

Abstract
The mammary gland develops through several distinct stages. The first transpires in the embryo as the ectoderm forms a mammary line that resolves into placodes. Regulated by epithelial/mesenchymal interactions, the placodes descend into the underlying mesenchyme and produce the rudimentary ductal structure of the gland present at birth. Subsequent stages of development – pubertal growth, pregnancy, lactation and involution – occur postnatally under the regulation of hormones. Puberty initiates branching morphogenesis, which requires growth hormone and estrogen, as well as IGF1, to create a ductal tree that fills the fat pad. Upon pregnancy the combined actions of progesterone and prolactin generate alveoli, which secrete milk during lactation. Lack of demand for milk at weaning initiates the process of involution whereby the gland is remodeled back to its pre-pregnancy state. These processes require numerous signaling pathways that have distinct regulatory functions at different stages of gland development. Signaling pathways also regulate a specialized subpopulation of mammary stem cells that fuel the dramatic changes in the gland occurring with each pregnancy. Our knowledge of mammary gland development and mammary stem cell biology has significantly contributed to our understanding of breast cancer and has advanced the discovery of therapies to treat this disease.
INTRODUCTION
The mammary gland (breast) distinguishes mammals from all other animals with its unique anatomical structure that secretes milk for the nourishment of the newborn. Mammary glands are epidermal appendages that evolved over 300 million years ago, most likely from apocrine sweat glands 1. They are complex secretory organs composed of a number of different cell types: epithelial cells that grow from the nipple into a fat pad, formed by adipocytes and infiltrated by vascular endothelial cells, fibroblasts and immune cells. This article focuses primarily on changes occurring in the epithelial compartment over the lifetime of the animal (Fig. 1). During embryogenesis, these changes are directed by signals from the mesenchyme, but during puberty and in adulthood, circulating hormones released from the pituitary and ovary provide additional instructive input. Two main cell types comprise the mammary epithelium: basal and luminal. The basal epithelium consists of myoepithelial cells, which generate the outer layer of the gland, and a small population of stem cells, which supply the different cell types. The luminal epithelium forms ducts and secretory alveoli and contains populations of cells defined by their hormone receptor status. Together with the myoepithelium, the luminal epithelium generates a bi-layered, tubular structure that allows form to meet function during lactation when the outer myoepithelial cells contract to squeeze milk from the inner alveolar luminal cells.
Figure 1. Illustration depicting the stages of postnatal mammary gland development.
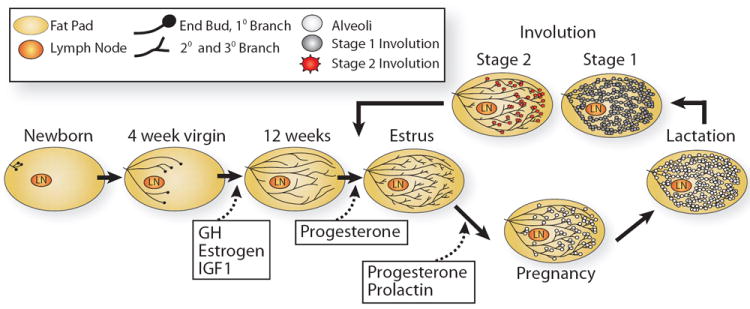
At birth, the mammary epithelium is rudimentary, consisting of only a few small ducts that grow allometrically until puberty (4-weeks in mice). With the onset of puberty, comes expansive growth in a process called ductal morphogenesis that fills the fat pad with the epithelial mammary tree. This growth is influence by growth hormone (GH) and estrogen, and a growth factor, insulin-like growth factor-1 (IGF1). In the mature virgin, short tertiary branches form under the influence of progesterone, but alveologenesis only occurs upon pregnancy with the induction of prolactin (PRL), which together with progesterone, fuels the growth of alveolar cells. PRL stimulation continues into the stage of lactogenesis, culminating in milk production that continues until a lack of demand at weaning signals involution and the mammary gland is remodeled back to its original adult state.
There are three major stages of breast development – embryonic, pubertal, and reproductive. Our knowledge of these stages is derived primarily from studies performed in mice, providing insight into the biology of the human breast. Although there are architectural and hormonal differences between mouse and human mammary glands, many researchers use the mouse mammary gland as a model system to explore developmental mechanisms because the gland is amenable to sophisticated in vivo and in vitro manipulations. These techniques have allowed researchers to investigate mechanisms underlying epithelial/mesenchymal interactions, hormonal regulatory controls and cell/cell communication. Here, we review how these basic processes shape the structure and function of the gland at each stage of development.
EMBRYONIC MAMMARY GLAND DEVELOPMENT
In the embryo there are two cellular compartments of the mammary gland, the epithelial compartment and the surrounding stromal compartment. These tissues are derived embryologically from ectoderm and mesoderm, respectively. The development of the murine gland begins at embryonic day (E) 10 with the formation of bilateral stripes (milk lines) of multilayered ectoderm that run anterior-to-posterior from the forelimb bud to hindlimb bud on the ventral surface of the embryo. By E11.5, the mammary line resolves into five pairs of placodes at reproducible locations. These 5 pairs develop asynchronously with 3 emerging first, followed by 4, then 1 and 5 simultaneously, followed finally by 2. The pairs are not identically determined as evidenced by loss or supernumerary formation of different pairs as a consequence of specific genetic mutations. Histologically, placodes appear as a thickened plate of ectoderm consisting of several layers of columnar-shaped cells that arise, not from cell proliferation 2, but instead from the migration and subsequent aggregation of ectodermal cells into surface clusters at the mammary line 3. In humans, the mammary lines form during the first trimester and give rise to a single pair of placodes 4.
Mammary line specification
The embryonic mammary mesenchyme underlying the mammary line provides key inductive signals that specify mammary epithelial cell differentiation. This was demonstrated in tissue recombination experiments in which E13 mouse mammary mesenchyme was combined with E13 midventral or dorsal epidermis and then grafted into host animals. These tissue recombinants yielded mammary ducts containing both luminal and myoepithelial cells and, if the hosts were pregnant upon grafting, lobuloalveolar structures that expressed milk proteins 5. The influence of mesenchyme on mammary epithelial development was also demonstrated in reciprocal tissue recombination experiments in which E12 and E16 mammary epithelium (ME) was combined with salivary mesenchyme (SM) and grown either in culture or under the kidney capsule 6,7. These tissue recombinants produced an epithelium that was morphologically reminiscent of salivary gland. However, during pregnancy and lactation of hosts harboring the older (E16 ME/SM) recombinants, the grafts displayed lobular-alveolar development and milk production 7. These and other studies show that mesenchyme contains critical inductive signals that produce, through a series of reciprocal epithelial-mesenchymal interactions, epithelium that is progressively restricted along the mammary lineage 8. In turn, the epithelium influences mesenchymal maturation such that by E14 the mammary mesenchyme has condensed to form a few layers of fibroblast-rich cells, closely surrounding the epithelial rudiment and distinct from the fat pad precursor mesenchyme, which develops from more deeply located subcutaneous mesenchymal cells 9.
The mammary line has been marked molecularly with Wnt10b and also the Wnt signaling reporter TOP (TCF optimal promoter)GAL (beta-galactosidase), which reports the transcriptional activity of the Tcf3/Lef1 target genes (Fig 2). Interestingly, TOPGAL activity appears one day earlier than Wnt10b expression in both the ectoderm and mesenchyme 10, indicating that an earlier Wnt signal emanates from the mesenchyme and initiates specification of the mammary lines. The essential nature of this Wnt signal in mammogenesis was demonstrated by the overexpression of a secreted Wnt inhibitor, Dickkopf (DKK1), in the ectoderm. This completely blocked the formation of mammary placodes and antagonized the local expression of Wnt10b, TOPGAL and other early markers such as the T-box transcription factor, Tbx3 10. Despite the importance of this initial signal in mammary gland specification, the identity of the Wnts that generate this initial signal is unknown, although there are several candidates. Wnt3, Wnt6 and Wnt10b are all expressed in the broad band of ectoderm flanking the mammary line 10, 11, and Wnt5a and Wnt11, which signal in a non-canonical fashion, are expressed in the underlying flank mesenchyme (Fig. 2) 10. This pattern of expression suggests that noncanonical Wnt signaling may restrict canonical signaling, limiting the response to Wnts to the ectodermal ridge that becomes the mammary line. Lef1-/- mice display a milder phenotype compared to mice overexpressing Dkk1. Instead of the absence of placode development, as is the case with Dkk1 overexpression, three pairs of small placodes form in Lef1-/- mice 12. This suggests that DKK1 quenches signaling from multiple TCF/LEF1 transcription factors that mediate the response to Wnts during mammary line specification and placode development, underscoring the complexity of the signaling required to pattern these specialized regions of ectoderm.
Figure 2. Illustration depicting the generation of the mammary milk line.
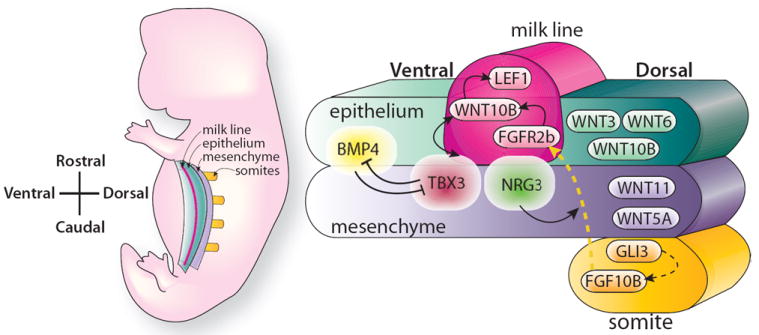
The position and specification of the multilayered epithelial ridge, the milk line, in an embryo at E10.5. The milk line is apparent between the forelimbs and hindlimbs that demarcate the rostral and caudal extension of the line. Specification of the line requires early Wnt signaling in the epithelium and in the mesenchyme that flanks the milk line. Tbx3 expression is required at early time points vertically under the line and it controls subsequent Wnt signaling within the line that is required for the development of mammary placodes. Tbx3 expression is regulated by FGF10 that emanates from somites underlying the line, as well as Wnt signaling in the flank and BMP4 signaling localized at the ventral border. For details, see text. (Adapted from Cowin and Wysolmerski 2010, with permission)
Placode development
Once the mammary line has been delineated, signals from the mesenchyme regulate the dorsal/ventral patterning of the placodes. The normal developmental outcome of numerous signaling cascades is 5 pairs (1 in humans) of symmetrically localized placodes, which have a button-like appearance on the surface of the ectoderm. TBX3 is a factor expressed very early, at E10.25, in a restricted region of mesenchyme underlying the presumptive mammary line 13, and later accumulating in the placodes. Mice homozygous for a null allele of Tbx3 exhibit an almost complete failure to develop mammary placodes, whereas the heterozygous deficiency results in poorly developed or missing glands 13. In humans, a similar defect is caused by haploinsufficiency for TBX3, resulting in ulnar-mammary syndrome characterized by upper limb deficiency and severe mammary hypoplasia 14. These phenotypes show that Tbx3 plays a fundamental role during mammary gland development, and other studies have demonstrated that its effects are exerted through the regulation of Wnt signaling. Wnt10b and Lef1 expression are expanded in mice ectopically expressing Tbx3 15, whereas their expression is absent in Tbx3-/- mice 16. These data show that TBX3 has the ability to position Lef1 expression and thereby define the mammary line. This raises the question as to the identity of the signal responsible for localizing Tbx3. Bone morphogenetic protein 4, BMP4, which is expressed along the ventral border of the mammary line, is one candidate for regulating the ventral extent of Tbx3 expression (Fig. 2)15. BMP4 antagonizes Tbx3 expression and vice versa, leading to a model of reciprocal antagonism, whereby the mammary line is delineated, at least in part, by the actions of these two factors restricting the expression of Wnt10b.
BMP4 and TBX3, however, do not work alone because fibroblast growth factors (FGFs) also pattern the dorsal/ventral placement of placodes by regulating the expression of Wnt10b and Lef1 (Fig. 2). Fgf10 is expressed at E10.5 in the central and ventral regions of thoracic somites underlying the mammary line 17. Identification of the potential role for this cue and its cognate receptor, FGFR2b, in mammary line formation came from the analysis of their knockout mice, which develop only the #4 placode 18. Since then, additional mutations have been discovered that similarly affect mammary placode development by inhibiting Fgf10 expression, either directly by affecting its expression, or indirectly by disrupting thoracic somite development. For example, Fgf10 expression is abolished in the Pax3 mutant, which fails to undergo ventral extension of thoracic somites, eliminating the somatic source of FGF10 and generating a corresponding dorsal shift in lateral line formation 17. A similar birth defect, Poland syndrome, occurs in humans and is characterized by the underdevelopment of the somite-derived pectoral muscle on one side of the body, and a corresponding hypoplasia of the overlying breast on that side 19. These defects arise from disruption in FGF10 signaling, rather than loss of the somite, because somitic development is normal in Fgf10 heterozygote animals, whereas Fgf10+/- glands are characterized by the absence of Wnt10b and Lef1 expression, reduced thickening of the ectoderm along the mammary line and subsequent loss of buds 3 and 5 17, 20. A similar phenotype occurs in mutant mice with no functional GLI3 (extratoes, Gli3Xt/Xt mutants). GLI3 is a transcription factor that generally functions as a transcriptional repressor in the hedgehog signaling pathway, although a hedgehog-independent role as a transcriptional activator has also been reported 21. The similarities between the Gli3Xt/Xt and Fgf10-/- phenotypes suggest that GLI3 positively regulates Fgf10 expression, similar to its regulation of other FGF family members in different developmental contexts (Fig. 2) 22. This idea is supported by the rescue of both Wnt10b expression, albeit at low frequency, and ectodermal thickening that occurs when FGF10 beads are implanted into the flanks of Gli3xt/xt mutants 17.
These data have allowed researchers to sketch a rudimentary signaling cascade in which GLI3 regulates FGF10 expression in the somite (Fig. 2). The FGF10 signal is transduced through the mesenchyme to regulate Wnt10b expression in the presumptive mammary epithelium. There are likely intervening factors that relay the signals from Gli3 to Fgf10, and, an as yet unidentified mechanism, which may facilitate the interaction between FGF10, expressed by the underlying somite and FGFR2b, expressed in the mammary line. One model proposes that FGF10-expressing cells are brought in direct contact with the mammary line by changes in cell adhesion that allow somitic cells, expressing FGF10, to delaminate, migrate and directly contact placode epithelium expressing FGFR2b 17. Neuregulin-3 (NRG3), a member of the epidermal growth factor (EGF) family, is a candidate regulator of cell-cell contacts. It is expressed in the mesenchyme underlying the mammary line and regulates the expression of cell adhesion receptors and extracellular matrix components such as tenascin, which is involved in tissue remodeling 23. In hypomorphic Nrg3Ska mutants, placode #3 is small or absent, and there is ectopic placodal development near placode #4 24. There is no loss of Fgf10 or mesenchymal Tbx3 expression in Nrg3Ska animals, indicating that NRG3 acts downstream of these factors. However, Wnt10b and Lef1 are altered, displaying decreased or absent expression at placode #3 and ectopic expression at the supernumerary buds, data that suggest an upstream role for NRG3 in regulating these factors. Concordantly, NRG3 coated beads or overexpression of Nrg3 in basal epithelia under the control of a keratin 14 (Krt14) promoter induces Wnt signaling and expression in the mammary line and generates supernumerary placodes that develop nipples and glandular epithelia 23, 25. Together, these studies suggest a role for NRG3 in signaling to its epithelial ERBB4 receptor and modulating cell adhesion that promotes transduction of somatic FGF10 signals and development of placodal epithelium.
