

Abstract
Seven-transmembrane receptors (7TMRs), also called G protein–coupled receptors (GPCRs), represent the largest class of drug targets, and they can signal through several distinct mechanisms including those mediated by G proteins and the multifunctional adaptor proteins β-arrestins. Moreover, several receptor ligands with differential efficacies toward these distinct signaling pathways have been identified. However, the structural basis and mechanism underlying this ‘biased agonism’ remains largely unknown. Here, we develop a quantitative mass spectrometry strategy that measures specific reactivities of individual side chains to investigate dynamic conformational changes in the β2-adrenergic receptor occupied by nine functionally distinct ligands. Unexpectedly, only a minority of residues showed reactivity patterns consistent with classical agonism, whereas the majority showed distinct patterns of reactivity even between functionally similar ligands. These findings demonstrate, contrary to two-state models for receptor activity, that there is significant variability in receptor conformations induced by different ligands, which has significant implications for the design of new therapeutic agents.
GPCRs are involved, directly or indirectly, in nearly every physiological process in the human body and are activated by a diverse array of sensory and chemical stimuli including odorants, biogenic amines and peptides1–3. Upon agonist stimulation, conformational changes in the receptor lead to the recruitment and activation of heterotrimeric G proteins that lead to the generation of second messengers such as cyclic AMP (cAMP), G protein– coupled receptor-kinases (GRKs) that recognize and phosphorylate the receptor, and the β-arrestins4, which are multifunctional adaptor proteins that signal through a number of pathways and ultimately terminate G protein–mediated signaling by the receptor1,2,5,6. Classic receptor theory posits that the active conformation of the receptor (denoted R*) is responsible for interacting with and activating all of these downstream partners (G proteins, GRKs and β-arrestins, among others) and that agonists of different efficacies change the equilibrium between active and inactive (denoted R) receptor conformations. Accordingly, full agonists stabilize the active state completely, partial agonists shift the equilibrium toward the active state less effectively than full agonists, and antagonists decrease signaling either by stabilizing the inactive state (that is, inverse agonists7) or by occupying the orthosteric ligand-binding pocket and having no effect on the relative proportion of each state (that is, neutral antagonists). Traditionally, the active receptor has been thought to signal with equal intrinsic efficacies to all downstream signaling pathways, and alterations in signaling to these pathways are thought to be caused by their inherent differences in coupling efficiencies or amplifications.
However, multiple lines of evidence have challenged this classical view, suggesting instead that ligand activity at 7TMRs is indeed nonlinear (that is, ‘collateral efficacy’8) and that multiple receptor conformations signal with different intrinsic efficacies to various signaling pathways8–12, a phenomenon known as biased agonism or functional selectivity8,10–12. For example, a number of 7TMRs, including the β2-adrenergic receptor (β2AR), are known to signal through not only G proteins but also β-arrestins13–16, thus supporting the existence of multiple independent arms of signaling pathways downstream of these receptors9–12,17. Moreover, treatment with ‘biased ligands’ can selectively activate only one or a subset of these pathways and have different effects than endogenous balanced agonists13–15,18,19. Elucidating the structural elements associated with these distinct signaling mechanisms and pathways could facilitate the design of safer and more efficacious therapeutic agents1,11,18–20.
Previous attempts to assess the possible existence of different 7TMR conformations used cell-based biosensors and in vitro fluorescent labels on engineered receptor constructs21–23. Though such studies provide insight into agonist-induced receptor conformational changes, they have neither been definitive nor systematic. Moreover, although recent X-ray crystal structures24–30 of 7TMRs, including the prototypic β2AR31, have provided valuable insight into static atomic-level structural details, they suffer from several inherent limitations. For example, to obtain diffraction-quality crystals of these intractable and dynamic molecules, specific strategies to reduce the flexibility of the receptors were used, such as replacing the highly dynamic third intracellular loop (ICL3) with T4 lysozyme26,27,29, thermostabilization by alanine-scanning mutagenesis30 and cocrystallization with antibodies25,29 that stabilize specific receptor conformations. Therefore, these structures are unable to capture the full complexity and dynamics of the unmodified 7TMRs, in which structural flexibility appears to be important in assuming different conformational states that allow the generation of distinct signaling outputs in response to different ligands. Hence, complementary approaches to characterize ligand-dependent and dynamic conformational changes of 7TMRs in solution will be crucial to revealing new insights into the mechanism of 7TMR signaling and activation.
Here, we describe a powerful approach that examines site-specific protein conformational changes using MS-based quantitative analysis, and we apply it to investigate β2AR structural dynamics associated with different ligand–receptor complexes. The method presented here allows precise quantitative measurement of the changes in the labeling (via protiated, or ‘light’, and deuterated, or ‘heavy’, reagents) of reactive residues as a function of time. Using this method, we demonstrate in a definitive and systematic manner the presence of both agonist- and ligand-dependent conformational changes induced by a wide panel of drugs at the β2AR. These results provide important insights into the role of ligands in promoting distinct receptor conformations and the mechanistic basis of multiple signaling states of 7TMRs.
