

Abstract
TAR DNA binding protein-43 (TDP-43) immunoreactive neuronal inclusions are detected in 20–30% of Alzheimer disease (AD) brains, but the distribution of this pathology has not been rigorously studied. In this report we describe region-specific distribution and density of TDP-43 positive neuronal cytoplasmic inclusions (NCIs) in clinically demented individuals with high probability AD pathology, all with Braak neurofibrillary tangle stages of V or VI. Sections of hippocampus, amygdala, as well as temporal, frontal and parietal neocortex were analyzed with TDP-43 immunohistochemistry, and the density of NCIs was assessed using a semiquantitative scoring method. Of the 29 cases, 6 had TDP-43 positive NCIs in the amygdala only, and 7 had TDP-43 inclusions restricted to amygdala and hippocampus. In 16 cases TDP-43 immunoreactivity was more widespread, affecting temporal, frontal or parietal neocortex. These findings indicate that medial temporal lobe limbic structures are vulnerable to TDP-43 pathology in advanced AD, and that the amygdala appears to be the most vulnerable region. The distribution of the lesions in this cross-sectional analysis may suggest a progression of TDP-43 pathology in AD, with limbic structures in the medial temporal lobe affected first followed by higher order association cortices.
Keywords: Amygdala, FTLD-U, FTLD-MND, frontotemporal dementia, motor neuron disease
INTRODUCTION
TAR DNA binding protein-43 (TDP-43) has been identified as a major ubiquitinated protein in frontotemporal lobar degeneration with ubiquitin-positive, tau- and alpha-synuclein negative inclusion bodies (FTLD-U), frontotemporal lobar degeneration with motor neuron disease (FTLD-MND), and amyotrophic lateral sclerosis (ALS) [6, 12, 22]. Subsequent studies have indicated that TDP-43 immunoreactive inclusion bodies can be detected in other neurodegenerative disorders, including Alzheimer disease (AD) [1, 13], Parkinson’s disease dementia and dementia with Lewy bodies [21], and in parkinsonism-dementia complex and ALS of Guam [9]. TDP-43 immunoreactive inclusion bodies can be classified as neuronal intranuclear inclusions (NIIs), neuronal cytoplasmic inclusions (NCIs), and dystrophic neurites (DNs) [6]. The relative abundance and distribution of ubiquitin-immunoreactive NCIs, DNs and NII have been used in several schemes to subtype FTLD-U [14, 18, 23], but their significance in non-FTLD neurodegenerative disorders remains unclear. We have previously identified a subgroup of patients with clinical dementia and advanced AD who had TDP-43 immunoreactivity in the hippocampus and characterized the clinical phenotypes associated with these patients [13]. While the presence of hippocampal TDP-43 immunoreactivity readily identifies a select group of patients [1], the region-specific distribution of TDP-43 immunoreactivity beyond the hippocampus has only been examined in one previous study [1]. In that study cases were classified a limbic predominant and diffuse according to the distribution of TDP-43 in limbic or neocortical areas, analogous to classification of α-synuclein pathology in Lewy body disorders [16]. Involvement of the amygdala was not addressed in that study [1]. Furthermore, it is not known if involvement of the hippocampus is always present in cases with TDP-43 immunoreactivity. Here we characterize the distribution of TDP-43 immunoreactivity in frontal, temporal and parietal neocortical regions, and identify cases in which the amygdala is the only region affected.
MATERIALS AND METHODS
The Mayo Clinic Autopsy Database from January 1, 2000 to December 31, 2006 was searched for patients with clinical dementia and a pathologic diagnosis of high probability AD based on NIA-Reagan criteria [11] and Braak neurofibrillary tangle stages of V or VI [4]). All cases had been previously examined by an experienced neuropathologist (JEP). Other pathologic processes were excluded with immunohistochemistry for neurofilament protein, ubiquitin, α-synuclein and phospho-tau. In each case, Braak staging was performed and AD was diagnosed.
