

Summary
Each microenvironment is controlled by a specific set of regulatory elements that have to be finely and constantly tuned to maintain local homeostasis. These environments could be site specific, such as the gut environment, or induced by chronic exposure to microbes. Various populations of dendritic cells are central to the orchestration of this control. In this review, we discuss some new findings associating dendritic cells from defined compartments with the induction and control of regulatory T cells in the context of exposure to both commensal and pathogenic microbes.
Keywords: dendritic cell, regulatory T cell, Foxp3+, microbe, commensal
Introduction
Humans have coevolved with microbial partners, and for the most part, this coexistence can be considered beneficial or neutral. This is clearly the case for the flora composed of commensals and symbiotes that invade our lung, skin, or gut soon after birth. However, in some instances, interactions with microorganisms can be detrimental and lead to pathological consequences. Despite these disparate outcomes, the distinction between a commensal and a pathogenic microorganism is not always obvious, and many of the strategies we have developed to coexist peacefully with our positive partners can be hijacked or manipulated by potentially harmful microorganisms to ensure their own survival. Our work focuses on understanding the bases of these common strategies and how deregulation of host-microbe dialogue leads to pathogenic consequences.
Although a large number of mechanisms, including natural Foxp3+ regulatory T cells (Tregs) contribute to the maintenance of peripheral tolerance, in defined situations or specific sites the body requires generation of Tregs to preserve its integrity. Each microenvironment is controlled by a specific set of regulatory elements that have to be finely and constantly tuned to maintain local homeostasis. These environments could be specific sites, such as the gut or skin, or be induced by chronic exposure to microbes. In this review, we discuss how dendritic cells (DCs) of various origin, status, activation, or location can differentially control the induction and function of Tregs in the context of microbial exposure.
Induction of Foxp3+ Tregs by DCs
Aside from evidence that natural forkhead box protein 3 (Foxp3)+ Tregs arise and mature in the thymus (1-3), there is clear evidence that Foxp3+ Tregs can develop extrathymically under certain conditions (4-7). Despite a growing body of literature documenting a potential role for these converted cells in the control of autoimmune or inflammatory diseases (8), the nature of the antigen-presenting cells (APCs) involved in this conversion process has only recently been examined. Several reports support the idea that immature DCs may be more efficient at inducing Foxp3+ Tregs in the presence of transforming growth factor-β (TGF-β) than activated DCs. For instance, targeting of antigens to immature DCs via the regulatory receptor DEC205 can favor the induction of Foxp3+ T cells de novo (7). While virtually all APCs under steady state conditions may have the capacity to induce antigen specific Tregs, DCs appear to be more efficient at this process than other APCs (9). Spleen DCs are more potent than DC-depleted APCs for the induction of Tregs and required lower doses of peptide antigen (9). In the absence of exogenous interleukin-2 (IL-2), endogenous IL-2 production by T cells favoring Treg conversion can be efficiently triggered by DCs expressing CD80/CD86 but not by other APCs (9). However, another study proposed that B cells are actually more efficient at inducing Foxp3+ Tregs than splenic DCs in the presence of TGF-β (10). This discrepancy is likely to be associated with the level of activation of the APCs in these different settings.
The notion that some DC subsets from lymphoid tissues could be more efficient at inducing Tregs than others came from a recent study showing that CD8+ DCs induce higher conversion than other spleen DC subsets in the presence of TGF-β (11). Several molecules contribute to the neo-induction of Foxp3+ Tregs. For instance, the B7/cytotoxic T-lymphocyte antigen-4 (CTLA-4) axis is important to favor the induction of these cells (12-14). A role for programmed death ligand 1 (PD-L1) expressed by DCs in supporting Treg induction has been recently reported (11), adding another potential layer to the role of this molecule in the control of peripheral tolerance (15).
Role of tissue-specific DCs in the induction of Tregs
The gut is a major mucosal barrier, constantly exposed to both pathogenic and commensal microorganisms along with ingested dietary antigens. In fact, no other tissue is subjected to as great a level of antigenic pressure as the gut. The adult human intestine contains up to 100 trillion microorganisms (16). At birth, the massive exposure to these neoantigens as well as the shift in gut flora composition following infection (17) imposes a unique challenge to this environment. Immune reactivity against non-pathogenic gut elements is not only wasteful but is also known to lead to severe tissue damage (e.g. inflammatory bowel disease). However, the development of active immunity is required to protect the host against invasive pathogens. Different subsets of regulatory T cells have been shown to be instrumental in the maintenance of this complex homeostasis. Additionally, several subsets of DCs with regulatory properties have been described with the capacity to induce IL-10 secretion from T cells or induce oral tolerance under steady state conditions (18-20, reviewed in 21). DCs from the small intestinal lamina propria (LP) display several immunoregulatory features, for example constitutive expression of IL-10 (20). Some of these features may have been influenced by conditioning signals received from non-inflammatory cytokines constitutively produced by the intestinal epithelia. These cytokines include TGF-β and the T-helper 2 (Th2)-response driving thymic stromal lymphopoietin (TSLP) (22, 23, reviewed in 21).
We and others have recently shown that the gut-associated lymphoid tissue is a preferential site for the peripheral induction of Foxp3+ Treg cells (24-27) (Fig. 1A). A role for local DCs in this conversion process is supported by the observation that DCs from the LP of the small intestine and from mesenteric lymph node (MLN) are noticeably better than splenic DCs at inducing the expression of Foxp3 in naive T cells in the presence of exogenous TGF-β (24, 25). Similarly, LP macrophages can efficiently induce Foxp3+ T cells (28). In particular, DCs expressing CD103 in these two compartments can induce Foxp3+ T cells in the absence of any exogenous factors (24, 25). This conversion process was associated with their capacity to release bioactive TGF-β which could be linked with their ability to activate latent TGF-β (25). This model is supported by the observation that DCs lacking the TGF-β-activating integrin αvβ8 or αv fail to induce Foxp3+ Tregs in vitro (29, 30). Furthermore, mice in which myeloid cells do not express αv or DCs do not express αvβ8 have reduced colonic Foxp3+Treg and develop colitis (29, 30). Loss of v-integrin expression by myeloid cells led to the development of intestinal inflammation, probably through the combined effects of a failure to remove apoptotic cells and a loss of TGF-β activation (30).
Fig.1. Control of regulatory T cells in the GI tract.