RESULTS
Development of chemical labeling strategy for the β2AR
To develop an approach that would allow us to quantitatively analyze protein conformational changes, we used stable isotope–labeled (that is, light and heavy versions of) N-ethylmaleimide (NEM) and succinic anhydride reagents to selectively label thiol groups of cysteines and primary ε-amine groups of lysine side chains, respectively. Derivatization of thiol groups of cysteines with either NEM-H5 or NEM-D5 occurs via a Michael-type addition reaction at the α,β-unsaturated bond, whereas in ε-amine groups of lysines with either light (SA-H4) or heavy succinic anhydride (SA-D4), derivatization occurs at any of the two chemically equivalent electrophilic carbons of the carbonyl groups (Fig. 1a,b, top). Additionally, the mass difference observed during MS analysis between paired peptide peaks (the light-heavy peaks, also known as ‘doublets’) is 5 Da in Cys-NEM and 4 Da in lysine–succinic anhydride, consistent with the mass difference between their respective stable isotope reagent pairs (Fig. 1a,b, bottom; and Supplementary Results, Supplementary Fig. 1). These differences allowed us to accurately quantify, during MS analysis, the changes in the labeling of reactive residues as a function of time via measurement of the light or heavy (that is, protiated or deuterated) peptide mass peak intensity ratios. Reactivity of amino acid side chains with such specific reagents is a function of changes in their pKa and protonation state, which are in turn associated with variations in residue microenvironment within the protein structure.
Figure 1. Labeling of cysteines and lysines in the β2AR to monitor conformational changes.

(a) top, reaction of thiolate anion of cysteine side chain with N-ethylmaleimide (neM-H5 or neM-d5) by nucleophilic addition at the double bond of the maleimide ring. bottom, representative isotope-peak pair (doublet) corresponding to a chymotryptic peptide ( 125cviAvdRYF133) modified at cys125 by a light and heavy neM (m/z 1210.6 and 1215.6, respectively; ~Δm/z = 5). (b) top, reaction of the ε-nH2 group of the lysine side chain with succinic anhydride (SA-H4 or SA-d4) by nucleophilic addition at one of the carbonyl groups. bottom, representative spectrum of doublet peaks corresponding to a chymotryptic peptide ( 259RRSSKF264) modified at lys263 by light and heavy succinic anhydride (m/z 880.5 and 884.5, respectively; ~Δm/z = 4).
Having investigated the optimal derivatization conditions, we next performed covalent labeling reactions using specific stable-isotope labeled reagents (NEM-H5 and NEM-D5 or SA-H4 and SA-D4) to determine the susceptibility of native β2AR cysteines and lysines to their respective reagents. Under optimal experimental conditions, we found doublets (either labeled with NEM-H5 and NEM-D5, as in Fig. 1a, bottom, or with SA-H4 and SA-D4, as in Fig. 1b, bottom) that gave detectable signals corresponding to the labeling of four cysteines and five lysines, whereas the signals for the remaining residues were weak and thus excluded from this study (Supplementary Table 1). Furthermore, by taking one of longest peptide isotope peak pairs among the selected peptides, we evaluated the precision of our peptide quantification and observed an excellent linear correlation between observed signal intensities and mole fractions of differentially labeled β2AR (slope = 0.9785 ± 0.13; R2 = 0.9996; P < 0.0001), demonstrating that the MALDI-TOF MS analytical platform used here is direct, error-controlled and quantitative with a linear dynamic range spanning over two orders of signal magnitude (Supplementary Fig. 2). The nine residues that were found to be suitable for quantitative site-specific conformational change studies are located throughout the β2AR (Fig. 2) and are present in the second (Lys1403.59) and third (Lys2355.74, Lys2636.25 and Cys2656.27) intracellular loops (ICLs), the extracellular surface at TM7 (Lys 3057.32) and the intracellular domains of TM2 (Cys772.48), TM3 (Cys1253.44), TM5 (Lys2275.66) and TM7 (Cys3277.54) (superscripts refer to Ballesteros–Weinstein residue numbering system32). Labeling of the β2AR at these sites with the appropriate group specific reagent— that is, NEM or succinic anhydride—did not affect the pharmacological properties of the receptor as assessed by radio-ligand binding experiments (Supplementary Fig. 3) and other studies33.
Figure 2. Schematic two-dimensional topology of human β2AR showing location of sites of study.

A total of nine amino acids in single-letter code are highlighted. cysteines are shown in orange and lysines are shown in blue. the numbers indicate positions of the amino acid sequence.
Pharmacological properties of ligands at the β2AR
To identify ligand-specific β2AR conformations, we used nine structurally and functionally distinct ligands, as shown in Table 1 (and in more detail in Supplementary Table 2), each of which had differential abilities to signal via a Gαs-coupled cAMP pathway, recruit β-arrestins and activate extracellular signal–regulated kinase 1/2 (ERK1/2). Based on their ability to produce receptor-mediated second messenger cAMP, these ligands were classified as full agonists (isoproterenol and THRX-144877), partial agonists (salbutamol and salmeterol), weak partial agonist (pindolol), antagonists or inverse agonists (propranolol, carazolol, ICI-118551 and carvedilol). Among the antagonists, both carvedilol and carazolol are able to activate ERK1/2 signaling via G protein–independent mechanisms, whereas carvedilol, but not carazalol, is able to weakly promote β-arrestin recruitment15. Structurally, the first four ligands are grouped as catechol (isoproterenol) or noncatechol (salbutamol, salmeterol, and THRX-144877) arylethanolamine-based agonists, and the last five are classified as aryloxypropanolamine-based antagonists, commonly referred to as ‘β-blockers’ (Table 1).
Table 1.
Summary of pharmacological properties of β2AR ligands
| Ligand | Chemical structure | G protein activation (cAMP accumulation) |
β-arrestin recruitment |
ERK1/2 activation |
|---|---|---|---|---|
| Isoproterenol |
|
++++ | +++ | ++++ |
| THRX-144877 |
|
++++ | ++++ | ++++ |
| Salbutamol |
|
+++ | ++ | +++ |
| Salmeterol |
|
+++ | ++ | +++ |
| Propranolol |
|
– | 0 | + |
| Pindolol |
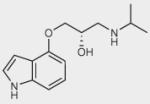
|
+ | + | ++ |
| Carazolol |
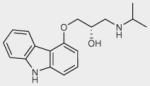
|
– | 0 | + |
| ICI-118551 |
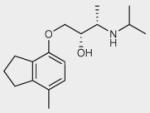
|
– | 0 | – |
| Carvedilol |
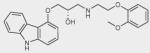
|
– | + | + |
Qualitative functional properties of ligands expressed relative to maximal stimulations (++++). Quantitative data, estimation of ligand efficacy and further description can be found in Supplementry Table 2.
