did not show an increase in Ltf or Muc4 gene expression. The examination of PR target genes, Indian hedgehog (Ihh), cyclin-dependent kinase inhibitor 1 (p21), c-fos, follistatin (Fst), and mitogen-induced gene 6 (Mig6), indicated none differed across groups (data not shown), as would be expected, because this model lacks circulating progesterone. Krt18, a marker for epithelial cells (41), increased in all groups but the αERKO to WT group after estradiol treatment. This result correlates with the increase in epithelial cell proliferation observed histologically (Fig. 3).
Because βERKO to WT and WT to βERKO lesions exhibited similar phenotypes to WT, additional studies examined αERKO-mediated differences. The lesions fixed for histology were stained by H&E and immunohistochemically for CD3 and F4/80 to assess degree of inflammation. The average scores are depicted in Fig. 6A, and CD3 and F4/80 IHC are shown in Supplemental Fig. 2. Minimal inflammation was seen in the WT to WT group with significant increases in inflammation in groups WT to αERKO and αERKO to WT. The inflammation score in the WT to αERKO group decreased with estradiol treatment, but the decrease in inflammation score did not occur in the αERKO to WT group with estradiol treatment. Transcript levels of inflammatory cytokines, TNFα, interferon γ (Ifnγ), regulated upon activation normal T cell expressed and secreted (Rantes), and IL-12 from the lesions were assessed (Fig. 6B). For WT to WT minimal expression of Tnfα, Ifnγ, Rantes, and Il-12 were observed, and the gene expression remained unchanged after treatment with estradiol. The expression of Tnfα, Ifnγ, and Il-12 was elevated in the WT to αERKO group, and Tnfα, Ifnγ, Rantes, and Il-12 all increased in a host-dependent manner in the αERKO to WT group. The levels of Tnfα, Ifnγ, Rantes, and Il-12 decreased in response to estradiol in the WT to αERKO group; however, a ligand-dependent decrease was not observed for Tnfα, Ifnγ, or Il-12 when the lesion lacked ERα (αERKO to WT group), suggesting a lesion-dependent response. In a similar manner, the same gene expression pattern was observed for Il-1β, Il-16, granulocyte colony-stimulating factor receptor (Gcsfr), and macrophage inflammatory protein 1β (Mip1b) (data not shown). The gene expression changes correspond with the inflammation scores assigned to the lesion. These data suggest both the host and donor have roles in the inflammatory response, but ERα is essential in the inflammatory properties of the endometriosis-like lesions.
Fig. 6.
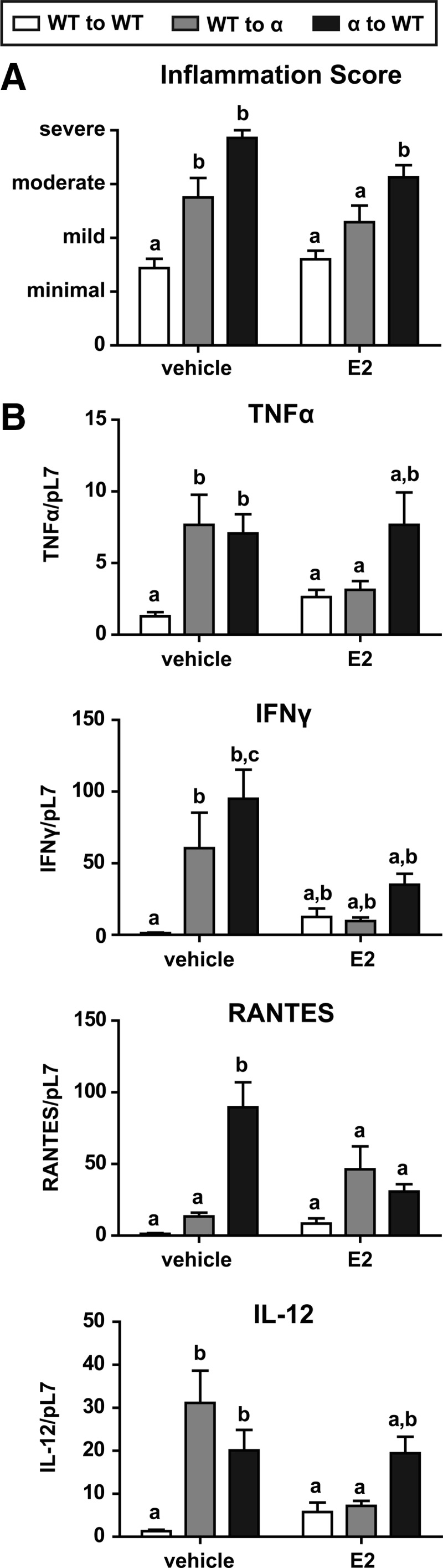
Endometriosis-like lesions from WT to αERKO and αERKO to WT vehicle-treated mice have higher inflammation scores, with the WT to αERKO increased inflammation score resolved with estradiol (E2) treatment. A, Inflammation score of endometirosis-like lesions. Lesions fixed for histology were stained by H&E and immunohistochemically for CD3 and F4/80 to assess degree of inflammation. When the inflammatory cells involved less than 10, 11–30, 31–60, and more than 60% of the tissue section, the inflammation was graded as minimal (1), mild (2), moderate (3), and severe (4), respectively. B, Gene expression analysis of inflammatory markers. Total RNA was isolated from WT to WT, WT to αERKO, WT to βERKO, αERKO to WT, and βERKO to WT endometriosis-like lesions. Mice were treated with vehicle (corn oil) or estradiol (2.5 μg/mouse·wk) for 3 wk. Transcripts were quantified by real-time PCR as described under Materials and Methods and were normalized relative to RpL7. Letters indicate P < 0.05 by one way ANOVA with Tukey's multiple-comparison posttest.
