

Abstract
The DF-4 connector is a novel industry standard for the connection of a defibrillator lead to the generator. It aims at reducing the bulk created by two or three pins at the proximal end of the defibrillator lead and its corresponding ports at the header of the device. Having only one connection port between the lead and the device reduces the material in the pocket, the risk of lead-to-port mismatch, may lower the risk of lead abrasion, and probably makes the implantation procedure a little easier since only one set screw is required. However, all these conceived benefits are related to convenience rather than to a medical need.
After the recent experiences with the possible negative clinical impact of ‘minor’ changes like simply downsizing a defibrillator lead, a word of caution is warranted. The lead is the weakest part of the defibrillator system, complex in design and undergoing constant stress through movement. It is very hard to predict which issues may evolve over time with the changes in lead design. Does the perceived benefit really outweigh an unpredictable risk in a sensitive medical product like a defibrillator? This article tries to address the possible issues of the new spring contacts instead of set screws, the proximity of the low- and high-voltage connections as well as the inability of adding a pace/sense or an additional shock lead without a special adaptor, and advocates a measured speed in the introduction of this technology.
Keywords: Implantable cardioverter defibrillator, Artificial pacemaker, Cardiac resynchronization therapy, Implantable electrodes, Biventricular pacing, Benefits and risks, Risk–benefit assessment, Consumer product safety, Lead malfunction
Introduction
Pacemaker or defibrillator leads connect the myocardial site of sensing or stimulation with the respective cardiac implantable electronic devices (CIEDs). When replacing a CIED, the leads usually remain in place and are mounted individually to the header of the new device. Occasionally, additional leads are placed to address sensing or capture issues with existing leads. Since the late 1980s and early 1990s, the unified industry standards IS-1 and DF-1 ensure interchangeability of generators and leads from different manufacturers, thus allowing the device system to be tailored to the individual patient's needs. Defibrillator leads hence possess a bifurcation (single-coil lead; one IS-1, one DF-1) or a trifurcation (dual-coil leads; one IS-1, two DF-1) at their proximal end. In view of the reduced battery size, high-voltage capacitors and electronics, the size of a header with 3–5 plugs contributes more to the total size of the device. During replacement, it can be difficult to free all two or three components of the lead in a scarred pocket, which can lead to prolonged procedure times and increase the risk of surgical lead damage. In dual-coil leads, there also exists a risk for inadvertently connecting the right ventricular (RV) coil and the superior vena cava (SVC)-coil DF-1 plugs to the wrong port. Since many centres do not perform defibrillation threshold testing, this could easily go unnoticed.
Hence, there are numerous reasons to consider a simpler industry standard that ideally results in only one connection between lead and device.
The new four-pole connector standard
Work on the new four-pole connector began in 1998 with the objective to increase system reliability by reducing its complexity. The idea was to design a connector system that has the seals placed inside a single port and not on the lead connector anymore. The new standard was formalized in March 2010, when the International Organization for Standardization (ISO) published its new standard Active implantable medical devices—four-pole connector system for implantable cardiac rhythm management devices—Dimensional and test requirements ISO 27186.1 Its specifications apply to both low-energy (IS-4) and high-energy (DF-4) leads. It has been finally endorsed by the Association for the Advancement of Medical Instrumentation in August 2011.2 International Organization for Standardization-directed interchangeability testing using prototypes from three different manufacturers was completed in June 2009 in order to ensure that IS-4/DF-4 would be compatible with future implanted devices. However, many requirements in the standard are not mandatory but recommendations only (e.g. need for clinical studies). Also, the standard does not specify all connector features and aspects of functional compatibility, safety, or reliability of leads and generators. This raises concerns regarding a rushed and unmanaged introduction of this new technology in the market without appropriate clinical studies.