Sections from the medial temporal lobe at the level of the lateral geniculate body (including hippocampus, entorhinal cortex, occipitotemporal gyrus and usually inferior temporal gyrus) were previously analyzed with TDP-43 immunohistochemistry (1:2000, ProteinTech Group, Chicago, IL) [13]. AD cases with TDP-43 immunoreactivity in the hippocampus were then analyzed for TDP-43 pathology in multiple brain regions, including neocortical regions (frontal, temporal, and parietal) and amygdala. When available, sections from the medulla containing hypoglossal nucleus were also analyzed. In addition to these cases, 22 cases of high probability AD, previously found to be negative for hippocampal TDP-43 immunoreactivity were also screened with TDP-43 immunohistochemistry of amygdala as well as frontal, temporal and parietal neocortices. 5 cases of Pick disease, 6 cases of corticobasal degeneration, 1 case of frontotemporal dementia with parkinsonism linked to chromosome 17, and 3 cases of Creutzfeldt-Jakob disease were included as control cases. TDP-43 immunoreactivity was considered to be positive only if NCIs were identified. The intensity of TDP-43 immunoreactivity was graded semi-quantitatively by the density of NCIs with the following scheme: 0 = no NCIs identified in the entire section; 0.5 = 1–5 NCIs in the entire section; 1 = an average of 1 NCI identified per 5 high power fields (400 ×); 2 = an average of 1 NCI in every high power field; 3 = multiple NCIs identified in each high power field. The presence of any TDP-43 immunoreactivity in DNs and NII also was noted. For all cases, the density of neurofibrillary tangles and neuritic plaques were also analyzed semi-quantitatively in the amygdala according to CERAD guidelines [19]
Statistical Analysis
Statistical analyses were performed using the JMP computer software (JMP Software, version 6.0.0; SAS Institute Inc, Cary, NC, USA) with statistical significance set at p < 0.05. Student’s t-test was used to compare the average density of cortical NCIs between temporal cortex and extra-temporal cortex (the higher score between frontal and parietal cortex was used for analysis). Linear regression was used for correlational analysis between the density of temporal NCIs and the number of extra-temporal cortical region involvement. Chi-square test and Fisher’s exact test were used to analyze the difference in additional pathology (hippocampal sclerosis and Lewy bodies).
RESULTS
Twenty three patients with advanced AD were initially identified to have TDP-43 positive NCIs in the hippocampal dentate gyrus. In this group, the median age at death was 88 years (Table 1), with 9 patients at or over the age of 90 years at death. Clinically, 22 patients (96%) carried the antemortem diagnosis of clinically probable AD, and one was given the diagnosis of amnestic mild cognitive impairment five years prior to autopsy. All cases of advanced AD with hippocampal NCIs had prominent TDP-43 positive NCIs in the amygdala (Figure 1A), but a variable degree of TDP-43 positive NCIs in the hippocampal dentate gyrus [13]. All but three cases also had prominent NCIs in the entorhinal cortex (Table 1). Semi-quantitative analysis of TDP-43-positive cases showed the amygdala to have the highest density of NCIs of all regions examined (Table 1). Among the 23 cases, 7 cases had TDP-43 positive NCIs restricted to the amygdala and hippocampus. An additional 16 cases had TDP-43 positive NCIs in neocortex, limited to the temporal cortex in 9 cases (Figure 1B) and affecting frontal or parietal or both in addition to the temporal lobe in 7 cases (Figure 1C). The temporal lobe had a higher density of NCIs than any other neocortical regions (p < 0.001). There was a positive association between the semi-quantitative density of TDP-43 positive NCIs in the temporal cortex and the number of additional neocortical regions involved (p < 0.01, R2=0.30). None of the cases in which medulla was examined (N=10) were any TDP-43 positive NCIs detected in the hypoglossal nucleus.
Table 1.