The presence of unattached floaters in the αERKO to WT group may be a result of the inability to establish a blood supply; therefore, we examined levels of angiogenic marker transcripts. Angiogenic markers were detected 21 d after tissue injection; however, no differences in transcript levels were observed across the different lesion groups. Markers of angiogenesis, Vegfa, Mmp7, and Timp1 were examined in uterine tissue from WT and αERKO mice treated with estradiol (10 μg/kg) for 2 h. An estradiol-dependent increase in gene expression of vascular endothelial growth factor (VEGF) a (Vegfa), matrix metalloproteinase (MMP) 7 (Mmp7), and tissue inhibitor of matrix metalloproteinases (TIMP) 1 (Timp1) was seen in the WT uteri; however, no changes in gene expression were seen in the αERKO uteri (Fig. 7). These data emphasize the critical nature of ERα in uterine angiogenesis and of the development of endometriosis-like lesions.
Fig. 7.

Key angiogenic markers are not increased with estradiol (E2) treatment in αERKO uteri. Total uterine RNA was isolated from WT and αERKO mice treated with vehicle (0.85% saline/0.25% ethanol) or estradiol (250 ng/mouse) for 2 h. Transcripts were quantified by real-time PCR as described under Materials and Methods and were normalized relative to RpL7. *, P < 0.05, unpaired one-tailed t test.
Discussion
Using a mouse model of endometriosis, we demonstrate the importance of ERα and ERβ activity and estradiol-mediated signaling in multiple components of the development of endometriosis-like lesions (Fig. 8). First, establishing lesions distal to the injection site requires ERα because no lesions are formed distally from injecting αERKO uterine tissue. We find, as shown previously (42–44), that estradiol is not required for establishment of endometriosis-like lesions. Second, we demonstrated the lack of attachment is likely due to the inability of αERKO uteri to secrete necessary mediators/chemotactic agents for neoangiogenesis in response to estradiol. Third, ERα and ERβ and estradiol are involved in the establishment of a pattern for lesion growth. Lesions from or in hosts lacking ERα or ERβ are much less organized than the WT to WT vehicle-treated controls, but after estradiol treatment, WT lesions become organized (WT to αERKO and WT to βERKO). Fourth, ERα and estradiol are involved in modulation of lesion inflammation and/or paracrine signaling. WT tissue into αERKO host (WT to αERKO) has lesions that have increased inflammation scores that decrease with estradiol treatment, but the classical epithelial to stromal PR relocalization does not occur. These findings suggest the important role of paracrine-mediated responses in lesion responsiveness. Finally, endometriosis-like lesion growth is mediated predominately by estradiol signaling via ERα to increase lesion size, fluid volume, increased epithelial cell height, and epithelial cell proliferation.
Fig. 8.