Additional FGF signaling, mediated through FGFR1b and/or FGFR2c in the ectoderm, is required to induce and maintain Tbx3 expression, first in the mesenchyme underlying the mammary line and later in the placode 26. Positioning of the TBX3 signal in the region underlying the nascent placode is accomplished by the positive input of FGF10 17, and the negative regulation of BMP4 in the ventral mesenchyme 15. Once Tbx3 expression becomes localized, it generates a second wave of Wnt signaling, mediated by WNT10b that ultimately specifies the fate of the mammary epithelium by promoting the pseudo stratification of ectodermal cells to generate placodes. The signaling pathways that lie downstream of Lef activation in the mammary placode are poorly understood, but likely include the target Edar, a receptor for ectodysplasin-A (EDA), which is a member of the TNF superfamily 27. Overexpression of Eda results in the formation of supernumerary placodes located between the normal 3rd and 4th placodes 28. Mutations in the corresponding gene in humans causes hypohidrotic ectodermal dysplasia (HED), characterized by the lack of breast and other tissues that develop from the ectoderm (e.g. skin, hair, nails, teeth and sweat glands) 29. Together, these studies illustrate the important insights into placode development that have been derived from the study of mouse mutants. However, it also clear from these studies that our understanding is incomplete and that each pair of placodes in mice appear to have both shared and distinct genetic requirements for their formation.
Mammary bud and rudimentary duct formation
During the next stage of development, mammary placodes expand to form a round ball of cells that descends into the underlying mesenchyme by E14 (Fig. 3) 30. This sphere of cells continues to descend as a stalk is formed connecting the mammary bud to the epidermis. Surrounding the epithelium is the condensed mesenchyme comprising a thin layer of fibroblastic cells. At E16, a solid cord of epithelial cells extends from the mammary bud and grows through this condensed mesenchyme into the fat pad precursor mesenchyme, which at this stage is a small collection of preadipocytes. Once the epithelial sprout reaches the fat pad, it begins to branch by equal division of the terminal bud. This dichotomous branching yields a rudimentary ductal system of 10-15 branches that is generated without hormonal input and remains largely quiescent until puberty. In humans, instead of a single epithelial sprout extending from the mammary bud, several sprouts form, creating multiple mammary trees that unite at the nipple.
Figure 3. A schematic representation portraying embryonic mammary gland development.
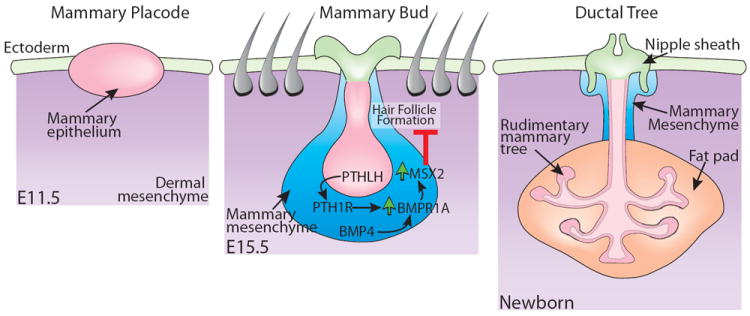
Mammary placodes expand into a ball of cells that descends into the underlying mesenchyme. Parathyroid hormone-related protein (PTHLH) signals from the epithelium to the mesenchyme to increase the expression of bone morphogenetic protein receptor-1A (BMPR1A). Bone morphogenetic protein 4 (BMP4) expressed in the mesenchyme signals through BMPR1A to MSX2 and inhibits hair follicle formation at the developing nipple sheath. The mammary epithelium grows into a small, simple tree-like structure containing an open lumen and remains in this form until birth. For details, see text. (Adapted from Cowin and Wysolmerski 2010, with permission)
Before embryonic development concludes, three additional morphological processes occur, formation of the ductal lumen, generation of the nipple structure and, in mice, specification of male versus female mammary glands. Lumen formation is initially observed at E16 when intercellular spaces develop within the cords. These spaces increase in size and number until a distinct lumen is apparent at E18 31. Studies performed in cultured mammary cells grown in a 3-dimensional matrix have implicated three processes in mammary lumen formation: apoptosis, autophagy and cellular remodeling 32-34; however, the precise mechanisms giving rise to open luminal space in the embryonic epithelial rudiments have not yet been delineated. Nipple generation occurs by modifications of the skin overlying the primary mammary mesenchyme and involves thickening of the epidermis, suppression of hair follicles and generation of a nipple sheath from keratinocytes at the site where the primary duct connects to the skin surface 35. Sexual dimorphism of the mammary gland is also achieved during this stage of embryonic development in mice, but later in humans. In male mice, the mesenchyme surrounding the stalk continues to condense until it severs the bud resulting in a greatly diminished ductal system 36. These latter two processes, nipple generation and sexual dimorphism, require signaling that is mediated by the parathyroid hormone-related protein (PTHLH) (described below). In humans, sexual dimorphism is accomplished via a different mechanism and the bud is not severed during embryogenesis. Instead, male and female glands undergo comparable development until puberty when their regulation by different hormones specifies their size 4. Thus, human male breasts retain a normal growth potential as evidenced by the incidence of gynecomastia, the benign enlargement of breast tissue in males as a consequence of altered estrogen-androgen balance.
The parathyroid hormone-related protein (PTHLH) plays a central role in regulating the transition from budding to branching. PTHLH is produced by the mammary epithelium. It signals through the G-protein coupled receptor, Type1 PTH/PTHLH receptor (PTH1R) 37, expressed in the mammary mesenchyme 38. The absence of either PTHLH or PTH1R in knockout mice, results in chondrodystrophy leading to neonatal death and characterized by numerous embryonic defects, including a lack of nipples and mammary ducts. A similar phenotype is also observed in humans suffering from loss-of-function mutations in the PTH1Rgene, resulting in a rare, lethal form of dwarfism known as Blomstrand chondrodysplasia 39, characterized by severe defects in endochondral bone formation and the absence of nipple or duct development 40. Careful examination of Pthlh-/- and Pth1r-/- mutant glands reveal normal development throughout the mammary bud stage, but subsequent failure of the bud to undergo the initial phase of branching at E16 41. This results in degeneration of mammary epithelial cells, such that by birth only the scattered remnants of ductal structures remain, although the fat pad and vasculature remain intact. Moreover, mutant glands lack nipple structures, which failed to develop on the ventral epidermal surface.
Further investigation into PTHLH signaling has revealed that the pathway maintains epithelial cell fate and triggers nipple differentiation, in part by orchestrating events mediated by other signaling systems, notably Wnt and Bmp. As discussed in the section above, Wnt signaling plays an essential role in mammary gland specification during placode development, but by E14, Lef1 expression is downregulated in both the epithelium and the epidermis, whereas it is upregulated in the mammary mesenchyme. In Pthlh-/- and Pth1r-/- glands this developmental regulation of Lef1 does not occur. Instead, expression of Lef1 is retained in the epithelium and in the epidermal cells surrounding the presumptive nipple; yet it is absent in the mesenchyme 42. These studies suggest that PTHLH, expressed in the epithelium, signals to mesenchymal PTH1R to modulate Wnt signaling and, consequently, cell fate. This idea is supported by experiments in which Pthlh expression is expanded into basal keratinocytes by driving its expression under the regulation of a Krt14 promoter. In these animals, Lef1 expression is retained in the ventral epidermis, which becomes in essence hairless nipple skin 42. These effects of PTHLH appear to require BMP4 signaling, which is augmented through the upregulation of the BMP receptor-1A, BMPR1A, by PTHLH in the mesenchyme (Fig. 3) 35. BMP4 rescues the outgrowth of Pthlh-/- mammary buds in culture 35. It also increases the expression of transcription factor Msx2 in mesenchyme, a regulatory event that is required for PTHLH suppression of hair follicle formation upon the nipple, as demonstrated by the recovery of hair follicle development in Krt14-Pthlh transgenic mice that have been crossed with Msx2-/- mice 35.
Altogether, these studies on PTHLH signaling underscore the importance of epithelial/mesenchymal interactions in regulating processes such as mammary bud formation. PTHLH, produced by the epithelia, acts on its receptor, expressed in the surrounding mesenchyme to modulate Wnt and BMP signaling and induce the production of a specialized condensed mesenchyme that maintains mammary epithelial cell fate. This triggers nipple sheath formation in the overlying epidermis and initiates ductal outgrowth. By the end of embryonic development, these and other regulatory processes generate a rudimentary ductal system that provides the framework for mammary outgrowth during puberty.
PUBERTAL MAMMARY GLAND DEVELOPMENT
At birth the gland is just a rudimentary ductal system, yet it is competent to produce milk. Fetal exposure to maternal hormones can cause milk expression in human babies, which is sometimes referred to as witch’s milk. As these endocrine influences subside, the gland undergoes a period of allometric growth, keeping up with overall body development, until puberty when expansive proliferation occurs, filling the fat pad under the influence of hormones and growth factors (Fig. 1). Terminal end buds (TEBs) are club-shaped structures at the tips of growing ducts that penetrate the fat pad, driven by the proliferation of a single layer of cap cells at the tip of the TEB and by the underlying prelumenal epithelium (Fig. 4). The primary ductal structure is generated by TEB bifurcation and is regulated by the surrounding stroma. Cap cells of the TEB differentiate into myoepithelial cells, forming the outer layer of the tubular ductal bilayer that encircles inner luminal cells 43. Secondary branches sprout laterally from the primary ducts until a tree-like pattern of ducts occupies up to 60% of the available fatty stroma, leaving abundant space for infilling that will occur under the influence of pregnancy hormones. Under cyclical ovarian stimulation, short tertiary branches will form, but full blossoming of the alveolar buds into units capable of milk secretion occurs only under the influence of pregnancy hormones (Fig. 1). In human, the pubertal breast contains a similarly extensive mammary tree, but the lateral branches lead to terminal ducts that give rise to terminal ductal-lobular units comprising numerous blind-ended ductules, called acini 4. These acini are embedded in fibroblastic, intralobular stroma that is far more pronounced in the human breast than the adipocytes-rich stroma surrounding the branches of the rodent mammary tree.
Figure 4. Schematized view of the events occurring during pubertal development.
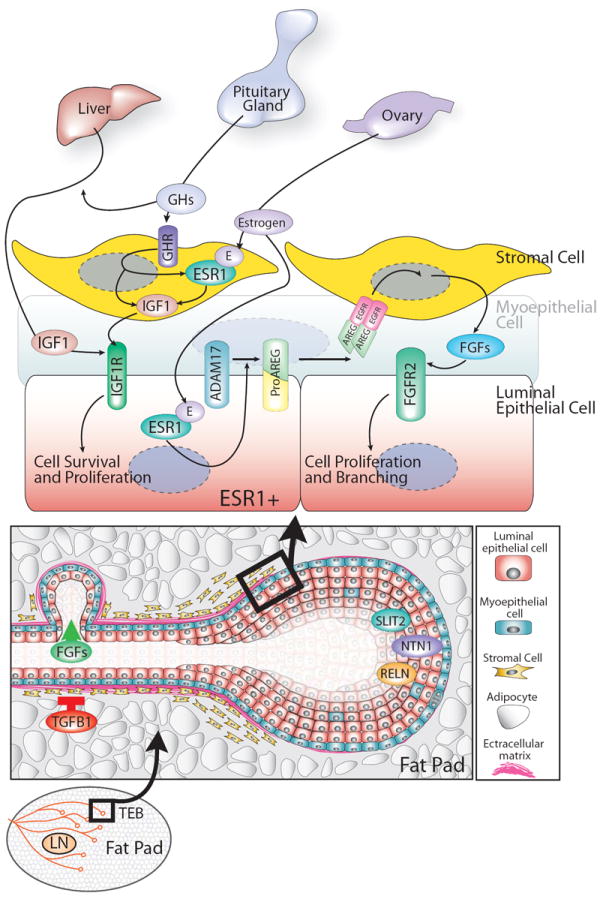
Terminal end buds (TEBs) grow through the mammary fat pad, fueled by cell proliferation (diagram at the bottom). Growth hormone (GH) regulates cell proliferation by inducing the expression of insulin-like growth factor-1 (IGF1) in both the liver and mammary stroma. IGF1 acts, together with estrogen secreted from the ovary, to induce epithelial cell proliferation (diagram at the top). Estrogen signaling through its receptor (ESR1) acts via a paracrine fashion to stimulate the release of epidermal growth factor (EGF) family member, AREG, which proceeds to bind its receptor on stromal cells and induce expression of FGFs. FGFs, in turn, to stimulate luminal cell proliferation. Other factors, such as TGFB1, Reelin (RELN), Slit2 and Netrin1 (NTN1) contribute to mammary architecture by either positively or negatively regulating cell proliferation or maintaining cell-cell interactions. For details, see text.
Hormones and growth factors control ductal morphogenesis
Studies, performed as early as the 1930s, demonstrated that extracts of the pituitary gland regulate mammary gland function, with researchers observing enhanced mammogenesis and lactogenesis upon its administration 44. Two hormones were identified that are responsible for these effects: growth hormone (GH) and prolactin (PRL). Later studies showed that mammary pubertal development was disrupted in mice lacking Gh, insulin-like growth factor 1 (Igf1) or estrogen receptor (alpha) (Esr1), genes that mediate pathways regulating ductal outgrowth and morphogenesis. In contrast, development of the adolescent gland occurs normally in mice lacking Prl or progesterone receptor (Pgr), genes that mediate signaling pathways regulating alveologenesis (described in the next section).
Growth hormone and IGF1 are global directors of postnatal development
Growth hormone, secreted from the pituitary gland, is an important global regulator of mammary gland development, but current evidence suggests that its effect on the mammary gland is mediated largely, or even entirely, through IGF1 whose production it stimulates in the stroma (Fig. 4) 45, 46. Growth hormone receptor (Ghr) knockout mice are small for their age, creating a murine model of the human Laron syndrome, a hereditary dwarfism disorder resulting from defects in GHR 47. Ghr-/- mice also display a dramatic (90%) reduction in serum IGF1 levels 47 and delayed mammary gland development with eventual outgrowth of only a sparse tree 45. To evaluate the compartment of the mammary gland (epithelial or stromal) that is affected by the loss of Ghr, tissue recombination experiments were performed in which Ghr-/- KO and WT epithelium were grafted into WT stromal fat pads. In these experiments, the endogenous WT epithelium is pre-cleared from the host fat pad to prevent confusion arising from a fat pad containing both host WT and graft KO epithelium. Recombinants containing Ghr-/- epithelium/WT stroma looked identical to WT/WT recombinants, demonstrating that GH signaling is required in the mammary stroma, but not the mammary epithelium 45. The current model is that GH signals through GHR in stromal fibroblasts, inducing IGF1, which then signals to the mammary epithelium (Fig. 5). This model is supported by studies on Igf1-/- mice, which exhibit greatly diminished ductal development, similar to the phenotype observed in Ghr-/- mice 46. Furthermore, Igf1-/-mice produce normal levels of GH and their defects in mammary development can be rescued by IGF1 treatment, demonstrating the essential nature of IGF1 on mammary gland development downstream of GH.