Clinical and pathological characterization of cases of Alzheimer disease with hippocampal TDP-43 NCIs according to increasing degree of TDP-43 pathology.
| TDP-43 Pattern of Distribution |
Patient | Age of Death |
Primary Clinical Diagnosis |
Secondary Clinical Diagnosis |
Braak Staging |
Amygdala | Entorhinal Cortex |
Temporal Cortex |
Frontal Cortex |
Parietal Cortex |
|---|---|---|---|---|---|---|---|---|---|---|
| Amygdala Plus Hippocampus | 1 | 79 | AD | 6 | 3+ | 3+ | 0 | 0 | 0 | |
| 2 | 97 | AD | 5 | 3+ | 3+ | 0 | 0 | 0 | ||
| 3 | 99 | AD | B12 deficiency | 5 | 3+ | 3+ | 0 | 0 | 0 | |
| 4 | 92 | AD | 6 | 3+ | 3+ | 0 | 0 | 0 | ||
| 5 | 82 | AD | Depression | 5 | 3+ | 3+ | 0 | 0 | 0 | |
| 6 | 56 | AD | 6 | 3+ | 3+ | 0 | 0 | 0 | ||
| Temporal | 7 | 89 | Possible AD | 5 | 3+ | 3+ | 0 | 0 | 0 | |
| 8 | 79 | AD | 5 | 3+ | 3+ | 0.5+ | 0 | 0 | ||
| 9 | 93 | AD | 5 | 3+ | 3+ | 1+ | 0 | 0 | ||
| 10 | 97 | AD | Depression | 5 | 3+ | 1+ | 1+ | 0 | 0 | |
| 11 | 96 | AD | 5 | 3+ | 2+ | 1+ | 0 | 0 | ||
| 12 | 100 | AD | 6 | 3+ | 3+ | 1+ | 0 | 0 | ||
| 13 | 79 | aMCI | 6 | 3+ | 3+ | 1+ | 0 | 0 | ||
| 14 | 83 | AD | 6 | 3+ | 3+ | 1+ | 0 | 0 | ||
| 15 | 98 | AD | Parkinsonism | 6 | 3+ | 3+ | 2+ | 0 | 0 | |
| 16 | 88 | AD | 6 | 3+ | 3+ | 2+ | 0 | 0 | ||
| Temporal-Plus | 17 | 88 | AD | 6 | 3+ | 2+ | 1+ | 0.5+ | 0 | |
| 18 | 85 | AD | 6 | 3+ | 3+ | 2+ | 0 | 0.5+ | ||
| 19 | 87 | AD | 6 | 3+ | 3+ | 0.5+ | 0 | 1+ | ||
| 20 | 82 | AD | Head injury | 6 | 3+ | 3+ | 1+ | 1+ | 0.5+ | |
| 21 | 89 | AD | 6 | 3+ | 3+ | 2+ | 0.5+ | 0.5+ | ||
| 22 | 90 | AD | 6 | 3+ | 3+ | 2+ | 0.5+ | 0.5+ | ||
| 23 | 82 | AD | 6 | 3+ | 3+ | 3+ | 0.5+ | 0.5+ |
(AD = probable Alzheimer disease; aMCI = amnestic mild cognitive impairment)
Figure 1.