Estrogen signaling through ERα is required for endometriosis-like lesion growth and responsivity. A schematic representation is shown of the roles of ERα, ERβ, and estradiol in endometriosis-like disease pathology in an immunocompetent mouse model. Symbols denote where ERα, ERβ, or estradiol are important for endometriosis-like lesions.
Evaluation of human endometriosis lesions demonstrates increased ERβ expression and increased ERβ promoter methylation (23); accordingly, we anticipated a more robust effect on lesion development using the βERKO as either donor or host. Human endometrial xenografts implanted into immunocompromised mice and treated with the ERβ-selective agonist ERb-041 prevented lesion formation in 40–75% of mice (45). In our immunocompetent mouse model, the slight differences seen in the βERKO to WT group regarding a minor reduction in estrogen-induced increase in lesion weight and subtle differences in lesion tissue organization and cell proliferation suggests a primary role for ERα. However, the results indicate a potential role for ERβ in the endometriosis-like lesions for forming proper tissue structures and a maximal mitogenic response to estradiol, compared with the WT to WT endometriosis-like lesions. In humans, ERβ is expressed throughout the menstrual cycle and is highest during the preovulatory period, whereas minimal ERβ is detected in the mouse uterus (46); this difference may account for the discrepancy in the role of ERβ between our study and previous reports.
In women, endometrial lesions colonize the peritoneal cavity and attach to organs in the abdomen such as the bowel, bladder, fallopian tubes, ligaments, ovaries, and cul-de-sac areas of the uterus (5). In our mouse model, we found endometriosis-like lesions in these same locations suggestive of the involvement of paracrine, inflammatory, or chemoattractive factors in lesion placement. The influence of estradiol on inflammation is observed in women with endometriosis (5, 47). Estradiol affects subpopulations of cytokines and promotes endothelial healing and angiogenesis in the cardiovascular system (48). In our model, estradiol signaling through ERα shows increased mitogenesis and decreases the transcript levels of Ifnγ, Tnfα, and Il-12. Estradiol has both proinflammatory and antiinflammatory roles depending on context, which in turn affects neoangiogenesis, a process critical for establishment of endometriosis lesions (9). The normal uterus requires angiogenesis to undergo vascular and glandular proliferation, differentiation, and regeneration during each menstrual cycle (49). Neoangiogenesis is a cascade of events tightly regulated by factors including VEGF family members, MMP family members and TIMP family members which culminates in the initiation of blood flow (49). Estrogen regulates VEGF, and expression levels are increased in eutopic and ectopic endometrium of women with endometriosis and mouse models of endometriosis (50–53). Consistent with our findings, mouse models of endometriosis show angiogenic inhibitors decrease endometriosis-like lesion formation (54, 55). MMP family members, which promote invasion, are infrequently expressed in healthy adult tissue; however, MMP are dysregulated in patients with endometriosis, and in a rat model of endometriosis, neutralization of TIMP-1 restores fecundity (56–60). We demonstrate the lack of establishment of αERKO lesions distal to the injection site is linked to the inability of estradiol via ERα to induce angiogenic factor responses needed to provide vasculature to the αERKO uterine tissue. Validating to our study, the production of leptin, a cytokine that possesses both immune and angiogenic properties, is modulated by estradiol, and in an endometriosis mouse model, blocking leptin impairs the establishment and development of endometriosis-like lesions (30, 61). In addition, the requirement for ERα for lesion establishment in our model is consistent with the use of various hormone or antihormone treatments and their partial effectiveness for endometriosis treatments (5).