The connector port cavity design of the IS-4/DF-4 connector consists of an epoxy header with a cylindrical bore opening. This means that instead of three separate cavities to connect to the RV-implantable cardioverter defibrillator (ICD) lead, the new port provides one combined cavity with four contacts, of which two can be of high voltage. An IS-4 or DF-4 lead now has only one plug providing four poles. Overall, the new design reduces the number of connections between the leads and the device, and has no trifurcation yoke. In the new connector design, the seals insulating the different contacts are mounted in the device header (see Figure 1).
Figure 1.
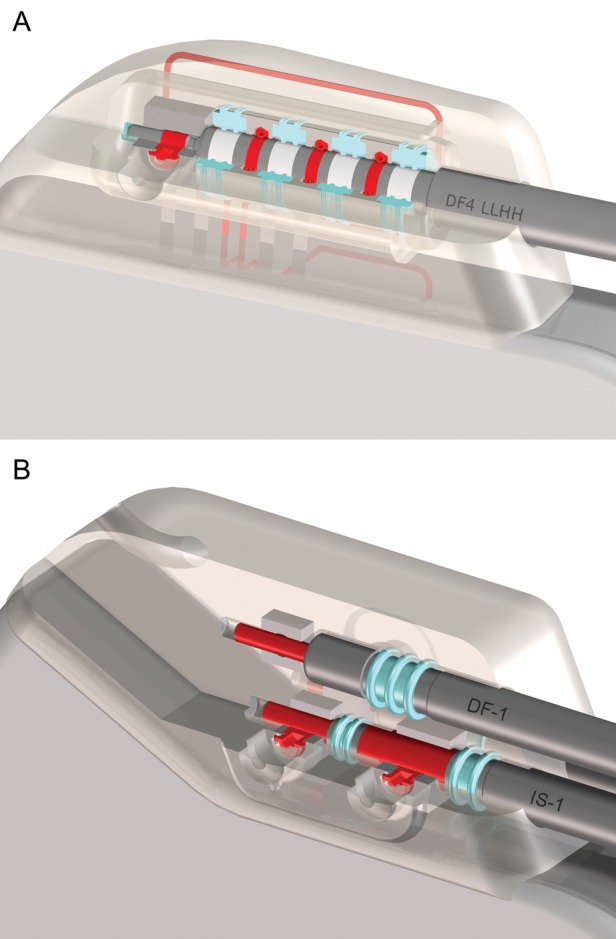
(A) Single-port DF-4 cavity for both high and low voltage (DF4 LLHH). The two poles on the right are high-voltage poles, and the other two poles including the tip pole are for low-voltage connections. A single distal set screw to the tip electrode holds the lead in place. An IS-4 connector would look similar but could take in up to four low-voltage poles. (B) DF-1 (top) and IS-1 (bottom) connectors. The DF-1 port has a single distal set screw, whereas the IS-1 port may have two set screws (as shown) or a distal set screw with a proximal spring contact. In contrast to the DF-4 system, the seal rings are part of the lead and not the connector. Seal rings are shown in light blue and the poles in red. (Images reprinted with the permission of Biotronik SE, Berlin, Germany.)
Risk–benefit considerations
There are a number of potential advantages for the implanting physician and for the patient with devices following the new standard. An overview about the expected benefits and risks of the new design is given in Table 1. Importantly, most of the presented arguments are related to convenience rather than an unmet clinical or safety need. One of the proposed advantages relates to shorter and easier implantation procedures, which is particularly attractive for cardiac resynchronization therapy defibrillators (CRT-Ds) using three leads and requiring multiple electrical contacts.3 Via a reduced risk of lead-to-can abrasion (due to fewer connections) the new design is expected to increase device reliability. The reduction in the number of lead-to-device connections is also expected to reduce pin-to-port mismatch. Furthermore, the smaller devices and less material needed for the leads are considered a benefit for patients, although the actual reduction only applies to the size of the header (−0.4 to +1.4 cc). For the currently marketed DF-4 ICD and CRT-D devices, there is actually no significant reduction in total volume (Table 2).
Table 1.