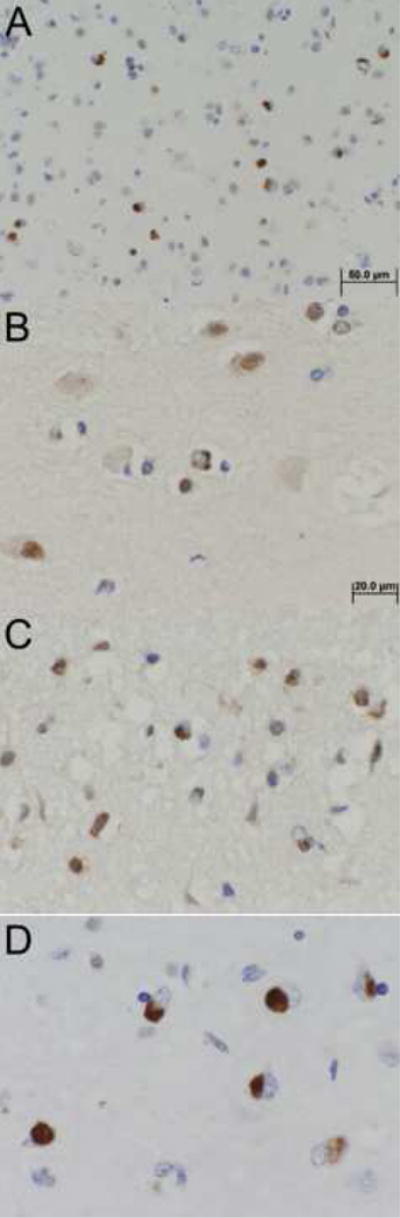
TDP-43 positive neuronal cytoplasmic inclusions (NCIs) in representative cases - A: amygdala section in amygdala plus hippocampus case (200×); B: temporal neocortex in a temporal case (400×); C: frontal neocortex in a temporal-plus case (400×); D: amygdala section in an amygdala-only case (400×).
Of the 22 additional cases without TDP-43 immunoreactivity in the hippocampus, 6 cases had TDP-43 positive NCIs limited to the amygdala (Figure 1D). The density of NCIs in the amygdala was highly variable for amygdala-only cases compared with the uniformly abundant NCIs in the amygdala of cases that also had TDP-43 immunoreactivity in the hippocampus (Table 2). None of the 15 control cases had TDP-43 positive NCIs in the amygdala or elsewhere.
Table 2.
Clinical and pathological characterization of cases of Alzheimer disease without hippocampal TDP-43 NCIs. All cases of hippocampal-negative cases also had negative TDP-43 immunoreactivity in the frontal, temporal, and parietal cortical regions.
| TDP-43 Pattern of Distribution | Patient | Age of Death | Primary Clinical Diagnosis | Secondary Clinical Diagnosis | Braak Staging | Amygdala TDP-43 | Entorhinal Cortex |
|---|---|---|---|---|---|---|---|
| Amygdala Only | 101 | 87 | AD | 5 | 1+ | 0 | |
| 102 | 79 | AD | 6 | 3+ | 0 | ||
| 103 | 82 | AD | Depression | 6 | 3+ | 0.5+ | |
| 104 | 98 | AD | 6 | 3+ | 1+ | ||
| 105 | 95 | AD | 5 | 3+ | 1+ | ||
| 106 | 79 | AD | 5 | 3+ | 3+ | ||
| None | 107 | 94 | AD | Depression | 6 | 0 | 0 |
| 108 | 91 | AD | 6 | 0 | 0 | ||
| 109 | 89 | AD | B12 deficiency | 6 | 0 | 0 | |
| 110 | 86 | AD | 6 | 0 | 0 | ||
| 111 | 85 | AD | 6 | 0 | 0 | ||
| 112 | 83 | AD | 6 | 0 | 0 | ||
| 113 | 80 | AD | Depression | 6 | 0 | 0 | |
| 114 | 79 | AD | Depression | 6 | 0 | 0 | |
| 115 | 66 | AD | DLB | 6 | 0 | 0 | |
| 116 | 91 | AD | 5 | 0 | 0 | ||
| 117 | 89 | AD | 5 | 0 | 0 | ||
| 118 | 88 | Multi-infarct dementia | 5 | 0 | 0 | ||
| 119 | 87 | AD | 5 | 0 | 0 | ||
| 120 | 86 | AD | Single stroke | 5 | 0 | 0 | |
| 121 | 81 | AD | 5 | 0 | 0 | ||
| 122 | 79 | Multi-infarct dementia | 5 | 0 | 0 |
(AD = probable Alzheimer disease; aMCI = amnestic mild cognitive impairment; DLB = dementia with Lewy bodies)
The 6 cases of amygdala-only TDP-43 were added to the 23 cases in which TDP-43 immunoreactivity was detected in the hippocampus. The 29 cases fell into four main groups based on the distribution of TDP-43 immunoreactivity: amygdala-only (n = 6), amygdala plus hippocampus (n = 7), temporal (including amygdala, hippocampus and temporal neocortex, n = 9), and temporal-plus (additional extra-temporal involvement in the frontal or parietal neocortex, n = 7). Semi-quantitative counts of neuritic plaques and neurofibrillary tangles in the amygdala of these cases were always frequent, regardless of the presence or absence of TDP-43 positive NCIs. No correlation was found between the semi-quantitative count of AD pathology and TDP-43 positive NCIs in the amygdala, although a ceiling effect of AD pathology could not be excluded.