Estrogen replacement antagonizes the onset of inflammation caused by the lack of endogenous estrogens (62). Along with our model, in which estradiol suppresses inflammatory factors, this occurs in other disease models such as adjuvant-induced arthritis and cutaneous wound healing (63). A decrease in macrophage recruitment by estradiol subsequently suppresses monocyte recruitment/macrophage activation at the inflammation site, which allows estradiol to have a beneficial effect on inflammatory disease (62). In the uterus, macrophage recruitment is hormone dependent with uterine epithelium and stroma being responsive to antigenic challenges (64, 65). An endometriosis mouse model blocking p38 MAPK decreased the severity of endometriosis due to a decrease in inflammatory markers found in the peritoneal cavity (66). In our model, the inflammatory response is minimal/mild in the WT to WT group, which has large cystic estradiol-responsive lesions. The WT to αERKO vehicle group has a moderate inflammatory response but is resolved with estradiol, presumably due to the estradiol response through ERα expression in the lesions. In the αERKO to WT group, the immune response is severe in the injection site lesions, and the severity of inflammation is not resolved with estradiol treatment. The unattached floaters of injected uterine tissue were seen only in the αERKO to WT groups; therefore, we hypothesize the lack of clearance and attachment of the αERKO minced uterine tissue is due to a lack of chemotactic and angiogenic factors as they attract the necessary monocytes, macrophages, T cells, B cells, and other immune cell types required for neovascularization (26). Additionally, with estradiol selectively altering the secretion of chemokines (64), we suggest, and a focus of our future studies, that the lack of PR epithelial to stromal relocalization in WT to αERKO estradiol-treated lesions is likely due to the lack of secretion of a paracrine factor or that the necessary paracrine factors are not regulated properly by the αERKO host. The disruption in paracrine signaling does not allow for the proper stimulation to initiate the switch of PR from the epithelial to stromal components in this group, demonstrating the importance of the host peritoneal environment for lesion responsiveness. Additionally, as seen with other endometriosis-model systems, the WT to αERKO lesions do not have increased PR transcript levels after estradiol treatment or altered gene expression of tested PR target genes (67, 68). Our results demonstrate the important role for the host estradiol, ERα-mediated signaling on lesion responsiveness.
The effects of estradiol are dependent on multiple criteria including the immune stimulus, the cell types involved during different phases of disease, the amount of estradiol, and the microenvironment (26). The roles of estradiol on inflammation and neoangiogenesis via ERα oppose each other in endometriosis, and this is seen in our model system. The WT to WT estradiol group, having increased mitogenesis and a decrease in inflammation results in larger, cystic, proliferative lesions. This opposing role of estradiol in this model and in women with endometriosis requires in-depth focus on the mechanistic actions of ERα for the development of selective ER modulators that will allow for the uncoupling of these mechanisms of action to focus efforts for disease treatment.
Supplementary Material
Acknowledgments
We thank Drs. Bo Rueda and Jill Attaman for invaluable discussion regarding the induction of endometriosis-like disease. We thank James Clark for amazing surgical assistance. We thank Dr. Carmen Williams for helpful discussion and Drs. Kymberly Gowdy and Miranda Bernhardt for critical review of this manuscript.
This work was supported by National Institutes of Health Grant Z01ES70065.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ER
- Estrogen receptor
- αERKO
- ERα-knockout
- H&E
- hematoxylin and eosin
- IHC
- immunohistochemistry
- MMP