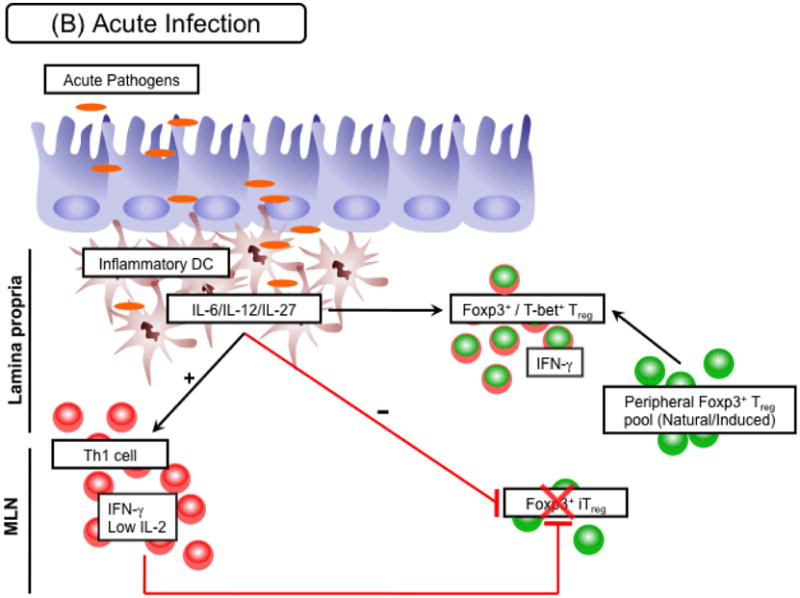
(A) Steady state/chronic infection. At steady state, several subsets of small intestine DCs are exposed to luminal antigens derived from gut flora or food. Of particular importance, CD103+ DCs metabolize vitamin A to retinoic acid (RA), which is able to favor conversion of Foxp3+ Tregs by directly acting on naive T cells and limiting effector cytokine release by activated T cells. Expansion of neoconverted Foxp3+ Tregs, along with other induced Foxp3+ Treg (iTregs) and natural Foxp3+ Tregs that make up the peripheral Treg pool, is further supported by the availability of IL-2 or other common γ chain cytokines (e.g. IL-7, IL-15). Subsequently, this compartment acts as one layer of regulation within the gut environment to limit local immune activation stimulated by luminal antigens. A similar scenario may occur in the context of chronic infection in which conversion needs to be maintained in the face of an ongoing immune response. As a consequence chronic pathogens may have co-evolved to release factors that enhance the conversion process, such as TGF-β. (B) Acute infection. In the presence of an acute infection, such as infection with Toxoplasma gondii, peripheral Foxp3+ Treg conversion is inhibited. This inhibition is in part mediated by the influx of inflammatory DC subsets into tissue sites. By releasing pro-inflammatory cytokines, including IL-12 or IL-27, these DCs support the expansion of IFN-γ secreting Th1 cells. IFN-γ directly inhibits the Foxp3+ Treg conversion process. In addition limitation of IL-2 in Th1 polarized environments hinders the development of this compartment. Moreover, IL-12 from inflammatory DC subsets can drive expression of T-bet within previously Foxp3+ Treg allowing them to adapt to the inflammatory within the tissue. All these mechanisms act together to favor a controlled effector response towards the invading pathogen. However, in highly inflammatory circumstances expression of T-bet may eventually drive IFN-γ production in regulatory T cells. In this situation the breakdown of the conversion process along with expression of effector cytokines by Foxp3+ Tregs may potentially result in severe pathogenesis.
Recent evidence demonstrated that this specialized function of CD103+ DCs can be also traced to their distinct origin. Indeed, DCs expressing CD103 arise from macrophage–DC precursors through a Flt-3 ligand growth factor mediated pathway, while CD11b+CD14+CXCR1+ LP DCs derived from grafted Ly6Chi monocytes under the control of granulocyte-macrophage colony-stimulating factor (GM-CSF) (31, 32). Using infectious agents and colitis models, several groups reported that in the gut, inflammation, and IL-17 induction can be usually associated with DCs that do not express CD103 (31-33).
Gut DCs also have an important role in dictating the homing potential of lymphocytes. DCs isolated from the Peyer's patches, small-intestinal LP, and MLN promote the expression of the gut-homing receptors α4β7-integrin and CCR9 by CD4+ and CD8+ T cells (34-37). The molecule CCR9 binds to CCL25 produced by epithelial cells of the small intestine and α4β7-integrin binds to mucosal vascular addressin cell-adhesion molecule 1 (MADCAM1), which is expressed by the vascular endothelium of the gastrointestinal tract. It is becoming clear that nutrient status can impact an individual's susceptibility to intestinal pathologies (38). In the case of vitamin A, and in particular, its transcriptionally active metabolite, retinoic acid (RA), prolonged insufficiency not only disrupts the integrity of the intestinal epithelial barrier but also prevents the proper deployment of effector lymphocytes into the gut-associated lymphoid tissue (GALT) following priming. Indeed, the capacity of GALT DCs to imprint gut homing receptors on lymphocytes is associated with their capacity to release RA (38-40). More recently, it was demonstrated that RA and cytokines produced by DCs in the Peyer's Patches synergized to promote IgA secretion by gut activated B cells (40). Addition of RA to naturally occurring Treg in vitro can also promote their expression of gut tropic receptors and favor their migration to the GALT (41).
We and others found that another effect of RA on the immune regulation of the GI tract is associated with its capacity to enhance the TGF-β-mediated generation of Foxp3+ Tregs (10, 24-26, 28, 42-44) (Fig. 1A). The induction of Foxp3 observed in the presence of small-intestinal LP DCs and CD103+ MLN DCs can be inhibited by a retinoic-acid receptor (RAR) antagonist (24, 25). Conversely, incubation of splenic or CD103− MLN DCs with both TGF-β and RA enhanced their capacity to induce Foxp3 Tregs (24-26). RA produced (or acquired) by intestinal macrophages can also synergize with TGF-β to induce Foxp3+ Tregs (28). Importantly, RA can also induce the conversion of naive CD4+ T cells purified from human cord blood into Foxp3+ Treg cells (42). Reciprocally, RA can inhibit the generation of Th-17 cells (26, 42-44), suggesting that RA may play an important role in maintaining the balance between effector and regulatory populations in the gastrointestinal tract.
The actions of RA are mediated by nuclear receptors which are ligand-inducible transcriptional regulators and belong to two distinct families: RAR and RXR. Each family consists of the three genetic isotypes (RAR- or RXRα, β and γ). We found that normal conversion occurred with RARβ and RARγ deficient T cells but was abolished with RARα-deficient T cells (45). Conversely, these cells produced elevated levels of both IL-17 and interferon-γ (IFN-γ). Thus, in contrast to the functional redundancy observed for other roles of RA, only one receptor, RARα, is involved in the induction of Foxp3+ Tregs.
At least two mechanisms have been identified by which RA can enhance the capacity of TGF-β to induce Tregs. Recent evidence demonstrated that one of these mechanisms was associated with the capacity of RA to shut down effector cytokines that interfere with Foxp3 expression (45-47). Additionally, RA can also act directly on naive T cells to favor TGF-β-mediated Foxp3+ Treg induction (48).
Although the capacity of GALT DCs or macrophages to imprint gut-homing receptors and induce Foxp3+ Tregs is associated with their capacity to release RA, the relevant producers of this metabolite have not been formally identified. Synthesis of RA from stored or dietary retinol depends on the expression of the appropriate enzymes, which can be expressed directly by GALT DC. DC form Peyer's patch and MLN express Aldh1a1 and Aldh1a2, respectively (25, 39). We found that CD103+ DCs are the only subset of resident LP APC able to exert a RLDH activity (Hall, unpublished observation). However, other cells including epithelial cells or MLN stromal cells can express enzymes associated with vitamin A metabolism (49, 50), suggesting that other APC subsets may acquire RA from alternative sources and store it. Indeed, monocyte-derived DCs pretreated with RA can take on several attributes characteristic of mucosal DCs, such as their capacity to induce mucosal homing receptor expression and favor immunoglobulin A (IgA) responses. These gut-derived features acquired by DCs were associated with the capacity of DCs to become carriers and not producers of RA (49).