DISCUSSION
In this study of patients with clinical dementia and advanced pathologic changes of AD, we demonstrate the distribution of TDP-43 immunoreactive NCIs and show that the amygdala is the most frequently affected region. In addition, the amygdala had the most abundant TDP-43 pathology for cases in which TDP-43 immunoreactivity was detected in other regions. In a small number of cases (n=6), TDP-43 immunoreactivity was detected only in the amygdala. In the 23 cases with TDP-43 immunoreactivity in the hippocampus as well as the amygdala, involvement of cortical areas was most often detected in temporal lobe, followed by frontal and parietal lobes. Moreover, there was a correlation between the semi-quantitative score of temporal neocortical TDP-43 pathology and the extent of extra-temporal neocortical involvement. Therefore, analysis of the temporal lobar structures, especially the amygdala, remains critical in the identification of TDP-43-associated pathology in AD. Survey of hippocampal or neocortical regions alone would most likely underestimate the number of cases with TDP-43 pathology.
The prominent involvement of medial temporal lobe structures with less often spread to other cortical regions has been noted previously in AD and classified as “limbic” or “diffuse”[1], with the diffuse type being more frequently associated with pathology that is unique to FTLD-U such as NIIs. Such a step-wise spread may reflect differences in intrinsic susceptibility to TDP-43 associated pathology between neuronal populations, staged degeneration of neurons in associated networks, non-specific vulnerability to inclusion body formation in neurons affected by a primary neurodegenerative process such as Alzheimer disease, or a combination of the processes. As only a small percentage of TDP-43 positive inclusions were positive for phospho-tau[1], this pattern likely does not represent non-specific immunoreactivity in AD-associated intranuclear pathology. It is not known if this pattern of TDP-43 co-pathology holds true in neurodegenerative disorders with only TDP-43 pathology such as FTLD-U and ALS. An analogous situation exists for α-synuclein in Parkinson’s disease compared to α-synuclein pathology occurring in the setting of advanced AD. In the former there is a predictable progression of pathology from brainstem to limbic lobe to cortex [5], while in AD some cases have α-synuclein pathology relatively restricted to the medial temporal lobe structures [25]. The cellular co-localization of tau and TDP-43 originally noted in AD [1] has now been shown to extend to AD cases with Lewy bodies, and some neurons show co-deposition of all three proteins [10]. It remains to be determined whether a similar pattern of TDP-43 pathologic distribution we reported here exists in Lewy body disease.
While a staging system of TDP-43 pathology has not been proposed for the primary TDP-43-proteinopathies (FTLD-U and ALS), several lines of evidence point to possible early involvement of amygdala in these disorders. Clinically, severe atrophy of the medial temporal lobe structures, including amygdala and hippocampus, is common in clinically diagnosed behavioral variant frontotemporal dementia and in semantic dementia [7, 8]. Amygdala atrophy is also common in pathologically confirmed FTLD-U [3, 17]. At autopsy some cases of ALS also have ubiquitin-positive pathology and neuronal loss in the amygdala [2]. The extent of ubiquitin-immunoreactive inclusions and neuronal loss also correlate with the severity of clinical cognitive impairment in ALS [15, 20, 26] Thus, TDP-43 pathology in the amygdala may represent the one of the earliest stages of TDP-43-associated neurodegeneration.