- matrix metalloproteinase
- PR
- progesterone receptor
- SMA
- smooth muscle actin
- TIMP
- tissue inhibitor of matrix metalloproteinases
- VEGF
- vascular endothelial growth factor
- vWF
- von Willebrand factor
- WT
- wild type.
References
- 1. Rawson JM. 1991. Prevalence of endometriosis in asymptomatic women. J Reprod Med 36:513–515 [PubMed] [Google Scholar]
- 2. Galle PC. 1989. Clinical presentation and diagnosis of endometriosis. Obstet Gynecol Clin North Am 16:29–42 [PubMed] [Google Scholar]
- 3. Sampson JA. 1927. Metastatic or Embolic Endometriosis, due to the Menstrual Dissemination of Endometrial Tissue into the Venous Circulation. Am J Pathol 3:93–110.43 [PMC free article] [PubMed] [Google Scholar]
- 4. Bulun SE, Imir G, Utsunomiya H, Thung S, Gurates B, Tamura M, Lin Z. 2005. Aromatase in endometriosis and uterine leiomyomata. J Steroid Biochem Mol Biol 95:57–62 [DOI] [PubMed] [Google Scholar]
- 5. Giudice LC, Kao LC. 2004. Endometriosis. Lancet 364:1789–1799 [DOI] [PubMed] [Google Scholar]
- 6. Sharpe-Timms KL. 2001. Endometrial anomalies in women with endometriosis. Ann NY Acad Sci 943:131–147 [DOI] [PubMed] [Google Scholar]
- 7. Capellino S, Montagna P, Villaggio B, Sulli A, Soldano S, Ferrero S, Remorgida V, Cutolo M. 2006. Role of estrogens in inflammatory response: expression of estrogen receptors in peritoneal fluid macrophages from endometriosis. Ann NY Acad Sci 1069:263–267 [DOI] [PubMed] [Google Scholar]
- 8. Bukulmez O, Hardy DB, Carr BR, Word RA, Mendelson CR. 2008. Inflammatory status influences aromatase and steroid receptor expression in endometriosis. Endocrinology 149:1190–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weiss G, Goldsmith LT, Taylor RN, Bellet D, Taylor HS. 2009. Inflammation in reproductive disorders. Reprod Sci 16:216–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Taylor HS, Bagot C, Kardana A, Olive D, Arici A. 1999. HOX gene expression is altered in the endometrium of women with endometriosis. Hum Reprod 14:1328–1331 [DOI] [PubMed] [Google Scholar]
- 11. Bischoff F, Simpson JL. 2004. Genetic basis of endometriosis. Ann NY Acad Sci 1034:284–299 [DOI] [PubMed] [Google Scholar]
- 12. Kulak J, Jr, Fischer C, Komm B, Taylor HS. 2011. Treatment with bazedoxifene, a selective estrogen receptor modulator, causes regression of endometriosis in a mouse model. Endocrinology 152:3226–3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Couse JF, Korach KS. 1999. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev 20:358–417 [DOI] [PubMed] [Google Scholar]
- 14. Mendelson CR. 2009. Fetal-maternal hormonal signaling in pregnancy and labor. Mol Endocrinol 23:947–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wen DX, Xu YF, Mais DE, Goldman ME, McDonnell DP. 1994. The A and B isoforms of the human progesterone receptor operate through distinct signaling pathways within target cells. Mol Cell Biol 14:8356–8364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Condon JC, Hardy DB, Kovaric K, Mendelson CR. 2006. Up-regulation of the progesterone receptor (PR)-C isoform in laboring myometrium by activation of nuclear factor-κB may contribute to the onset of labor through inhibition of PR function. Mol Endocrinol 20:764–775 [DOI] [PubMed] [Google Scholar]
- 17. Hewitt SC, Kissling GE, Fieselman KE, Jayes FL, Gerrish KE, Korach KS. 2010. Biological and biochemical consequences of global deletion of exon 3 from the ERα gene. FASEB J 24:4660–4667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Couse JF, Curtis SW, Washburn TF, Lindzey J, Golding TS, Lubahn DB, Smithies O, Korach KS. 1995. Analysis of transcription and estrogen insensitivity in the female mouse after targeted disruption of the estrogen receptor gene. Mol Endocrinol 9:1441–1454 [DOI] [PubMed] [Google Scholar]
- 19. Couse JF, Yates MM, Sanford R, Nyska A, Nilson JH, Korach KS. 2004. Formation of cystic ovarian follicles associated with elevated luteinizing hormone requires estrogen receptor-β. Endocrinology 145:4693–4702 [DOI] [PubMed] [Google Scholar]