The precise factors that govern the activation of some of these enzymes as well as how inflammation or infections modify the metabolism of vitamin A remain to be characterized. A recent report demonstrated that stimulation of DCs via Toll-like receptor 2 (TLR2) induced vitamin A metabolizing enzymes suggesting that these enzymes are tightly controlled by interaction with microbes (51). Furthermore, IL-4 and GM-CSF can synergistically trigger DCs to acquire RA-producing capacity (52). An important future area of study will be to understand the timing necessary for DCs migrating in the GALT to acquire RA from epithelial cells and how these processes are modified during inflammatory responses.
Defined microenvironments may thus have evolved self-containing strategies in which local mediators can imprint homing properties while at the same time favoring the induction or function of Treg. Site-specific cells (e.g. neurons) or factors (e.g. hormones) can also favor the induction of Foxp3+ Tregs (53, 54). It is therefore tempting to speculate that a link between homing and regulatory function induction may represent a more general mechanism. Such strategy could allow the constant generation and migration of Treg to defined compartments. These Tregs are expected to have the prerequisite antigen specificities (e.g. flora antigens), status of activation, and survival requirement, allowing them to regulate a defined microenvironment.
Microbially induced Tregs
To coexist with a host, persistent microbes must sustain their life cycle while at the same time delaying or preventing host destruction. Microbe-mediated modulation of innate and acquired immune responses must meet these requirements in order to maintain a homeostatic environment. This is clearly the case for microflora that invade the gut or skin as well as for ‘pathogenic’ microbes that establish chronic infections. All persistent microbes obey the same principle: the immune system constitutes part of their ecological niche, and they have co-evolved with their host to learn how to manipulate APC function in order to dictate an immune response appropriate to ensure their survival. For instance, microbes have been shown to induce a large array of cells with regulatory properties to favor their own survival (55). To achieve an appropriate balance between effector and regulatory responses over the course of an infection, a number of layers of regulation will be required. One of these is likely provided by infection-driven conversion of Foxp3+ Tregs. This idea is supported by the observation that release of anti-inflammatory cytokines, in particular TGF-β, is frequently a downstream consequence of initiating an immune response towards an invading pathogen. Moreover, many microbes target niches within the host that are highly enriched for TGF-β production, such as the gastrointestinal tract, the skin, and the eye.
In some infection settings, TGF-β can even be released directly by infected cell populations. For example, the trypomastigote stage of Trypanosoma cruzi induced TGF-β and IL-10 secretion by DCs (56). Compelling data in a mouse model of malaria suggest that TGF-β and Tregs are central regulators of immunopathology and parasite expansion (57). During late infection with Plasmodium yoelii infection, DCs migrate to the spleen of infected mice and secrete TGF-β together with IL-10 and prostaglandin E2 (PGE2) (58). Following experimental malaria infection of human volunteers, enhanced TGF–β and Foxp3+ Treg responses in peripheral blood mononuclear cells correlate with a faster parasitic growth rate (57). Cells with natural Treg cell characteristics are rapidly induced following blood stage infection and are associated with a decrease of pro-inflammatory cytokines and antigen-specific responses. Monocytes are a likely source of the early TGF-β production in this infection (57).
Foxp3+ Treg conversion may be of particular importance in the emergence of an immunologically hyporesponsive state to chronic infections allowing for the re-establishment of homeostasis (Figure A). One situation in which this outcome is observed is helminth infection, where an immune competent host can be parasitized for decades often with poor development of protective immunity.
It is apparent from mouse models that augmentation of the Foxp3+ Treg compartment is a feature common to a number of these parasites (59-63). Using anti-CD25 depletion, it has been possible to show that in these settings Foxp3+ Tregs can both favor parasite persistence (63) and limit infection-induced immunopathology (62). Mechanisms that enhance Foxp3+ Treg conversion could therefore be favorable to helminth survival and transmission.
A key mechanism by which helminths are thought to regulate the host immune response is through the release of a complex mixture of excreted/secreted products (ES) (reviewed in 64). Both specific molecules identified from ES and total ES have the potential to target DC and T cells to support Foxp3+ Treg conversion. For example, treatment of bone marrow-derived DCs with ES from the gut-dwelling helminths Heligmosomoides polygyrus and Nippostrongylus brasiliensis limits their activation profile upon subsequent exposure to a variety of microbial products (65, 66). In this modified state of responsiveness the DC maintains an immature phenotype characterized by a lack of IL-12 production and a failure to upregulate co-stimulatory molecules. Immature DCs, as previously discussed, are more competent to drive Foxp3+ Treg conversion, and, although not yet tested, it can be hypothesized that manipulating DCs to remain in this state may be a powerful mechanism to bias towards Foxp3+ Treg conversion in the face of an ongoing immune response.
In addition to affecting DC function, an alternative tactic that helminths may use to favor conversion is release of factors capable of directly enhancing this process, such as TGF-β. TGF-β homologues have been identified from a number of helminth genomes (67-69), are excreted/secreted, and can trigger activation of mammalian TGF-β receptors (69). Furthermore, total ES from H. polygyrus has recently been shown to induce Foxp3 expression in naive T cells, in vitro, via a mechanism that is dependent upon in tact TGF-β signaling (Grainger et al., manuscript submitted). This provides the first direct evidence of a parasite manipulating the conversion process. However, although TGF-β homologues have been cloned from the H. polygyrus genome (67), it has not been possible to demonstrate that these molecules are responsible for the ability of H. polygyrus ES to drive de novo Foxp3 expression. As well as increasing levels of TGF-β, availability of vitamin A may also be altered by helminth ES. Retinoid-binding proteins are known to be present in several helminth ES (70, 71) and could, therefore, act to enhance its uptake and processing by host tissues.
Much work still needs to be done to establish the relative contribution of converted Treg cells to peripheral tolerance and the outcome of infection, as well as how different pathogens can utilize or interfere with this pathway to favor their own survival. Currently, in the absence of definitive markers to distinguish endogenous versus converted Foxp3+ Tregs, these questions will remain difficult to answer.