Figure 5. Schematized view of the events that generate lactation competence during pregnancy.
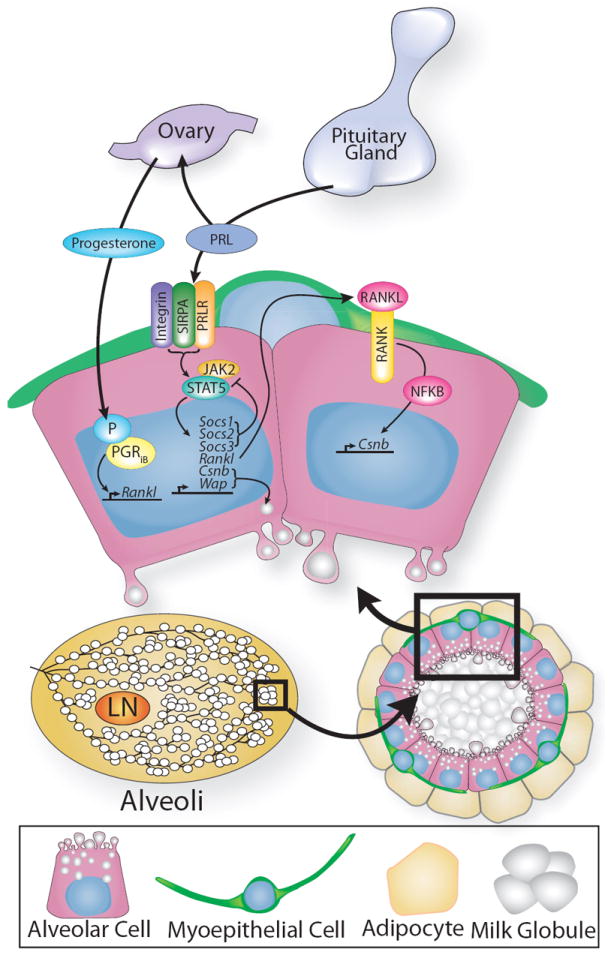
Alveoli develop into milk-secreting lobules regulated by prolactin (PRL) that works together with progesterone. Both hormones regulate transcriptional programs that include the control of Rankl TNFSF11), which signals in a paracrine fashion to stimulate proliferation by upregulating expression of target genes such as Cyclin delta 1 (Ccnd1) through the RANK receptor on neighboring cells. Progesterone stimulates secondary and tertiary branching, while PRL integrates many signals, including those from the extracellular matrix (ECM) by interacting with integrin through transmembrane, signal regulatory protein alpha (SIRPA). PRL transduces this information through pathways, such as those mediated by JAK2/STAT5, whose downstream targets include milk genes casein beta (Csnb) and whey acidic protein (Wap). For details, see text.
In the last decade, researchers have tried to determine the relative importance in mammary gland development of IGF1 produced locally versus that produced systemically by the liver. Initial results suggested that local IGF1, functioning in a paracrine, rather than endocrine, manner, plays a central role. This was inferred from studies on mice that carry an insertional mutation in the Igf1 gene, Igf1tm2Ts also known as the Igf1 midi allele, resulting in a 70%, rather than 100%, reduction in Igf1 expression 48. These mice display reduced mammary ductal branching, confirming the importance of Igf1. In contrast, a liver specific deletion, which reduces the levels of circulating IGF1 by 75%, displays normal ductal morphogenesis, suggesting that circulating IGF1 is nonessential 48. However, neither of these mouse models directly address the effects of circulating IGFI in the presence or absence of local IGFI. To accomplish this, mice were generated that produce only circulating IGF1 by overexpressing an Igf1 transgene specifically in the liver of Igf1-/- mice 49. These conditions rescued not only body size, but also mammary gland development, showing that systemic IGF1 can compensate for the loss of local IGF1 production. Nevertheless, there appears to be interplay between systemic and local sources because control mice, expressing both normal levels of mammary IGF1 and transgenic levels of serum IGF1, displayed increased cellular proliferation and enhanced mammary development 49. These results suggest that an overabundance of IGF1 can inappropriately drive mammary epithelial cell growth, for example in the tumor setting. Indeed, high levels of IGF1 have been associated with an increased risk of malignancy 50, 51, but the relative contribution of local versus circulating IGF1 to tumorigenesis has not yet been determined.
Estrogen manages local cell growth during puberty
The ovarian hormone, estrogen, is another critical regulator of pubertal mammary development and is responsible for the tremendous surge in growth occurring during this period that generates a functional mammary gland (Fig. 4). Estrogen, a membrane-soluble ligand, is released from the ovary and activates gene expression through intracellular receptors. For a long time, it was unclear whether hormones such as estrogen had direct effects on mammary gland development or whether, instead, they functioned indirectly to stimulate the release of hormones such as prolactin from the pituitary 52. The first studies to show the direct influence of estrogen on mammary gland development involved the delivery of estrogen or anti-estrogen directly to the gland using Elvax pellets, a biologically compatible polymer that can be implanted into tissue 53. These studies unequivocally demonstrated that estrogen stimulates mammary ductal growth in both ovariectomized and intact animals and that this stimulation is blocked by the estrogen receptor antagonist, tamoxifen 54, 55. These studies were the first to identify local actions of estrogen on estrogen receptors, which broadly expressed in both epithelial and stromal compartments of the gland.
Estrogen acts together with IGF1 to regulate ductal morphogenesis, as evidenced by the synergistic enhancement of ductal outgrowth in Igf1-/- mice treated with estrogen together with IGF1 46. There are two forms of intracellular estrogen receptors, alpha and beta, encoded by different genes, Esr1 and Esr2. ESR1 is the major receptor operating during ductal morphogenesis as determined by the analysis of knockout phenotypes 56, 57. Mice lacking Esr1, similar to mice lacking Igf1, only form a rudimentary ductal system that fails to undergo the estrogen-mediated growth spurt that accompanies puberty 56. Yet only a subpopulation of luminal epithelial cells express ESR1, with fewer than 40% of these cells incorporating the cell proliferation marker, BrdU 58. One explanation for this observation is that ESR1-positive cells drive proliferation by signaling in a paracrine fashion and, in this case, only a few cells would be required to initiate signaling. Alternatively, ESR1 expression may be rapidly downregulated in response to estrogen and, as a result, ESR1-positive cells would have been missed in an experiment looking for co-expressed ESR1 and BrdU 58. To investigate these possibilities, researchers again turned to the technique of transplantation, but this time they generated mosaic epithelial outgrowths by mixing Esr1-/- cells with WT (Esr1-positive) cells and grafting them into precleared fat pads 59. These studies demonstrated that a subpopulation of WT cells rescues the knockout phenotype when mixed together with Esr1-/- cells, supporting the notion that ESR1-positive cells signal in a paracrine fashion to stimulate the proliferation of neighboring ESR1-negative. Amphiregulin (AREG), a member of the EGF family, is currently the leading candidate for the factor that is released by ESR1-positive cells and signals cell proliferation (see section on growth factors). In addition to this role in stimulating cell growth during ductal morphogenesis, estrogen also functions during pregnancy in the growth and maintenance of alveolar cells as demonstrated over fifty years ago in hormone replacement studies on ovariectomized mice 60, 61. These observations were confirmed by conditional ablation of Esr1 in alveoli following ductal elongation, resulting in defective lobuloalveolar development and inadequate milk production 62.
Altogether, these studies on GH, IGF1 and estrogen have led to a model in which pituitary GH induces the production of IGF1 by the liver (Fig. 4). These factors, together with locally produced IGF1 in the mammary stroma and epithelium, act via their receptors in the mammary gland to stimulate TEB formation and ductal branching. Estrogen acts in concert with IGF1 to generate the burst of proliferation required for ductal morphogenesis. ESR1 operates in a subset of epithelial cells and induces the release of AREG that signals through the stroma, generating additional growth factors (e.g. FGFs) that contribute to the rapid period of mammary gland growth occurring at puberty (see below).
Growth factors sculpt ductal tree architecture
Members of the EGF and FGF family of growth factors signal through their respective tyrosine kinase receptors to positively influence mammary gland ductal morphogenesis. In terms of the EGF family, AREG is the only obligatory ligand, although several EGF family members can affect ductal outgrowth. For example, transforming growth factor alpha (TGFA), NRG1 and EGF are all capable of rescuing ductal development in ovariectomized and Esr1-/- mice 63, 64, but only mice lacking Areg have a mammary gland phenotype that closely matches that of the Esr1 KO 65, 66. The expression of Areg is strongly induced in mammary epithelial cells by estrogen at puberty, but this transmembrane protein binds to the epidermal growth factor receptor (EGFR) that is located at a distance on stromal cells across the basement membrane. Studies have shown that the transmembrane metalloproteinase Adam 17 (formerly known as TACE, TNFα converting enzyme) releases AREG from mammary epithelial cells, allowing for the paracrine activation of stromal EGFR 67. Evidence supporting this sequence of events include the observation that defects in ductal outgrowth observed in Adam17-/- glands resemble those in Areg-/- and Esr1-/- glands and the fact that local administration of soluble AREG rescues the development of Adam17-/- glands 67. Nevertheless, despite the delineation of these events, the growth factor that actually drives cell proliferation at puberty has not been definitively identified. AREG signals to its receptor on stromal cells and can, therefore, only indirectly affect mammary epithelial cells. Indeed, tissue recombination experiments have confirmed that epithelial deletion of EGFR does not disrupt ductal development, while deletion of stromal EGFR results in dramatically reduced ductal outgrowth 68, further supporting the notion that paracrine circuitry of the estrogen response occurs via cross-talk with the stroma. FGFs are attractive candidates to mediate the mitogenic signal since they are upregulated in the stroma during puberty 69, and their cognate receptor, FGFR2, is required on epithelial cells for ductal elongation 70.
While positive factors that elicit growth are clearly important for creating the ductal architecture, negative regulators are equally important and particularly crucial in the mammary gland, which has an open ductal architecture, characterized by ample space between branches for pregnancy-induced alveolar infilling. Transforming growth factor beta1 (TGFB1) has been identified as a major negative regulator of mammary branching and ductal elongation and it restricts end bud bifurcation and branch formation by limiting epithelial proliferation 71-73. Although some of the anti-proliferative effects of TGFB1 are mediated through stromal receptors 74, 75, numerous studies have demonstrated significant inhibitory signaling occurring within the epithelium. For example, transplantation of either Tgfb1-/-epithelium 73 or Tgfb1+/- epithelium, which has dramatically reduced TGFB1 levels due to the absence of positive feedback 72, into WT mammary fat pads results in increased epithelial proliferation and ductal extension, while overexpression of dominant negative TGF beta receptor II (TGFBR2) in the epithelium leads to alveolar hyperplasia 76. Studies using 3-dimensional micropatterned collagen gel assays that control the shape of mammary epithelial tubules in culture demonstrate that TGFB1 can be released in gradients capable of inhibiting the sites of branching 77. WNT5A is one downstream mediator of these growth inhibitory effects and it acts by regulating proliferation through the LEF/TCF pathway 78. These and other studies demonstrate that mammary branching morphogenesis requires a balance of positive and negative factors that generate, by still largely unknown mechanisms, the stochastic pattern of widely-spaced branch outgrowth observed in mammary trees.
In addition to members of the major growth factor families, a host of additional secreted proteins have been identified that regulate mammary morphogenesis. Examples include members of neurogenic families. These factors were originally discovered in the nervous system, but also regulate developmental events in other organs. Netrin1 (NTN1), Slit2 (SLIT2) and Reelin (RELN) are all proteins that regulate axon guidance and cell migration during neural development. These factors also regulate mammary ductal morphogenesis. Ntn1 and Slit2 are expressed in both luminal and myoepithelial cells, but their receptors are restricted to the outer myoepithelial layer. A loss-of-function mutation in these genes, individually, results in TEB abnormalities and, together, in ductal defects characterized by delamination of the epithelial cell layers 79, 80. In contrast, RELN is expressed by the periductal stromal and its receptor is expressed in both cell types of the epithelium. Reln knockouts contain only rudimentary ductal structures that exhibit decreased terminal branching and a disorganized luminal layer 81. Other proteins, such as milk fat globule-EGF factor 8 (MFGE8) and matrix metalloproteinases (MMPs), function during branching morphogenesis, but also have roles in other stages of mammary gland development such as involution. MFGE8 is expressed in the epithelium, regulating cell proliferation during ductal outgrowth 82 and the elimination of apoptotic cells during involution 83. In contrast, stromal MMPs function similarly at both developmental time points and are involved in remodeling the basement membrane to assist with architectural changes required for morphogenesis.
Altogether, these lines of investigation have led to a model in which estrogen binding to ESR1 in sensor cells induces the expression of transmembrane AREG, which is released from the cell surface by Adam17 (Fig. 4). Cleaved AREG makes its way, by unknown mechanisms, across the basement membrane, to stromal fibroblasts expressing its receptor, EGFR. Activation of EGFR induces the expression of FGFs that, in turn, stimulate cell proliferation through FGFR2 expressed on the mammary epithelium. TGFB1 functions as a negative regulator of ductal morphogenesis by inhibiting cell proliferation and restricting branch formation. Other factors also play important roles and additional information needs to be gathered on the many, diverse extracellular signals that contribute to the morphogenesis of the adolescent gland. An important question raised by these studies is how do quiescent ESR1-positive cells become proliferative in hormone-dependent, ESR1-positive human breast tumors. The answer may be through the robust crosstalk that occurs between epithelial ERα and stromal EGFR signaling. Studies on hyperplastic enlarged lobular units (HELUs), considered the earliest lesion with premalignant potential, have shown that both Esr1, and Areg are upregulated and, together, they may generate a self-propagating growth-regulatory loop that fuels tumorigenesis 84. Despite the clinical success of estrogen receptor antagonist, tamoxifen, there is great interest in adding to the arsenal of drugs that will slow the growth of ER-positive tumors. Recent studies suggest that AREG may be an attractive therapeutic target for breast cancer intervention 85.
ADULT MAMMARY GLAND DEVELOPMENT
Pregnancy and lactation are accompanied by major changes as the mammary tree prepares to bear fruit
The mammary gland must undergo numerous changes to prepare for lactation. These changes require both gland maturation and alveologenesis and are primarily under the control of progesterone and prolactin. The first transformation of pregnancy is a tremendous increase in secondary and tertiary ductal branching, providing ductal arbors for the second transformation, alveolar development. Proliferating epithelial cells generate alveolar buds that progressively cleave and differentiate into distinct alveoli, which become milk-secreting lobules during lactation. Interstitial adipose tissue disappears as the proliferating epithelial cells occupy the interductal spaces. Increased vascularization occurs and by mid-pregnancy, each alveolus is surrounded by a basket-like network of capillaries 86. By late pregnancy the alveoli encompass the majority of the fat pad and show some secretory activity as pregnancy approaches term. Some of these changes also occur during estrus cycles when the gland exhibits mild proliferation and differentiation that includes the limited expression of milk proteins, followed by involution.