It remains unknown if the presence of TDP-43 immunoreactivity in advanced AD has any clinical significance. Preliminary studies on this same cohort suggests that it is associated with poorer cognitive performance [13], although it is difficult to determine the cognitive burden of region-specific TDP-43 pathology such as amygdala-only TDP-43 NCIs. It is unlikely that TDP-43 pathology in AD is merely related to severity of AD pathology, as the AD cases in this study that lacked TDP-43 immunoreactivity had very similar AD pathology based upon CERAD plaque and tangle scores and Braak neurofibrillary tangle staging. It may be difficult to unequivocally assign a clinical significance to a particular type of pathology in cases with mixed pathology, and this is true not only for TDP-43 immunoreactivity, but also for ischemic-vascular lesions, Lewy bodies and hippocampal sclerosis [24]. What can be said is that cases with mixed pathology usually have more severe clinical phenotype [13, 24]. An approach that might be successful at parsing the contribution of TDP-43 pathology to the clinical syndrome is to examine clinical presentations of patients with and without TDP-43 immunoreactivity who have intermediate- or low-likelihood AD type pathology.
In summary, we present a study on regional distribution of TDP-43-associated pathology in advanced AD. We find that the amygdala is the most frequently affected regions and usually the most severely affected region, suggesting that it may be the earliest affected region in such cases. We hypothesize that TDP-43 associated cortical pathology may spread from limbic structures in the medial temporal lobe (amygdala and hippocampus) to multimodal association cortices in temporal lobe and then to frontal and parietal lobes. How subcortical structures (e.g., basal ganglia, thalamus and brainstem) play into this scheme will require additional studies.
Acknowledgments
This study was supported by NIH grants P50 AG16574, U01 AG06786, and K12 HD49078 (to KAJ) and the generous support of the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer’s Disease Research Program of the Mayo Foundation, U.S.A.
References
- 1.Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol. 2007;61:435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson VE, Cairns NJ, Leigh PN. Involvement of the amygdala, dentate and hippocampus in motor neuron disease. J Neurol Sci. 1995;129 Suppl:75–78. doi: 10.1016/0022-510x(95)00069-e. [DOI] [PubMed] [Google Scholar]
- 3.Barnes J, Whitwell JL, Frost C, Josephs KA, Rossor M, Fox NC. Measurements of the amygdala and hippocampus in pathologically confirmed Alzheimer disease and frontotemporal lobar degeneration. Arch Neurol. 2006;63:1434–1439. doi: 10.1001/archneur.63.10.1434. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 5.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 6.Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL, 3rd, Schneider JA, Kretzschmar HA, Carter D, Taylor-Reinwald L, Paulsmeyer K, Strider J, Gitcho M, Goate AM, Morris JC, Mishra M, Kwong LK, Stieber A, Xu Y, Forman MS, Trojanowski JQ, Lee VM, Mackenzie IR. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007;171:227–240. doi: 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan D, Fox NC, Scahill RI, Crum WR, Whitwell JL, Leschziner G, Rossor AM, Stevens JM, Cipolotti L, Rossor MN. Patterns of temporal lobe atrophy in semantic dementia and Alzheimer’s disease. Ann Neurol. 2001;49:433–442. [PubMed] [Google Scholar]
- 8.Galton CJ, Patterson K, Graham K, Lambon-Ralph MA, Williams G, Antoun N, Sahakian BJ, Hodges JR. Differing patterns of temporal atrophy in Alzheimer’s disease and semantic dementia. Neurology. 2001;57:216–225. doi: 10.1212/wnl.57.2.216. [DOI] [PubMed] [Google Scholar]
