

Abstract
Ischemic acute kidney injury (AKI) triggers an inflammatory response which exacerbates injury that requires increased expression of endothelial adhesion molecules. To study this further, we used in situ hybridization, immunohistology, and isolated endothelial cells, and found increased Toll-like receptor 4 (TLR4) expression on endothelial cells of the vasa rectae of the inner stripe of the outer medulla of the kidney 4 h after reperfusion. This increase was probably due to reactive oxygen species, known to be generated early during ischemic AKI, because the addition of hydrogen peroxide increased TLR4 expression in MS1 microvascular endothelial cells in vitro. Endothelial TLR4 may regulate adhesion molecule (CD54 and CD62E) expression as they were increased on endothelia of wild-type but not TLR4 knockout mice in vivo. Further, the addition of high-mobility group protein B1, a TLR4 ligand released by injured cells, increased adhesion molecule expression on endothelia isolated from wild-type but not TLR4 knockout mice. TLR4 was localized to proximal tubules in the cortex and outer medulla after 24 h of reperfusion. Thus, at least two different cell types express TLR4, each of which contributes to renal injury by temporally different mechanisms during ischemic AKI.
Keywords: acute kidney injury, endothelium, inflammation, ischemiat–reperfusion
Acute renal ischemia triggers an inflammatory response that exacerbates injury.1–4 This inflammation requires increased expression of endothelial adhesion molecules that facilitate diapedis of leukocytes into renal tissues.5 This requirement was demonstrated when these molecules were inactivated by antibodies, transgenic knockout, or antisense, and the inactivation not only prevented inflammation but also ameliorated ischemic injury.6–10 Despite their importance in the inflammatory response to ischemic injury, the regulation of these endothelial adhesion molecules is not well understood.
We now suggest that endothelial Toll-like receptor 4 (TLR4) regulates these molecules early after ischemia. We report that TLR4 appears on the endothelia of the vasa rectae of the inner stripe of the outer medulla (ISOM) within 4 h of reperfusion. This increased TLR4 may be important because, in macrophages, in which the function of TLR4 is best understood, HMGB1 and other molecules released by injured and dying cells bind to TLR4 and trigger production of proinflammatory cytokines.11–15 We suggest that endothelial TLR4 also triggers inflammation, but through stimulation of unique endothelial functions—the increased expression of endothelial adhesion molecules that allow diapedesis of leukocytes from the blood into injured renal tissues. Consistent with this suggestion, we find that adhesion molecules are expressed by wild-type, but not TLR4 (−/−), endothelia in vivo. Furthermore, we find that HMGB1 increases adhesion molecule expression on wild-type, but not TLR4 (−/−), endothelia in vitro.
Our observations are consistent with and build on previous reports that TLR4 has a maladaptive role in ischemic acute kidney injury (AKI). Thus, there was a beneficial effect of transgenic knockout of TLR4 on ischemic AKI.16,17 Furthermore, TLR4 was found on radioresistant tubular cells,16,18,19 and stimulation of tubular TLR4 by HMGB1 released by injured renal cells resulted in tubular apoptosis and cytokine/chemokine production. These tubular events would contribute significantly to injury after 24 h.
However, the effects of TLR4 on ischemic AKI are likely to be complex and involve more than tubules. Tubules did not express TLR4 until 24 h and later after reperfusion and may contribute to injury known to occur after the first 24 h. Our results suggest that endothelial TLR4 would contribute to early injury by regulating adhesion molecule expression and thus inflammation. To our knowledge, this is the first demonstration of renal endothelial TLR4 and its regulatory role on adhesion molecule expression during ischemic AKI. Altogether, our data is consistent with the following hypothesis: during the first 24 h of AKI, acute ischemic injury results in the release of HMGB1; HMGB1 interacts with endothelial TLR4; this activates endothelial cells to express adhesion molecules; these endothelial adhesion molecules allow the maladaptive inflammation that exacerbates ischemic AKI.
The contribution of TLR4 to ischemic AKI is complex. At least two different cell types—renal tubules, as previously reported,16,18,19 and renal endothelia, as we now report—express TLR4. Each cell type contributes to renal injury by means of different mechanisms and at a different time after reperfusion.
RESULTS
At 4 h reperfusion, increased endothelial TLR4 on the vasa rectae of the ISOM
After 23 min of clamping, but no reperfusion, TLR4 mRNA already increased by 1.8-fold compared with sham (Figure 1). TLR4 expression further increased to a maximum at 4 h reperfusion, gradually decreased over the next 18 h reperfusion, but was still elevated at 24 h.
Figure 1. Toll-like receptor 4 (TLR4) mRNA abundance in ischemic kidney peaks at 4 h reperfusion.

The mean values and standard deviation errors of four kidneys per point are shown. The y axis is the TLR4/GAPDH mRNA determined by quantitative reverse transcription-PCR and analyzed by the comparative Ct method. At each time point, the TLR4 Ct is normalized to the GAPDH Ct. The calibrator gene is TLR4 for the sham kidney at time zero, when the renal arterial clamp was removed. P<0.05 between sham and AKI at each time point.
Because TLR4 had not previously been found in the ischemic kidney before 24 h reperfusion, we used four independent techniques to confirm and localize the increased expression of TLR4 at 4 h reperfusion: dissection of the various sections of the kidney, in situ hybridization, immunohistology, and analysis of freshly isolated endothelial cells. All four techniques, discussed below, are consistent with increased TLR4 on endothelia of the vasa rectae of the ISOM during ischemic AKI.
(1) Localization of increased TLR4 to the ISOM by dissection. Gross changes in the kidney after ischemia and 4 h of reperfusion allow its dissection into three separate compartments;20–22 there is congestion of red cells in the ISOM that allows its easy identification and dissection as a single compartment (Figure 2a). The (cortex + OSOM (outer stripe of the outer medulla)) is identified as the pale structure outside the ISOM, and the inner medulla is identified by its characteristic papillary shape. We compared TLR4 mRNA in each of these compartments in Figure 2b and 2c; most of the TLR4 is localized to the ISOM.
Figure 2. Toll-like receptor 4 (TLR4) expression in regions of the kidney.