Measuring labeling kinetics and conformational dynamics
One major advantage of using residue-specific, stable-isotope labeling followed by mass MS analysis is that this technique can accurately and precisely quantify the extent of residue side chain labeling in proteins34,35. We assessed the effects of the set of β2AR-ligands on the time course of the reaction of each of the four cysteines and five lysines with their specific stable isotope labeled reagents (NEM or succinic anhydride). Labeling the receptor at these different sites allowed us to probe the conformational dynamics of its associated structural elements upon ligand binding. The reactivity of each site in the different β2AR–ligand complexes was measured by monitoring the sequential increase in the signal intensity of the heavy peak (NEM-D5- or SA-D4-labeled peptide) relative to a reference light peak (NEM-H5- or SA-H4-labeled peptide), allowing us to accurately quantify the extent of labeling in the form of ‘percent site labeled’ (%F) as a function of time for each site-ligand pair (Fig. 3, Supplementary Fig. 4 and Supplementary Methods). In the transients (Fig. 3d and Supplementary Fig. 5), the measurable differences we observed in the reactivities of different residues with various ligands were suggestive of changes in structural rearrangements upon ligand binding. Notably, only one of these transients (Lys2275.66) in the presence of propranolol) could be well fit (R2 = 0.988) to a single exponential function. The remainder of these time-course datasets were well fitted by a double exponential function with fast and slow relaxation times, with 0.991 being the lowest R2 value (fitting functions and the best fit parameters are described in Supplementary Methods and Supplementary Table 3). Fits that included a burst-phase component demonstrated the absence of any significant burst-phase amplitudes. On the basis of these data, it can be concluded that the sampling scheme captured the vast majority of the amplitude in the fast and slow phases of the labeling process. The observation of such biphasic reaction kinetics requires a mechanism involving at least three distinct reactive species, a reasonable interpretation of which is that there are at least three distinct conformational ensembles with different reactivities in each set of conditions36.
Figure 3. Schematic illustration of time-dependent, residue-specific labeling experiment designed to monitor conformational changes in the β2AR.

(a,b) Strategy for labeling of cysteines in purified β2AR, initiated in two pools by adding either neM-H5 as in (a) or neM-d5 as in (b). equal amounts of the two pools are mixed, subjected to proteolysis, and MS-analyzed to determine peptide fragments that have been modified. (c) Representative doublets with singly charged ion ([M+H]+) peaks at m/z 1336.6 and 1341.7 that correspond to peptide 327cRSpdFRiAF336 modified at cys327 by neM and exhibit a mass difference of 5 da following modification with either neM-H5 or neM-d5 (details are listed in Supplementary Methods and Supplementary Fig. 4). (d) Representative time-course curves of the extent of neM reactivity at cys327, expressed as percent of sites labeled (%F) plotted versus labeling time in minutes on a logarithmic scale, after treatment with carrier solvent or indicated ligands. the solid lines in each plot are the best fit obtained after fitting to double exponential function. data represent the average of at least three independent experiments ± s.e.m.
To quantitatively compare the labeling kinetics at the different sites, we calculated the negative logarithm of the relaxation times (–log τ) for each ligand, which we refer to as the labeling reactivity factor (L-factor) (Figs. 4 and 5). For the majority of residues, except Lys3057.32, the fast relaxation times were chosen for structural analysis because in some cases the slow relaxation times were on a time scale similar to the longest time point of the experiment. For each site, we used receptor without ligand (receptor-only) as a reference control for subsequent analysis, and thus, for any given site, a difference in the L-factors between the receptor without ligand and a ligand-bound complex reflects ligand-dependent conformational changes (that is, a large L-factor reflects higher reactivity relative to receptor-only samples, and vice versa). To further validate the accuracy of our analytical methodology, we compared the L-factor values determined for the nine β2AR sites in the receptor-only sample using two separate stable-isotope labeling strategies, conventional and inverse (where protiated and deuterated reagents are swapped in the scheme shown in Fig. 3a,b), and found excellent linear correlation between them (Supplementary Fig. 6).
Figure 4. Reactivities of residues in β2AR featuring conformational rearrangements of classic receptor activation.
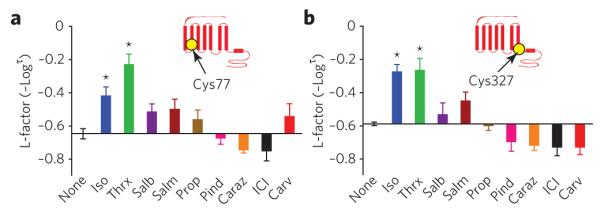
(a,b) effects of nine β2AR ligands on neM reactivity at cys77 (a) and at cys327 (b). insets indicate position of labeled residue in the β2AR snake-like diagram. bar graphs depict the reactivity of each site with the different ligands bound to the β2AR, indicated on the y-axis by l-factor values relative to the value for receptor without ligand. bars extending below the x-axis indicate lower l-factors, and bars extending above it indicate higher l-factors relative to the receptor without ligand. All l-factors shown are for the fast phase. data correspond to the means ± standard errors of at least three independent experiments. Asterisks indicate statistical significance (*P < 0.05) compared to control receptor alone by one-way AnovA.
Figure 5. Reactivities of residues in β2AR featuring ligand-specific conformational rearrangements.

