

Abstract
Although arguably the most important reaction in glycoscience, chemical glycosylations are among the least well understood of organic chemical reactions resulting in an unnecessarily high degree of empiricism and a brake on rational development in this critical area. To address this problem primary 13C kinetic isotope effects now have been determined for the formation of β- and α-manno- and glucopyranosides by a natural abundance NMR method. In contrast to the common current assumption, for three of the four cases studied the experimental values concur with those computed for associative displacement of the intermediate covalent glycosyl trifluoromethanesulfonates. For the formation of the α-mannopyranosides the experimentally determined KIE differs significantly from that computed for an associative displacement, which is strongly suggestive of a dissociative mechanism that approaches the intermediacy of a glycosyl oxocarbenium ion. The application of comparable experiments to other glycosylation systems should shed further light on their glycosylation mechanisms and thus assist in the design of better reactions conditions with improved stereoselectivity.
The perceived importance of oligosaccharides in biology and the potential for their exploitation in medicine has made glycoscience one of the most vibrant and dynamic fields at the interface of chemistry and biology.1–4 Because of the difficulties in obtaining pure, homogeneous samples of oligosaccharides from natural sources, glycochemistry is a critical and fundamental component of glycoscience.5–8 Glycochemistry turns on the critical hub of glycosidic bond formation,9, 10 yet, paradoxically, the art of glycosidic bond formation is one of the least well understood11,12 and most empirical13 in the science of organic chemistry. Excepting so-called alternative methods,10 essentially all discussions of glycosidic bond formation invoke the glycosyl oxocarbenium ion,14,15 but this oft computed16–18 key intermediate has yet to be observed directly.14,19 In reality, glycosylation reactions are probably best considered as taking place by a range of mechanisms with more or less tightly associated transition states between the two formal extremes of the SN1 and SN2 pathways, with the position of any particular example on this spectrum determined by a number of factors including protecting groups, solvents, leaving group and promoter.14,15 In the absence of any direct observation of glycosyl oxocarbenium ions we fall back on indirect methods and reaction kinetics, but these are typically complicated by the (at least) three-component nature of most glycosylation systems that involve a glycosyl donor, acceptor, and a promoter. We report here on the use of primary 13C kinetic isotope effect (KIE) measurements in which the extent of 12C/13C isotopic fractionation at the anomeric position in the course of a glycosylation reaction is determined and, with the aid of quantum chemical calculations, used to assign more or less tightly associated transition states to particular cases. To avoid the need for synthesis of isotopically enriched substrates the experiment is conducted at natural abundance by very high field NMR spectroscopy (200 MHz operating frequency for 13C measurement). Overall, the technique provides insight into the mechanism of an important glycosylation reaction and its dependence on substitution pattern.
Results and Discussion
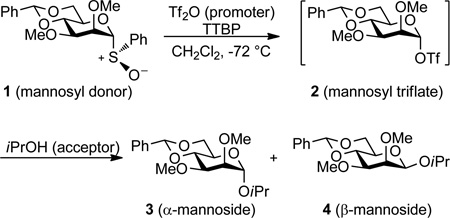
Our demonstration that the 4,6-O-benzylidene-directed β-mannopyranosylation reaction20–22 proceeds via the coupling of a highly reactive α-mannosyl triflate23,24 with an acceptor alcohol without the need for a promoter provided a rare opportunity to study an important class of glycosylation reaction in detail. The rapidity of the reaction at low temperatures and the absence of a suitable chromophore excluded the possibility of simple kinetic measurements for the determination of reaction order and focused our attention on the measurement of KIEs by NMR methods,25–27 preferably at natural abundance so as to avoid the time-consuming and expensive synthesis of labeled compounds.28,29 Problems of sensitivity and resolution (500 MHz 1H operating frequency) limited our earlier work to the observation of secondary 2H KIEs and even then required the laborious synthesis of samples enriched in deuterium.30 With an 800 MHz (1H operating frequency) NMR spectrometer and cryogenic probe technology at our disposal, we have now succeeded in determining primary 13C KIE’s at natural abundance for benzylidene-directed glycosylation reactions in the glucose and mannose series and show that mechanism is a function of donor stereochemistry and in the mannose series differs for the two anomeric products.