(a) Ischemic kidney. The kidney has been dissected along its long axis and the dark lines show how the (OSOM + cortex) may be dissected from the congested (red) inner stripe of the outer medulla (ISOM), and the ISOM from the inner medulla (papilla) as described in the text. The inset shows a kidney cut along its short axis and the dotted lines show how the (OSOM + cortex) was dissected from the ISOM from this view. (b) Ischemia increases TLR4 mRNA in the (ISOM + OSOM + cortex) but not in the inner medulla. The x axis is the TLR4 mRNA determined by quantitative reverse transcription (qRT)-PCR with the Ct normalized to GAPDH Ct. This value is further adjusted to take into account the fact that the inner medulla comprised 3±1% (n =3, mean±s.e.) of the total mass of the kidney. We assumed uniform distribution of GAPDH throughout the kidney; therefore, the values displayed on the x axis for the inner medulla are 3 × dCT (TLR4/GAPDH), and that for the ISOM + OSOM + cortex is 97 × dCT (TLR4/GAPDH). (c) Greater TLR4 expression in the ischemic ISOM compared with the (OSOM + cortex). The x axis is the TLR4 mRNA determined by qRT-PCR and the comparative Ct method. TLR4 Ct is normalized to the GAPDH Ct. The calibrator gene is TLR4 in the ischemic OSOM + cortex. Each value shown in b and c is the mean ± s.e. of six determinations. P<0.05 between sham and ischemic kidneys in b, and between ISOM and (OSOM + cortex) in c. OSOM, outer stripe of the outer medulla.
To compare TLR4 expression in ischemic and sham kidneys, a modified dissection strategy was used because, in contrast to ischemic kidneys, there is little vascular congestion in the ISOM of sham kidneys, and the ISOM cannot be readily differentiated from the (OSOM + cortex). Therefore, these three regions must be dissected as a single compartment, that is, the (ISOM + OSOM + cortex); the remaining compartment is the inner medulla. Figure 2b shows that TLR4 mRNA in the inner medulla did not change after ischemia. In contrast, ischemia/reperfusion increased TLR4 mRNA in the (ISOM + OSOM + cortex) by 2.5-fold. As discussed in the previous paragraph, most of this increase is in the ISOM (Figure 2c).
(2) Localization of TLR4 mRNA in the ISOM to the ischemic vasa rectae by in situ hybridization. Although at 4 h, TLR4 was found on both the inner medulla and the ISOM, we focus the remainder of this study on events in the ISOM, because this is the region of greatest injury and inflammation during ischemic AKI (see reviews by Lieberthal et al.,23 Thadhani et al.,24 Brezis and Rosen25). Thus, understanding TLR4 expression at this site may result in understanding the mechanisms at the major site of structural injury during ischemic AKI.
By in situ hybridization, we found TLR4 mRNA in the ISOM of ischemic kidneys at 4 h reperfusion (Figure 3a), but not in sham kidneys (Figure 3c). Control radiolabeled sense TLR4 RNA did not reveal staining of the ischemic kidney (Figure 3b). The area of the red box in Figure 3a was studied in greater detail in Figure 3d–f. These panels suggest that the TLR4 mRNA was localized to endothelial cells of the vasa rectae. In serial sections, the same structures (denoted by white arrows) were positive by staining with antibodies to von Willebrand’s factor (Figure 3e) and TLR4 mRNA (Figure 3f), and had the morphology of endothelial cells on brightfield examination of hematoxylin-and-eosin-counter-stained slides (Figure 3d). This technique measures silver grains precipitated by radiation emitted from the S35 antisense probe bound to TLR4 mRNA. These grains may be some distance from the source. Therefore, our conclusion that endothelia express TLR4 rests not only on in situ hybridization alone but also on immunohistology and on the study of isolated endothelial cells discussed below.
Figure 3. Toll-like receptor 4 (TLR4) mRNA localized to inner stripe of the outer medulla (ISOM) by in situ hybridization.

Wild-type kidneys were studied at 4 h reperfusion. (a) Ischemic antisense. The darkfield photomicrograph shows in situ hybridization for TLR4 mRNA in ischemic kidney using antisense S35 TLR4 mRNA. White dots denote aggregates of silver grains overlying TLR4 mRNA. Red arrows denote increased expression of TLR4 in the vascular bundles of the ISOM of the ischemic kidney. One of these vascular bundles is outlined by a red box that is further studied in d–f. (b) Ischemic sense. This negative control shows the absence of staining of the ischemic kidney hybridized with sense S35 TLR4 mRNA. (c) Sham antisense. This negative control shows the absence of TLR4 mRNA in the non-ischemic kidney. (d) Bright field hematoxylin and eosin. Serial sections of the area delineated by the red box in a were studied. Arrows point to the same vasa rectae in the serial sections d–f. (e) Immunostaining with anti-vWF (von Willebrand’s factor), which stains the endothelium. (f) In situ hybridization for TLR4. White dots indicate in situ hybridization with S35-labeled antisense TLR4 RNA.
(3) Localization of TLR4 proteins in the ISOM to the ischemic vasa rectae by immunohistology. We use three different protocols and two different anti-TLR4 antibodies to demonstrate endothelial TLR4. In Figure 4, we use double immunostaining with a phycoerythrin-conjugated monoclonal anti-TLR4 and von Willebrand’s factor at 4 h reperfusion to also show that vasa rectae are positive for TLR4. In Figure 5, we use a rat monoclonal anti-TLR4, a biotinylated goat antirat secondary antibody, and Texas red streptavidin to localize TLR4 on endothelium. Semiserial sections suggest that TLR4 (Figure 5a) is located on the same interstitial structures as CD31 (Figure 5b), which is an endothelial marker. In Supplementary Figure S5 online, we use a polyclonal anti-TLR4, and an alkaline-phosphatase-labeled secondary antibody. This figure shows staining of interstitial structures that are likely endothelium (Supplementary Figure S5A online), and also shows the absence of nonspecific staining (Supplementary Figure S5B online).
Figure 4. Immunofluorescence shows colocalization of Toll-like receptor 4 (TLR4) and von Willebrand’s factor on the vasa rectae of the ischemic inner stripe of the outer medulla (ISOM).

The ischemic kidney was studied at 4 h reperfusion. The same tissue section is shown in all panels, but different input and output light filters were used. Blue is 4,6-diamidino-2-phenylindole. (a) TLR4 visualized with phycoerythrin-conjugated monoclonal rat anti-mouse TLR4 (Santa Cruz, sc 13591). (b) Endothelia visualized with anti-von Willebrand’s antibody and then goat anti-rabbit IgG-Cy3. (c) Overlap of a and b.
Figure 5. Localization of Toll-like receptor 4 (TLR4) and the endothelial marker CD31 in the ischemic outer medulla.