(a–g) bar graphs summarizing the effects of various ligands on the changes in the l-factors of seven different sites of the β2AR, expressed relative to the receptor without ligand: cys125 (a), lys140 (b), lys227 (c), lys235 (d), lys263 (e), cys265 (f) and lys305 (g). All l-factors shown are for the fast phase except for lys305. l-factor for lys227 is not shown, as no detectable amplitude for the fast phase was observed. data correspond to the means ± standard errors from at least three independent experiments. Asterisks indicate statistical significance (*P < 0.05) compared to control receptor alone by one-way AnovA.
Conformational rearrangements of classic 7TMR activation
To gain insights into the effects of different ligands on the conformational rearrangements around each residue in the various ligand-bound β2AR complexes, we examined specific L-factor values for each site-ligand pair. At only two of the nine residues analyzed did we observe changes in reactivity that correlated with classical levels of agonism. These residues were Cys772.48, located in the intra cellular portion of TM2, and Cys3277.54, located at the cytoplasmic end of TM7, both of which displayed similar L-factor patterns that correlate with the rank order of agonist efficacy for G protein– mediated effects (Fig. 4a,b). Of the panel of β2AR ligands tested relative to the unbound receptor, THRX-144877 (Thrx), a full agonist, had the highest reactivity of Cys772.48 (Fig. 4a) with the highest L-factor (P < 0.001, Thrx versus ICI, one-way ANOVA). For the same site, this L-factor pattern was followed by the other full agonist, isoproterenol (P < 0.01, isoproterenol versus ICI), and to a lesser extent by the two partial agonists, salbutamol and salmeterol. Notably, inverse agonists such as carazolol and ICI-118551 caused opposite L-factor effects at this site. The reactivity of Cys3277.54 also showed a similar pattern, though the difference in reactivity between agonists and antagonists was more pronounced (P < 0.001, isoproterenol or Thrx versus ICI) (Fig. 4b). Thus, the L-factor pattern at these two positions correlates well with classic activation of Gαs.
Conformational rearrangements specific to ligands
In contrast to our observations at Cys772.48 and Cys3277.54, the reactivities at Cys1253.44, Lys1403.59, Lys2275.66, Lys2355.74, Lys2636.25, Cys2656.27 and Lys3057.32 revealed intriguing patterns that do not reflect the known efficacy profile of the ligands. For example, at Cys1253.44 treatment with antagonists resulted in relatively higher L-factor values than treatment with the noncatechol full agonist THRX-144877 and partial agonists salbutamol and salmeterol (Fig. 5a). Notably, the reactivity of this site in the presence of isoproterenol, a catechol-based full agonist, reached a level comparable to that displayed by the antagonists. Furthermore, at Lys 1403.59, isoproterenol showed an increase in reactivity, whereas the remaining ligands showed few changes or none at all (Fig. 5b). The reactivities of Lys2275.66 and Lys2355.74 residues, located at the cystoplasmic end of TM5, also differed in the presence of pharmacologically similar ligands (Fig. 5c,d). At Lys2275.66, increased L-factors were observed in the presence of isoproterenol, salmeterol and ICI-118551 relative to those measured in the receptor without ligand (P < 0.05, none versus salmeterol; P < 0.01, carazolol versus salmeterol or ICI). At Lys 2636.25, an L-factor that was markedly higher than those for all other ligands (P < 0.05, carvedilol versus none) was observed in carvedilol-bound β2AR complex (Fig. 5e). Notably, carazolol, which is functionally very similar to carvedilol in terms of inverse agonism at Gαs and weak agonism in β-arrestin signaling, showed no effect in reactivity at this site (P < 0.01, carvedilol versus carazolol). On the other hand, the L-factor for Cys2656.27 in the carvedilol-bound β2AR complex was very small and was opposite to that observed at Lys2636.25 (P < 0.05, none versus carvedilol) (Fig. 5f). At Lys3057.32, on the extracellular surface region of the β2AR, the highest reactivity was observed in the presence of THRX-144877, whereas only relatively small changes in reactivity were observed with the isoproterenol, salbutamol, and salmeterol bound-β2AR complexes (P < 0.05, Thrx versus isoproterenol or ICI) (Fig. 5g). These results demonstrate that the majority of the conformational changes induced by ligand binding are ligand-specific in nature and do not correlate with classical agonism for G protein–signaling.
DISCUSSION
Understanding the structural mechanism for 7TMR signaling and biased agonism is currently a central issue in receptor biology that could have significant implications for the design of new therapeutic agents. With the recent high-resolution structures from X-ray crystallography24–30, considerable insight has been gained into possible mechanisms for 7TMR activation, although such studies can only yield a static picture of the ligand–receptor complex. In this context, we have developed a quantitative MS-based approach to directly monitor the conformational changes and dynamics that occur upon ligand binding to the β2AR in an effort to unravel the complex interplay between the receptor and different ligands. This approach provides residue-specific structural information to identify ligand-induced changes in receptor conformations. The conformational changes can be elucidated quantitatively and in solution, unlike other related techniques37 that are limited to providing only qualitative structural information. In the course of analysis of our data in the presence of functionally and structurally different ligands, we observed two patterns of conformational rearrangements of the β2AR: one that correlates with classical levels of agonism and another that appeared to be specific for certain ligands. These results thus support multistate dynamic behavior of these receptors and argue for greater complexity in ligand–receptor conformations than those that lead to G protein activation alone.