Progesterone makes crucial contributions to alveologenesis
Progesterone, like estrogen, is a membrane soluble, ovarian hormone that signals through intracellular receptors to generate epithelial growth in the mammary gland (Fig. 5). Progesterone is responsible for the extensive side-branching and alveologenesis required to create a lactation-competent gland. In combination with prolactin, it also promotes the differentiation of specialized structures, alveoli, which synthesize and secrete milk during lactation. The importance of progesterone in both these processes was revealed by the analysis of progesterone receptor knockout mice. Pgr-/- glands produce only a simple epithelial tree, even following the surge of growth at puberty, and upon pregnancy they do not undergo the associated ductal proliferation or lobuloalveolar differentiation 87, 88. Transplantation studies were performed to address whether PGR is required in the epithelial, stromal or both compartments and revealed that only epithelial Pgr expression is required for side branching and alveologenesis 88. In the epithelium, the pattern of Pgr expression changes from a uniform pattern of expression in the juvenile to a scattered expression pattern in a subset of epithelial cells in the adult 89. This suggests that, similar to estrogen signaling, progesterone signaling occurs in a paracrine fashion. This was evaluated by using mosaic analysis in which Pgr-/- cells were mixed with WT (Pgr-positive) cells and grafted into pre-cleared fat pads, similar to the experiments performed with Esr1-/- cells 88. The Prg-/-/WT mammary gland outgrowths appeared normal, demonstrating that WT cells rescue the KO phenotype and supporting the idea that progesterone signals via a paracrine mechanism.
Studies on progesterone receptors have been complicated by the fact that there are two receptor isoforms (PGRiA and PGRiB), produced by alternate promoter usage from a single gene. They are identical except that PGRiB contains 164 additional N-terminal residues. Both isoforms of PGR are expressed in the mammary gland of the virgin mouse and during pregnancy 90, with PGRiA levels exceeding those of PGRiB. Each of these isoforms has been knocked out individually and they have distinct physiological functions in some reproductive tissues. However, in the mammary gland loss of Pgria does not produce a phenotype, whereas loss of Pgrib results in significantly reduced side branching and alveologenesis during pregnancy 91, 92. These data show that PGRiB is uniquely required in the mammary gland, but they do not rule out a distinct function for PGRiA, which may be compensated by the presence of PGRiB. Indeed expression profiling studies on human breast cancer cell lines engineered to express one or the other isoform show that PGRiA and PGRiB regulate distinct and overlapping sets of genes 93. This pattern of regulation may have clinical significance since human breast cancers expressing altered ratios of the two PGR isoforms have different, ratio-specific clinical outcomes 94.
There is still much to be learned about signaling downstream of progesterone receptors, but recent data identify tumor necrosis factor ligand superfamily, member 11 (TNFSF11), also known as RANKL (receptor activator of NFKB1 ligand), as a key paracrine-mediator of progesterone-induced proliferation (Fig. 5). RANKL signals through Tumor necrosis factor receptor superfamily, member 11a (TNFRSF11A), also known as RANK, to regulate a broad range of physiological processes, including osteoclastogenesis and bone-remodeling, by stimulating NFKB1 in neighboring cells. Given this role in regulating skeletal calcium release, perhaps it is not surprising that RANKL should play such an essential role in regulating alveologenesis, the first step in transmitting maternal calcium to infant mammals. Mice lacking Rankl or Rank fail to undergo alveologenesis during pregnancy 95, a phenotype that is very similar to that described in Pgrib-/- glands. Moreover, the phenotype of such glands can be rescued by the ectopic expression of Rankl, an experiment that was performed by both viral infection of bulk mammary epithelial cells 96, and in a more refined manner by using an inducible bigenic system that drives Rankl expression specifically in the cells that normally express it 97. Numerous studies have shown that RANKL is induced by progesterone 98, specifically in PGR-positive luminal epithelial cells 91 and recent data suggest that it is the paracrine signal that tells neighboring PGR-negative cells to proliferate 96 (Fig. 5). This central role of RANKL in generating the pro-growth response to progesterone is illustrated by recent studies demonstrating its responsibility in driving cell proliferation in progestin-dependent breast cancers. This has led to the idea that RANKL inhibitors, a therapy currently being pursued for arthritis, periodontal disease and osteoporosis, should also be considered for breast cancer treatment 99, 100.
Prolactin collaborates with progesterone to generate a lactation-competent gland
PRL is the major generator of lactational competence during pregnancy, and it functions both indirectly, through its regulation of ovarian progesterone production, and directly via its affect on mammary epithelial cells. PRL is a small polypeptide hormone produced by the pituitary, as well as other sites, such as mammary epithelium. It binds to a member of the class I cytokine receptor superfamily, resulting in the activation of a number of signaling pathways, including Jak/Stat, Map Kinase and phosphoinositide (PI) 3 Kinase (Fig. 5). The role of PRL during mammary gland morphogenesis has been investigated using Prl and Prolactin Receptor (Prlr) knockout mice 101, 102. Embryonic and postnatal development appears to be normal in these mice and there is typical outgrowth of the mammary tree until puberty, whereupon a lack of side-branching and alveolar budding results in a sparse tree, similar to that observed in Pgr-/- mice. To determine whether this defect is intrinsic to the mammary epithelium or due to systemic effects, transplantation experiments were performed in which Prl-/- epithelium was grafted into pre-cleared WT fat pads 103, 104. These experiments revealed normal development in the transplanted tissue, suggesting that defects in knockout glands are due to the lack of PRL/PRLR signaling in other tissues. This requirement for systemic PRL was confirmed by experiments in which the side-branching and alveolar defects of Prl-/- mice were rescued by engrafting Prl+/- pituitaries under the kidney capsules 105. One of the systemic consequences of losing PRL signaling is low serum levels of hormones due to impaired secretion from the ovary. Researchers found that simply restoring progesterone levels in ovariectomized Prl-/- mice restored ductal side branching 105, providing further affirmation that PRL and progesterone work together to generate lobuloalveolar growth. Progesterone treatment also rescued the infertility observed in Prl-/- mice, allowing researchers to examine the effect of Prl loss, alone, on mammary lobuloalveolar development. Surprising, alveologenesis was normal in these animals, but this was explained by the presence of a prolactin family 3, subfamily d, member 1 (PRL3D1), also known as placental lactogen, which can also bind and activate PRLR. Consequently, definitive demonstration of a requirement for PRL signaling in alveologenesis was achieved by transplantation experiments in which Prlr-/- epithelium was engrafted into WT mice. Ductal morphogenesis appeared normal in these mice, since progesterone levels were normal, but alveologenesis failed, revealing an essential, epithelial intrinsic role for PRL/PRLR signaling in lobular alveolar development and milk production.
The JAK2/STAT5 signaling pathway is activated downstream of PRL/PRLR and it plays an essential role in establishing alveologenesis (Fig. 5). This was demonstrated by the analysis of Jak2-/- and Stat5-/- mice whose mammary gland defects phenocopy those observed in Prlr-/- glands. Moreover, conditional loss of Stat5 following PRL-induced differentiation of alveolar cells showed that this pathway is required for maintenance of the pregnancy program 106-108. Signaling via the PRLR/JAK2/STAT5 pathway culminates in the expression of milk protein genes, including casein beta (Csn2) and whey acidic protein (Wap), which contain STAT5 responsive elements in their promoters. The pathway is subject to precise control by a number of positive and negative regulators, including integrins, which are the major class of extracellular matrix (ECM) receptors and required for full STAT5 activation 109. Cross-talk between integrin and PRLR is mediated by signal regulatory protein alpha (SIRPA), a transmembrane glycoprotein. SIRPA integrates the gland’s response to the ECM during lactation, generating a complex that includes JAK2 and regulates the response of PRLR as a function of integrin interaction with various ECM components 110. Downstream of PRLR signaling is RANKL and, as in the case of PGR signaling, it is a paracrine-acting target of PRL 111. The regulation of Rankl by both progesterone and PRL explains how the homozygous deletion of each gene results in a similar phenotype: decreased side-branching and disrupted alveologenesis. Moreover, transgenic expression of Rankl alone, in the virgin gland is sufficient to elicit the characteristics of pregnancy, producing both ductal side-branching and alveolar budding, functions ascribed to PRL and progesterone 112. Negative regulation of PRLR signaling is achieved by members of the suppressors of cytokine signaling (SOCS) family via a classical negative feedback loop. PRL induces the expression of SOCS1, which in turn binds to JAK2 and inhibits its association with STAT5. Socs1-/- mice display enhanced alveolar development and milk production during pregnancy. Thus, SIRPA, RANKL and SOCS are three examples of many that illustrate the complexity of the signaling pathways that regulates the response of cells to PRL 113. This web of pathways results in a finely tuned transcriptional program that, in turn, regulates many signaling networks and ultimately controls the cellular differentiation of alveolar cells and their production of milk.
In sum, by helping mothers cope with unreliable food stores, lactation has made a critical contribution to mammalian evolution. Progesterone and PRL orchestrate this essential reproductive event by engaging in a complicated and intertwined relationship (Fig. 5). Each regulates the other’s expression, and each controls overlapping and unique transcriptional regulatory networks that govern both the proliferation and differentiation of mammary epithelial cells. RANKL serves as one point of cross-talk between these to two signaling pathways, but there are likely additional pathways that function in cooperative and antagonist ways to transform progesterone and PRL signals into the structural and functional changes required to generate and deliver milk.
Involution remodels the tree back to its original adult state
The lack of demand at weaning causes milk to stagnate in the mammary epithelium, initiating the process of involution that removes milk-producing epithelial cells and remodels the epithelial tree back to a simple ductal architecture (Figs. 1, 6). Research on involution has been facilitated by forced weaning protocols that involve the removal of suckling mouse pups at the height of lactation. This induces synchronous involution, allowing the elucidation of morphological and molecular events underlying the process. Two phases of involution have been defined (Fig. 6). The first is reversible, such that resumption of suckling reestablishes the milk supply. This step is accompanied by apoptosis, alveolar cell detachment and, within 12 hours, the accumulation of shed cells into the lumen. Remarkably, no major morphological changes occur with this phase of involution, aside from changes in alveolar shape due to milk engorgement. However, at 48 hours, a transition into phase two of involution begins and alveoli start to collapse. This phase is irreversible and the milk supply is lost, regardless of whether or not suckling is reinitiated. During this phase, breakdown of the ECM and activation or proteases induces a second wave of apoptosis. This results in a massive period of tissue remodeling, which by six days leads to the removal of most of the secretory epithelium and its subsequent replacement by adipocytes. Although a few remaining alveoli may persist, the remodeled gland is morphologically very similar to the virgin gland, even though it is molecularly distinct with respect to gene expression 114, 115.
Figure 6. Illustration depicting the two stages of mammary gland involution.
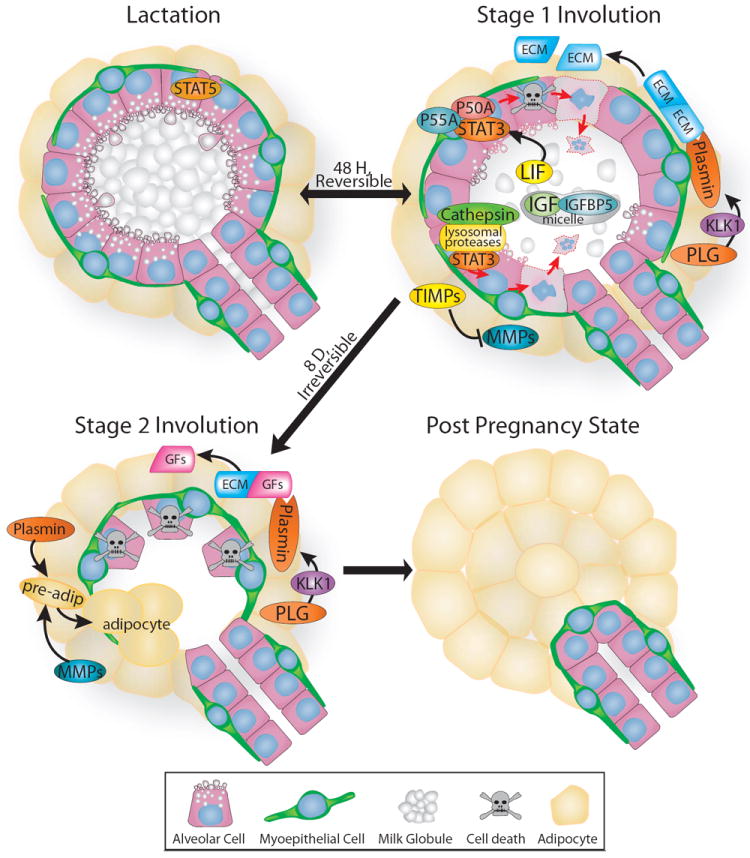
Upon weaning, the gland is remodeled back to its pre-pregnancy state. Stage one is reversible and is regulated largely by STAT3, which is induced by leukemia inhibitory factor (LIF) and opposes pro-survival STAT5 signaling by upregulating the expression of numerous proteins including: lysosomal proteases, cathepsins; insulin-like growth factor binding protein5 (IGFBP5); and two regulatory isoforms of phosphatidylinositol 3 kinase, p50alpha (P50A) and p55alpha (P55A). Cell death and limited proteolysis of the extracellular matrix (ECM) occurs during this stage as plasminogen (PLG) is converted to plasmin through the actions of plasma kallikrein (KLK1), yet the alveoli largely retain their shape. This changes during stage two, which is irreversible and characterized by alveolar collapse and adipocyte differentiation. MMPs are released from their inhibitors, tissue inhibitors of metalloproteinases (TIMPs), and collaborate with plasmin to return the mammary gland to its pre-lactation state by releasing growth factors (GFs) and remodeling the ECM. For details, see text.
Local cues regulate the first phase of involution
Various approaches in mice (e.g. teat sealing and inducible expression of T-antigen) have been used to demonstrate the role of local versus systemic factors in the first phase of involution. In each case, lactation is disrupted in a different way: teat sealing physically prevents milk release, whereas T-antigen expression triggers apoptosis 116, 117. The teat sealing model demonstrated that apoptosis is induced by local factors because it occurred despite the presence of suckling from unsealed control teats, which maintains the systemic level of lactation hormones. In the T-antigen overexpression model, glands exhibit early involution, with alveolar apoptosis resulting in lactation failure. Yet, constant suckling in this model, generated by serial litter replacement, protects the alveoli, leaving open lumens and intact lobules, demonstrating that systemic hormones can preserve lobuloalveolar structure, even in the midst of significant programmed cell death. Other studies have identified both intrinsic (mitochondrial) and extrinsic (death receptor) apoptotic pathways as those responsible for generating the massive cell death occurring during this initial phase of involution 118-120. Recent studies, however, show that the earliest steps are independent of these apoptotic processes and rely instead on lysosomal membrane permeabilization 121. Despite these many studies, the actual trigger of apoptosis is still unknown. Alveolar stretch, the build-up of apoptotic factors in static milk and/or disruption of cell-ECM contacts are all likely to play a role. Altogether these studies demonstrate that local cues regulate the first phase of involution, whereas the second phase depends on the fall of systemic hormones and can be blocked by the administration of glucocorticoids116.