(a) TLR4 staining (bright red). Arrowhead shows one of many TLR4-positive peritubular endothelia. Arrow shows a nucleus (4,6-diamidino-2-phenylindole positive) that has the elongated shape typical of endothelial nuclei and that is surrounded by staining for TLR4. (b) CD31 staining. Semiserial section to a. Arrowhead shows one of many CD31-positive endothelia. Arrows show two endothelial nuclei (blue) surrounded by CD31 staining (bright green). ‘T’ shows several of many tubules in a and b. Anti-TLR4 appears to stain CD31-positive endothelia.
All together, the experiments in this section use three different techniques—gross dissection, in situ hybridization, and also immunostaining with three different protocols with two different anti-TLR4 antibodies. Data using all three techniques are consistent with the presence of TLR4 on endothelia in the ISOM of ischemic kidneys at 4 h reperfusion. In addition, later in this paper, we discuss a fourth technique, analysis of isolated endothelial cells from ischemic versus sham kidneys. This also confirmed the increased TLR4 expression on ischemic renal endothelia (Figure 8).
Figure 8. Toll-like receptor 4 (TLR4) mRNA expression on CD31+ cells of wild-type sham versus acute kidney injury (AKI) kidneys.

TLR4 mRNA on CD31+ cells was measured by quantitative reverse transcription-PCR and analyzed by the comparative Ct method. The calibrator gene is the sham kidney at 0 h reperfusion. P<0.05 between sham and AKI kidneys at each time point.
Reactive oxygen species stimulate endothelial TLR4 expression
The above studies show that renal endothelial TLR4 increases at 4 h reperfusion in vivo. To determine whether this increase was a direct result of endothelial stress occurring during ischemia/reperfusion, we used an in vitro model of ischemic AKI, stress by H2O2.26 H2O2 and other reactive oxygen species are produced during both ischemia and reperfusion27–29 and should stress endothelia in vivo. We exposed MS1 microvascular endothelial cells to H2O2 for 30 min. The cells were then allowed to recover in complete medium containing FCS and no H2O2. This treatment stimulated TLR4 expression (Figure 6a). The kinetics of TLR4 expression were similar to that in vivo and previously shown in Figure 1. Thus, maximum TLR4 expression occurred after cells had recovered from H2O2 for 4 h, and diminished by 24 h.
Figure 6. Endothelial Toll-like receptor 4 (TLR4) is directly increased by stress in vitro.

Confluent MS1 murine endothelial cells were treated with H2O2 (a) or tunicamycin in Earl’s Balanced Solution (EBSS) (b) for 30 min, and assayed for TLR4 mRNA immediately (zero time) or recultured in complete medium without H2O2 or tunicamycin and assayed after 4 or 24 h. The y axis is the TLR4/GAPDH mRNA determined by quantitative reverse transcription-PCR and analyzed by the comparative Ct method (see Materials and Methods section). The calibrator gene is TLR4 expression in the untreated control in EBSS for 30 min and in complete medium at the given recovery time. Means and standard errors are shown; *P<0.05.
Another in vitro model of ischemic AKI is stress by tunicamycin.30 This agent inhibits glycosylation of proteins and thus prevents appropriate protein folding in the endoplasmic reticulum. This triggers endoplasmic reticulum stress and an ‘unfolded protein response’ that ultimately regulates gene transcription and other cell functions. Similar endoplasmic reticulum stress occurs during AKI because ischemia disrupts the adenosine triphosphate concentrations and the cellular oxidative state required for appropriate protein folding.31–34 We found that endoplasmic reticulum stress (tunicamycin) also stimulates MS1 microvascular endothelial cells to express TLR4 (Figure 6b).
Endothelial activation during ischemic AKI requires TLR4
If endothelial TLR4 is important in the pathophysiology of ischemic AKI, then expression of critical endothelial adhesion molecules should depend on a functional TLR4 gene. We took two approaches to address this question. First, we determined the effect of a TLR4 deletion mutation on these endothelial adhesion molecules during ischemic AKI in vivo. Second, we determined the effect of this TLR4 deletion mutation on the ability of HMGB1 to stimulate endothelial adhesion molecule expression in vitro. HMGB1 is a damaged associated molecule11–14 released by injured cells during ischemic AKI.16
Effect of a TLR4 deletion mutation on endothelial adhesion molecules during ischemic AKI in vivo
We compared the effect of TLR4 genotype on endothelial genes required for the inflammatory response to ischemic AKI; these include endothelial adhesion molecules such as E-selectin (CD62E) and ICAM1 (CD54).5–10,35 For this comparison, we developed a quantitative assay to assess the role of TLR4 in regulating these genes. We used anti-CD31-conjugated Dynabeads (Invitrogen, Carlsbad, CA) to retrieve endothelial cells from enzymatically dispersed kidneys. Before using this technique in the ischemic AKI system, we validated it. Figure 7 shows that this technique did indeed isolate endothelial cells. The CD31-positive cells from both sham (Figure 7a) and AKI (Figure 7b) kidneys expressed endothelial markers CD31, CD54, and CD106. In addition to these commonly used markers for endothelia, we also used ESM1 (endothelial-specific molecule 1, or endocan), which is only expressed by endothelia.36 Cells not binding to the anti-CD31 beads expressed much lower, often undetectable, levels of these endothelial markers. Therefore, the anti-CD31 beads may be used to isolate and study CD31 endothelial cells from both sham and AKI kidneys. Note that this experiment was designed to compare CD31-positive versus CD31-negative cells in either sham or AKI kidneys. It was not designed to compare CD31 cells in sham versus AKI kidneys.
Figure 7. Validation of anti-CD31 Dynabeads as an isolation method for renal endothelial cells.

(a) Endothelial markers on isolated CD31+ cells from sham wild-type kidneys. (b) Endothelial markers on isolated CD31+ cells from acute kidney injury (AKI) wild-type kidneys. The y axis is the gene of interest/GAPDH mRNA measured by quantitative reverse transcription-PCR and analyzed by the comparative Ct method. The calibrator gene is the gene of interest in the anti-CD31 Dynabead-purified population. ‘CD31+’ cells are those binding to anti-CD31 beads. ‘Non-CD31+’ cells are those not binding to anti-CD31 beads. P<0.05 for each pairwise comparison between CD31+ and non-CD31+ for each endothelial cell marker.
Using this technique, we confirmed the increased expression of endothelial TLR4 at 4 h reperfusion in wild-type kidneys. TLR4 expression on isolated CD31-positive cells is increased by 2.28±0.13-fold in AKI compared with sham kidneys (Figure 8). This increased TLR4 on endothelial cells confirms our in situ hybridization (Figure 3) and immunohistochemistry results (Figures 4, 5, and Supplementary Figure S5 online).