Summary of potential benefits and risk of newly designed IS-4/DF-4 connectors and leads
| Potential benefits | Potential risks |
|---|---|
| Treatment related | |
| • Shorter and more convenient implantation procedure | |
| • Enables multipolar P/S leads (e.g. for LV pacing) | |
| Safety related | |
| • Reduced risk of lead-to-port mismatch | • Spring contacts (instead of set screws) may be prone to connection problems |
| • Reduced risk of unset set screws | • Complex connector design may give rise to sealing failures |
| • Reduced risk of abrasion | • DF-4 connector does not allow additional pace/sense leads in case of sensing problems (only by use of special adaptor, not available yet) |
| • New set of seals at device replacement | • DF-4 connector does not allow additional shock lead (e.g. subcutaneous array) in case of elevated defibrillation threshold (only by use of a special adaptor, not available yet) |
| • Requires specially-designed cables for intra-operative testing. Higher risk of incompatibility in case of device replacement | |
| • Inability to add an additional shock or pace/sense lead without an adaptor | |
| Patient-related | |
| • Less material in the device pocket | |
| • Reduced size of device header |
Table 2.
Volume (cm3) of the currently marketed DF-1 devices and the matching DF-4 model
| Model | DF-1 | DF-4 |
|---|---|---|
| MDT Protecta VR | 37 | 37 |
| MDT Consulta CRT-D | 38 | 40 |
| SJM Fortify VR | 35 | 35 |
| SJM Unify CRT-D | 36 | 36 |
| BS Teligen VR | 31.5 | 30.5 |
| BS Cognis CRT-D | 32.5 | 32 |
MDT, Medtronic; SJM, St Jude Medical; BS, Boston Scientific
However, despite these anticipated advantages, first calls to treat IS-4/DF-4 as an ‘investigational technology’ were voiced shortly after the draft standard became known.3
Conceivable hardware risks are mainly related (i) to the use of spring contacts, (ii) to short circuits caused by the complex connector design, and (iii) to perceived gaps in the ISO standard.
The current IS-1/DF-1 connectors use set screws for all electrical connections between lead and device which ensure high and constant contact pressure for a reliable connection. For the sensing functionality, the new spring contacts used in IS-4/DF-4 devices could cause noise signals, potentially leading to oversensing. Since the spring contacts are also used for the transmission of high-energy shock treatments, limited contact could result in a compromised defibrillator functionality of ICD and CRT-D devices.
In the current IS-1/DF-1 devices, low- and high-energy contacts are in separate cavities, embedded in non-conducting material that provides sufficient electrical insulation; the sealing systems to insulate the different contacts from each other are mounted on the lead. With the new standard, the high- and low-voltage applications are placed in the same cavity, with limited space for insulation and sealing. Contact pressure of the seals and the spring contacts has to be limited to allow for easy insertion and retraction of the lead connector. In IS-4/DF-4 connectors, the lead plug has to go through four seals with three intermediate spring contacts; DF-1 connectors have one seal and no spring contacts; and IS-1 connectors two seals, plus occasionally one spring contact. In the new connector design, a single seal insulates the high- from the low-voltage contacts and also the high-voltage contacts from each other. Sealing failure may cause sensing problems or short circuits. This is of particular concern for the high-voltage contacts, as device damage or inefficient shocks may result.
The standard alligator clips currently used for lead testing during the implantation procedure should not be used, as the sharp clips may damage the lead surface area underneath the seals. Instead, new cables have been designed (that may also be used with IS-1 leads).
The compact design of the new connector provides limited system flexibility that might pose significant drawbacks in clinical practice. Should sensing problems or elevated pacing or defibrillation thresholds occur, additional pace/sense-(P/S-) or shock leads may only be implanted by using a special adaptor (to connect an IS-1/DF-1 lead). However, currently no such adaptor is available, meaning that currently the only available option is lead and device replacement.4
Finally, there is a higher potential for logistical problems. Before the wide-spread application of the IS-1 standard, physicians were confronted with 3.2, 5, and 6 mm connectors.5,6 In the early phase of CRT, one manufacturer also had its own LV-1 standard for the left ventricular lead. In the case of a generator change, one has to know the type of connector in order to avoid a mismatch between the lead and the generator. It is worth mentioning that all the ICD manufacturers test their system with their own lead, and that cross-manufacturer replacement may result in problems. The addition of a new standard complicates this issue and the likelihood for starting an operation before realizing that the connector and the header do not fit inadvertently rises.7 This would result in prolonged operation times, use of adaptors, and probably an increased risk of infection during a replacement.