- 9.Geser F, Winton MJ, Kwong LK, Xu Y, Xie SX, Igaz LM, Garruto RM, Perl DP, Galasko D, Lee VM, Trojanowski JQ. Pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol. 2008;115:133–145. doi: 10.1007/s00401-007-0257-y. [DOI] [PubMed] [Google Scholar]
- 10.Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K, Togo T, Katsuse O, Uchikado H, Furukawa Y, Kosaka K, Arai H. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 2007;1184:284–294. doi: 10.1016/j.brainres.2007.09.048. [DOI] [PubMed] [Google Scholar]
- 11.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:1095–1097. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR, Parisi JE, Dickson DW. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology. 2006;66:41–48. doi: 10.1212/01.wnl.0000191307.69661.c3. [DOI] [PubMed] [Google Scholar]
- 13.Josephs KA, Whitwell JL, Knopman DS, Hu WT, Stroh DA, Baker M, Rademakers R, Boeve BF, Parisi JE, Smith GE, Ivnik RJ, Petersen RC, Jack CR, Jr, Dickson DW. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology. 2008;70:1850–1857. doi: 10.1212/01.wnl.0000304041.09418.b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katsuse O, Dickson DW. Ubiquitin immunohistochemistry of frontotemporal lobar degeneration differentiates cases with and without motor neuron disease. Alzheimer Dis Assoc Disord. 2005;19 Suppl 1:S37–43. doi: 10.1097/01.wad.0000183889.61421.a8. [DOI] [PubMed] [Google Scholar]
- 15.Kawashima T, Dohura K, Kikuchi H, Iwaki T. Cognitive dysfunction in patients with amyotrophic lateral sclerosis is associated with spherical or crescent-shaped ubiquitinated intraneuronal inclusions in the parahippocampal gyrus and amygdala, but not in the neostriatum. Acta Neuropathol (Berl) 2001;102:467–472. doi: 10.1007/s004010100398. [DOI] [PubMed] [Google Scholar]
- 16.Kosaka K, Yoshimura M, Ikeda K, Budka H. Diffuse type of Lewy body disease: progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree--a new disease? Clin Neuropathol. 1984;3:185–192. [PubMed] [Google Scholar]
- 17.Kril JJ, Halliday GM. Clinicopathological staging of frontotemporal dementia severity: correlation with regional atrophy. Dement Geriatr Cogn Disord. 2004;17:311–315. doi: 10.1159/000077161. [DOI] [PubMed] [Google Scholar]
- 18.Mackenzie IR, Baborie A, Pickering-Brown S, Du Plessis D, Jaros E, Perry RH, Neary D, Snowden JS, Mann DM. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol. 2006;112:539–549. doi: 10.1007/s00401-006-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 20.Mitsuyama Y. Presenile dementia with motor neuron disease in Japan: clinico-pathological review of 26 cases. J Neurol Neurosurg Psychiatry. 1984;47:953–959. doi: 10.1136/jnnp.47.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE, Arnold SE, Siderowf A, Grossman M, Leverenz JB, Woltjer R, Lopez OL, Hamilton R, Tsuang DW, Galasko D, Masliah E, Kaye J, Clark CM, Montine TJ, Lee VM, Trojanowski JQ. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007;114:221–229. doi: 10.1007/s00401-007-0261-2. [DOI] [PubMed] [Google Scholar]
- 22.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 23.Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, Bruce J, Grossman M, Trojanowski JQ, Lee VM. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169:1343–1352. doi: 10.2353/ajpath.2006.060438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- 25.Uchikado H, Lin WL, DeLucia MW, Dickson DW. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol. 2006;65:685–697. doi: 10.1097/01.jnen.0000225908.90052.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wightman G, Anderson VE, Martin J, Swash M, Anderton BH, Neary D, Mann D, Luthert P, Leigh PN. Hippocampal and neocortical ubiquitin-immunoreactive inclusions in amyotrophic lateral sclerosis with dementia. Neurosci Lett. 1992;139:269–274. doi: 10.1016/0304-3940(92)90569-s. [DOI] [PubMed] [Google Scholar]