In the past decade, numerous signaling pathways have been identified that regulate the developmental switch from lactation to involution, but the STAT family of proteins were found to play particularly important roles in this transition, by regulating the flow of survival signals through the PI3-kinase/AKT pathway. STAT5A and STAT5B positively regulate AKT signaling, whereas STAT3 inhibits it. Within 3–6 h of milk stasis, a change in the phosphorylation status of STAT5 proteins alters their activity state, inactivating STAT5A and STAT5B, but activating STAT3 116. This toggle-like nature of STAT proteins in the lactation/involution switch was demonstrated in a series of gain and loss-of-function experiments. For example, overexpressing either Stat5a or a constitutively activated form of Akt1 delays mammary gland involution 122, 123, while knocking out these genes has the opposite effect and accelerates involution 107, 124. STAT5 binds to consensus sequences within the Akt1 locus in a PRL-dependent manner and initiates transcription of Akt1 from a mammary specific promoter, generating a strong cell survival signal 122. In contrast, STAT3 antagonizes pro-survival STAT5 signaling and promotes pro-apoptotic pathways 125, as evidenced by conditional deletion of Stat3 in the mammary gland, resulting in decreased apoptosis and a dramatic delay in involution 126. The primary activator of STAT3 in the gland is the cytokine leukemia inhibitory factor (LIF), and in its absence Stat3 is dephosphorylated, resulting in delayed involution, similar to the delay observed in the Stat3-/- glands 127. Once activated, STAT3 directly regulates the expression of two inhibitory subunits of the class IA PI3 kinase, p50alpha and p55alpha, which are generated by alternative splicing of the Pik3r3 gene. This alters the subunit composition of PI3 kinase, restricting its activity and, consequently, the activity of AKT 128. Another mechanism by which STAT3 antagonizes pro-survival signaling is upregulation of the insulin-like growth factor binding protein-5 (IGFBP5). IGFBP5 is thought to sequester IGF1 in casein micelles and, as a result, interfere with pro-proliferative IGF1 signaling 129. Overexpression of IGFBP5 in the mammary gland results in accelerated involution that can be reversed by exogenous IGF1 treatment, whereas, deletion of IGFBP5 leads to delayed mammary gland involution 130. Similarly, STAT3 promotes apoptotic pathways through the transcriptional control of numerous targets. For example, it upregulates the expression of lysosomal proteases cathepsin B and cathepsin L, while downregulating their endogenous inhibitor Serpin2A 121. Upregulation of these proteases results in lysosomal membrane permeabilization, which induces programmed cell death at the earliest stages of involution without the involvement of the classical apoptotic pathways. Taken together, these data illustrate that specific activation of STAT3 or STAT5 alone is sufficient to promote or suppress apoptosis, respectively, and that these transcription factors elicit some of their many actions by regulating the AKT pathway.
The second phase of involution is controlled by circulating factors
The second phase of involution is accompanied by dramatic changes in mammary gland architecture, including the remodeling of the basement membrane, collapse of alveoli and differentiation of adipocytes (Fig. 6). Important regulators of these processes are members of the serine protease and matrix metalloproteinase (MMP) families that are responsible for activating plasminogen and breaking down the ECM, respectively. The expression of these factors in the mammary gland stroma begins during the first phase of involution when serine proteases are activated and mediate epithelial apoptosis. At the same time, MMPs are kept in check by the coordinate expression of their negative regulators, tissue inhibitors of metalloproteases (TIMPS).
The serine protease, plasminogen, is synthesized in the liver and circulates as a zymogen through blood. Its activation to plasmin is controlled locally in the mammary gland by the serine protease Kallikrein 1 (KLK1). Mammary glands lacking plasminogen fail to involute after weaning and are marked by the accumulation of fibrotic stroma and the abnormal differentiation of adipocytes 131. A similar phenotype is observed by inhibiting KLK1 through intraperitoneal injection of an inhibitor, ecotin, engineered to be highly specific for this enzyme 132. During the first phase of involution, plasmin contributes to apoptotic process by disrupting the interactions between cells and the ECM. In the second phase of involution, plasmin is joined by the upregulation and activation of MMPs, such as MMP3, whose overexpression disrupts the basal lamina and causes premature involution 133. Similarly, premature activation of MMP2, which occurs in TIMP3-deficient glands, results in accelerated involution that is irreversible, even during the first phase 134. These studies demonstrate that balanced protease activity, controlled by both regulated activation and direct inhibition, is required for the gland to properly transition through the phases of involution. Moreover, these enzymes influence the differentiation of adipocytes that is required to replace tissue loss due to apoptosis of epithelial cells. One of the ways they contribute to adipogenesis is by releasing growth and differentiation factors from the cell surface and ECM. Thus, by giving the gland the ability to remodel in response to fluctuating local and hormonal environments, MMPs and serine proteases play critical roles in mammary gland biology. While their importance in tissue restructuring during involution is highlighted here, they also participate in other processes such as milk production during pregnancy 135 and branching morphogenesis during puberty 136, underscoring their central position in regulating mammary gland development.
Involution and its connection to cancer
There is a great deal of interest in understanding the signaling processes involved in postlactational and age-related lobular involution, and the connection these processes share with cancer. Postlactational involution is a process subject to much regulation, yet transcriptional profiling studies have shown that this rapid and extensive period of tissue remodeling mimics wound healing and pathological conditions such as tumorigenesis 137. This raises the possibility that involution creates a tumor microenvironment that promotes further transformation of preneoplastic mammary cells, providing an explanation for the transient increase in breast cancer risk observed following pregnancy 138. This possibility has been validated by experiments in which ECM was isolated from postlactational mammary glands and shown to contain tumorigenic ECM fragments that facilitate the invasiveness of breast cancer cells in culture and increase the metastasis of breast cancer in animal models 139, 140.
In contrast, age-related lobular involution is a distinct process that involves the replacement of glandular epithelium and interlobular connective tissues with fat. Eventually in the aged breast, all that remains are a few acini and ducts embedded in thin strands of collagen. This reduction in epithelial tissue has beneficial consequences for the diagnosis and development of breast cancer. First, the increased ratio of fat to epithelium increases the effectiveness of mammography 141. Second, this reduction in epithelial tissue may physiologically protect the breast from cancer, as evidenced by a recent clinical study that correlated lobular involution with reduced breast cancer risk 142. Premenopausal women whose breasts had undergone partial or complete lobular involution were found to have significantly decreased risk of breast cancer, while postmenopausal women, whose breast exhibited delayed lobular involution, had an elevated breast cancer risk. These results suggest that a fundamental protective event occurs with lobular involution. Recent studies suggest this protection may be due, at least in part, to the depletion of mammary stem cells in the aging breast.
MAMMARY STEM CELLS
Stem cells are specialized cells with the capacity to both self-renew and generate daughter cells that can differentiate down several lineages to produce all cell types found in mature tissue. Over 50 years ago, researchers postulated the existence of multipotent mammary stem cells (MaSCs) due to the observation that mammary epithelium is able to undergo multiple rounds of proliferation, differentiation and apoptosis with pregnancy. In vivo serial transplantation assays were designed to test this hypothesis. In this assay, a small tissue fragment is transplanted into pre-cleared fat pads and allowed to grow into a mature mammary tree, before another small fragment is harvested and transplanted. This can be performed serially for up to 7 generations until senescence, which is thought to occur due to depletion of the original stem cells contained in the initial fragment 143. Later experiments revealed that neither the developmental state nor reproductive history of the gland had a significant impact on the longevity of the mammary transplants 144, 145. It was not until 2006, however, that self-renewing multipotent stem cells were isolated from mouse mammary gland by fluorescent activated cell sorting (FACS) to detect cell surface markers CD24 (heat stable integrin) and either CD29 (β1-integrin) or CD49f (α6-integrin) 146, 147. Similarly, a mammary stem cell has also been identified in human breast, although different markers were used for its identification 148, 149. These studies show that most, if not all, stem cells express α6 and β1 integrins, positioning them in a basal position in the gland, possibly corresponding with a basal-positioned small light cell (SLC) that was identified by electron microscopy and occurs at a frequency of 1–3% within the epithelium 150, 151. Other estimates of stem cell frequency are derived from calculating the number of mammary repopulating units (MRUs) in freshly dissociated mammary cells using a limiting dilution transplantation assay. These studies have generated a wide range of estimates from 1 MRU in 100 total cells to <1 in 4,900 total cells, presumably due to the differences in techniques used to dissociate and transplant cells 146, 147, 152.
This range of estimated stem cell frequencies illustrates how difficult it has been for researchers to achieve an understanding of mammary stem cell biology. There are many impediments, including the technical complexity of dissociating solid tissue into single cell suspensions for FACS and the lack of markers to specifically label these cells. Nevertheless, there is currently a tremendous effort to delineate the stem cell hierarchy of the gland. The current model is that a MaSC gives rise to committed progenitors, which proceed to differentiate along the myoepithelial and luminal epithelial lineages. It seems likely that there is at least one intermediate cell, a bipotent progenitor, which lies between the MaSC and the committed progenitor, but its identity has remained elusive. The identification of the myoepithelial progenitor has also remained elusive, hindered by the fact that the MaSCs themselves express basal markers. A luminal progenitor population, however, has been identified 153, and there is evidence that it gives rise to cells committed to either ductal or alveolar cell fates 154.
How a MaSC gives rise to progenitor cells, while regenerating itself, is also a subject of much investigation. In nonvertebrate organisms and some mammalian organs, stem cells undergo asymmetric cell division in which the cell reproduces itself and generates a daughter cell with a different cell fate 155. However, asymmetric cell divisions have not been definitively demonstrated in the mammary gland. An alternate mechanism of stem cell renewal and division is symmetric cell division, whereby a MaSC simply divides to give rise to two progenitors or divides to replenish itself. How these symmetric and asymmetric cell divisions are controlled raises additional questions; where do stem cells reside and what is the nature of the stem cell niche? Studies have shown that MRUs are distributed throughout the mammary epithelial tree, since transplantation of tissue fragments from any part of the tree can result in mammary outgrowth 156. Yet, tissue fragments harvested from the periphery of ductal outgrowths result in earlier senescence compared to tissue transplanted from the center of the outgrowth 157. These studies suggested that peripheral transplants contain MaSCs that have been aged by having undergone more cell divisions than their counterparts in the center. Additionally, there may also be a difference in MaSC concentration in different locations of the gland as a recent multi-scale in situ analysis has shown that putative MaSCs are distributed in a graded manner with more toward the nipple and fewer at the opposite end of the mammary gland, with large ducts containing a reservoir of these cells 158. Signaling pathways, such as the Notch 159, Hedgehog 160 and Wnt pathways 161, regulate these proliferative behaviors and niche characteristics and appear to be similar to those pathways directing stem cells in other systems, although many details concerning the control of these individual signaling pathways and their cross-talk remain unknown.
This ongoing research on mammary gland development and stem cells is significantly contributing to our understanding of breast cancer. Breast cancers have been divided into six distinct subtypes on the basis of gene expression profiling: luminal A, luminal B, basal-like, claudin-low, Her2/ErbB2-overexpressing, and normal-breast-like subtypes 162. In 2003, breast cancer stem cells were identified in patient samples 163, echoing the identification over a decade ago of stem cells in the hematopoietic system that are the source of acute myeloid leukemia (AML) 164. These and other findings have led to the cancer stem cell (CSC) hypothesis, positing that at least some tumors are initiated by one or more self-renewing CSCs that differentiate into large populations of non self-renewing, yet rapidly dividing, cells responsible for generating the tumor mass 165. In the hematopoietic system, there is strong evidence that resident stem cells are the likely target of oncogenic mutation in some diseases 164, but in solid tumors the origin of the CSC remains unresolved. They could arise from mammary stem/progenitor cells or be generated de novo as part of the multi-step mutational process that gives rise to a tumor. At least for some breast tumors, evidence is growing that tissue-resident stem/progenitor cells are the likely targets of transformation because some of the breast tumor subtypes share molecular profiles with stem/progenitor cells 166. This finding supports the notion that mutations in stem/progenitor cells generate a self-renewing CSC capable of producing cells that constitute the tumor mass. This self-renewal could occur at the same time that a subpopulation of CSCs maintain a quiescent program, allowing them to escape traditional therapies such as radiation and chemotherapy that target rapidly dividing cells 167. Current challenges are in developing strategies to identify all the cell types in the mammary hierarchy with specific markers and in defining the mutations that transform stem/progenitor cells into CSCs. With knowledge of the parent cells and the transformation processes, novel therapies, such as the use of small interfering (si)RNAs, can be further developed to eradicate cancers by targeting the cells from which they arise.
CONCLUSION
Tremendous progress has been made in delineating the signaling pathways controlling the three distinct stages constituting mammary gland development: embryonic, pubertal and reproductive. These stages have many unique characteristics, as well as many common attributes. Occurring over the lifespan of an individual, each stage is separated temporally, as well as by hormonal requirement. Embryonic development proceeds in the absence of hormonal regulation, whereas the pubertal and reproductive stages are governed by customized hormonal inputs that prepare for and then result in milk production. During embryogenesis, local epithelial/mesenchymal interactions direct many developmental processes and are responsible for determining the location of the gland and controlling cell-fate, such that by birth the tissue compartments are precisely positioned and the nascent structure is correctly established. Epithelial/mesenchymal interactions also govern development during postnatal stages of mammary gland morphogenesis, but after puberty, regulation by hormones and growth factors profoundly changes the nature of these interactions. Hormones generate complex signaling networks that influence epithelial/mesenchymal interactions by regulating the production of secondary signaling pathways, which drive cross-talk between and within compartments. In all these complex processes, however, one message is clear: Mother Nature is parsimonious and the same pathways are deployed over and over again. This is evident as STAT proteins are involved in branching, lactation and involution, FGFs in embryogenesis and branching, MMPs in branching and involution; the list goes on. These same pathways are also hijacked in cancer cells and our growing understanding of them has fueled tremendous progress in breast cancer research. These gains in our understanding have ushered in exciting times, and as this knowledge is translated into molecule-based diagnostics and therapies, we are currently witnessing the tangible benefits basic research has brought to society.
Figure 7. A schematic representation of the stem cell hierarchy within the mammary gland.
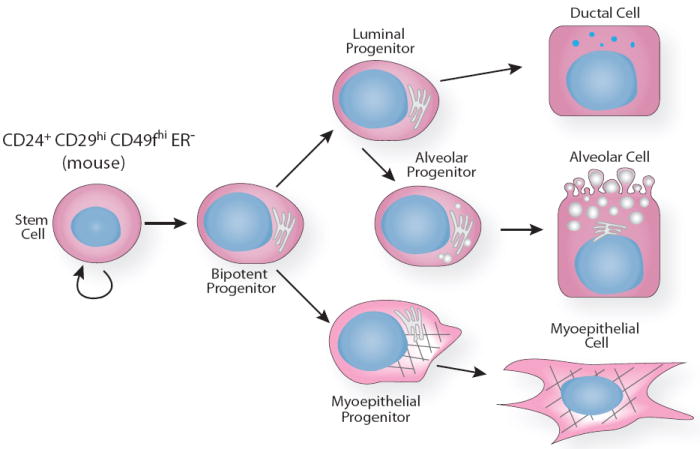
A stem cell divides symmetrically or asymmetrically to generate a bipotent progenitor cell that gives rise to both a luminal and myoepithelial cell progenitor. Studies suggest that luminal progenitors differentiate into cells that are restricted to either ductal or alveolar lineages and as yet unidentified intermediate cells may be required for terminal differentiation. In contrast, myoepithelial progenitors are thought to differentiate directly into myoepithelial cells. This illustration represents one interpretation of available data and the true hierarchy is likely to be much more complex. For details, see text.
Acknowledgments
We apologize to authors whose work was not cited due to space limitations. We thank Drs. Jennifer Compton and Gary Silberstein for comments. We acknowledge the NIH (RO1 CA-128902), Congressionally Directed Medical Research Program (W81XWH-08-1-0380), Santa Cruz Cancer Benefit Group, Initiative for Maximizing Student Diversity (NIH GM058903) (HM) and Center for Biomolecular Science and Engineering (5P41HG002371-09) (HM).