We noted a very small increase in TLR4 in the whole sham kidney at 4 h (Figure 1), and suspected that this increase was a response to cytokine release from muscle and skin injured during the sham surgery, which included laparotomy, right nephrectomy, and dissection to expose the left renal artery. This small amount of TLR4 mRNA was below the limit of detection in the sham kidney by in situ hybridization (Figure 3), and we detected no statistically significant increase or decrease in TLR4 on endothelial cells isolated from sham kidneys (Figure 8). The source of the small increase in TLR4 mRNA in the sham kidney of Figure 1 is not known.
In addition, Figure 8 shows that endothelial TLR4 had decreased toward baseline levels at 24 h reperfusion. This is in contrast to the sustained high levels of TLR4 mRNA seen at 24 h reperfusion in the whole kidney (Figure 1). These results are explained by the appearance of TLR4 on tubules (Figure 9). We identified these as proximal tubules by colocalization with Lotus tectragonolobus lectin37,38 in the cortex and OSOM (Figure 10). Thus, the total amount of TLR4 remains elevated for at least 24 h after reperfusion, but TLR4 is expressed on different cells at different times. At 4 h, TLR4 is expressed on endothelial cells; at 24 h, expression is on tubular cells.
Figure 9. Immunohistochemistry of Toll-like receptor 4 (TLR4) at 24 h reperfusion shows tubular, not endothelial, TLR4 expression in wild-type kidneys.

(a) Sham wild-type kidneys; TLR4 at 24 h reperfusion. (b) Acute kidney injury (AKI) wild-type kidneys; TLR4 at 24 h reperfusion. ‘t’ shows several tubules that express TLR4 at 24 h reperfusion. 4,6-Diamidino-2-phenylindole (blue) and anti-TLR4 (red).
Figure 10. Proximal tubular Toll-like receptor 4 (TLR4) at 24 h reperfusion.
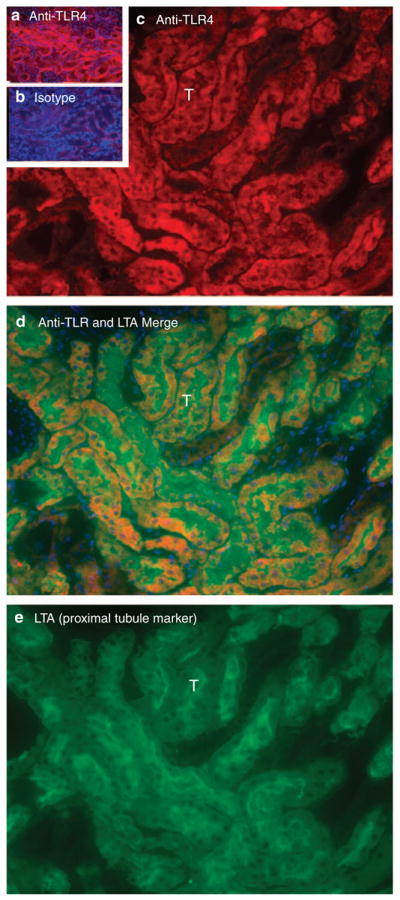
(a) Outer stripe of the outer medulla (OSOM) stained for TLR4 (red). (b) OSOM stained with isotype control primary antibody. Exposure times for a and b are identical. (c) High-power view of OSOM stained for TLR4. ‘T’ is one of many tubules in c–e. (d) Merged lotus tetragonolobus lectin (LTA) and TLR4 of same section. Blue is 4,6-diamidino-2-phenylindole (DAPI) staining of nuclei. DAPI staining reveals nuclei. LTA marks proximal tubule cells. (e) High-power view of same section stained for LTA.
If TLR4 is required for the expression of endothelial activation markers, then these markers should appear on endothelial cells during AKI of wild-type, but not TLR4 (−/−), kidneys. Figure 11 confirms this prediction for CD54, CD106, and CD62E at 4 h reperfusion.
Figure 11. Endothelial markers on CD31+ cells from sham versus acute kidney injury (AKI) kidneys of wild-type versus Toll-like receptor 4 (TLR4) (−/−) mice.

Kidneys clamped for 23 min and then studied at 4 and 24 h reperfusion. Endothelial cells isolated by anti-CD31 Dynabeads. The y axis is the gene of interest/GAPDH mRNA measured by quantitative reverse transcription-PCR and analyzed by the comparative Ct method. The calibrator gene is the gene of interest in sham kidneys. (a) Wild-type kidneys at 4 h reperfusion. (b) TLR4 (−/−) kidneys at 4 h reperfusion. P<0.05 for pairwise comparisons between sham and AKI kidneys for each marker in a.
Effect of a TLR4 deletion mutation on the ability of HMGB1 to stimulate endothelial adhesion molecule expression in vitro
The above data show a critical role for TLR4 in endothelial activation in vivo, but does not show whether this role is directly mediated by TLR4 on endothelia or indirectly through cytokines released by other cells. To address this question, we tested the ability of wild-type versus TLR4 (−/−) endothelia to be stimulated by HMGB1 in vitro. This molecule is a ‘damage-associated-molecular pattern molecule’3,15,39 that is released by damaged cells during ischemic AKI16 and that is a ligand for TLR4. We found that HMGB1 increased endothelial adhesion molecules (CD54, CD106, and CD62E) in wild-type renal endothelia, isolated using anti-CD31 Dynabeads (Figure 12a). In contrast, renal endothelia isolated from TLR4 (−/−) mice did not respond to HMGB1 in vitro (Figure 12b).
Figure 12. HMGB1 activates wild-type, but not Toll-like receptor 4 (TLR4) (−/−), endothelial cells in vitro.

Endothelial cells were isolated from unmanipulated wild-type (a) or TLR4−/− (b) kidneys using anti-CD31 Dynabeads. These cells were stimulated by media, HMGB1, or lipopolysaccharide. The y axis is the gene of interest/GAPDH mRNA measured by quantitative reverse transcription-PCR and analyzed by the comparative Ct method. The calibrator gene is the gene of interest isolated from cells stimulated by media only. *P<0.05 between indicated groups by pairwise multiple comparison procedure (Tukey’s test) after Kruskal–Wallis analysis of variance on ranks.
Note that HMGB1 increases CD62E expression in wild-type endothelia, but lipopolysaccharide does not. This illustrates the known difference in signaling by different TLR4 ligands; each ligand may activate its own TLR4 receptor complex and intracellular signaling pathways.40 Another relevant example is the TLR4-dependent abilities of lipopolysaccharide to decrease, but HMGB1 to increase, the carcinogenicity of croton oil.41
All together, our in vivo and in vitro data suggest that direct stimulation of TLR4 on endothelial cell surfaces results in expression of endothelial adhesion molecules that are important for the inflammatory response to AKI.