Control of gut homeostasis via crosstalk with commensals
Paradoxically, commensal and pathogenic microbes interact with the host immune system through similar conserved ligands that are cardinal features of microorganisms (72). Many of these ligands signal through the TLRs (72). TLRs are widely expressed by cells of hematopoietic origin as well as non-hematopoietic cells, including the epithelial cells lining the intestinal tract (73). The initial identification of TLRs in the splenic and peripheral blood leukocyte compartments paved our understanding of how engagement of these molecules by invasive pathogens can activate inflammatory cascades that initiate adaptive immunity (74). However, mucosal tissues via their interaction with commensals are the only environments in constant contact with TLR ligands. Yet, the purpose of TLR signaling by the commensal flora has only recently begun to be elucidated. For example, it is now clear that TLR signaling in the intestinal epithelial compartment is crucially involved in the maintenance of intestinal homeostasis and tissue repair (75). Further, these signals also positively regulate the sampling of luminal contents by DCs from the underlying LP compartment (76). Commensal floral interactions with TLRs have also been shown to mediate tolerance to food antigens (77). Previous work has demonstrated that TLR signaling can influence both the function and expansion of Tregs (78). In some instances, this control is exerted via direct TLR engagement on Treg, whereas in others it occurs through T effector (Teff) cells and/or APC stimulation. Although some interactions have been proposed to increase Treg activity, including TLR4 and TLR5 (79, 80), others involving TLR2, TLR8, and TLR9 have been shown to limit Treg cell function (81-84). TLR stimulation of Tregs may also influence their proliferation (83, 84). Because of the constant exposure of the intestinal immune system to the flora, TLR signaling is likely to play a major role in maintaining the presence and function of Tregs in the GALT. TLR9 recognizes unmethylated cytosine phosphate guanosine (CpG) dinucleotides, which are abundant in prokaryotic DNA found in intestinal flora. Using synthesized sequences containing CpG, previous studies have shown that engagement of TLR9 expressed on DCs, Tregs, and conventional T cells can limit Treg suppressive function (81, 85). We have also demonstrated that the presence of CpG at the time of Leishmania infection can limit accumulation of Treg in the infected dermis (86). Previous work identified an association between Crohn's disease and a promoter polymorphism in the TLR9 gene in humans (87). Such association supports the idea of a role for gut floral DNA (gfDNA) sensing in the pathophysiology of inflammatory bowel diseases (IBD). We have investigated how constitutive interaction between gfDNA and TLR9 in the gut can act as immunological adjuvants and critically controls the balance between Treg and Teff and found that gut flora derived DNA (gfDNA) plays a major role in intestinal homeostasis through toll like receptor 9 (TLR9) engagement (88). Naïve TLR9 deficient (TLR9−/−) mice displayed a striking increase in the frequency of Foxp3+ Treg within intestinal effector sites, accompanied by a significant reduction in constitutive IL-17 and IFN-γ production by Teff. Complementing this, gfDNA but not other TLR agonists strongly constrained the capacity of LP DCs to induce Treg conversion in vitro. Further, Treg/Teff disequilibrium in TLR9−/− mice led to impaired immune responses to oral infection with microsporidia and to oral vaccination, which could be rescued through neutralization of Tregs. Impaired intestinal immune responses could be recapitulated in wildtype mice treated with broad-spectrum antibiotics and was reversible following reconstitution with purified gfDNA. Together these data point to gfDNA as a natural adjuvant for priming intestinal responses via modulation of Treg/Teff equilibrium. This reveals a previously unidentified aspect of gut flora in the regulation of mucosal immunity. Such control appears to be essential for development of effective immune responses against invading microorganisms as well as mucosally administered vaccines and correlates with restricted peripheral Treg conversion.
The gut flora, by virtue of a vast repertoire of bacterial species, can engage TLR9 through differential expression of CpG motifs within its bacterial genomes (89). Yet it is still unclear by what means gfDNA becomes accessible for interaction with TLR9. Epithelia in the GI tract at steady-state express TLR9 on their surface (both apical and basolateral) (90, 91). However, TLR9 signaling on the apical side can mediate anti-inflammatory effects, linking lumenal signals from gfDNA to intestinal homeostasis (90). Our observation that Treg frequencies were unaffected in the large intestine (88) suggests a compartmentalization of the effect of TLR9 signaling on the control of gut homeostasis. Indeed, the colon contains the highest microbial concentration and requires further regulation than the small intestine. In this site the adjuvant effect of bacteria DNA may be lost to prevent detrimental inflammation and could in some instances become regulatory. In support of this, oral administration of CpG was shown to ameliorate the severity of dextran sodium sulfate (DSS)-induced colitis via TLR9 (92). Along these lines, the absence of TLR9 exacerbated the severity of DSS‐induced colitis (90). Thus in the context of a highly inflammatory setting, TLR9 signaling on epithelial cells can mediate a protective function. Thus, gfDNA makes essential contributions to intestinal homeostasis through two seemingly disparate pathways with the establishment of this dichotomy potentially resting on the cellular target (e.g. epithelial cells versus APCs), the intensity of the tissue disruption or even the gut compartment involved (small versus large intestine).
In addition to promoting development of the immune system and control of metabolic functions, intestinal microflora play a major protective role by displacing pathogens and enhancing barrier fortification (93, 94). In particular, gut flora interactions with specific TLRs can protect against gut injury (75, 92, 95-97) or mediate oral tolerance against dietary antigens (77).
Alteration to the structural integrity of TLR signaling components is often associated with profound clinical outcome and susceptibility to various infections or autoimmune disorders (98). The structure and composition of the gut flora reflect natural selection at both the microbial and host levels. One possibility could be that the absence of TLR9 would alter the gut flora composition. Modification of gut flora was shown in areas of inflamed gut in IBD patients (99-105). Furthermore, the presence of certain bacteria can aggravate small intestinal immunopathology following oral infection (17). It is tempting to speculate that alteration of Treg homeostasis mediated by TLR9 signaling, either because of genetic polymorphism or changes in gut flora composition, could also have consequences on development of gut inflammatory disorders. Thus, control of Treg ratio and Teff function in the gastrointestinal tract is likely to be differentially regulated by specific gut flora species. Lending to this hypothesis, we found that in contrast to DNA from E. coli, DNA from the probiotic Lactobacillus paracasei cannot restore effector responses in the gastrointestinal tract (Bouladoux, unpublished observation). Previous work showed that in vitro, gut flora bacteria are not all equal in their capapacity to stimulate TLR9 and do so with various levels of efficiency that correlate with their frequency of [CG] dinucleotides (89). An alternative explanation could be that the DNA of certain commensal bacteria may be more enriched in suppressive motifs. These suppressive ODN, are rich in poly G or GC sequences, tend to be methylated and are present in the DNA of mammals and certain virus (106-108). These motifs can interfere with DNA stimulatory effects and have been shown to block the production of Th1 and pro-inflammatory cytokines (106-108). Thus we could speculate that motif differences at the DNA level may also be responsible for the differential capacity of commensals to control gut homeostasis.
Negative control of Treg induction by pathogens
It is becoming clear that the status of activation of DCs as well as inflammatory mediators modulates the capacity of these cells to induce Tregs de novo (5, 109-113). For instance, IL-6 and TGF-β in tandem can direct the production of IL-17-secreting T cells over Tregs (5, 109) and Th1 and Th2 effector cytokines have an antagonistic effect on Treg conversion (47). In addition, exposure to activated DCs or strong costimulation limit the induction of Foxp3+ Tregs in favor of the induction of effector responses (10, 11, 114). A correlate of this observation would be that inflammation triggered by infection can antagonize Treg induction. Indeed, acute infection with Listeria monocytogenes in mice failed to induce Foxp3 by conventional CD4+ T cells (115). Similarly, we found that the constitutive induction of Tregs in the gastrointestinal tract following exposure to oral antigens is inhibited following oral infection with the protozoan T. gondii (Fig. 1B). Upon infectious challenge, the gut-resident APCs are replaced by inflammatory cells that have not been conditioned by the gut environment (116). Indeed, we found that LP DCs from infected mice are less efficient at inducing Treg in vitro via their capacity to induce IFN-γ (117). Similarly, gut DCs exposed to Toxoplasma antigen in vitro are impaired in their capacity to induce Tregs, demonstrating that tolerogenic DCs can lose their capacity to induce Tregs when activated by defined commensal products or pathogens in favor of the induction of effector cells (117, 118). Previous reports examining both gut and lung inflammation support the idea that restricted or defective Treg conversion can enhance immunopathology (8, 119), supporting the idea that interference with constitutive Treg induction during infection may directly contribute to the pathological process. Furthermore, our findings also raise the possibility that exposure to antigen at a time of acute infection may impair the acquisition of tolerance against innocuous antigens.