References
- 1.Oftedal OT. The mammary gland and its origin during synapsid evolution. J Mammary Gland Biol Neoplasia. 2002;7:225–252. doi: 10.1023/a:1022896515287. [DOI] [PubMed] [Google Scholar]
- 2.Balinsky BI. On the prenatal growth of the mammary gland rudiment in the mouse. J Anat. 1950;84:227–235. [PMC free article] [PubMed] [Google Scholar]
- 3.Propper AY. Wandering epithelial cells in the rabbit embryo milk line. A preliminary scanning electron microscope study. Dev Biol. 1978;67:225–231. doi: 10.1016/0012-1606(78)90311-1. [DOI] [PubMed] [Google Scholar]
- 4.Howard BA, Gusterson BA. Human breast development. J Mammary Gland Biol Neoplasia. 2000;5:119–137. doi: 10.1023/a:1026487120779. [DOI] [PubMed] [Google Scholar]
- 5.Cunha GR, Young P, Christov K, Guzman R, Nandi S, Talamantes F, Thordarson G. Mammary phenotypic expression induced in epidermal cells by embryonic mammary mesenchyme. Acta Anat (Basel) 1995;152:195–204. doi: 10.1159/000147698. [DOI] [PubMed] [Google Scholar]
- 6.Kratochwil K. Organ specificity in mesenchymal induction demonstrated in the embryonic development of the mammary gland of the mouse. Dev Biol. 1969;20:46–71. doi: 10.1016/0012-1606(69)90004-9. [DOI] [PubMed] [Google Scholar]
- 7.Sakakura T, Nishizuka Y, Dawe CJ. Mesenchyme-dependent morphogenesis and epithelium-specific cyto differentiation in mouse mammary gland. Science. 1976;194:1439–1441. doi: 10.1126/science.827022. [DOI] [PubMed] [Google Scholar]
- 8.Robinson GW, Karpf AB, Kratochwil K. Regulation of mammary gland development by tissue interaction. J Mammary Gland Biol Neoplasia. 1999;4:9–19. doi: 10.1023/a:1018748418447. [DOI] [PubMed] [Google Scholar]
- 9.Sakakura T, Sakagami Y, Nishizuka Y. Dual origin of mesenchymal tissues participating in mouse mammary gland embryogenesis. Dev Biol. 1982;91:202–207. doi: 10.1016/0012-1606(82)90024-0. [DOI] [PubMed] [Google Scholar]
- 10.Chu EY, Hens J, Andl T, Kairo A, Yamaguchi TP, Brisken C, Glick A, Wysolmerski JJ, Millar SE. Canonical WNT signaling promotes mammary placode development and is essential for initiation of mammary gland morphogenesis. Development. 2004;131:4819–4829. doi: 10.1242/dev.01347. [DOI] [PubMed] [Google Scholar]
- 11.Veltmaat JM, Van Veelen W, Thiery JP, Bellusci S. Identification of the mammary line in mouse by Wnt10b expression. Dev Dyn. 2004;229:349–356. doi: 10.1002/dvdy.10441. [DOI] [PubMed] [Google Scholar]
- 12.van Genderen C, Okamura RM, Farinas I, Quo RG, Parslow TG, Bruhn L, Grosschedl R. Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in LEF-1-deficient mice. Genes Dev. 1994;8:2691–2703. doi: 10.1101/gad.8.22.2691. [DOI] [PubMed] [Google Scholar]
- 13.Jerome-Majewska LA, Jenkins GP, Ernstoff E, Zindy F, Sherr CJ, Papaioannou VE. Tbx3, the ulnar-mammary syndrome gene, and Tbx2 interact in mammary gland development through a p19Arf/p53-independent pathway. Dev Dyn. 2005;234:922–933. doi: 10.1002/dvdy.20575. [DOI] [PubMed] [Google Scholar]
- 14.Bamshad M, Lin RC, Law DJ, Watkins WC, Krakowiak PA, Moore ME, Franceschini P, Lala R, Holmes LB, Gebuhr TC, et al. Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nat Genet. 1997;16:311–315. doi: 10.1038/ng0797-311. [DOI] [PubMed] [Google Scholar]
- 15.Cho KW, Kim JY, Song SJ, Farrell E, Eblaghie MC, Kim HJ, Tickle C, Jung HS. Molecular interactions between Tbx3 and Bmp4 and a model for dorsoventral positioning of mammary gland development. Proc Natl Acad Sci U S A. 2006;103:16788–16793. doi: 10.1073/pnas.0604645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davenport TG, Jerome-Majewska LA, Papaioannou VE. Mammary gland, limb and yolk sac defects in mice lacking Tbx3, the gene mutated in human ulnar mammary syndrome. Development. 2003;130:2263–2273. doi: 10.1242/dev.00431. [DOI] [PubMed] [Google Scholar]
- 17.Veltmaat JM, Relaix F, Le LT, Kratochwil K, Sala FG, van Veelen W, Rice R, Spencer-Dene B, Mailleux AA, Rice DP, et al. Gli3-mediated somitic Fgf10 expression gradients are required for the induction and patterning of mammary epithelium along the embryonic axes. Development. 2006;133:2325–2335. doi: 10.1242/dev.02394. [DOI] [PubMed] [Google Scholar]
- 18.Mailleux AA, Kelly R, Veltmaat JM, De Langhe SP, Zaffran S, Thiery JP, Bellusci S. Fgf10 expression identifies parabronchial smooth muscle cell progenitors and is required for their entry into the smooth muscle cell lineage. Development. 2005;132:2157–2166. doi: 10.1242/dev.01795. [DOI] [PubMed] [Google Scholar]
- 19.Baban A, Torre M, Bianca S, Buluggiu A, Rossello MI, Calevo MG, Valle M, Ravazzolo R, Jasonni V, Lerone M. Poland syndrome with bilateral features: case description with review of the literature. Am J Med Genet A. 2009;149A:1597–1602. doi: 10.1002/ajmg.a.32922. [DOI] [PubMed] [Google Scholar]
- 20.Hatsell SJ, Cowin P. Gli3-mediated repression of Hedgehog targets is required for normal mammary development. Development. 2006;133:3661–3670. doi: 10.1242/dev.02542. [DOI] [PubMed] [Google Scholar]
- 21.Wang C, Ruther U, Wang B. The Shh-independent activator function of the full-length Gli3 protein and its role in vertebrate limb digit patterning. Dev Biol. 2007;305:460–469. doi: 10.1016/j.ydbio.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aoto K, Nishimura T, Eto K, Motoyama J. Mouse GLI3 regulates Fgf8 expression and apoptosis in the developing neural tube, face, and limb bud. Dev Biol. 2002;251:320–332. doi: 10.1006/dbio.2002.0811. [DOI] [PubMed] [Google Scholar]
- 23.Panchal H, Wansbury O, Parry S, Ashworth A, Howard B. Neuregulin3 alters cell fate in the epidermis and mammary gland. BMC Dev Biol. 2007;7:105. doi: 10.1186/1471-213X-7-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Howard BA, Gusterson BA. The characterization of a mouse mutant that displays abnormal mammary gland development. Mamm Genome. 2000;11:234–237. doi: 10.1007/s003350010043. [DOI] [PubMed] [Google Scholar]
- 25.Howard B, Panchal H, McCarthy A, Ashworth A. Identification of the scaramanga gene implicates Neuregulin3 in mammary gland specification. Genes Dev. 2005;19:2078–2090. doi: 10.1101/gad.338505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eblaghie MC, Song SJ, Kim JY, Akita K, Tickle C, Jung HS. Interactions between FGF and Wnt signals and Tbx3 gene expression in mammary gland initiation in mouse embryos. JAnat. 2004;205:1–13. doi: 10.1111/j.0021-8782.2004.00309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fliniaux I, Mikkola ML, Lefebvre S, Thesleff I. Identification of dkk4 as a target of Eda-A1/Edar pathway reveals an unexpected role of ectodysplasin as inhibitor of Wnt signalling in ectodermal placodes. Dev Biol. 2008;320:60–71. doi: 10.1016/j.ydbio.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 28.Mustonen T, Pispa J, Mikkola ML, Pummila M, Kangas AT, Pakkasjarvi L, Jaatinen R, Thesleff I. Stimulation of ectodermal organ development by Ectodysplasin-A1. Dev Biol. 2003;259:123–136. doi: 10.1016/s0012-1606(03)00157-x. [DOI] [PubMed] [Google Scholar]
- 29.Monreal AW, Ferguson BM, Headon DJ, Street SL, Overbeek PA, Zonana J. Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat Genet. 1999;22:366–369. doi: 10.1038/11937. [DOI] [PubMed] [Google Scholar]
- 30.Cowin P, Wysolmerski J. Molecular mechanisms guiding embryonic mammary gland development. Cold Spring Harb Perspect Biol. 2010;2:a003251. doi: 10.1101/cshperspect.a003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hogg NA, Harrison CJ, Tickle C. Lumen formation in the developing mouse mammary gland. J Embryol Exp Morphol. 1983;73:39–57. [PubMed] [Google Scholar]
- 32.Blatchford DR, Quarrie LH, Tonner E, McCarthy C, Flint DJ, Wilde CJ. Influence of microenvironment on mammary epithelial cell survival in primary culture. J Cell Physiol. 1999;181:304–311. doi: 10.1002/(SICI)1097-4652(199911)181:2<304::AID-JCP12>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 33.Ewald AJ, Brenot A, Duong M, Chan BS, Werb Z. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev Cell. 2008;14:570–581. doi: 10.1016/j.devcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 2002;111:29–40. doi: 10.1016/s0092-8674(02)01001-2. [DOI] [PubMed] [Google Scholar]
- 35.Hens JR, Dann P, Zhang JP, Harris S, Robinson GW, Wysolmerski J. BMP4 and PTHrP interact to stimulate ductal outgrowth during embryonic mammary development and to inhibit hair follicle induction. Development. 2007;134:1221–1230. doi: 10.1242/dev.000182. [DOI] [PubMed] [Google Scholar]
- 36.Kratochwil K, Schwartz P. Tissue interaction in androgen response of embryonic mammary rudiment of mouse: identification of target tissue for testosterone. Proc Natl Acad Sci U S A. 1976;73:4041–4044. doi: 10.1073/pnas.73.11.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Juppner H, Abou-Samra AB, Freeman M, Kong XF, Schipani E, Richards J, Kolakowski LF, Jr, Hock J, Potts JT, Jr, Kronenberg HM, et al. A G protein-linked receptor for parathyroid hormone and parathyroid hormone-related peptide. Science. 1991;254:1024–1026. doi: 10.1126/science.1658941. [DOI] [PubMed] [Google Scholar]
- 38.Wysolmerski JJ, McCaughern-Carucci JF, Daifotis AG, Broadus AE, Philbrick WM. Overexpression of parathyroid hormone-related protein or parathyroid hormone in transgenic mice impairs branching morphogenesis during mammary gland development. Development. 1995;121:3539–3547. doi: 10.1242/dev.121.11.3539. [DOI] [PubMed] [Google Scholar]
- 39.Jobert AS, Zhang P, Couvineau A, Bonaventure J, Roume J, Le Merrer M, Silve C. Absence of functional receptors for parathyroid hormone and parathyroid hormone-related peptide in Blomstrand chondrodysplasia. J Clin Invest. 1998;102:34–40. doi: 10.1172/JCI2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wysolmerski JJ, Cormier S, Philbrick WM, Dann P, Zhang JP, Roume J, Delezoide AL, Silve C. Absence of functional type 1 parathyroid hormone (PTH)/PTH-related protein receptors in humans is associated with abnormal breast development and tooth impaction. J Clin Endocrinol Metab. 2001;86:1788–1794. doi: 10.1210/jcem.86.4.7404. [DOI] [PubMed] [Google Scholar]
- 41.Wysolmerski JJ, Philbrick WM, Dunbar ME, Lanske B, Kronenberg H, Broadus AE. Rescue of the parathyroid hormone-related protein knockout mouse demonstrates that parathyroid hormone-related protein is essential for mammary gland development. Development. 1998;125:1285–1294. doi: 10.1242/dev.125.7.1285. [DOI] [PubMed] [Google Scholar]
- 42.Foley J, Dann P, Hong J, Cosgrove J, Dreyer B, Rimm D, Dunbar M, Philbrick W, Wysolmerski J. Parathyroid hormone-related protein maintains mammary epithelial fate and triggers nipple skin differentiation during embryonic breast development. Development. 2001;128:513–525. doi: 10.1242/dev.128.4.513. [DOI] [PubMed] [Google Scholar]
- 43.Williams JM, Daniel CW. Mammary ductal elongation: differentiation of myoepithelium and basal lamina during branching morphogenesis. Dev Biol. 1983;97:274–290. doi: 10.1016/0012-1606(83)90086-6. [DOI] [PubMed] [Google Scholar]
- 44.Trott JF, Vonderhaar BK, Hovey RC. Historical perspectives of prolactin and growth hormone as mammogens, lactogens and galactagogues--agog for the future! J Mammary Gland Biol Neoplasia. 2008;13:3–11. doi: 10.1007/s10911-008-9064-x. [DOI] [PubMed] [Google Scholar]
- 45.Gallego MI, Binart N, Robinson GW, Okagaki R, Coschigano KT, Perry J, Kopchick JJ, Oka T, Kelly PA, Hennighausen L. Prolactin, growth hormone, and epidermal growth factor activate Stat5 in different compartments of mammary tissue and exert different and overlapping developmental effects. Dev Biol. 2001;229:163–175. doi: 10.1006/dbio.2000.9961. [DOI] [PubMed] [Google Scholar]
- 46.Ruan W, Kleinberg DL. Insulin-like growth factor I is essential for terminal end bud formation and ductal morphogenesis during mammary development. Endocrinology. 1999;140:5075–5081. doi: 10.1210/endo.140.11.7095. [DOI] [PubMed] [Google Scholar]
- 47.Zhou Y, Xu BC, Maheshwari HG, He L, Reed M, Lozykowski M, Okada S, Cataldo L, Coschigamo K, Wagner TE, et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse) Proc Natl Acad Sci U S A. 1997;94:13215–13220. doi: 10.1073/pnas.94.24.13215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richards RG, Klotz DM, Walker MP, Diaugustine RP. Mammary gland branching morphogenesis is diminished in mice with a deficiency of insulin-like growth factor-I (IGF-I), but not in mice with a liver-specific deletion of IGF-I. Endocrinology. 2004;145:3106–3110. doi: 10.1210/en.2003-1112. [DOI] [PubMed] [Google Scholar]
- 49.Cannata D, Lann D, Wu Y, Elis S, Sun H, Yakar S, Lazzarino DA, Wood TL, Leroith D. Elevated circulating IGF-I promotes mammary gland development and proliferation. Endocrinology. 2010;151:5751–5761. doi: 10.1210/en.2010-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bohlke K, Cramer DW, Trichopoulos D, Mantzoros CS. Insulin-like growth factor-I in relation to premenopausal ductal carcinoma in situ of the breast. Epidemiology. 1998;9:570–573. [PubMed] [Google Scholar]
- 51.Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, Rosner B, Speizer FE, Pollak M. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet. 1998;351:1393–1396. doi: 10.1016/S0140-6736(97)10384-1. [DOI] [PubMed] [Google Scholar]
- 52.Lieberman ME, Maurer RA, Gorski J. Estrogen control of prolactin synthesis in vitro. Proc Natl Acad Sci U S A. 1978;75:5946–5949. doi: 10.1073/pnas.75.12.5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Silberstein GB, Daniel CW. Elvax 40P implants: sustained, local release of bioactive molecules influencing mammary ductal development. Dev Biol. 1982;93:272–278. doi: 10.1016/0012-1606(82)90259-7. [DOI] [PubMed] [Google Scholar]
- 54.Daniel CW, Silberstein GB, Strickland P. Direct action of 17 beta-estradiol on mouse mammary ducts analyzed by sustained release implants and steroid autoradiography. Cancer Res. 1987;47:6052–6057. [PubMed] [Google Scholar]