A key structural element in 7TMRs that has been implicated in receptor activation mechanisms and internalization processes38–40 upon ligand binding is the highly conserved NPxxY motif. Interestingly, one of the two residues that showed L-factor patterns in accordance with classic G protein activation, Cys3277.54, is located adjacent to this conserved NPxxY motif at the cytoplasmic end of TM7 (residues 322–326 in the β2AR). In addition, examination of this region in the crystal structures of carazolol-bound β2AR27 and rhodopsin24,41 shows ordered water molecules that mediate interhelical hydrogen bond networks and other interactions between different polar and charged side chains, including Asp792.50 of TM2 and Asn3227.49 of the NPxxY motif from TM7 (Fig. 6a, blue box). Notably, these water-mediated hydrogen-bond networks in 7TMRs have also been suggested to play an important role in the receptor activation process by maintaining the stability of the inactive state and by propagating activation upon agonist binding41. It is therefore conceivable from the high degree of similarity in the L-factor patterns at Cys772.48 of TM2 and Cys3277.54 of TM7 that the conformational transitions between the structural elements around these two residues are linked structurally and are mediated by a cluster of conserved residues and ordered water molecules across 7TMRs. For G protein activation in these receptors, it is likely that substantial reorientation and weakening of these polar interhelical interactions occur that could result in changes in the microenvironment of Cys772.48 as well as pronounced structural rearrangements at the cytoplasmic end of TM7 (Fig. 6a, black box), as observed in opsin28 and the putative β2AR active state29. Therefore, the reactivity of side chains in this region, such as those at Cys3277.54, may represent a measure of efficacy for a given ligand in promoting a receptor conformation responsible for Gαs coupling.
Figure 6. Proposed models Illustrating the conformational rearrangements observed in different structural elements of the β2AR.

(a) A ribbon diagram of carazolol-bound β2AR-t4l (pdb: 2RH1) is shown on the left. top right, cys77 and cys327 are shown as yellow spheres, Asp79 in tM2 and NPxxY in tM7 are shown as sticks, water molecules as red spheres and hydrogen bonds as dashed red lines. bottom right, cytoplasmic end view of tM7 of the superposition of structures of opsin (magenta; pdb: 3dQb), rhodopsin (pale blue; pdb: 1GZM) and carazolol–β2AR–t4l highlighting structural rearrangement at the cytosolic end of tM7 of the β2AR (red arrow). (b) interactions of cys125 in tM3 with pro211 in tM5 and potential ligand-specific rearrangements of the side chain illustrated by the arrow in red. (c) locked conformation of icl2 (lys140 in blue sphere), pointing toward the 7tM-helical bundle in the carazolol–β2AR-t4l structure and alternative ligand-specific structural rearrangements of icl2 region (dashed red lines). (d) Residues (lys227, lys235, lys263 and cys265) in the intracellular loop (icl3) region and adjoining tMs 5 and 6 of β2AR. the side chains of lys263 and cys265 are oriented opposite to each other on the loop. the proposed structural rearrangement of icl3 (dashed red lines) is indicated by red arrow. (e) extracellular surface view of β2AR showing location of salt bridge between Asp192 (red spheres) and lys305 (blue sphere). tMs 1–7 are green, and ecls 1–3 are cyan. Figures were prepared with pyMol (delano Scientific).
A common interaction conserved among 7TMRs is the ionic lock, a salt bridge between Arg1313.50 of the highly conserved E/DRY motif and Glu2686.30, whose disruption is a key feature of receptor activation as demonstrated by mutational and biophysical analyses22,42,43. Although the agonists used in our study appear to show an increased reactivity relative to receptor alone at the residues located in the vicinity of Arg1313.50 and Glu2686.30 (that is, Cys1253.44, Lys2636.25 and Cys2656.27), the L-factor pattern in the presence of antagonists is surprising. For example, the relative increase in the reactivity of Cys1253.44 in the presence of antagonists is consistent with ligand-induced changes in its microenvironment, which may include stabilization or destabilization of interactions involving the side chains of TM3 and TM5 associated with inactivation or activation of the receptor (Fig. 6b). In the crystal structure of carazolol-bound β2AR, ICL2 adopts a locked conformation by inserting itself into the 7TM-helical bundle, mainly via hydrogen bonds between the amino acid side chains of ICL2, such as Lys1403.59 and Tyr1413.60, with Arg1313.50, Gln2295.67 and Glu2686.30 (Fig. 6c). The relatively low reactivity observed at Lys1403.59 in the carazolol-bound β2AR complex correlates well with an inaccessible, locked conformation of the loop. In contrast, the L-factor value observed at this residue in the isoproterenol-bound β2AR complex is markedly high relative to that in other agonists and suggests that ICL2 can adopt distinct functional ligand-dependent conformational rearrangements (Fig. 6c). Such conformational states of ICL2 may include α-helical and non-helical conformations29,44, which may be associated with the differential ability of activated receptors to recognize and bind to G proteins and β-arrestins45,46.
Other illustrations of functional ligand-dependent conformational rearrangements are the reactivities observed at Lys2636.25 and Cys2656.27 in the cytoplasmic end of TM6 in the third intracellular loop (ICL3)42,43 and on the extracellular surface region at Lys3057.32 of TM7 of the β2AR. Notably, the reactivities of the carvedilol-bound β2AR complex at both Lys2636.25 and Cys2656.27 suggest the existence of significant ligand-specific side chain reorganization in the neigh-boring regions of these residues. These structural rearrangements in β2AR may include exposure of the loop and its associated residues toward the intracellular surface and are unique conformational changes that none of the other ligands, including carazolol (with relatively similar function and chemical structure), are able to stabilize (Fig. 6d). Whether this reflects the ability of carvedilol, unique among all the antagonists tested, to promote receptor interaction with β-arrestin (and thus serve as a biased ligand), its ability to bind at a putative allosteric site or some other as-yet-undiscovered function remains to be determined. Lastly, the observed variation in the reactivity at Lys3057.32 in the different ligand-bound β2AR complexes could reflect a range of distances in the D192–K305 salt bridge and conformational rearrangements of the extracellular region of TM7. Conformational coupling of this salt bridge to the orthosteric site and its ligand-dependent regulation in the β2AR has also been previously demonstrated by two-dimensional nuclear magnetic resonance spectroscopy47. Our observation of higher reactivity at Lys3057.32 in the THRX-144877–bound β2AR complex may therefore suggest ligand-specific conformational rearrangement of the region that would likely involve conformational alterations of the salt bridge (Fig. 6e). These data in aggregate therefore support the concept that the β2AR exists in multiple conformations that are induced by functionally different ligands.