Control of Treg plasticity by DCs
Recent findings suggest that acquisition of transcription factors in addition to Foxp3 can confer unique functional characteristics to Tregs (120). For instance, expression of T-bet by Tregs was recently shown to favor their proper control of Th1 environments (121). We found that in the context of virulent T. gondii infection, Tregs express high levels of the Th1 transcription factor T-bet (Fig. 1B). When isolated from the primary site of T. gondii infection, LP DCs can readily induce T-bet expression by Tregs via, in part, their capacity to produce IL-12. As LP DCs gain the capacity to produce IL-12 in this environment, we found that T-bet expression was associated with acquisition of responsiveness to IL-12 via enhanced signal transducer and activator of transcription 4 (Stat4) phosphorylation (117). Other factors, such as IL-27, which was found to be highly expressed in LPDC from infected mice, are also likely to contribute to this imprinting (Fig. 1B). Based on recent findings that T-bet favors Treg tropism to sites of infection during Mycobacterium tuberculosis via upregulation of CXCR3 (121). We could speculate that expression of T-bet by Tregs may confer an advantage to these cells in terms of homing and exerting regulation in a Th1 environment. Thus, the appropriation of Teff lineage transcription factors by Treg suggests that under non-pathogenic situations, acquisition of these elements controlled by local DCs may represent a necessary layer of their regulation.
In contrast, we found that Tregs do not survive or accumulate preferentially at sites of acute infection. Further, we found that T-bet expression led to production of IFN-γ, a cytokine responsible for both effector and pathogenic responses during T. gondii infection. Although T-cell lineage subsets were initially believed to represent stable populations of cells, recent evidence shows that lymphocytes maintain a certain degree of plasticity with respect to their capacity to produce cytokines (122, 123). Cells expressing both Foxp3 and IL-17 can be found in mucosal tissue or in vitro cultures (122, 124, 125). Genome-wide mapping of H3K4me3 and H3K27me3 performed in ex vivo Tregs revealed markers of both repression and induction at the tbx21 locus. However, the Ifng locus did not shown any sign of induction or repression (123), suggesting that it is poised for transcriptional activation. A previous report demonstrated that Tregs expressing both Foxp3 and T-bet were induced by CD8α+ DCs and could protect against airway hyperactivity (126). A role for IFN-γ in mediating Treg function has been reported in a model of graft transplants (127) and recent evidence demonstrates that, in vitro, Tregs can acquire expression of this cytokine (123); however, the capacity of Tregs to express IFN-γ under more physiologic settings and in particular during infection remained unclear. We propose that such effects may be associated with, or arise as a consequence of pathology. Indeed, we only detected IFN-γ production by Treg in situations leading to death of the infected host (117). This would suggest that in the presence of high levels of inflammatory mediators, T-bet expression may reach a threshold that superimposes an effector program on Tregs. Given their high degree of self-reactivity, it is plausible that Tregs can contribute to tissue damage or lose suppressive capacity if armed with effector cytokines (128). Indeed, a recent report highlighted that Foxp3 instability and acquisition of IFN-γ can favor the development of autoimmune diabetes (129).
Control of endogenous Tregs during infections
Overcoming regulation is a prerequisite to the expression of effector responses. Mechanisms associated with negative control of Tregs include direct Treg stimulation as well as strong costimulation of effector T cells mediated by local DCs. We recently found that effector T cells can also control Tregs via an alternative mechanism. Over the past few years, a strong body of literature suggests that a primary function of IL‐2 is to promote the expansion and survival of Treg (130). Indeed, previous work showed that the number of Tregs can be indexed to the number of IL-2-producing effector cells (131). Intriguingly, IL-2 production declines upon differentiation into Teff cells, implying that in the context of a highly polarized Th1 response, IL-2 may become limiting. Accordingly, we found that following oral infection with T. gondii, the emergence of a Th1 response was associated with a dramatic reduction of IL-2 production by CD4+ T cells in the gastrointestinal tract and in the periphery. Previous work demonstrated that T-bet acquisition as well as exposure of T cells to IL-27, IL-12, or IL-2 itself can limit IL-2 production (132, 133). In addition, Teff cells, via their consumption of growth factor, may also contribute to deprive the host from exogenous IL-2. A link between defective IL-2 production and Treg dysfunction has been previously proposed as one of the mechanisms associated with break down of tolerance in nonobese diabetic mice (134). In this particular study, enhanced apoptosis by Tregs was proposed as the primary mechanism of Treg control (134). It is tempting to speculate that under most circumstances, Tregs would be the primary bystander targets of IL-2 withdrawal during a Th1 response, thus providing an opportunity for effector responses to control infection. Indeed, we found that following oral infection with T. gondii, shutdown of IL-2 in a highly Th1-polarized environment triggered by infection directly contributes to the incapacity of Tregs to parallel effector responses and consequently leads to immunopathogenesis (117). These data could suggest a novel mechanism for Th1 pathogenicity that extends beyond their proinflammatory program to limit Treg survival. It would be important to assess how other Treg survival factors, such as IL-15 (135), are controlled during various acute infections and how this control could contribute to the regulation of local responses against pathogens.
Regulation has to be shaped in a manner specific to the microenvironment targeted. Because a large numbers of studies evaluating the mechanism of Treg induction were performed using mitogenic stimuli or using DCs from lymphoid organ, our understanding of tissue-specific DC, the complexity of the local subsets, and the peculiarity of their function remains sparse and clearly requires further exploration.
Acknowledgments
This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health. We apologize to those authors whose work we were unable to cite because of space limitations.