After considering all of these potential new safety issues and the limited availability of clinical data for this new connector technology, one might conclude that the balance of benefits and risks is unclear regarding the new DF-4 standard. On the other hand, the risk/benefit ratio appears to be in favour of the new IS-4 standard applied to left ventricular lead technology. The new pacing concept with a quadripolar lead may improve response to therapy, overcome high capture thresholds, and avoid phrenic nerve stimulation. The absence of high-voltage components may also translate into fewer risks compared with the DF-4 connector. In other words, in the case of the IS-4 standard the much higher potential clinical benefit may more likely outweigh the risk.
Targeting the right issue
While the new connector standard may potentially provide additional benefits for physicians and/or patients, it does not address the technical issues with leads, which are considered to be the Achilles' heel of CIEDs. In fact, the modifications involve the component of the generator/lead system that has caused little trouble in the past: Alter et al.8 prospectively studied 440 consecutive patients who were implanted with an ICD between 1994 and 2005. The mean follow-up time was 46 ± 37 months. The most frequently detected complication during this study were inappropriate shocks and lead-related problems (each in 12% of the patients), followed by implantation-related problems (10%) and generator-related problems (6%). About half of the 52 patients affected by lead-related events had lead fractures or insulation defects (27 patients, representing 6% of the total sample). Overall in this study, problems related to the connector affected only five patients representing 1.1% of the total sample.5 It could be argued that the new connector and lead standard may potentially exacerbate any existing problems with the leads available and introduce connector problems that are currently of no concern.
Recent ICD-lead problems
Unfortunately, even minor changes in lead design, such as a reduction in lead size, have been shown to have the potential to result in major unexpected problems. In 2007, the Food and Drug Administration (FDA) issued a Class I recall for the Medtronic Sprint Fidelis® leads because of an increased lead fracture rate.9 A Class I recall is issued if there is ‘reasonable probability that the use of or exposure to a violative product will cause serious adverse health consequences or death’.10 The manufacturer withdrew the Sprint Fidelis® leads from the market after evaluating data from 25 000 patients and determining that their failure rate had reached 2.3% after 30 months. Although not statistically significant at that time, this failure rate was higher than the 0.9% failure rate for one of its older, standard diameter Quattro lead models. At the time that the lead was put under advisory, a recommendation was issued to closely observe patients with Sprint Fidelis® leads, but not to perform prophylactic extraction, as this by itself poses a substantial risk to patients. Considering that ∼268 000 Sprint Fidelis® leads have been implanted worldwide, significant healthcare resources have been invested, and will continue to be needed, to address the issues stemming from a new lead that was easier to implant.11 As can be seen from the latest data, the cumulative failure rate for the Sprint Fidelis® lead continues to increase over time and, according to the manufacturer, has reached close to 10% at 66 months for the most frequently implanted 6949 model (1866/21 500 leads being followed up).12
Also in 2007, first reports of another newly designed small-diameter lead emerged.13,14 While the Riata® lead has not been recalled, it seems to be associated with complication rates that also increase over time (16% at 6 years). A rare insulation defect at around the tricuspid valve and at the level of the SVC (that cannot be detected by regular device interrogation) was reported (n= 7, 20% of all lead failures).15 This is yet another example of how modifications in hardware design may cause unexpected problems.