DISCUSSION
Acute renal ischemia elicits an inflammatory response that exacerbates injury; this inflammation requires the increased expression of endothelial adhesion molecules that facilitate diapedesis of leukocytes into renal tissues. 1–10 However, the regulation of these endothelial adhesion molecules is not well understood.
On the basis of observations made in this study, we now suggest that endothelial TLR4 contributes to this regulation. First, we show increased expression of endothelial TLR4 of the ISOM at 4 h reperfusion. Because TLR4 had previously been reported only on tubules at 24 h and later, we used four different techniques to confirm our new findings: analysis of dissected renal tissues, in situ hybridization, immunohistology, and analysis of isolated renal endothelial cells. Second, we developed a technique for isolating renal endothelia and quantitatively assaying their expression of adhesion molecules. We find increased expression of adhesion molecules during ischemic AKI only in wild-type, but not TLR4 (−/−), endothelia. Third, we find that HMGB1, released by injured renal cells during ischemic AKI, interacts with endothelial TLR4 to activate adhesion molecule expression only by wild-type, but not TLR4 (−/−), endothelia in vitro. Finally, transgenic knockout of TLR4 ameliorates ischemic injury and inflammation;16,17 this shows a role for TLR4 in ischemic AKI. We propose that one role is the regulation of endothelial adhesion molecule expression. All together, these observations are consistent with the idea that endothelial TLR4 regulates the expression of the endothelial adhesion molecules required for the inflammatory response to injury early after ischemia/reperfusion.
Although our proposed role for endothelial TLR4 has not previously been demonstrated in ischemic AKI, such a role has been shown during ischemic injury of the cremaster muscle,42 and during endotoxin injury of the lung.43
Further support for our hypothesis is the localization of TLR4 to endothelia of the ISOM. The medullary thick ascending limb and the S3 straight proximal tubule that reside in this area are the tubules most vulnerable to ischemic injury in both rodents and humans.23,44–48 During ischemic AKI, injury to these cells should result in the release of HMGB1, and this HMGB1 should stimulate TLR4 on the adjacent endothelium. Because of endothelial activation in the ISOM, this is the area of greatest inflammation during ischemic AKI.5
At 24 h, but not 4 h, reperfusion, we found TLR4 protein on proximal tubules. To our knowledge, these are the first studies to use double immunofluorescence for TLR4 and tubule-specific markers to localize TLR4. At even later reperfusion times (days 3–5), TLR4 has also been reported on distal tubules and medullary thick ascending limb cells by using anti-Tamm Horsfall antibodies and in situ hybridization for TLR418 and on proximal tubules and distal nephrons by using anti-TLR4 antibodies; tubules were identified by morphology alone.19 Although we see strong, consistent expression of TLR4 on proximal tubules, we did occasionally see inconsistent staining of more distal segments. This study is focused on endothelial expression of TLR4 and events during the first 4 h of reperfusion; ongoing studies in our laboratory pursue the localization and functions of TLR4 on tubules and other cells after 24 h of reperfusion.
Although tubular TLR4 would regulate injury after 24 h, it would not affect earlier injury and inflammation. We suggest that this earlier injury results from TLR4 expression on renal endothelia, on the TLR4-dependent expression of endothelial adhesion molecules and on the ensuing inflammation. The different timing of TLR4 expression by tubules and endothelia reflects the different regulation of this molecule on these two cell types. During ischemic AKI, reactive oxygen species is released early during ischemia/reperfusion.26–28 We found that this reactive oxygen species increased endothelial TLR4 expression in vitro. This is consistent with the early expression of TRL4 on endothelia. On the other hand, tubular TLR4 is stimulated by interferon-γ and tumor necrosis factorα.18 This is consistent with the late expression of these cytokines.49–51
All together, our observations support the maladaptive role of endothelial activation in ischemic AKI,5,6,35,52–55 and also support the following hypothesis: at 4 h reperfusion, acute ischemic injury results in the release of HMGB1; HMGB1 interacts with endothelial TLR4; this activates endothelial cells to express adhesion molecules; these endothelial adhesion molecules allow the maladaptive inflammation that exacerbates ischemic AKI.
In conclusion, the contribution of TLR4 to ischemic AKI is complex, and just beginning to be understood. At least two different cell types—renal tubules, as previously reported,16,18,19 and renal endothelia, as now reported—express TLR4. Each cell type contributes to renal injury in a different way and at a different time after reperfusion.
MATERIALS AND METHODS
Animal model
Male C57BL/6J or C57BL/10J, 6–8 weeks of age, were from the Jackson Laboratory (Bar Harbor, ME). These differ with respect to only three genes (see Jackson Laboratory, Website: http://www.informatics.jax.org/external/festing/mouse/docs/C57BL.shtml.); we found no difference in their response to ischemia. TLR4 (−/−) mice were C57BL/10ScN/J mice (Jackson Laboratory), which were always compared with congenic (TLR4- +/+) C57BL/10J. Mice were anesthetized using isoflurane at 37°C by a TR-200 system with a rectal probe (Fine Science Tools, Foster City, CA). After a right nephrectomy, the left pedicle was occluded with a microaneurysm clip (Codman, Rayham, MA) for 23 min. Sham mice underwent the same procedure, except that the clip was put underneath the pedicle. The protocol was approved by our Institutional Animal Care and Use Committee.
In situ hybridization
The TLR4 fragment was amplified using published primers,18,56 subcloned into pDrive cloning Vector (Qiagen, Valencia, CA). S35 labeled sense probes were prepared after linearization of the plasmid with BamHI and transcribed with SP6 polymerase; antisense probes were prepared after linearization with NotI and transcription with T7 polymerase. Hybridization was performed on paraffin-embedded sections.