- 20. Rodriguez KF, Couse JF, Jayes FL, Hamilton KJ, Burns KA, Taniguchi F, Korach KS. 2010. Insufficient luteinizing hormone-induced intracellular signaling disrupts ovulation in preovulatory follicles lacking estrogen receptor-β. Endocrinology 151:2826–2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bulun SE, Monsavais D, Pavone ME, Dyson M, Xue Q, Attar E, Tokunaga H, Su EJ. 2012. Role of estrogen receptor-β in endometriosis. Semin Reprod Med 30:39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Trukhacheva E, Lin Z, Reierstad S, Cheng YH, Milad M, Bulun SE. 2009. Estrogen receptor (ER) beta regulates ERalpha expression in stromal cells derived from ovarian endometriosis. J Clin Endocrinol Metab 94:615–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xue Q, Lin Z, Cheng YH, Huang CC, Marsh E, Yin P, Milad MP, Confino E, Reierstad S, Innes J, Bulun SE. 2007. Promoter methylation regulates estrogen receptor 2 in human endometrium and endometriosis. Biol Reprod 77:681–687 [DOI] [PubMed] [Google Scholar]
- 24. Mulac-Jericevic B, Mullinax RA, DeMayo FJ, Lydon JP, Conneely OM. 2000. Subgroup of reproductive functions of progesterone mediated by progesterone receptor-B isoform. Science 289:1751–1754 [DOI] [PubMed] [Google Scholar]
- 25. Attia GR, Zeitoun K, Edwards D, Johns A, Carr BR, Bulun SE. 2000. Progesterone receptor isoform A but not B is expressed in endometriosis. J Clin Endocrinol Metab 85:2897–2902 [DOI] [PubMed] [Google Scholar]
- 26. Straub RH. 2007. The complex role of estrogens in inflammation. Endocr Rev 28:521–574 [DOI] [PubMed] [Google Scholar]
- 27. Sinaii N, Cleary SD, Ballweg ML, Nieman LK, Stratton P. 2002. High rates of autoimmune and endocrine disorders, fibromyalgia, chronic fatigue syndrome and atopic diseases among women with endometriosis: a survey analysis. Hum Reprod 17:2715–2724 [DOI] [PubMed] [Google Scholar]
- 28. Kelly RW, King AE, Critchley HO. 2001. Cytokine control in human endometrium. Reproduction 121:3–19 [DOI] [PubMed] [Google Scholar]
- 29. Punnonen J, Teisala K, Ranta H, Bennett B, Punnonen R. 1996. Increased levels of interleukin-6 and interleukin-10 in the peritoneal fluid of patients with endometriosis. Am J Obstet Gynecol 174:1522–1526 [DOI] [PubMed] [Google Scholar]
- 30. Styer AK, Sullivan BT, Puder M, Arsenault D, Petrozza JC, Serikawa T, Chang S, Hasan T, Gonzalez RR, Rueda BR. 2008. Ablation of leptin signaling disrupts the establishment, development, and maintenance of endometriosis-like lesions in a murine model. Endocrinology 149:506–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khan KN, Kitajima M, Hiraki K, Fujishita A, Sekine I, Ishimaru T, Masuzaki H. 2008. Immunopathogenesis of pelvic endometriosis: role of hepatocyte growth factor, macrophages and ovarian steroids. Am J Reprod Immunol 60:383–404 [DOI] [PubMed] [Google Scholar]
- 32. Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. 2000. Effect of single and compound knockouts of estrogen receptors α (ERα) and β (ERβ) on mouse reproductive phenotypes. Development 127:4277–4291 [DOI] [PubMed] [Google Scholar]
- 33. Winuthayanon W, Piyachaturawat P, Suksamrarn A, Ponglikitmongkol M, Arao Y, Hewitt SC, Korach KS. 2009. Diarylheptanoid phytoestrogens isolated from the medicinal plant Curcuma comosa: biologic actions in vitro and in vivo indicate estrogen receptor-dependent mechanisms. Environ Health Perspect 117:1155–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Couse JF, Curtis SW, Washburn TF, Eddy EM, Schomberg DW, Korach KS. 1995. Disruption of the mouse oestrogen receptor gene: resulting phenotypes and experimental findings. Biochem Soc Trans 23:929–935 [DOI] [PubMed] [Google Scholar]
- 36. Anaf V, Simon P, Fayt I, Noel J. 2000. Smooth muscles are frequent components of endometriotic lesions. Hum Reprod 15:767–771 [DOI] [PubMed] [Google Scholar]