The lack of clinical evidence
In general, the introduction of a new class III medical device (life-supporting, life-sustaining, and implantable devices, or devices which pose a potentially unreasonable risk of illness or injury) requires the clinical evaluation of safety and efficacy information of a device, which, according to the FDA can either be existing scientific literature, a clinical study, or a combination of both.16 For minor modifications, engineering tests (‘bench testing’) and tests in animals can be acceptable alternatives for demonstrating the safety and efficacy of a newly developed medical device.17,18 However, when introducing significant changes to technology, human clinical trial data might be necessary, either pre- or post-regulatory approval.18 The first DF-4 implants were approved in the US in April 2009, followed shortly thereafter by CE-mark approval in Europe. As a condition of FDA approval of its first DF-4 device and lead, St Jude Medical started a post-approval study including up to 1700 patients and followed up for 5 years, in order to collect data on the new connector system (clinical trial number NCT00940888).19 The estimated study completion date is December 2016. At present, this is the only study specifically designed to investigate the field performance of devices and leads using the DF-4 connector standard.
Another post-surveillance study using devices with the new DF-4 standard (Cogent-4, clinical trial number NCT00606710) has been completed but not reported yet. However, this study targeted outcome variables other than connector-related safety, and it will probably not contribute relevant data on the reliability of the new connector.
In its most recent product performance report, St Jude Medical reports on the relative performance of its DF-4 products compared with its IS-1/DF-1 products. The report claims a 45% reduction of connector-related complaints for DF-4, based on data collected between August 2009 and December 2010.20 From past experience it can be seen that true failure rates occur 3–4 years after implantation. Therefore, reporting a risk reduction of connector-related complaints with DF-4 may be premature.
First experience as reported in Manufacturer and User Facility Device Experience
A number of adverse event (AE) reports for devices with DF-4 connectors can already be found in the Manufacturer and User Facility Device Experience (MAUDE) database run by the FDA. Since reporting to this database is voluntary, the actual frequency of such AEs is unknown. Although one should be cautious regarding the interpretation of case reports, two cases were selected from the MAUDE database, based on an apparent connection to the DF-4 connector technology. One event report for a CRT-D at 1 month after implantation refers to the explantation of the device due to noise and inappropriate therapy. No noise or anomaly was detected during bench testing in the automated testing equipment, and thus the cause of the noise remained undetermined. It was suspected that the set screw was damaged during implantation, causing a lead connection issue.21 Another report refers to inappropriate high-voltage therapy by an ICD 14 months after implantation. The electrograms showed non-physiological noise, but the set screw was found tight during surgery. The device was considered having a connection issue and hence was explanted and the RV lead capped.22
Quantifying an expected failure rate
These experiences, with increasing failure rates and unusual clinical and technical presentation of problems caused by newly designed leads, warrant long-term performance data of the new IS-4/DF-4 connector, which appropriately describe its associated risks. Only with such data can widespread introduction of this new sophisticated connector technology be justified.
If one wanted to perform a post-marketing surveillance study to rule out a situation comparable with the Sprint Fidelis experience, how many of the new IS-4/DF-4 connector pairs (port and lead) would have to be observed and for how long?
An exact trigger rate for a potential recall could not be identified from published sources, but historic event rates indicate a maximum acceptable failure rate of 1.5% during 24 months. Assuming a maximum failure rate of 0.9% in the population, a study would have to include ∼2900 patients to have a statistical power of 90% to confirm non-inferiority (using the 1.5% safety margin mentioned above) at the one-sided 5% level of significance.23 One manufacturer is currently conducting a post-marketing surveillance study that will enrol up to 1700 patients.19
Summary
Cardiac implantable electronic devices are based on sophisticated technology that has proven to save many patient lives over the past 20 years. The implantation of a cardiac device, its programming, and analysis of the diagnostic data require a deep understanding of the technology by both clinicians and technicians. Available features vary between device models from different manufacturers, and every new product generation is associated with changes and extensions in functionality that the users have to get accustomed to. Technological advances should be encouraged, but should balance clinical needs against additional risks resulting from changes in design.
In the past, leads have been much more often incriminated than generators for causing technical problems that inconvenience both patients and physicians. Significant changes in lead design introduced during the last years have backfired. Compared with leads, connectors have lower failure rates. To the best of our knowledge, no generator was ever put under advisory because of a connector problem.