Immunohistology
In Figure 4, kidneys were fixed in Immunohistofix, embedded in Immunohistowax (Intertiles, Brussels, Belgium),57 sections stained with phycoerythrin-conjugated monoclonal rat anti-mouse TLR4 (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-mouse von Willebrand’s factor antibody (Abcam, Cambridge, MA), and then goat anti-rabbit IgG-Cy3(Jackson ImmunoResearch Laboratories, West Grove, PA). In Supplementary Figure 5 online, renal tissue was fixed by 10% formalin, embedded in paraffin, and immunostained with primary rabbit anti-mouse polyclonal TLR4 (SC-10741, Santa Cruz Biotechnology) and secondary biotinylated goat anti-rabbit antibody. Endogenous alkaline phosphatase was inactivated with levamisole, and endogenous biotin was blocked by the streptavidin–biotin block system. Sections were stained with Fast Red (Vector Laboratories, Burlingame, CA). Normal rabbit immunoglobulin G (IgG) was used as control. In Figures 5, 9, and 10, the left kidney was in situ perfused with cold phosphate-buffered saline, followed by cold 4% paraformaldehyde, fixed in 4% paraformaldehyde at 4°C for 1 h, transferred into 20% sucrose, embedded with OCT (Tissue-Tek, Sacura, Torrance, CA), snap-frozen in isopentane, fixed in 4% paraformaldehyde, and blocked with 10% normal goat serum. Sections were incubated with rat anti-mouse TLR4 (R&D Systems, Minneapolis, MN) for 2 h, followed by biotinylated goat anti-rat IgG (Santa Cruz Biotechnology) for 30 min, then Texas red-streptavidin (Vector Laboratories) for 30 min. Rat IgG2a was the control. For double staining, fluorescein-lotus tetragonolobus lectin (Vector Laboratories) was applied for 20 min. Sections were mounted with VECTASHIELD Mounting Medium with 4,6-diamidino-2-phenylindole (Vector Laboratories) and visualized using a Carl Zeiss Axioplan2 Imaging microscope (Carl Zeiss MicroImaging, Thornwood, NY).
Endothelial cells in vitro
The murine pancreatic endothelial cell line MS1 was purchased from ATCC (Manassas, VA). Cells were cultured in complete media—Dulbecco’s Modified Eagle Medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum (Hyclone, Logan, Utah), 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen), and incubated in a humidified atmosphere at 37°C (5% CO2). Confluent MS1 monolayers were washed with Dulbecco’s phosphate-buffered saline three times. Cells were then treated with H2O2 (500 μM, Sigma, St Louis, MO) or tunicamycin (4 μg/ml, Sigma) for 30 min in Earle’s Balanced Salt Solution at 37°C. After stress exposure, cells were replenished with complete medium containing 10% fetal calf serum and incubated for an additional 4 or 24 h at 37°C.
Endothelial cell isolation
Kidney tissues were minced into 1–2 mm fragments and dissociated in phosphate-buffered saline with Liberase Blendzyme 3 (Roche, Indianapolis, IN) (0.18 Wunsch Units/ml) at 37°C for 25 min with gentle shaking. Dynabeads Sheep anti-rat IgG (Invitrogen) (25 μl) was coated with 1.5 μg of rat anti-mouse CD31 (MEC13.3, BD Pharmingen, San Jose, CA). Tissue digest was filtered through 40 μm cell strainer, incubated with Dynabeads at 4°C for 20 min, and washed.
HMGB1 treatment of primary renal endothelial cells
Endothelial cells isolated from wild-type B10 or TLR4-null kidneys were resuspended in complete medium and incubated at 37°C (5% CO2). Cells were treated with recombinant human HMGB1 (R&D Systems) at 5 μg/ml or 10 μg/ml for 2 h or with an ultrapure form of lipopolysaccharide that activates only TLR4 (Escherichia coli 0111:B4, InvivoGen, San Diego, CA).
RNA isolation and real-time reverse transcription-PCR
Kidney tissues were snap-frozen in liquid nitrogen, stored at −80°C, disrupted using PowerGen 700 homogenizer (Fisher Scientific, Pittsburgh, PA), and total RNA extracted by an RNeasy Midi kit (Qiagen). Total RNA from endothelial cells was extracted with an RNeasy Mini kit (Qiagen). RNA was treated with a Turbo DNA-free kit (Applied Biosystems, ABI, Carlsbad, CA). 1 μg of total RNA was reverse transcribed using a High Capacity cDNA reverse transcription kit (ABI). Confirmation by melting point analysis and relative quantitation of each gene were performed using SYBR Green PCR Reagents and an ABI PRISM 7000 Sequence Detection System. The comparative Ct (cycle threshold) of each gene of interest was normalized to the GAPDH Ct determined under the same conditions.58 The following are the PCR primers: GAPDH forward 5′-AGGTCGGTGTGAACGGATTTG-3′, reverse 5′-TGTAGACCATGTAGTTGAGGTCA-3′; TLR4 forward 5′-AAACGGCAACTTGGACCTGA-3′, reverse 5′-AGCTTAGCAGCCATGTGTTCCA-3′; ESM1 forward 5′-ATTGCCAGTCGGGCATATGTGA-3′, reverse 5′-TTCTCTCACAGCGTTGCCAT-3′; CD31 forward 5′-AGGAAAGCCAAGGCCAAACA-3′, reverse 5′-AATGGCTGTTGGCTTCCACACT-3′; ICAM1 (CD54) forward 5′-AGCCAATTTCTCATGCCGCACA-3′, reverse 5′-AGCTTTGGGATGGTAGCTGGAA-3′; VCAM1 (CD106) forward 5′-TTCGGTTGTTCTGACGTGTGCT-3′, reverse 5′-AGAGCTCAACACAAGCGTGGAT-3′; E-selectin (CD62E) forward 5′-AGCCTGCCATGTGGTTGAATGT-3′, reverse 5′-AACGTGCATGTCGTGTTCCA-3′.
Supplementary Material
{kind=link}
Acknowledgments
Supported by NIH RO-1 DK069633 grant to CYL, NIH F32DK084701 and T32DK07257 to JRH, a grant from the Beecherl Foundation to CYL, and by NIH DK079328 UT Southwestern O’Brien Kidney Research Core Center. We thank Ms Kathy Trueman for assistance in preparing the paper and figures, Martin Senitko and Patricia Cobo-Stark for advanced core technical assistance, and our animal care technicians.
Footnotes
DISCLOSURE
All the authors declared no competing interests.
Figure S5. Immunoalkaline phosphatase shows TLR4 on endothelia of the ischemic outer medulla.