In conclusion, by using a quantitative MS-based strategy that measures specific reactivities of multiple native residues in the same protein simultaneously, we have demonstrated that structurally and functionally distinct ligands show complex patterns of labeling at different residues in the β2AR, thus providing direct evidence for the presence of multiple ligand-specific conformations. This study therefore provides a definitive and systematic demonstration that different ligands induce qualitatively different receptor conformations. This is consistent with the ability of several structural elements within the receptor to adopt distinct conformations in response to ligands of different chemical structure, which may underlie the recently studied phenomenon of biased agonism and functional selectivity. More broadly, the findings presented here are incompatible with the widely held notion that all ligands of similar functional capabilities stabilize or destabilize similar sets of interactions in a given receptor and with the general concept of ‘two state’ models for receptor activity. Thus, our results open new avenues for further therapeutic development in targeting the residues or in the discovery of associated structural elements that may be associated with the activation of specific signaling pathways. This general approach should be applicable to the study of the conformational changes and dynamics of a wide range of soluble and membrane proteins and could also be applied to drug screening against protein targets that undergo function-dependent conformational changes.
METHODS
Expression and purification of the human β2AR
Human β2AR was expressed in baculovirus-infected Sf9 insect cells as N-terminal FLAG-tagged and C-terminal 6xHis-tagged proteins and was subsequently purified in n-dodecyl-β-D-maltoside (DDM) detergent using three-step affinity-chromatographic procedures. Purity of the protein was assessed by Coomassie blue staining resolved on SDS–PAGE. Complete details are described in Supplementary Methods.
Differential labeling of β2AR and MS analysis
Differential labeling reactions of cysteines and lysines of the β2AR with their specific stable isotope–labeled reagents were done under identical conditions using purified β2AR (2.5 μM) in 50 mM potassium phosphate buffer (pH 7.5) containing 50 mM NaCl and 0.02% (m/v) DDM, and incubation used carrier solvent or indicated ligand (50 μM) for 30 min at 25 °C in a 110-μl total reaction volume. Briefly, the kinetics of reaction of NEM (2 mM) with β2AR cysteine residues were measured by mixing a reference reaction mixture made with NEM-H5 with time-point samples taken from a reaction made with NEM-D5 at 25 °C. For experiments involving differential labeling of lysines of β2AR on ligand binding with protiated and deuterated succinic anhydride (SA-H4 and SA-D4), reactions were set up similarly with the exception that a final 100-mM concentration of L-lysine in 200 mM Tris-Cl (pH 8.0) was used instead of DTT (as in NEM labeling) for quenching. Equally sized aliquots of the protiated (light) and deuterated (heavy) reagent-labeled samples were then mixed and subjected to proteolytic digestion. The digests were analyzed using an ABI-4700 MALDI-TOF/TOF MS equipped with a nitrogen laser operating at 337 nm and 4000 Series Explorer software. The ratios of intensities of monoisotopic peaks of the deuterated (heavy) to protiated (light) reagent-labeled peptides for each time point and site-ligand pair were calculated to obtain percent intensity ratios (%R). Each percent intensity ratio (%R) data point was corrected by a specific site-ligand pair reactivity ratio (Rr), which was obtained by performing independent differential reactivity experiments (details are listed in Supplementary Methods). Both %R and Rr were used to compute the final percentage of sites labeled (%F) as a function of time via the following equation:
| (1) |
where %F(t) is the percent of site labeled, %R(t) is the ratio of the heavy/light signal intensity for time point t sample and Rr is the heavy/light reactivity ratio from the reactivity experiment. Further details of these procedures as well as those of quantification and inverse labeling experiments are described in Supplementary Methods.
Quantitation of site-specific labeling kinetics
To determine the appropriate fitting model, the progression curves (percent of sites labeled, %F, versus time in minutes) for each site-ligand pair were fit to each of the following three different models. The first model has a single phase with a relaxation time defined as:
| (2) |
where t is the labeling time, τ1 is the relaxation time, A is the amplitude and 100 – A is the burst-phase amplitude. The second model has two distinct observable phases but does not allow for a burst phase. This model is:
| (3) |
where τ1 and τ2 are the time constants for each phase; and A is the fractional amplitude of the τ1 phase. The third model allows for a burst phase and two additional phases and has the form:
| (4) |
where A and B are the amplitudes of the phases with relaxation times τ1 and τ2, respectively, and the burst-phase amplitude is (100 – A – B). All three models force F(∞) = 100, which is expected from the experimental design.
The models used to extract kinetic parameters from the data were chosen on the basis of the simplest model that produced a goodness of fit equal to or better than the more complicated model. On this basis, the single-exponential model was chosen for only the combination of Lys227 and propranolol. All other datasets required double-exponential fits. When these data were fitted with the third model, equation 4 (4), the quality of the fits were not significantly better than the fits with no burst phase. Therefore, the second model, equation 3 (3), was used to determine best-fit parameter values for all datasets except K227-propranolol. Supplementary Table 3 lists numerical values of the kinetic parameters, and Supplementary Methods describes complete details on this section.