References
- 1.Fontenot JD, Dooley JL, Farr AG, Rudensky AY. Developmental regulation of Foxp3 expression during ontogeny. J Exp Med. 2005;202:901–906. doi: 10.1084/jem.20050784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bensinger SJ, Bandeira A, Jordan MS, Caton AJ, Laufer TM. Major histocompatibility complex class II-positive cortical epithelium mediates the selection of CD4(+)25(+) immunoregulatory T cells. J Exp Med. 2001;194:427–438. doi: 10.1084/jem.194.4.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jordan MS, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- 4.Chen W, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 6.Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172:5149–5153. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 7.Knoechel B, Lohr J, Kahn E, Bluestone JA, Abbas AK. Sequential development of interleukin 2-dependent effector and regulatory T cells in response to endogenous systemic antigen. J Exp Med. 2005;202:1375–1386. doi: 10.1084/jem.20050855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Curotto de Lafaille MA, Kutchukhidze N, Shen S, Ding Y, Yee H, Lafaille JJ. Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity. 2008;29:114–126. doi: 10.1016/j.immuni.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 9.Yamazaki S, Bonito AJ, Spisek R, Dhodapkar M, Inaba K, Steinman RM. Dendritic cells are specialized accessory cells along with TGF- for the differentiation of Foxp3+ CD4+ regulatory T cells from peripheral Foxp3 precursors. Blood. 2007;110:4293–4302. doi: 10.1182/blood-2007-05-088831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204:1765–1774. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci USA. 2008;105:9331–9336. doi: 10.1073/pnas.0710441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang S, Alard P, Zhao Y, Parnell S, Clark SL, Kosiewicz MM. Conversion of CD4+ CD25- cells into CD4+ CD25+ regulatory T cells in vivo requires B7 costimulation, but not the thymus. J Exp Med. 2005;201:127–137. doi: 10.1084/jem.20041201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng SG, Wang JH, Stohl W, Kim KS, Gray JD, Horwitz DA. TGF-beta requires CTLA-4 early after T cell activation to induce FoxP3 and generate adaptive CD4+CD25+ regulatory cells. J Immunol. 2006;176:3321–3329. doi: 10.4049/jimmunol.176.6.3321. [DOI] [PubMed] [Google Scholar]
- 14.Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med. 2003;9:1202–1208. doi: 10.1038/nm924. [DOI] [PubMed] [Google Scholar]
- 15.Latchman YE, et al. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci USA. 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 17.Heimesaat MM, et al. Gram-negative bacteria aggravate murine small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii. J Immunol. 2006;177:8785–8795. doi: 10.4049/jimmunol.177.12.8785. [DOI] [PubMed] [Google Scholar]
- 18.Iwasaki A, Kelsall BL. Unique functions of CD11b+, CD8 alpha+, and double-negative Peyer's patch dendritic cells. J Immunol. 2001;166:4884–4890. doi: 10.4049/jimmunol.166.8.4884. [DOI] [PubMed] [Google Scholar]
- 19.Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol. 2003;3:331–341. doi: 10.1038/nri1057. [DOI] [PubMed] [Google Scholar]
- 20.Chirdo FG, Millington OR, Beacock-Sharp H, Mowat AM. Immunomodulatory dendritic cells in intestinal lamina propria. Eur J Immunol. 2005;35:1831–1840. doi: 10.1002/eji.200425882. [DOI] [PubMed] [Google Scholar]
- 21.Coombes JL, Powrie F. Dendritic cells in intestinal immune regulation. Nat Rev Immunol. 2008;8:435–446. doi: 10.1038/nri2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barnard JA, Warwick GJ, Gold LI. Localization of transforming growth factor beta isoforms in the normal murine small intestine and colon. Gastroenterology. 1993;105:67–73. doi: 10.1016/0016-5085(93)90011-z. [DOI] [PubMed] [Google Scholar]
- 23.Rimoldi M, et al. Intestinal immune homeostasis is regulated by the crosstalk between epithelial cells and dendritic cells. Nat Immunol. 2005;6:507–514. doi: 10.1038/ni1192. [DOI] [PubMed] [Google Scholar]
- 24.Sun CM, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coombes JL, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-b- and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mucida D, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 27.Mucida D, Kutchukhidze N, Erazo A, Russo M, Lafaille JJ, Curotto de Lafaille MA. Oral tolerance in the absence of naturally occurring Tregs. J Clin Invest. 2005;115:1923–1933. doi: 10.1172/JCI24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8:1086–1094. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 29.Travis MA, et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature. 2007;449:361–365. doi: 10.1038/nature06110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lacy-Hulbert A, et al. Ulcerative colitis and autoimmunity induced by loss of myeloid alphav integrins. Proc Natl Acad Sci USA. 2007;104:15823–15828. doi: 10.1073/pnas.0707421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Varol C, et al. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity. 2009;31:502–512. doi: 10.1016/j.immuni.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 32.Bogunovic M, et al. Origin of the lamina propria dendritic cell network. Immunity. 2009;31:513–525. doi: 10.1016/j.immuni.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fortin G, et al. A role for CD47 in the development of experimental colitis mediated by SIRPalpha+CD103- dendritic cells. J Exp Med. 2009;206:1995–2011. doi: 10.1084/jem.20082805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johansson-Lindbom B, et al. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J Exp Med. 2005;202:1063–1073. doi: 10.1084/jem.20051100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stagg AJ, Kamm MA, Knight SC. Intestinal dendritic cells increase T cell expression of alpha4beta7 integrin. Eur J Immunol. 2002;32:1445–1454. doi: 10.1002/1521-4141(200205)32:5<1445::AID-IMMU1445>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 36.Johansson-Lindbom B, Svensson M, Wurbel MA, Malissen B, Marquez G, Agace W. Selective generation of gut tropic T cells in gut-associated lymphoid tissue (GALT): requirement for GALT dendritic cells and adjuvant. J Exp Med. 2003;198:963–969. doi: 10.1084/jem.20031244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mora JR, et al. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 38.Ziegler TR, Evans ME, Fernandez-Estivariz C, Jones DP. Trophic and cytoprotective nutrition for intestinal adaptation, mucosal repair, and barrier function. Annu Rev Nutr. 2003;23:229–261. doi: 10.1146/annurev.nutr.23.011702.073036. [DOI] [PubMed] [Google Scholar]
- 39.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21:527–538. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 40.Mora JR, et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science. 2006;314:1157–1160. doi: 10.1126/science.1132742. [DOI] [PubMed] [Google Scholar]
- 41.Siewert C, et al. Induction of organ-selective CD4(+) regulatory T cell homing. Eur J Immunol. 2007;37:978–989. doi: 10.1002/eji.200636575. [DOI] [PubMed] [Google Scholar]
- 42.Kang SG, Lim HW, Andrisani OM, Broxmeyer HE, Kim CH. Vitamin A metabolites induce gut-homing FoxP3+ regulatory T cells. J Immunol. 2007;179:3724–3733. doi: 10.4049/jimmunol.179.6.3724. [DOI] [PubMed] [Google Scholar]
- 43.Schambach F, Schupp M, Lazar MA, Reiner SL. Activation of retinoic acid receptor-alpha favours regulatory T cell induction at the expense of IL-17-secreting T helper cell differentiation. Eur J Immunol. 2007;37:2396–2399. doi: 10.1002/eji.200737621. [DOI] [PubMed] [Google Scholar]
- 44.Elias KM, et al. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–1020. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hill JA, et al. Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi Cells. Immunity. 2008;29:758–770. doi: 10.1016/j.immuni.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cantorna MT, Nashold FE, Chun TY, Hayes CE. Vitamin A down-regulation of IFN-gamma synthesis in cloned mouse Th1 lymphocytes depends on the CD28 costimulatory pathway. J Immunol. 1996;156:2674–2679. [PubMed] [Google Scholar]