Supplementary material is linked to the online version of the paper at http://www.nature.com/ki
References
- 1.Sutton TA, Fisher CJ, Molitoris BA. Microvascular endothelial injury and dysfunction during ischemic acute renal failure. Kidney Int. 2002;62:1539–1549. doi: 10.1046/j.1523-1755.2002.00631.x. [DOI] [PubMed] [Google Scholar]
- 2.Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatory disease? Kidney Int. 2004;66:480–485. doi: 10.1111/j.1523-1755.2004.761_2.x. [DOI] [PubMed] [Google Scholar]
- 3.Jang HR, Rabb H. The innate immune response in ischemic acute kidney injury. Clin Immunol. 2009;130:41–50. doi: 10.1016/j.clim.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kinsey GR, Li L, Okusa MD. Inflammation in acute kidney injury. Nephron Exp Nephrol. 2008;109:e102–e107. doi: 10.1159/000142934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Greef KE, Ysebaert DK, Persy V, et al. ICAM-1 expression and leukocyte accumulation in inner stripe of outer medulla in early phase of ischemic compared to HgCl2-induced ARF. Kidney Int. 2003;63:1697–1707. doi: 10.1046/j.1523-1755.2003.00909.x. [DOI] [PubMed] [Google Scholar]
- 6.Kelly KJ, Williams WW, Jr, Colvin RB, et al. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J Clin Invest. 1996;97:1056–1063. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelly KJ, Williams WW, Jr, Colvin RB, et al. Antibody to intercellular adhesion molecule 1 protects the kidney against ischemic injury. Proc Natl Acad Sci USA. 1995;91:812–816. doi: 10.1073/pnas.91.2.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rabb H, Mendiola CC, Dietz J, et al. Role of CD11a and CD11b in ischemic acute renal failure in rats. Am J Physiol. 1994;267:F1052–F1058. doi: 10.1152/ajprenal.1994.267.6.F1052. [DOI] [PubMed] [Google Scholar]
- 9.Kiew LV, Munavvar AS, Law CH, et al. Effect of antisense oligodeoxynucleotides for ICAM-1 on renal ischaemia-reperfusion injury in the anaesthetised rat. J Physiol. 2004;557:981–989. doi: 10.1113/jphysiol.2004.061788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haller H, Dragun D, Miethke A, et al. Antisense oligonucleotides for ICAM 1 attenuate reperfusion injury and renal failure in the rat. Kidney Int. 1996;50:473–480. doi: 10.1038/ki.1996.338. [DOI] [PubMed] [Google Scholar]
- 11.Oppenheim JJ, Tewary P, de la Rosa G, et al. Alarmins initiate host defense. Adv Exp Med Biol. 2007;601:185–194. doi: 10.1007/978-0-387-72005-0_19. [DOI] [PubMed] [Google Scholar]
- 12.Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol. 2008;3:99–126. doi: 10.1146/annurev.pathmechdis.3.121806.151456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barton GM. A calculated response: control of inflammation by the innate immune system. J Clin Invest. 2008;118:413–420. doi: 10.1172/JCI34431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat RevImmunol. 2004;4:469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- 15.Lu CY, Hartono J, Senitko M, et al. The inflammatory response to ischemic acute kidney injury: a result of the ‘right stuff’ in the ‘wrong place’? Curr Opin Nephrol Hypertens. 2007;16:83–89. doi: 10.1097/MNH.0b013e3280403c4e. [DOI] [PubMed] [Google Scholar]
- 16.Wu H, Chen G, Wyburn KR, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pulskens WP, Teske GJ, Butter LM, et al. Toll-like receptor-4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS ONE. 2008;3:e3596. doi: 10.1371/journal.pone.0003596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolfs TG, Buurman WA, van Schadewijk A, et al. In vivo expression of Toll-like receptor 2 and 4 by renal epithelial cells: IFN-gamma and TNF-alpha mediated up-regulation during inflammation. J Immunol. 2002;168:1286–1293. doi: 10.4049/jimmunol.168.3.1286. [DOI] [PubMed] [Google Scholar]
- 19.Kim BS, Lim SW, Li C, et al. Ischemia-reperfusion injury activates innate immunity in rat kidneys. Transplantation. 2005;79:1370–1377. doi: 10.1097/01.tp.0000158355.83327.62. [DOI] [PubMed] [Google Scholar]
- 20.Mason J, Torhorst J, Welsch J. Role of the medullary perfusion defect in the pathogenesis of ischemic renal failure. Kidney Int. 1984;26:283–293. doi: 10.1038/ki.1984.171. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto K, Wilson DR, Baumal R. Outer medullary circulatory defect in ischemic acute renal failure. Am J Pathol. 1984;116:253–261. [PMC free article] [PubMed] [Google Scholar]
- 22.Park KM, Kramers C, Vayssier-Taussat M, et al. Prevention of kidney ischemia/reperfusion-induced functional injury, MAPK and MAPK kinase activation, and inflammation by remote transient ureteral obstruction. J Biol Chem. 2002;277:2040–2049. doi: 10.1074/jbc.M107525200. [DOI] [PubMed] [Google Scholar]
- 23.Lieberthal W, Nigam SK, Bonventre JV, et al. Acute renal failure. I. Relative importance of proximal vs distal tubular injury. Am J Physiol. 1998;275:F623–F631. doi: 10.1152/ajprenal.1998.275.5.F623. [DOI] [PubMed] [Google Scholar]
- 24.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med. 1996;334:1448–1460. doi: 10.1056/NEJM199605303342207. [DOI] [PubMed] [Google Scholar]
- 25.Brezis M, Rosen S. Hypoxia of the renal medulla – its implications for disease. N Engl J Med. 1995;332:647–655. doi: 10.1056/NEJM199503093321006. [DOI] [PubMed] [Google Scholar]
- 26.Arany I, Faisal A, Nagamine Y, et al. p66shc inhibits pro-survival epidermal growth factor receptor/ERK signaling during severe oxidative stress in mouse renal proximal tubule cells. J Biol Chem. 2008;283:6110–6117. doi: 10.1074/jbc.M708799200. [DOI] [PubMed] [Google Scholar]
- 27.Li C, Jackson RM. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol Cell Physiol. 2002;282:C227–C241. doi: 10.1152/ajpcell.00112.2001. [DOI] [PubMed] [Google Scholar]
- 28.Chandel NS, Maltepe E, Goldwasser E, et al. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nath KA, Norby SM. Reactive oxygen species and acute renal failure. Am J Med. 2000;109:665–678. doi: 10.1016/s0002-9343(00)00612-4. [DOI] [PubMed] [Google Scholar]
- 30.Kawakami T, Inagi R, Takano H, et al. Endoplasmic reticulum stress induces autophagy in renal proximal tubular cells. Nephrol Dial Transplant. 2009;24:2665–2672. doi: 10.1093/ndt/gfp215. [DOI] [PubMed] [Google Scholar]