- 37. van Kaam KJ, Schouten JP, Nap AW, Dunselman GA, Groothuis PG. 2008. Fibromuscular differentiation in deeply infiltrating endometriosis is a reaction of resident fibroblasts to the presence of ectopic endometrium. Hum Reprod 23:2692–2700 [DOI] [PubMed] [Google Scholar]
- 38. Kitano T, Matsumoto T, Takeuchi H, Kikuchi I, Itoga T, Sasahara N, Kinoshita K. 2007. Expression of estrogen and progesterone receptors in smooth muscle metaplasia of rectovaginal endometriosis. Int J Gynecol Pathol 26:124–129 [DOI] [PubMed] [Google Scholar]
- 39. Condon JC, Jeyasuria P, Faust JM, Wilson JW, Mendelson CR. 2003. A decline in the levels of progesterone receptor coactivators in the pregnant uterus at term may antagonize progesterone receptor function and contribute to the initiation of parturition. Proc Natl Acad Sci USA 100:9518–9523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gollub EG, Waksman H, Goswami S, Marom Z. 1995. Mucin genes are regulated by estrogen and dexamethasone. Biochem Biophys Res Commun 217:1006–1014 [DOI] [PubMed] [Google Scholar]
- 41. Mo B, Vendrov AE, Palomino WA, DuPont BR, Apparao KB, Lessey BA. 2006. ECC-1 cells: a well-differentiated steroid-responsive endometrial cell line with characteristics of luminal epithelium. Biol Reprod 75:387–394 [DOI] [PubMed] [Google Scholar]
- 42. Lin YJ, Lai MD, Lei HY, Wing LY. 2006. Neutrophils and macrophages promote angiogenesis in the early stage of endometriosis in a mouse model. Endocrinology 147:1278–1286 [DOI] [PubMed] [Google Scholar]
- 43. D'Hooghe TM, Bambra CS, Raeymaekers BM, De Jonge I, Lauweryns JM, Koninckx PR. 1995. Intrapelvic injection of menstrual endometrium causes endometriosis in baboons (Papio cynocephalus and Papio anubis). Am J Obstet Gynecol 173:125–134 [DOI] [PubMed] [Google Scholar]
- 44. Hirata T, Osuga Y, Yoshino O, Hirota Y, Harada M, Takemura Y, Morimoto C, Koga K, Yano T, Tsutsumi O, Taketani Y. 2005. Development of an experimental model of endometriosis using mice that ubiquitously express green fluorescent protein. Hum Reprod 20:2092–2096 [DOI] [PubMed] [Google Scholar]
- 45. Harris HA, Bruner-Tran KL, Zhang X, Osteen KG, Lyttle CR. 2005. A selective estrogen receptor-beta agonist causes lesion regression in an experimentally induced model of endometriosis. Hum Reprod 20:936–941 [DOI] [PubMed] [Google Scholar]
- 46. Lecce G, Meduri G, Ancelin M, Bergeron C, Perrot-Applanat M. 2001. Presence of estrogen receptor beta in the human endometrium through the cycle: expression in glandular, stromal, and vascular cells. J Clin Endocrinol Metab 86:1379–1386 [DOI] [PubMed] [Google Scholar]
- 47. Nothnick WB. 2001. Treating endometriosis as an autoimmune disease. Fertil Steril 76:223–231 [DOI] [PubMed] [Google Scholar]
- 48. Arnal JF, Fontaine C, Billon-Galés A, Favre J, Laurell H, Lenfant F, Gourdy P. 2010. Estrogen receptors and endothelium. Arterioscler Thromb Vasc Biol 30:1506–1512 [DOI] [PubMed] [Google Scholar]
- 49. Jaffe CA, Ocampo-Lim B, Guo W, Krueger K, Sugahara I, Demott-Friberg R, Barkan AL. 2000. Growth hormone secretory dynamics over the menstrual cycle. Endocr J 47:549–556 [DOI] [PubMed] [Google Scholar]
- 50. Bourlev V, Volkov N, Pavlovitch S, Lets N, Larsson A, Olovsson M. 2006. The relationship between microvessel density, proliferative activity and expression of vascular endothelial growth factor-A and its receptors in eutopic endometrium and endometriotic lesions. Reproduction 132:501–509 [DOI] [PubMed] [Google Scholar]
- 51. Takehara M, Ueda M, Yamashita Y, Terai Y, Hung YC, Ueki M. 2004. Vascular endothelial growth factor A and C gene expression in endometriosis. Hum Pathol 35:1369–1375 [DOI] [PubMed] [Google Scholar]
- 52. McLaren J, Prentice A, Charnock-Jones DS, Millican SA, Müller KH, Sharkey AM, Smith SK. 1996. Vascular endothelial growth factor is produced by peritoneal fluid macrophages in endometriosis and is regulated by ovarian steroids. J Clin Invest 98:482–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hirota Y, Tranguch S, Daikoku T, Hasegawa A, Osuga Y, Taketani Y, Dey SK. 2008. Deficiency of immunophilin FKBP52 promotes endometriosis. Am J Pathol 173:1747–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hull ML, Charnock-Jones DS, Chan CL, Bruner-Tran KL, Osteen KG, Tom BD, Fan TP, Smith SK. 2003. Antiangiogenic agents are effective inhibitors of endometriosis. J Clin Endocrinol Metab 88:2889–2899 [DOI] [PubMed] [Google Scholar]