The IS-4 standard was introduced to improve patient management in the field of CRT, and is therefore likely to be of real clinical value. The DF-4 standard on the other hand, is more oriented towards the implanting physician's convenience. Thus, there seems to be no clinical reason to routinely replace four-pole ICD connectors with a system for which there is currently no proof of efficacy and safety. At this point in time, the claimed convenience benefits for the implanting physician and the patient do not seem to outweigh the potential risks, the magnitude of which are largely unknown at present. Proper risk assessment in the form of clinical studies is needed, as are repair options in case of lead issues. When applying the HRS/ACC/AHA recommendations for determining whether a lead is ‘new’, the IS-4/DF-4 connector standard clearly qualifies as such ‘new technology’. It thus belongs to an investigational setting first, followed by a cautious and responsible market introduction, rather than being widely introduced in a large uncontrolled patient population. An adequately powered clinical study capable of assessing the safety of this new but unproven connector standard will require time and resources. Remote monitoring may play an important role in such a study, ensuring that problems are detected as early as possible to increase patients' safety.
Eventually, the issue comes down to the question of how big a perceived advantage of a new technology has to be in order to justify the risk of a potential new problem, which can never be completely ruled out.
Funding
Funding to pay the Open Access publication charges was provided by Biotronik.
Acknowledgement
The authors wish to thank Ms Antje Smala (Global Reimbursement and Health Economics at BIOTRONIK SE & Co KG, Berlin, Germany) for her assistance during the preparation of this manuscript.
Conflict of interest: C.S. has received grants from Biotronik, Medtronic, Sorin, St. Jude Medical and Boston Scientific. H.B. has received grants from Biotronik, Boston Scientific, Medtronic, Sorin and St. Jude Medical.
References
- 1.International Organization for Standardization (ISO) ISO 27186:2010: Active Implantable Medical Devices—Four-pole Connector System for Implantable Cardiac Rhythm Management Devices—Dimensional and Test Requirements.http://www.iso.org/iso/iso_catalogue/catalogue_tc/catalogue_detail.htm?csnumber=44045 . [Google Scholar]
- 2.Association for the Advancement of Medical Instrumentation (AAMI) ANSI/AAMI/ISO 27186:2010—Active Implantable Medical Devices—Four-pole Connector System for Implantable Cardiac Rhythm Management Devices—Dimensional and Test Requirements.http://www.aami.org/applications/search/details.cfm?WebID=P993_D5889. (April 2011, date last accessed. [Google Scholar]
- 3.Hauser RG, Almquist AK. Learning from our mistakes ? Testing new ICD technology. N Engl J Med. 2008;24:2517–9. doi: 10.1056/NEJMp0805359. [DOI] [PubMed] [Google Scholar]
- 4.Cogert GA, Cameron CS, Sandler DA. Limitations of the DF-4 defibrillator connector necessitating device removal. Pacing Cardiac Electrophysiol. 2012;35:e24–6. doi: 10.1111/j.1540-8159.2010.02873.x. [DOI] [PubMed] [Google Scholar]
- 5.Calfee RV, Suason SH. A voluntary standard for 3.2 mm unipolar and bipolar pacemaker leads and connectors. Pacing Cardiac Electrophysiol. 1986;6:1181–5. doi: 10.1111/j.1540-8159.1986.tb06690.x. [DOI] [PubMed] [Google Scholar]
- 6.de Voogt WG. Pacemaker leads: performance and progress. Am J Cardiol. 1999;5B:187D–91D. doi: 10.1016/s0002-9149(98)01022-4. [DOI] [PubMed] [Google Scholar]
- 7.Seegers J, Zabel M, Lüthje L, Vollmann D. Ventricular oversensing due to manufacturer-related differences in implantable cardioverter-defibrillator signal processing a sensing lead properties. Europace. 2010;12:1460–6. doi: 10.1093/europace/euq269. [DOI] [PubMed] [Google Scholar]
- 8.Alter P, Waldhans S, Plachta E, Moosdorf R, Grimm W. Complications of implantable cardioverter defibrillator therapy in 440 consecutive patients. Pacing Cardiac Electrophysiol. 2005;28:926–32. doi: 10.1111/j.1540-8159.2005.00195.x. [DOI] [PubMed] [Google Scholar]
- 9.Food and Drug Administration (FDA) 2007. Medtronic Inc. Sprit Fidelis® Defibrillator Leads. http://www.fda.gov/MedicalDevices/Safety/RecallsCorrectionsRemovals/ListofRecalls/ucm062377.htm .