- 47.Wei J, Duramad O, Perng OA, Reiner SL, Liu YJ, Qin FX. Antagonistic nature of T helper 1/2 developmental programs in opposing peripheral induction of Foxp3+ regulatory T cells. Proc Natl Acad Sci USA. 2007;104:18169–18174. doi: 10.1073/pnas.0703642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mucida D, et al. Retinoic acid can directly promote TGF-beta-mediated Foxp3(+) Treg cell conversion of naive T cells. Immunity. 200930:471–472. doi: 10.1016/j.immuni.2009.03.008. author reply 472 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saurer L, McCullough KC, Summerfield A. In vitro induction of mucosa-type dendritic cells by all-trans retinoic acid. J Immunol. 2007;179:3504–3514. doi: 10.4049/jimmunol.179.6.3504. [DOI] [PubMed] [Google Scholar]
- 50.Hammerschmidt SI, et al. Stromal mesenteric lymph node cells are essential for the generation of gut-homing T cells in vivo. J Exp Med. 2008;205:2483–2490. doi: 10.1084/jem.20080039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manicassamy S, et al. Toll-like receptor 2-dependent induction of vitamin A-metabolizing enzymes in dendritic cells promotes T regulatory responses and inhibits autoimmunity. Nat Med. 2009;15:401–409. doi: 10.1038/nm.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yokota A, et al. GM-CSF and IL-4 synergistically trigger dendritic cells to acquire retinoic acid-producing capacity. Int Immunol. 2009;21:361–377. doi: 10.1093/intimm/dxp003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Y, Teige I, Birnir B, Issazadeh-Navikas S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med. 2006;12:518–525. doi: 10.1038/nm1402. [DOI] [PubMed] [Google Scholar]
- 54.Tai P, et al. Induction of regulatory T cells by physiological level estrogen. J Cell Physiol. 2008;214:456–464. doi: 10.1002/jcp.21221. [DOI] [PubMed] [Google Scholar]
- 55.Belkaid Y. Regulatory T cells and infection: a dangerous necessity. Nat Rev Immunol. 2007;7:875–888. doi: 10.1038/nri2189. [DOI] [PubMed] [Google Scholar]
- 56.Poncini CV, Alba Soto CD, Batalla E, Solana ME, Gonzalez Cappa SM. Trypanosoma cruzi induces regulatory dendritic cells in vitro. Infect Immun. 2008;76:2633–2341. doi: 10.1128/IAI.01298-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walther M, et al. Upregulation of TGF-beta, FOXP3, and CD4+CD25+ regulatory T cells correlates with more rapid parasite growth in human malaria infection. Immunity. 2005;23:287–296. doi: 10.1016/j.immuni.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 58.Ocana-Morgner C, Wong KA, Lega F, Dotor J, Borras-Cuesta F, Rodriguez A. Role of TGF-beta and PGE2 in T cell responses during Plasmodium yoelii infection. Eur J Immunol. 2007;37:1562–1574. doi: 10.1002/eji.200737068. [DOI] [PubMed] [Google Scholar]
- 59.Finney CA, Taylor MD, Wilson MS, Maizels RM. Expansion and activation of CD4(+)CD25(+) regulatory T cells in Heligmosomoides polygyrus infection. Eur J Immunol. 2007;37:1874–1886. doi: 10.1002/eji.200636751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wilson MS, Taylor MD, Balic A, Finney CA, Lamb JR, Maizels RM. Suppression of allergic airway inflammation by helminth-induced regulatory T cells. J Exp Med. 2005;202:1199–1212. doi: 10.1084/jem.20042572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McSorley HJ, Harcus YM, Murray J, Taylor MD, Maizels RM. Expansion of Foxp3+ regulatory T cells in mice infected with the filarial parasite Brugia malayi. J Immunol. 2008;181:6456–6466. doi: 10.4049/jimmunol.181.9.6456. [DOI] [PubMed] [Google Scholar]
- 62.D'Elia R, Behnke JM, Bradley JE, Else KJ. Regulatory T cells: a role in the control of helminth-driven intestinal pathology and worm survival. J Immunol. 2009;182:2340–2348. doi: 10.4049/jimmunol.0802767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taylor MD, et al. CTLA-4 and CD4+ CD25+ regulatory T cells inhibit protective immunity to filarial parasites in vivo. J Immunol. 2007;179:4626–4634. doi: 10.4049/jimmunol.179.7.4626. [DOI] [PubMed] [Google Scholar]
- 64.Hewitson JP, Grainger JR, Maizels RM. Helminth immunoregulation: the role of parasite secreted proteins in modulating host immunity. Mol Biochem Parasitol. 2009;167:1–11. doi: 10.1016/j.molbiopara.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Balic A, Harcus Y, Holland MJ, Maizels RM. Selective maturation of dendritic cells by Nippostrongylus brasiliensis-secreted proteins drives Th2 immune responses. Eur J Immunol. 2004;34:3047–3059. doi: 10.1002/eji.200425167. [DOI] [PubMed] [Google Scholar]
- 66.Segura M, Su Z, Piccirillo C, Stevenson MM. Impairment of dendritic cell function by excretory-secretory products: a potential mechanism for nematode-induced immunosuppression. Eur J Immunol. 2007;37:1887–1904. doi: 10.1002/eji.200636553. [DOI] [PubMed] [Google Scholar]
- 67.McSorley HJ, et al. daf-7-related TGF-beta homologues from Trichostrongyloid nematodes show contrasting life-cycle expression patterns. Parasitology. 2009:1–13. doi: 10.1017/S0031182009990321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gomez-Escobar N, van den Biggelaar A, Maizels R. A member of the TGF-beta receptor gene family in the parasitic nematode Brugia pahangi. Gene. 1997;199:101–109. doi: 10.1016/s0378-1119(97)00353-3. [DOI] [PubMed] [Google Scholar]
- 69.Gomez-Escobar N, Gregory WF, Maizels RM. Identification of tgh-2, a filarial nematode homolog of Caenorhabditis elegans daf-7 and human transforming growth factor beta, expressed in microfilarial and adult stages of Brugia malayi. Infect Immun. 2000;68:6402–6410. doi: 10.1128/iai.68.11.6402-6410.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kennedy MW, Allen JE, Wright AS, McCruden AB, Cooper A. The gp15/400 polyprotein antigen of Brugia malayi binds fatty acids and retinoids. Mol Biochem Parasitol. 1995;71:41–50. doi: 10.1016/0166-6851(95)00028-y. [DOI] [PubMed] [Google Scholar]
- 71.Hewitson JP, et al. The secretome of the filarial parasite, Brugia malayi: proteomic profile of adult excretory-secretory products. Mol Biochem Parasitol. 2008;160:8–21. doi: 10.1016/j.molbiopara.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 72.Sansonetti PJ, Di Santo JP. Debugging how bacteria manipulate the immune response. Immunity. 2007;26:149–161. doi: 10.1016/j.immuni.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 73.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 74.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 75.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 76.Chieppa M, Rescigno M, Huang AY, Germain RN. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J Exp Med. 2006;203:2841–2852. doi: 10.1084/jem.20061884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bashir ME, Louie S, Shi HN, Nagler-Anderson C. Toll-like receptor 4 signaling by intestinal microbes influences susceptibility to food allergy. J Immunol. 2004;172:6978–6987. doi: 10.4049/jimmunol.172.11.6978. [DOI] [PubMed] [Google Scholar]
- 78.Sutmuller RP, Morgan ME, Netea MG, Grauer O, Adema GJ. Toll-like receptors on regulatory T cells: expanding immune regulation. Trends Immunol. 2006;27:387–393. doi: 10.1016/j.it.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 79.Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197:403–411. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, Levings MK. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol. 2005;75:8051–8059. doi: 10.4049/jimmunol.175.12.8051. [DOI] [PubMed] [Google Scholar]
- 81.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 82.Peng G, et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309:1380–1384. doi: 10.1126/science.1113401. [DOI] [PubMed] [Google Scholar]
- 83.Sutmuller RP, et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485–494. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc Natl Acad Sci USA. 2006;103:7048–7053. doi: 10.1073/pnas.0601554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Larosa DF, Gelman AE, Rahman AH, Zhang J, Turka LA, Walsh PT. CpG DNA inhibits CD4+CD25+ Treg suppression through direct MyD88-dependent costimulation of effector CD4+ T cells. Immunol Lett. 2007;108:183–188. doi: 10.1016/j.imlet.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu W, Weigand L, Belkaid Y, Mendez S. Immunomodulatory effects associated with a live vaccine against Leishmania major containing CpG oligodeoxynucleotides. Eur J Immunol. 2006;36:3238–3247. doi: 10.1002/eji.200636472. [DOI] [PubMed] [Google Scholar]