Competitive radio-ligand binding experiments
Competition-binding assays of purified β2AR (treated with DMSO, 2 mM NEM or 2 mM succinic anhydride) using [125I]cyanopindolol were performed with various concentrations of competing ligands (THRX-144877 or carvedilol) followed by rapid filtration of receptor bound radioactivity on a Whatman GF/B filter. IC50 values for the ligands were determined by fitting the data to nonlinear regression analysis. Details are described in Supplementary Methods.
cAMP generation
Accumulation of second messenger cAMP was measured by an improved version of a fluorescence resonance energy transfer (FRET)– based cAMP biosensor (ICUE)48. HEK-293 cells stably expressing β2AR and ICUE2 were stimulated with various concentrations of β2AR ligands for 3 min. cAMP concentrations were quantified as a FRET ratio between CFP and YFP intensity.
β-arrestin recruitment
β-arrestin recruitment to ligand-occupied β2AR was assessed using a luciferase-based reporter gene assay known as ‘Tango’ in 96-well format, as previously described in reference 49.
ERK activation
ERK1/2 phosphorylation was assessed using a cellular activation of signaling ELISA kit (SA Biosciences), essentially according to the manufacturer’s recommendations.
Estimation of ligand efficacy
The operational model developed by Black and Leff50 was used to estimate efficacy by calculating the coupling coefficient, τ:
where Em is the maximal response of the system to a full agonist, KD is the agonist dissociation constant and τ is the ‘coupling efficiency’ between the agonist– receptor complex and its downstream signaling partners. Details on cAMP generation, β-arrestin recruitment, ERK activation and estimation of ligand efficacy are described in Supplementary Methods.
Statistical analysis
Statistical analysis and curve-fitting were done using Prism 5.01. For statistical comparison, ANOVA or a Student’s two-tailed unpaired t-test were used, with P values of <0.05 considered significant.
Supplementary Material
Acknowledgments
R.J.L. is an investigator with the Howard Hughes Medical Institute. This work was supported in part by grants from the US National Institutes of Health (HL16037 and HL70631) to R.J.L. T.G.O. is supported by a grant from the US National Institute of General Medical Sciences (5RO1GM081666). We gratefully acknowledge T. Haystead (Duke University) and D. Loiselle for valuable assistance with the mass spectrometry experiments, and Theravance, Inc. (South San Francisco, California, USA) for the supply of THRX-144877; we are also grateful to R.T. Strachan, J. Kovacs, R. Dror (D.E. Shaw Research, New York, New York, USA), C.H. Borchers (University of Victoria, Victoria, British Columbia, Canada), I.A. Kaltashov (University of Massachusetts), B. Donald (Duke University) and members of his laboratory for stimulating ideas and helpful discussions; we also thank X. Jiang and C.M. Lam for excellent technical assistance and D. Addison and Q. Lennon for secretarial assistance.
Footnotes
Accession codes. Protein Data Bank: The structure of human β2AR is deposited under accession code 2RH1, and the structures of the GPCRs opsin and rhodopsin are deposited under accession codes 3DQB and 1GZM, respectively.
Author contributions A.W.K., K.X., T.G.O. and R.J.L. designed the experiments; A.W.K., K.X., S.A. and A.K.S. conducted experiments; A.W.K., K.X., S.R., S.A., A.K.S., J.S., T.G.O. and R.J.L. analyzed data; A.W.K., K.X. and R.J.L. wrote the paper; all authors read, edited and discussed the paper.
Competing financial interests The authors declare no competing financial interests.
Additional information Supplementary information is available online at http://www.nature.com/naturechemicalbiology/. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Lefkowitz RJ. Seven transmembrane receptors: something old, something new. Acta Physiol. (Oxf.) 2007;190:9–19. doi: 10.1111/j.1365-201X.2007.01693.x. [DOI] [PubMed] [Google Scholar]
- 2.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 3.Lagerström MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 4.Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 5.Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 2002;115:455–465. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- 6.Ahn S, Nelson CD, Garrison TR, Miller WE, Lefkowitz RJ. Desensitization, internalization, and signaling functions of beta-arrestins demonstrated by RNA interference. Proc. Natl. Acad. Sci. USA. 2003;100:1740–1744. doi: 10.1073/pnas.262789099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samama P, Pei G, Costa T, Cotecchia S, Lefkowitz RJ. Negative antagonists promote an inactive conformation of the beta 2-adrenergic receptor. Mol. Pharmacol. 1994;45:390–394. [PubMed] [Google Scholar]
- 8.Kenakin T. Collateral efficacy in drug discovery: taking advantage of the good (allosteric) nature of 7TM receptors. Trends Pharmacol. Sci. 2007;28:407–415. doi: 10.1016/j.tips.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 9.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 10.Violin JD, Lefkowitz RJ. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol. Sci. 2007;28:416–422. doi: 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kenakin T. Functional selectivity and biased receptor signaling. J. Pharmacol. Exp. Ther. 2011;336:296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- 13.Wei H, et al. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. USA. 2003;100:10782–10787. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gesty-Palmer D, et al. Distinct beta-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J. Biol. Chem. 2006;281:10856–10864. doi: 10.1074/jbc.M513380200. [DOI] [PubMed] [Google Scholar]