- 55. Nap AW, Griffioen AW, Dunselman GA, Bouma-Ter Steege JC, Thijssen VL, Evers JL, Groothuis PG. 2004. Antiangiogenesis therapy for endometriosis. J Clin Endocrinol Metab 89:1089–1095 [DOI] [PubMed] [Google Scholar]
- 56. Osteen KG, Bruner-Tran KL, Keller NR, Eisenberg E. 2002. Progesterone-mediated endometrial maturation limits matrix metalloproteinase (MMP) expression in an inflammatory-like environment: a regulatory system altered in endometriosis. Ann NY Acad Sci 955:37–47; discussion 86–8, 396–406 [DOI] [PubMed] [Google Scholar]
- 57. Kyama CM, Mihalyi A, Simsa P, Falconer H, Fulop V, Mwenda JM, Peeraer K, Tomassetti C, Meuleman C, D'Hooghe TM. 2009. Role of cytokines in the endometrial-peritoneal cross-talk and development of endometriosis. Front Biosci (Elite Ed) 1:444–454 [DOI] [PubMed] [Google Scholar]
- 58. Bruner-Tran KL, Eisenberg E, Yeaman GR, Anderson TA, McBean J, Osteen KG. 2002. Steroid and cytokine regulation of matrix metalloproteinase expression in endometriosis and the establishment of experimental endometriosis in nude mice. J Clin Endocrinol Metab 87:4782–4791 [DOI] [PubMed] [Google Scholar]
- 59. Stilley JA, Birt JA, Nagel SC, Sutovsky M, Sutovsky P, Sharpe-Timms KL. 2010. Neutralizing TIMP1 restores fecundity in a rat model of endometriosis and treating control rats with TIMP1 causes anomalies in ovarian function and embryo development. Biol Reprod 83:185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Becker CM, Louis G, Exarhopoulos A, Mechsner S, Ebert AD, Zurakowski D, Moses MA. 2010. Matrix metalloproteinases are elevated in the urine of patients with endometriosis. Fertil Steril 94:2343–2346 [DOI] [PubMed] [Google Scholar]
- 61. Shimizu H, Shimomura Y, Nakanishi Y, Futawatari T, Ohtani K, Sato N, Mori M. 1997. Estrogen increases in vivo leptin production in rats and human subjects. J Endocrinol 154:285–292 [DOI] [PubMed] [Google Scholar]
- 62. Vegeto E, Ciana P, Maggi A. 2002. Estrogen and inflammation: hormone generous action spreads to the brain. Mol Psychiatry 7:236–238 [DOI] [PubMed] [Google Scholar]
- 63. Jansson L, Olsson T, Holmdahl R. 1994. Estrogen induces a potent suppression of experimental autoimmune encephalomyelitis and collagen-induced arthritis in mice. J Neuroimmunol 53:203–207 [DOI] [PubMed] [Google Scholar]
- 64. Sárvári M, Hrabovszky E, Kalló I, Solymosi N, Tóth K, Likó I, Széles J, Mahó S, Molnár B, Liposits Z. 2011. Estrogens regulate neuroinflammatory genes via estrogen receptors α and β in the frontal cortex of middle-aged female rats. J Neuroinflammation 8:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wira CR, Rossoll RM. 1995. Antigen-presenting cells in the female reproductive tract: influence of the estrous cycle on antigen presentation by uterine epithelial and stromal cells. Endocrinology 136:4526–4534 [DOI] [PubMed] [Google Scholar]
- 66. Yoshino O, Osuga Y, Koga K, Hirota Y, Hirata T, Ruimeng X, Na L, Yano T, Tsutsumi O, Taketani Y. 2006. FR 167653, a p38 mitogen-activated protein kinase inhibitor, suppresses the development of endometriosis in a murine model. J Reprod Immunol 72:85–93 [DOI] [PubMed] [Google Scholar]
- 67. Osteen KG, Igarashi TM, Bruner-Tran KL. 2003. Progesterone action in the human endometrium: induction of a unique tissue environment which limits matrix metalloproteinase (MMP) expression. Front Biosci 8:d78–d86 [DOI] [PubMed] [Google Scholar]
- 68. Osteen KG, Bruner-Tran KL, Ong D, Eisenberg E. 2002. Paracrine mediators of endometrial matrix metalloproteinase expression: potential targets for progestin-based treatment of endometriosis. Ann NY Acad Sci 955:139–146; discussion 157–158, 396–406 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.