- 87.Torok HP, Glas J, Tonenchi L, Bruennler G, Folwaczny M, Folwaczny C. Crohn's disease is associated with a toll-like receptor-9 polymorphism. Gastroenterology. 2004;127:365–366. doi: 10.1053/j.gastro.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 88.Hall JA, et al. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity. 2008;29:637–649. doi: 10.1016/j.immuni.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dalpke A, Frank J, Peter M, Heeg K. Activation of toll-like receptor 9 by DNA from different bacterial species. Infect Immun. 2006;74:940–946. doi: 10.1128/IAI.74.2.940-946.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee J, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol. 2006;8:1327–1336. doi: 10.1038/ncb1500. [DOI] [PubMed] [Google Scholar]
- 91.Ewaschuk JB, Backer JL, Churchill TA, Obermeier F, Krause DO, Madsen KL. Surface expression of Toll-like receptor 9 is upregulated on intestinal epithelial cells in response to pathogenic bacterial DNA. Infect Immun. 2007;75:2572–2579. doi: 10.1128/IAI.01662-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Katakura K, Lee J, Rachmilewitz D, Li G, Eckmann L, Raz E. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest. 2005;115:695–702. doi: 10.1172/JCI22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–1118. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- 94.O'Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. 2006;7:688–693. doi: 10.1038/sj.embor.7400731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rachmilewitz D, et al. Toll-like receptor 9 signaling mediates the anti-inflammatory effects of probiotics in murine experimental colitis. Gastroenterology. 2004;126:520–528. doi: 10.1053/j.gastro.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 96.Rachmilewitz D, et al. Immunostimulatory DNA ameliorates experimental and spontaneous murine colitis. Gastroenterology. 2002;122:1428–1441. doi: 10.1053/gast.2002.32994. [DOI] [PubMed] [Google Scholar]
- 97.Fukata M, et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1055–1065. doi: 10.1152/ajpgi.00328.2004. [DOI] [PubMed] [Google Scholar]
- 98.Uematsu S, Akira S. The role of Toll-like receptors in immune disorders. Expert Opin Biol Ther. 2006;6:203–214. doi: 10.1517/14712598.6.3.203. [DOI] [PubMed] [Google Scholar]
- 99.Ott SJ, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53:685–693. doi: 10.1136/gut.2003.025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Martin HM, et al. Enhanced Escherichia coli adherence and invasion in Crohn's disease and colon cancer. Gastroenterology. 2004;127:80–93. doi: 10.1053/j.gastro.2004.03.054. [DOI] [PubMed] [Google Scholar]
- 101.Barnich N, Boudeau J, Claret L, Darfeuille-Michaud A. Regulatory and functional co-operation of flagella and type 1 pili in adhesive and invasive abilities of AIEC strain LF82 isolated from a patient with Crohn's disease. Mol Microbiol. 2003;48:781–794. doi: 10.1046/j.1365-2958.2003.03468.x. [DOI] [PubMed] [Google Scholar]
- 102.Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol. 2005;43:3380–3389. doi: 10.1128/JCM.43.7.3380-3389.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Masseret E, et al. Genetically related Escherichia coli strains associated with Crohn's disease. Gut. 2001;48:320–325. doi: 10.1136/gut.48.3.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Seksik P, et al. Alterations of the dominant faecal bacterial groups in patients with Crohn's disease of the colon. Gut. 2003;52:237–242. doi: 10.1136/gut.52.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Darfeuille-Michaud A, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterology. 2004;127:412–421. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 106.Krieg AM, et al. Sequence motifs in adenoviral DNA block immune activation by stimulatory CpG motifs. Proc Natl Acad Sci USA. 1998;95:12631–12636. doi: 10.1073/pnas.95.21.12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen Y, et al. Identification of methylated CpG motifs as inhibitors of the immune stimulatory CpG motifs. Gene Ther. 2001;8:1024–1032. doi: 10.1038/sj.gt.3301482. [DOI] [PubMed] [Google Scholar]
- 108.Zhao H, Cheng SH, Yew NS. Requirements for effective inhibition of immunostimulatory CpG motifs by neutralizing motifs. Antisense Nucleic Acid Drug Dev. 2000;10:381–389. doi: 10.1089/oli.1.2000.10.381. [DOI] [PubMed] [Google Scholar]
- 109.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 110.Korn T, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nurieva R, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 112.Stumhofer JS, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 113.Zhou L, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 114.Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. 2005;6:1219–1227. doi: 10.1038/ni1265. [DOI] [PubMed] [Google Scholar]
- 115.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 116.Dunay IR, et al. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity. 2008;29:306–317. doi: 10.1016/j.immuni.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Oldenhove G, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hall JA, et al. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity. 2008;29:637–649. doi: 10.1016/j.immuni.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Izcue A, et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28:559–570. doi: 10.1016/j.immuni.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zheng Y, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458:351–356. doi: 10.1038/nature07674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:1–7. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lee YK, et al. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wei G, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725–6729. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- 125.Yang XO, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Stock P, Akbari O, Berry G, Freeman GJ, Dekruyff RH, Umetsu DT. Induction of T helper type 1-like regulatory cells that express Foxp3 and protect against airway hyper-reactivity. Nat Immunol. 2004;5:1149–1156. doi: 10.1038/ni1122. [DOI] [PubMed] [Google Scholar]
- 127.Sawitzki B, Kingsley CI, Oliveira V, Karim M, Herber M, Wood KJ. IFN-gamma production by alloantigen-reactive regulatory T cells is important for their regulatory function in vivo. J Exap Med. 2005;201:1925–1935. doi: 10.1084/jem.20050419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol. 2006;7:401–410. doi: 10.1038/ni1318. [DOI] [PubMed] [Google Scholar]
- 129.Zhou X, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Malek TR, Bayer AL. Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol. 2004;4:665–674. doi: 10.1038/nri1435. [DOI] [PubMed] [Google Scholar]
- 131.Almeida AR, Zaragoza B, Freitas AA. Indexation as a novel mechanism of lymphocyte homeostasis: the number of CD4+CD25+ regulatory T cells is indexed to the number of IL-2-producing cells. J Immunol. 2006;177:192–200. doi: 10.4049/jimmunol.177.1.192. [DOI] [PubMed] [Google Scholar]
- 132.Hwang ES, Hong JH, Glimcher LH. IL-2 production in developing Th1 cells is regulated by heterodimerization of RelA and T-bet and requires T-bet serine residue 508. J Exp Med. 2005;202:1289–1300. doi: 10.1084/jem.20051044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Villarino AV, et al. Helper T cell IL-2 production is limited by negative feedback and STAT-dependent cytokine signals. J Exp Med. 2007;204:65–71. doi: 10.1084/jem.20061198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tang Q, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity. 2008;28:687–697. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vang KB, Yang J, Mahmud SA, Burchill MA, Vegoe AL, Farrar MA. IL-2, -7, and -15, but not thymic stromal lymphopoeitin, redundantly govern CD4+Foxp3+ regulatory T cell development. J Immunol. 2008;181:3285–3290. doi: 10.4049/jimmunol.181.5.3285. [DOI] [PMC free article] [PubMed] [Google Scholar]