- 15.Wisler JW, et al. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc. Natl. Acad. Sci. USA. 2007;104:16657–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shukla AK, et al. Distinct conformational changes in beta-arrestin report biased agonism at seven-transmembrane receptors. Proc. Natl. Acad. Sci. USA. 2008;105:9988–9993. doi: 10.1073/pnas.0804246105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao K, et al. Global phosphorylation analysis of beta-arrestin-mediated signaling downstream of a seven transmembrane receptor (7TMR) Proc. Natl. Acad. Sci. USA. 2010;107:15299–15304. doi: 10.1073/pnas.1008461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gesty-Palmer D, et al. A beta-arrestin-biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci. Transl. Med. 2009;1:1ra1. doi: 10.1126/scitranslmed.3000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends. Mol. Med. 2010;17:126–139. doi: 10.1016/j.molmed.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Violin JD, et al. Selectively engaging beta-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J. Pharmacol. Exp. Ther. 2010;335:572–579. doi: 10.1124/jpet.110.173005. [DOI] [PubMed] [Google Scholar]
- 21.Reiner S, Ambrosio M, Hoffmann C, Lohse MJ. Differential signaling of the endogenous agonists at the beta2-adrenergic receptor. J. Biol. Chem. 2010;285:36188–36198. doi: 10.1074/jbc.M110.175604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yao X, et al. Coupling ligand structure to specific conformational switches in the beta2-adrenoceptor. Nat. Chem. Biol. 2006;2:417–422. doi: 10.1038/nchembio801. [DOI] [PubMed] [Google Scholar]
- 23.Ghanouni P, et al. Functionally different agonists induce distinct conformations in the G protein coupling domain of the beta 2 adrenergic receptor. J. Biol. Chem. 2001;276:24433–24436. doi: 10.1074/jbc.C100162200. [DOI] [PubMed] [Google Scholar]
- 24.Palczewski K, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 25.Rasmussen SG, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 26.Rosenbaum DM, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 27.Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheerer P, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 29.Rasmussen SG, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warne T, et al. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dixon RA, et al. Cloning of the gene and cDNA for mammalian beta-adrenergic receptor and homology with rhodopsin. Nature. 1986;321:75–79. doi: 10.1038/321075a0. [DOI] [PubMed] [Google Scholar]
- 32.Ballesteros JA, Weinstein H. Integrated methods for the construction of three dimensional models and computational probing of structure function relations in G protein-coupled receptors. Meth. Neurosci. 1995;25:366–428. [Google Scholar]
- 33.Stadel JM, Lefkowitz RJ. Multiple reactive sulfhydryl groups modulate the function of adenylate cyclase coupled beta-adrenergic receptors. Mol. Pharmacol. 1979;16:709–718. [PubMed] [Google Scholar]
- 34.Gygi SP, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 35.Ong SE, Mann M. Mass spectrometry-based proteomics turns quantitative. Nat. Chem. Biol. 2005;1:252–262. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- 36.Cantor CR, Schimmel PR. The Behavior of Biological Macromolecules##915–916. Freeman; 1980. [Google Scholar]
- 37.Kelly BL, Gross A. Potassium channel gating observed with site-directed mass tagging. Nat. Struct. Biol. 2003;10:280–284. doi: 10.1038/nsb908. [DOI] [PubMed] [Google Scholar]
- 38.Barak LS, Menard L, Ferguson SS, Colapietro AM, Caron MG. The conserved seven-transmembrane sequence NP(X)2,3Y of the G-protein-coupled receptor superfamily regulates multiple properties of the beta 2-adrenergic receptor. Biochemistry. 1995;34:15407–15414. doi: 10.1021/bi00047a003. [DOI] [PubMed] [Google Scholar]
- 39.Bouley R, et al. Functional role of the NPxxY motif in internalization of the type 2 vasopressin receptor in LLC-PK1 cells. Am. J. Physiol. Cell Physiol. 2003;285:C750–C762. doi: 10.1152/ajpcell.00477.2002. [DOI] [PubMed] [Google Scholar]
- 40.Fritze O, et al. Role of the conserved NPxxY(x)5,6F motif in the rhodopsin ground state and during activation. Proc. Natl. Acad. Sci. USA. 2003;100:2290–2295. doi: 10.1073/pnas.0435715100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okada T, et al. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proc. Natl. Acad. Sci. USA. 2002;99:5982–5987. doi: 10.1073/pnas.082666399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pei G, et al. A constitutively active mutant beta 2-adrenergic receptor is constitutively desensitized and phosphorylated. Proc. Natl. Acad. Sci. USA. 1994;91:2699–2702. doi: 10.1073/pnas.91.7.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J. Biol. Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- 44.Dror RO, et al. Identification of two distinct inactive conformations of the beta2-adrenergic receptor reconciles structural and biochemical observations. Proc. Natl. Acad. Sci. USA. 2009;106:4689–4694. doi: 10.1073/pnas.0811065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marion S, Oakley RH, Kim KM, Caron MG, Barak LS. A beta-arrestin binding determinant common to the second intracellular loops of rhodopsin family G protein-coupled receptors. J. Biol. Chem. 2006;281:2932–2938. doi: 10.1074/jbc.M508074200. [DOI] [PubMed] [Google Scholar]
- 46.Raman D, Osawa S, Gurevich VV, Weiss ER. The interaction with the cytoplasmic loops of rhodopsin plays a crucial role in arrestin activation and binding. J. Neurochem. 2003;84:1040–1050. doi: 10.1046/j.1471-4159.2003.01598.x. [DOI] [PubMed] [Google Scholar]
- 47.Bokoch MP, et al. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature. 2010;463:108–112. doi: 10.1038/nature08650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Violin JD, et al. beta2-adrenergic receptor signaling and desensitization elucidated by quantitative modeling of real time cAMP dynamics. J. Biol. Chem. 2008;283:2949–2961. doi: 10.1074/jbc.M707009200. [DOI] [PubMed] [Google Scholar]
- 49.Barnea G, et al. The genetic design of signaling cascades to record receptor activation. Proc. Natl. Acad. Sci. USA. 2008;105:64–69. doi: 10.1073/pnas.0710487105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Black JW, Leff P. Operational models of pharmacological agonism. Proc. R. Soc. Lond. B Biol. Sci. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.