- 10.Food and Drug Administration (FDA) Recalls, Market Withdrawals, & Safety Alerts—Background and Definitions. Last update 2009. http://www.fda.gov/Safety/Recalls/ucm165546.htm .
- 11.Medtronic Inc. 2007. Urgent Medical Device Information—Sprint Fidelis® Lead Patient Management Recommendations (‘Dear Dr letter’)http://www.medtronic.com/product-advisories/physician/sprint-fidelis/PROD-ADV-PHYS-OCT .
- 12.Medtronic Inc. Sprint Fidelis® Lead Performance—November 2010 Website Update.http://www.medtronic.com/product-advisories/physician/sprint-fidelis/6949-LEAD-PERFORMANCE .
- 13.Danik SB, Mansour M, Singh J, Reddy VY, Ellinor PT, Milan D, et al. Increased incidence of subacute lead perforation noted with one implantable cardioverter-defibrillator. Heart Rhythm. 2007;4:439–42. doi: 10.1016/j.hrthm.2006.12.044. [DOI] [PubMed] [Google Scholar]
- 14.Duray GZ, Israel CW, Schmitt J, Hohnloser SH. Implantable cardioverter-defibrillator lead disintegration at the level of the tricuspid valve. Heart Rhythm. 2008;8:1224–5. doi: 10.1016/j.hrthm.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 15.Erkapic D, Duray GZ, Bauernfeind T, De Rosa S, Hohnloser SH. Insulation defects of thin high-voltage ICD leads: an underestimated problem. J Cardiovasc Electrophysiol. 2011;22:1018–22. doi: 10.1111/j.1540-8167.2011.02055.x. [DOI] [PubMed] [Google Scholar]
- 16.Food and Drug Administration (FDA) 2009. Device Classificationhttp://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/PremarketSubmissions/PremarketApprovalPMA/ucm2007514.htm .
- 17.Shein MJ, Schultz DG. Testing new ICD technology. N Engl J Med. 2008;24:2610. doi: 10.1056/NEJMc086467. [DOI] [PubMed] [Google Scholar]
- 18.Heart Rhythm Society (HRS)/American College of Cardiology (ACC)/American Heart Association (AHA) Recommendations from the Heart Rhythm Society Task Force on lead performance policies and guidelines. Heart Rhythm. 2009;6:870–85. doi: 10.1016/j.hrthm.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 19.St Jude Medical. SJ4 Post Approval Study in Implantable Cardioverter-Defibrillator and Cardiac Resynchronization Therapy Defibrillator Patients. Clinical Trials Identifier NCT00940888. http://www.clinicaltrials.gov/ct2/show/NCT00940888?term=DF4&rank=1 .
- 20.St Jude Medical Inc. Product Performance Report—April 2011. http://www.sjmprofessional.com/Resources/product-performance-reports/Product-Performance-Report.aspx .
- 21.Food and Drug Administration (FDA) MAUDE—Manufacturer and User Facility Device Experience Databasehttp://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfmaude/detail.cfm?mdrfoi__id=1895808. 14 May 2011, last date accessed.
- 22.Food and Drug Administration (FDA) MAUDE—Manufacturer and User Facility Device Experience Databasehttp://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfmaude/detail.cfm?mdrfoi__id=1954687. 14 May 2011, date last accessed. [Google Scholar]
- 23.Bock J. Bestimmung des Stichprobenumfangs. München: Oldenbourg Verlag; 1998. pp. 68–70. (ISBN 978-3486245134) [Google Scholar]