- 55.Silberstein GB, Van Horn K, Shyamala G, Daniel CW. Essential role of endogenous estrogen in directly stimulating mammary growth demonstrated by implants containing pure antiestrogens. Endocrinology. 1994;134:84–90. doi: 10.1210/endo.134.1.8275973. [DOI] [PubMed] [Google Scholar]
- 56.Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci U S A. 1993;90:11162–11166. doi: 10.1073/pnas.90.23.11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, Sar M, Korach KS, Gustafsson JA, Smithies O. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci U S A. 1998;95:15677–15682. doi: 10.1073/pnas.95.26.15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zeps N, Bentel JM, Papadimitriou JM, D’Antuono MF, Dawkins HJ. Estrogen receptor-negative epithelial cells in mouse mammary gland development and growth. Differentiation. 1998;62:221–226. doi: 10.1046/j.1432-0436.1998.6250221.x. [DOI] [PubMed] [Google Scholar]
- 59.Mallepell S, Krust A, Chambon P, Brisken C. Paracrine signaling through the epithelial estrogen receptor alpha is required for proliferation and morphogenesis in the mammary gland. Proc Natl Acad Sci U S A. 2006;103:2196–2201. doi: 10.1073/pnas.0510974103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nandi S. Endocrine control of mammary-gland development and function in the C3H/He Crgl mouse. Journal of the National Cancer Institute. 1958;21:1039–1063. [PubMed] [Google Scholar]
- 61.Nandi S. Hormonal control of mammogenesis and lactogenesis in the C3H/He Crgl mouse. University of California Berkeley Publications in Zoology. 1959;65:1–128. [Google Scholar]
- 62.Feng Y, Manka D, Wagner KU, Khan SA. Estrogen receptor-alpha expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc Natl Acad Sci U S A. 2007;104:14718–14723. doi: 10.1073/pnas.0706933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Coleman S, Silberstein GB, Daniel CW. Ductal morphogenesis in the mouse mammary gland: evidence supporting a role for epidermal growth factor. Dev Biol. 1988;127:304–315. doi: 10.1016/0012-1606(88)90317-x. [DOI] [PubMed] [Google Scholar]
- 64.Kenney NJ, Bowman A, Korach KS, Barrett JC, Salomon DS. Effect of exogenous epidermal-like growth factors on mammary gland development and differentiation in the estrogen receptor-alpha knockout (ERKO) mouse. Breast Cancer Res Treat. 2003;79:161–173. doi: 10.1023/a:1023938510508. [DOI] [PubMed] [Google Scholar]
- 65.Luetteke NC, Qiu TH, Fenton SE, Troyer KL, Riedel RF, Chang A, Lee DC. Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development. 1999;126:2739–2750. doi: 10.1242/dev.126.12.2739. [DOI] [PubMed] [Google Scholar]
- 66.Ciarloni L, Mallepell S, Brisken C. Amphiregulin is an essential mediator of estrogen receptor alpha function in mammary gland development. Proc Natl Acad Sci U S A. 2007;104:5455–5460. doi: 10.1073/pnas.0611647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sternlicht MD, Sunnarborg SW, Kouros-Mehr H, Yu Y, Lee DC, Werb Z. Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulin. Development. 2005;132:3923–3933. doi: 10.1242/dev.01966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wiesen JF, Young P, Werb Z, Cunha GR. Signaling through the stromal epidermal growth factor receptor is necessary for mammary ductal development. Development. 1999;126:335–344. doi: 10.1242/dev.126.2.335. [DOI] [PubMed] [Google Scholar]
- 69.Chakravorti S, Sheffield L. Acidic and basic fibroblast growth factor mRNA and protein in mouse mammary glands. Endocrine. 1996;4:175–182. doi: 10.1007/BF02782762. [DOI] [PubMed] [Google Scholar]
- 70.Lu P, Ewald AJ, Martin GR, Werb Z. Genetic mosaic analysis reveals FGF receptor 2 function in terminal end buds during mammary gland branching morphogenesis. Dev Biol. 2008;321:77–87. doi: 10.1016/j.ydbio.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Silberstein GB, Daniel CW. Reversible inhibition of mammary gland growth by transforming growth factor-beta. Science. 1987;237:291–293. doi: 10.1126/science.3474783. [DOI] [PubMed] [Google Scholar]
- 72.Ewan KB, Shyamala G, Ravani SA, Tang Y, Akhurst R, Wakefield L, Barcellos-Hoff MH. Latent transforming growth factor-beta activation in mammary gland: regulation by ovarian hormones affects ductal and alveolar proliferation. Am J Pathol. 2002;160:2081–2093. doi: 10.1016/s0002-9440(10)61158-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ingman WV, Robertson SA. Mammary gland development in transforming growth factor beta1 null mutant mice: systemic and epithelial effects. Biol Reprod. 2008;79:711–717. doi: 10.1095/biolreprod.107.067272. [DOI] [PubMed] [Google Scholar]
- 74.Joseph H, Gorska AE, Sohn P, Moses HL, Serra R. Overexpression of a kinase-deficient transforming growth factor-beta type II receptor in mouse mammary stroma results in increased epithelial branching. Mol Biol Cell. 1999;10:1221–1234. doi: 10.1091/mbc.10.4.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cheng N, Bhowmick NA, Chytil A, Gorksa AE, Brown KA, Muraoka R, Arteaga CL, Neilson EG, Hayward SW, Moses HL. Loss of TGF-beta type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-alpha-, MSP-and HGF-mediated signaling networks. Oncogene. 2005;24:5053–5068. doi: 10.1038/sj.onc.1208685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gorska AE, Joseph H, Derynck R, Moses HL, Serra R. Dominant-negative interference of the transforming growth factor beta type II receptor in mammary gland epithelium results in alveolar hyperplasia and differentiation in virgin mice. Cell Growth Differ. 1998;9:229–238. [PubMed] [Google Scholar]
- 77.Nelson CM, Vanduijn MM, Inman JL, Fletcher DA, Bissell MJ. Tissue geometry determines sites of mammary branching morphogenesis in organotypic cultures. Science. 2006;314:298–300. doi: 10.1126/science.1131000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roarty K, Serra R. Wnt5a is required for proper mammary gland development and TGF-beta-mediated inhibition of ductal growth. Development. 2007;134:3929–3939. doi: 10.1242/dev.008250. [DOI] [PubMed] [Google Scholar]
- 79.Srinivasan K, Strickland P, Valdes A, Shin GC, Hinck L. Netrin-1/neogenin interaction stabilizes multipotent progenitor cap cells during mammary gland morphogenesis. Dev Cell. 2003;4:371–382. doi: 10.1016/s1534-5807(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 80.Strickland P, Shin GC, Plump A, Tessier-Lavigne M, Hinck L. Slit2 and netrin 1 act synergistically as adhesive cues to generate tubular bi-layers during ductal morphogenesis. Development. 2006;133:823–832. doi: 10.1242/dev.02261. [DOI] [PubMed] [Google Scholar]
- 81.Khialeeva E, Lane TF, Carpenter EM. Disruption of reelin signaling alters mammary gland morphogenesis. Development. 2011;138:767–776. doi: 10.1242/dev.057588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ensslin MA, Shur BD. The EGF repeat and discoidin domain protein, SED1/MFG-E8, is required for mammary gland branching morphogenesis. Proc Natl Acad Sci U S A. 2007;104:2715–2720. doi: 10.1073/pnas.0610296104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Atabai K, Fernandez R, Huang X, Ueki I, Kline A, Li Y, Sadatmansoori S, Smith-Steinhart C, Zhu W, Pytela R, et al. Mfge8 is critical for mammary gland remodeling during involution. Mol Biol Cell. 2005;16:5528–5537. doi: 10.1091/mbc.E05-02-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee S, Medina D, Tsimelzon A, Mohsin SK, Mao S, Wu Y, Allred DC. Alterations of gene expression in the development of early hyperplastic precursors of breast cancer. Am J Pathol. 2007;171:252–262. doi: 10.2353/ajpath.2007.061010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Baillo A, Giroux C, Ethier SP. Knock-down of amphiregulin inhibits cellular invasion in inflammatory breast cancer. J Cell Physiol. 2011 doi: 10.1002/jcp.22620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Djonov V, Andres AC, Ziemiecki A. Vascular remodelling during the normal andmalignant life cycle of the mammary gland. Microsc Res Tech. 2001;52:182–189. doi: 10.1002/1097-0029(20010115)52:2<182::AID-JEMT1004>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 87.Lydon JP, DeMayo FJ, Funk CR, Mani SK, Hughes AR, Montgomery CA, Jr, Shyamala G, Conneely OM, O’Malley BW. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995;9:2266–2278. doi: 10.1101/gad.9.18.2266. [DOI] [PubMed] [Google Scholar]
- 88.Brisken C, Park S, Vass T, Lydon JP, O’Malley BW, Weinberg RA. A paracrine role for the epithelial progesterone receptor in mammary gland development. Proc Natl Acad Sci U S A. 1998;95:5076–5081. doi: 10.1073/pnas.95.9.5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Seagroves TN, Lydon JP, Hovey RC, Vonderhaar BK, Rosen JM. C/EBPbeta (CCAAT/enhancer binding protein) controls cell fate determination during mammary gland development. Mol Endocrinol. 2000;14:359–368. doi: 10.1210/mend.14.3.0434. [DOI] [PubMed] [Google Scholar]
- 90.Shyamala G, Schneider W, Schott D. Developmental regulation of murine mammary progesterone receptor gene expression. Endocrinology. 1990;126:2882–2889. doi: 10.1210/endo-126-6-2882. [DOI] [PubMed] [Google Scholar]
- 91.Mulac-Jericevic B, Lydon JP, DeMayo FJ, Conneely OM. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc Natl Acad Sci U S A. 2003;100:9744–9749. doi: 10.1073/pnas.1732707100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mulac-Jericevic B, Mullinax RA, DeMayo FJ, Lydon JP, Conneely OM. Subgroup of reproductive functions of progesterone mediated by progesterone receptor-B isoform. Science. 2000;289:1751–1754. doi: 10.1126/science.289.5485.1751. [DOI] [PubMed] [Google Scholar]
- 93.Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem. 2002;277:5209–5218. doi: 10.1074/jbc.M110090200. [DOI] [PubMed] [Google Scholar]
- 94.Graham JD, Yeates C, Balleine RL, Harvey SS, Milliken JS, Bilous AM, Clarke CL. Characterization of progesterone receptor A and B expression in human breast cancer. Cancer Res. 1995;55:5063–5068. [PubMed] [Google Scholar]
- 95.Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA, Elliott R, Scully S, Voura EB, Lacey DL, et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell. 2000;103:41–50. doi: 10.1016/s0092-8674(00)00103-3. [DOI] [PubMed] [Google Scholar]
- 96.Beleut M, Rajaram RD, Caikovski M, Ayyanan A, Germano D, Choi Y, Schneider P, Brisken C. Two distinct mechanisms underlie progesterone-induced proliferation in the mammary gland. Proc Natl Acad Sci U S A. 2010;107:2989–2994. doi: 10.1073/pnas.0915148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mukherjee A, Soyal SM, Li J, Ying Y, He B, DeMayo FJ, Lydon JP. Targeting RANKL to a specific subset of murine mammary epithelial cells induces ordered branching morphogenesis and alveologenesis in the absence of progesterone receptor expression. FASEB J. 2010;24:4408–4419. doi: 10.1096/fj.10-157982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fernandez-Valdivia R, Mukherjee A, Creighton CJ, Buser AC, DeMayo FJ, Edwards DP, Lydon JP. Transcriptional response of the murine mammary gland to acute progesterone exposure. Endocrinology. 2008;149:6236–6250. doi: 10.1210/en.2008-0768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee HJ, Hanada R, Joshi PA, Aliprantis A, Glimcher L, et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature. 2010;468:98–102. doi: 10.1038/nature09387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gonzalez-Suarez E, Jacob AP, Jones J, Miller R, Roudier-Meyer MP, Erwert R, Pinkas J, Branstetter D, Dougall WC. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature. 2010;468:103–107. doi: 10.1038/nature09495. [DOI] [PubMed] [Google Scholar]
- 101.Ormandy CJ, Camus A, Barra J, Damotte D, Lucas B, Buteau H, Edery M, Brousse N, Babinet C, Binart N, et al. Null mutation of the prolactin receptor gene produces multiple reproductive defects in the mouse. Genes Dev. 1997;11:167–178. doi: 10.1101/gad.11.2.167. [DOI] [PubMed] [Google Scholar]
- 102.Horseman ND, Zhao W, Montecino-Rodriguez E, Tanaka M, Nakashima K, Engle SJ, Smith F, Markoff E, Dorshkind K. Defective mammopoiesis, but normal hematopoiesis, in mice with a targeted disruption of the prolactin gene. EMBO J. 1997;16:6926–6935. doi: 10.1093/emboj/16.23.6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Naylor MJ, Lockefeer JA, Horseman ND, Ormandy CJ. Prolactin regulates mammary epithelial cell proliferation via autocrine/paracrine mechanism. Endocrine. 2003;20:111–114. doi: 10.1385/ENDO:20:1-2:111. [DOI] [PubMed] [Google Scholar]
- 104.Brisken C, Kaur S, Chavarria TE, Binart N, Sutherland RL, Weinberg RA, Kelly PA, Ormandy CJ. Prolactin controls mammary gland development via direct and indirect mechanisms. Dev Biol. 1999;210:96–106. doi: 10.1006/dbio.1999.9271. [DOI] [PubMed] [Google Scholar]
- 105.Vomachka AJ, Pratt SL, Lockefeer JA, Horseman ND. Prolactin gene-disruption arrests mammary gland development and retards T-antigen-induced tumor growth. Oncogene. 2000;19:1077–1084. doi: 10.1038/sj.onc.1203348. [DOI] [PubMed] [Google Scholar]
- 106.Han Y, Watling D, Rogers NC, Stark GR. JAK2 and STAT5, but not JAK1 and STAT1, are required for prolactin-induced beta-lactoglobulin transcription. Mol Endocrinol. 1997;11:1180–1188. doi: 10.1210/mend.11.8.9952. [DOI] [PubMed] [Google Scholar]
- 107.Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, Robinson GW, Hennighausen L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wagner KU, Krempler A, Triplett AA, Qi Y, George NM, Zhu J, Rui H. Impaired alveologenesis and maintenance of secretory mammary epithelial cells in Jak2 conditional knockout mice. Mol Cell Biol. 2004;24:5510–5520. doi: 10.1128/MCB.24.12.5510-5520.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Naylor MJ, Li N, Cheung J, Lowe ET, Lambert E, Marlow R, Wang P, Schatzmann F, Wintermantel T, Schuetz G, et al. Ablation of beta1 integrin in mammary epithelium reveals a key role for integrin in glandular morphogenesis and differentiation. J Cell Biol. 2005;171:717–728. doi: 10.1083/jcb.200503144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Galbaugh T, Feeney YB, Clevenger CV. Prolactin receptor-integrin cross-talk mediated by SIRPalpha in breast cancer cells. Mol Cancer Res. 2010;8:1413–1424. doi: 10.1158/1541-7786.MCR-10-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Srivastava S, Matsuda M, Hou Z, Bailey JP, Kitazawa R, Herbst MP, Horseman ND. Receptor activator of NF-kappaB ligand induction via Jak2 and Stat5a in mammary epithelial cells. J Biol Chem. 2003;278:46171–46178. doi: 10.1074/jbc.M308545200. [DOI] [PubMed] [Google Scholar]