- 31.Kuznetsov G, Bush KT, Zhang PL, et al. Perturbations in maturation of secretory proteins and their association with endoplasmic reticulum chaperones in a cell culture model for epithelial ischemia. Proc Natl Acad Sci USA. 1996;93:8584–8589. doi: 10.1073/pnas.93.16.8584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santos CX, Tanaka LY, Wosniak JJ, et al. Mechanisms and Implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport and NADPH oxidase. Antioxid Redox Signal. 2009;11:2409–2427. doi: 10.1089/ars.2009.2625. [DOI] [PubMed] [Google Scholar]
- 33.Dickhout JG, Krepinsky JC. Endoplasmic reticulum stress and renal disease. Antioxid Redox Signal. 2009;11:2341–2352. doi: 10.1089/ars.2009.2705. [DOI] [PubMed] [Google Scholar]
- 34.Kitamura M. Endoplasmic reticulum stress and unfolded protein response in renal pathophysiology: Janus faces. Am J Physiol Renal Physiol. 2008;295:F323–F334. doi: 10.1152/ajprenal.00050.2008. [DOI] [PubMed] [Google Scholar]
- 35.Nemoto T, Burne MJ, Daniels F, et al. Small molecule selectin ligand inhibition improves outcome in ischemic acute renal failure. Kidney Int. 2001;60:2205–2214. doi: 10.1046/j.1523-1755.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- 36.Lassalle P, Molet S, Janin A, et al. ESM-1 is a novel human endothelial cell-specific molecule expressed in lung and regulated by cytokines. J Biol Chem. 1996;271:20458–20464. doi: 10.1074/jbc.271.34.20458. [DOI] [PubMed] [Google Scholar]
- 37.Laitinen L, Virtanen I, Saxen L. Changes in the glycosylation pattern during embryonic development of mouse kidney as revealed with lectin conjugates. J Histochem Cytochem. 1987;35:55–65. doi: 10.1177/35.1.3794309. [DOI] [PubMed] [Google Scholar]
- 38.Schulte BA, Spicer SS. Histochemical evaluation of mouse and rat kidneys with lectin-horseradish peroxidase conjugates. Am J Anat. 1983;168:345–362. doi: 10.1002/aja.1001680308. [DOI] [PubMed] [Google Scholar]
- 39.Kroemer G, Zitvogel L. Death, danger, and immunity: an infernal trio. Immunol Rev. 2007;220:5–7. doi: 10.1111/j.1600-065X.2007.00576.x. [DOI] [PubMed] [Google Scholar]
- 40.Midwood K, Sacre S, Piccinini AM, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15:774–780. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- 41.Mittal D, Saccheri F, Venereau E, et al. TLR4-mediated skin carcinogenesis is dependent on immune and radioresistant cells. EMBO J. 2010;29:2242–2252. doi: 10.1038/emboj.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rumbaut RE, Bellera RV, Randhawa JK, et al. Endotoxin enhances microvascular thrombosis in mouse cremaster venules via a TLR4-dependent, neutrophil-independent mechanism. Am J Physiol Heart Circ Physiol. 2006;290:H1671–H1679. doi: 10.1152/ajpheart.00305.2005. [DOI] [PubMed] [Google Scholar]
- 43.Andonegui G, Zhou H, Bullard D, et al. Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram-negative bacterial infection. J Clin Invest. 2009;119:1921–1930. doi: 10.1172/JCI36411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kashgarian M. Acute tubular necrosis an ischemic renal injury. In: Jennette JC, Olsen JC, Schwartz MM, Silva FG, editors. Heptinstall’s Pathology of the Kidney. 5. Vol. 5. Lippincott - Raven; Philadelphia, New York: 1999. pp. 863–889. [Google Scholar]
- 45.Lucke B. Lower nephron nephrosis. Mil Surg. 1946;99:371–396. [PubMed] [Google Scholar]
- 46.Oliver J, Mac DM, Tracy A. The pathogenesis of acute renal failure associated with traumatic and toxic injury; renal ischemia, nephrotoxic damage and the ischemic episode. J Clin Invest. 1951;30:1307–1439. doi: 10.1172/JCI102550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Solez K. Pathogenesis of acute renal failure. Int Rev Exp Pathol. 1983;24:277–333. [PubMed] [Google Scholar]
- 48.Brezis M, Rosen S, Silva P, et al. Renal ischemia: a new perspective. Kidney Int. 1984;26:375–383. doi: 10.1038/ki.1984.185. [DOI] [PubMed] [Google Scholar]
- 49.Daemen MA, van’t Veer C, Wolfs TG, et al. Ischemia/reperfusion-induced IFN-gamma up-regulation: involvement of IL-12 and IL-18. J Immunol. 1999;162:5506–5510. [PubMed] [Google Scholar]
- 50.Donnahoo KK, Meng X, Ayala A, et al. Early kidney TNF-alpha expression mediates neutrophil infiltration and injury after renal ischemia-reperfusion. Am J Physiol. 1999;277:R922–R929. doi: 10.1152/ajpregu.1999.277.3.R922. [DOI] [PubMed] [Google Scholar]
- 51.Daemen MARC, van de Ven MWCM, Heineman E, et al. Involvement of endogenous interleukin-10 and tumor necrosis factor-a in renal ischemia-reperfusion injury. Transplantation. 1999;67:792–800. doi: 10.1097/00007890-199903270-00003. [DOI] [PubMed] [Google Scholar]
- 52.Sutton TA. Alteration of microvascular permeability in acute kidney injury. Microvasc Res. 2009;77:4–7. doi: 10.1016/j.mvr.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Basile DP. The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function. Kidney Int. 2007;72:151–156. doi: 10.1038/sj.ki.5002312. [DOI] [PubMed] [Google Scholar]
- 54.Goligorsky MS, Patschan D, Kuo MC. Weibel-Palade bodies—sentinels of acute stress. Nat Rev Nephrol. 2009;5:423–426. doi: 10.1038/nrneph.2009.87. [DOI] [PubMed] [Google Scholar]
- 55.Friedewald JJ, Rabb H. Inflammatory cells in ischemic acute renal failure. Kidney Int. 2004;66:486–491. doi: 10.1111/j.1523-1755.2004.761_3.x. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Y, Woodward VK, Shelton JM, et al. Ischemia/reperfusion induces G-CSF gene expression by renal medullary thick ascending limb cells in vivo and in vitro. Am J Physiol Renal Physiol. 2004;286:F1193–F1201. doi: 10.1152/ajprenal.00379.2002. [DOI] [PubMed] [Google Scholar]
- 57.Pajak B, De Smedt T, Moulin V, et al. Immunohistowax processing, a new fixation and embedding method for light microscopy, which preserves antigen immunoreactivity and morphological structures: visualisation of dendritic cells in peripheral organs. J Clin Pathol. 2000;53:518–524. doi: 10.1136/jcp.53.7.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, et al. High-throughput real-time quantitative reverse transcription PCR. Curr Protoc Mol Biol. 2006;Chapter 15(Unit 15):18. doi: 10.1002/0471142727.mb1508s73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.