- 112.Fernandez-Valdivia R, Mukherjee A, Ying Y, Li J, Paquet M, DeMayo FJ, Lydon JP. The RANKL signaling axis is sufficient to elicit ductal side-branching and alveologenesis in the mammary gland of the virgin mouse. Dev Biol. 2009;328:127–139. doi: 10.1016/j.ydbio.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 113.Oakes SR, Rogers RL, Naylor MJ, Ormandy CJ. Prolactin regulation of mammary gland development. J Mammary Gland Biol Neoplasia. 2008;13:13–28. doi: 10.1007/s10911-008-9069-5. [DOI] [PubMed] [Google Scholar]
- 114.D’Cruz CM, Moody SE, Master SR, Hartman JL, Keiper EA, Imielinski MB, Cox JD, Wang JY, Ha SI, Keister BA, et al. Persistent parity-induced changes in growth factors, TGF-beta3, and differentiation in the rodent mammary gland. Mol Endocrinol. 2002;16:2034–2051. doi: 10.1210/me.2002-0073. [DOI] [PubMed] [Google Scholar]
- 115.Balogh GA, Heulings R, Mailo DA, Russo PA, Sheriff F, Russo IH, Moral R, Russo J. Genomic signature induced by pregnancy in the human breast. Int J Oncol. 2006;28:399–410. [PubMed] [Google Scholar]
- 116.Li M, Liu X, Robinson G, Bar-Peled U, Wagner KU, Young WS, Hennighausen L, Furth PA. Mammary-derived signals activate programmed cell death during the first stage of mammary gland involution. Proc Natl Acad Sci U S A. 1997;94:3425–3430. doi: 10.1073/pnas.94.7.3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Marti A, Feng Z, Altermatt HJ, Jaggi R. Milk accumulation triggers apoptosis of mammary epithelial cells. Eur J Cell Biol. 1997;73:158–165. [PubMed] [Google Scholar]
- 118.Marti A, Ritter PM, Jager R, Lazar H, Baltzer A, Schenkel J, Declercq W, Vandenabeele P, Jaggi R. Mouse mammary gland involution is associated with cytochrome c release and caspase activation. Mech Dev. 2001;104:89–98. doi: 10.1016/s0925-4773(01)00381-1. [DOI] [PubMed] [Google Scholar]
- 119.Song J, Sapi E, Brown W, Nilsen J, Tartaro K, Kacinski BM, Craft J, Naftolin F, Mor G. Roles of Fas and Fas ligand during mammary gland remodeling. J Clin Invest. 2000;106:1209–1220. doi: 10.1172/JCI10411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Baxter FO, Came PJ, Abell K, Kedjouar B, Huth M, Rajewsky K, Pasparakis M, Watson CJ. IKKbeta/2 induces TWEAK and apoptosis in mammary epithelial cells. Development. 2006;133:3485–3494. doi: 10.1242/dev.02502. [DOI] [PubMed] [Google Scholar]
- 121.Kreuzaler PA, Staniszewska AD, Li W, Omidvar N, Kedjouar B, Turkson J, Poli V, Flavell RA, Clarkson RW, Watson CJ. Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol. 2011 doi: 10.1038/ncb2171. [DOI] [PubMed] [Google Scholar]
- 122.Creamer BA, Sakamoto K, Schmidt JW, Triplett AA, Moriggl R, Wagner KU. Stat5 promotes survival of mammary epithelial cells through transcriptional activation of a distinct promoter in Akt1. Mol Cell Biol. 2010;30:2957–2970. doi: 10.1128/MCB.00851-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schwertfeger KL, Richert MM, Anderson SM. Mammary gland involution is delayed by activated Akt in transgenic mice. Mol Endocrinol. 2001;15:867–881. doi: 10.1210/mend.15.6.0663. [DOI] [PubMed] [Google Scholar]
- 124.Maroulakou IG, Oemler W, Naber SP, Klebba I, Kuperwasser C, Tsichlis PN. Distinct roles of the three Akt isoforms in lactogenic differentiation and involution. J Cell Physiol. 2008;217:468–477. doi: 10.1002/jcp.21518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Clarkson RW, Boland MP, Kritikou EA, Lee JM, Freeman TC, Tiffen PG, Watson CJ. The genes induced by signal transducer and activators of transcription (STAT)3 and STAT5 in mammary epithelial cells define the roles of these STATs in mammary development. Mol Endocrinol. 2006;20:675–685. doi: 10.1210/me.2005-0392. [DOI] [PubMed] [Google Scholar]
- 126.Chapman RS, Lourenco PC, Tonner E, Flint DJ, Selbert S, Takeda K, Akira S, Clarke AR, Watson CJ. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of Stat3. Genes Dev. 1999;13:2604–2616. doi: 10.1101/gad.13.19.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kritikou EA, Sharkey A, Abell K, Came PJ, Anderson E, Clarkson RW, Watson CJ. A dual, non-redundant, role for LIF as a regulator of development and STAT3-mediated cell death in mammary gland. Development. 2003;130:3459–3468. doi: 10.1242/dev.00578. [DOI] [PubMed] [Google Scholar]
- 128.Abell K, Bilancio A, Clarkson RW, Tiffen PG, Altaparmakov AI, Burdon TG, Asano T, Vanhaesebroeck B, Watson CJ. Stat3-induced apoptosis requires a molecular switch in PI(3)K subunit composition. Nat Cell Biol. 2005;7:392–398. doi: 10.1038/ncb1242. [DOI] [PubMed] [Google Scholar]
- 129.Tonner E, Barber MC, Travers MT, Logan A, Flint DJ. Hormonal control of insulin-like growth factor-binding protein-5 production in the involuting mammary gland of the rat. Endocrinology. 1997;138:5101–5107. doi: 10.1210/endo.138.12.5619. [DOI] [PubMed] [Google Scholar]
- 130.Ning Y, Hoang B, Schuller AG, Cominski TP, Hsu MS, Wood TL, Pintar JE. Delayed mammary gland involution in mice with mutation of the insulin-like growth factor binding protein 5 gene. Endocrinology. 2007;148:2138–2147. doi: 10.1210/en.2006-0041. [DOI] [PubMed] [Google Scholar]
- 131.Lund LR, Bjorn SF, Sternlicht MD, Nielsen BS, Solberg H, Usher PA, Osterby R, Christensen IJ, Stephens RW, Bugge TH, et al. Lactational competence and involution of the mouse mammary gland require plasminogen. Development. 2000;127:4481–4492. doi: 10.1242/dev.127.20.4481. [DOI] [PubMed] [Google Scholar]
- 132.Lilla JN, Joshi RV, Craik CS, Werb Z. Active plasma kallikrein localizes to mast cells and regulates epithelial cell apoptosis, adipocyte differentiation, and stromal remodeling during mammary gland involution. J Biol Chem. 2009;284:13792–13803. doi: 10.1074/jbc.M900508200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sympson CJ, Talhouk RS, Alexander CM, Chin JR, Clift SM, Bissell MJ, Werb Z. Targeted expression of stromelysin-1 in mammary gland provides evidence for a role of proteinases in branching morphogenesis and the requirement for an intact basement membrane for tissue-specific gene expression. J Cell Biol. 1994;125:681–693. doi: 10.1083/jcb.125.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fata JE, Leco KJ, Voura EB, Yu HY, Waterhouse P, Murphy G, Moorehead RA, Khokha R. Accelerated apoptosis in the Timp-3-deficient mammary gland. J Clin Invest. 2001;108:831–841. doi: 10.1172/JCI13171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Witty JP, Wright JH, Matrisian LM. Matrix metalloproteinases are expressed during ductal and alveolar mammary morphogenesis, and misregulation of stromelysin-1 in transgenic mice induces unscheduled alveolar development. Mol Biol Cell. 1995;6:1287–1303. doi: 10.1091/mbc.6.10.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wiseman BS, Sternlicht MD, Lund LR, Alexander CM, Mott J, Bissell MJ, Soloway P, Itohara S, Werb Z. Site-specific inductive and inhibitory activities of MMP-2 and MMP-3 orchestrate mammary gland branching morphogenesis. J Cell Biol. 2003;162:1123–1133. doi: 10.1083/jcb.200302090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Clarkson RW, Watson CJ. Microarray analysis of the involution switch. J Mammary Gland Biol Neoplasia. 2003;8:309–319. doi: 10.1023/b:jomg.0000010031.53310.92. [DOI] [PubMed] [Google Scholar]
- 138.Schedin P. Pregnancy-associated breast cancer and metastasis. Nat Rev Cancer. 2006;6:281–291. doi: 10.1038/nrc1839. [DOI] [PubMed] [Google Scholar]
- 139.Bemis LT, Schedin P. Reproductive state of rat mammary gland stroma modulates human breast cancer cell migration and invasion. Cancer Res. 2000;60:3414–3418. [PubMed] [Google Scholar]
- 140.McDaniel SM, Rumer KK, Biroc SL, Metz RP, Singh M, Porter W, Schedin P. Remodeling of the mammary microenvironment after lactation promotes breast tumor cell metastasis. Am J Pathol. 2006;168:608–620. doi: 10.2353/ajpath.2006.050677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ghosh K, Hartmann LC, Reynolds C, Visscher DW, Brandt KR, Vierkant RA, Scott CG, Radisky DC, Sellers TA, Pankratz VS, et al. Association between mammographic density and age-related lobular involution of the breast. J Clin Oncol. 2010;28:2207–2212. doi: 10.1200/JCO.2009.23.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Milanese TR, Hartmann LC, Sellers TA, Frost MH, Vierkant RA, Maloney SD, Pankratz VS, Degnim AC, Vachon CM, Reynolds CA, et al. Age-related lobular involution and risk of breast cancer. J Natl Cancer Inst. 2006;98:1600–1607. doi: 10.1093/jnci/djj439. [DOI] [PubMed] [Google Scholar]
- 143.Daniel CW, De Ome KB, Young JT, Blair PB, Faulkin LJ., Jr The in vivo life span of normal and preneoplastic mouse mammary glands: a serial transplantation study. Proc Natl Acad Sci U S A. 1968;61:53–60. doi: 10.1073/pnas.61.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Daniel CW, Young LJ, Medina D, DeOme KB. The influence of mammogenic hormones on serially transplanted mouse mammary gland. Exp Gerontol. 1971;6:95–101. doi: 10.1016/0531-5565(71)90053-2. [DOI] [PubMed] [Google Scholar]
- 145.Young LJ, Medina D, DeOme KB, Daniel CW. The influence of host and tissue age on life span and growth rate of serially transplanted mouse mammary gland. Exp Gerontol. 1971;6:49–56. doi: 10.1016/0531-5565(71)90048-9. [DOI] [PubMed] [Google Scholar]
- 146.Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, Wu L, Lindeman GJ, Visvader JE. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- 147.Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, Li HI, Eaves CJ. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- 148.Stingl J, Eaves CJ, Zandieh I, Emerman JT. Characterization of bipotent mammary epithelial progenitor cells in normal adult human breast tissue. Breast Cancer Res Treat. 2001;67:93–109. doi: 10.1023/a:1010615124301. [DOI] [PubMed] [Google Scholar]
- 149.Clarke RB. Isolation and characterization of human mammary stem cells. Cell Prolif. 2005;38:375–386. doi: 10.1111/j.1365-2184.2005.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chepko G, Smith GH. Three division-competent, structurally-distinct cell populations contribute to murine mammary epithelial renewal. Tissue Cell. 1997;29:239–253. doi: 10.1016/s0040-8166(97)80024-9. [DOI] [PubMed] [Google Scholar]
- 151.Kordon EC, Smith GH. An entire functional mammary gland may comprise the progeny from a single cell. Development. 1998;125:1921–1930. doi: 10.1242/dev.125.10.1921. [DOI] [PubMed] [Google Scholar]
- 152.Moraes RC, Zhang X, Harrington N, Fung JY, Wu MF, Hilsenbeck SG, Allred DC, Lewis MT. Constitutive activation of smoothened (SMO) in mammary glands of transgenic mice leads to increased proliferation, altered differentiation and ductal dysplasia. Development. 2007;134:1231–1242. doi: 10.1242/dev.02797. [DOI] [PubMed] [Google Scholar]
- 153.Asselin-Labat ML, Sutherland KD, Barker H, Thomas R, Shackleton M, Forrest NC, Hartley L, Robb L, Grosveld FG, van der Wees J, et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat Cell Biol. 2007;9:201–209. doi: 10.1038/ncb1530. [DOI] [PubMed] [Google Scholar]
- 154.Booth BW, Boulanger CA, Smith GH. Alveolar progenitor cells develop in mouse mammary glands independent of pregnancy and lactation. J Cell Physiol. 2007;212:729–736. doi: 10.1002/jcp.21071. [DOI] [PubMed] [Google Scholar]
- 155.Knoblich JA. Asymmetric cell division: recent developments and their implications for tumour biology. Nat Rev Mol Cell Biol. 2010;11:849–860. doi: 10.1038/nrm3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Smith GH, Medina D. A morphologically distinct candidate for an epithelial stem cell in mouse mammary gland. J Cell Sci. 1988;90(Pt 1):173–183. doi: 10.1242/jcs.90.1.173. [DOI] [PubMed] [Google Scholar]
- 157.Daniel CW, Young LJ. Influence of cell division on an aging process. Life span of mouse mammary epithelium during serial propagation in vivo. Exp Cell Res. 1971;65:27–32. doi: 10.1016/s0014-4827(71)80046-0. [DOI] [PubMed] [Google Scholar]
- 158.Fernandez-Gonzalez R, Illa-Bochaca I, Welm BE, Fleisch MC, Werb Z, Ortiz-de-Solorzano C, Barcellos-Hoff MH. Mapping mammary gland architecture using multi-scale in situ analysis. Integr Biol (Camb) 2009;1:80–89. doi: 10.1039/b816933k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Farnie G, Clarke RB. Mammary stem cells and breast cancer--role of Notch signalling. Stem Cell Rev. 2007;3:169–175. doi: 10.1007/s12015-007-0023-5. [DOI] [PubMed] [Google Scholar]
- 160.Visbal AP, Lewis MT. Hedgehog signaling in the normal and neoplastic mammary gland. Curr Drug Targets. 2010;11:1103–1111. doi: 10.2174/138945010792006753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wend P, Holland JD, Ziebold U, Birchmeier W. Wnt signaling in stem and cancer stem cells. Semin Cell Dev Biol. 2010;21:855–863. doi: 10.1016/j.semcdb.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 162.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 163.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 165.Visvader JE. Cells of origin in cancer. Nature. 2011;469:314–322. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 166.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin-Labat ML, Gyorki DE, Ward T, Partanen A, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 167.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]