The 4,6-O-benzylidene protected mannosylsulfoxide 1 was prepared as previously described23 and converted at −72 °C in dichloromethane solution to the α-glycosyltriflate 2 by the addition ofan excess of trifluoromethanesulfonic anhydride (Tf2O) in the presence of 2,4,6-tri-tert-butyl pyrimidine (TTBP)31 as a mild, hindered, non-nucleophilic base. The acceptor, 2-propanol, was then added in quantities so as to ensure partial conversion of triflate 2 to the α- and β-glycosides 3 and 4 before the reaction was quenched at −72 °C by the addition of saturated aqueous NaHCO3(Table 1). After work up, the extent of conversion was determined by integration of the 1H NMR spectrum of the crude reaction mixture (800 MHz) against the internal standard 4,4,5,5-tetramethyl-2-(1-naphthyl)-1,3-dioxolane, and the products isolated chromatographically. The 13C NMR spectra of the two anomeric products were then recorded and the integrals were determined for the remote benzylidene carbon, employed as an internal standard, and the anomeric carbon, in both the products, and compared with those previously determined in the same manner for the starting sulfoxide. Use of the standard equation for KIE determination based on the observation of reaction products (eq 1), where F1 is the fractional conversion and R0 and Rp the ratio of the anomeric and benzylidene carbon integrals in the substrate and product respectively,27 afforded the results presented in Table 1 for the α- and β-mannopyranoside products.
| Eq 1. |
Table 1.
Experimentally Determined Primary 13C KIE’s for α- and β-Mannopyranosides 3 (Entries 1–5) and 4 (Entries 6–10).
 | ||||
|---|---|---|---|---|
| Entry | Tf2O (equiv) |
Conversion (%) | α:β | KIE (25 °C)*,† |
| α-Mannopyranoside 3 | ||||
| 1 | 1.5 | 11.4 | 1:4 | 1.006 |
| 2 | 2.4 | 16.0 | 1:3.5§ | 1.004 |
| 3 | 2.4 | 8.9 | 1:2.7 | 1.006 |
| 4 | 2.5 | 13.5 | 1:2.2 | 1.006 |
| 5 | 2.5 | 20.9 | 1:1.7 | 1.002 |
| average | 1.005±0.002 | |||
| β-Mannopyranoside 4 | ||||
| 6 | 1.5 | 45.1 | 1:4 | 1.026 |
| 7 | 2.4 | 55.6 | 1:3.5§ | 1.019 |
| 8 | 2.4 | 24.1 | 1:2.7 | 1.022 |
| 9 | 2.5 | 30.3 | 1:2.2 | 1.021 |
| 10 | 2.5 | 34.7 | 1:1.7 | 1.025 |
| average | 1.023±0.003 | |||
Errors quoted in the average values are at 1σ (see Supplementary Information, Tables S1–S4 for details)
The higher α:β ratio for this entry with respect to entries 3 and 4, despite the comparable amount of Tf2O employed presumably reflects the reaction of adventitious water with Tf2O, which lowers its effective concentration; see text for a discussion of the effect of Tf2O concentration.
Comparison of the results presented in Tables 1 reveals that the primary 13C KIE values for the α- and β-mannoside products are distinctly different, suggesting different mechanisms for the formation of the two anomers. The values for the β-anomer 4 fall close to the range (1.03–1.08) expected for a bimolecular(SN2) reaction,25 while those for the α-anomer 3 are closer to the expected values (1.00–1.01) of a unimolecular (SN1) reaction.25 It is also evident from inspection of Table 1 that the ratio of the two products depends on the amount of Tf2O employed with lower β:α ratios occurring when more Tf2O was employed. We interpret this observation, which was deliberately exploited in order to obtain sufficient amounts of the minor α-anomer for the NMR experiments, as being due to the competing triflation of the alcohol, which reduces its concentration and so retards the bimolecular reaction. This is consistent with formation of the α-product being zero order in alcohol (SN1), while that of the β-product depends on the alcohol concentration (SN2), and therefore with the KIE measurements.
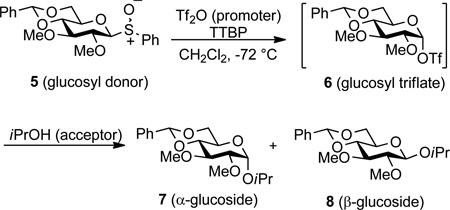
In contrast to mannopyranosylation,21,22 in the glucopyranose series the presence of a 4,6-O-benzylidene acetal results in preferential α-glycoside formation most likely reflecting a shift in mechanism. Hence, we examined the formation of the α- and β-glucosides 7 and 8 from the sulfoxides 5, via the triflate 6, by the same 13C NMR KIE technique leading to the data presented in Table 2.32 Unlike the case of the mannopyranosides, the observed 13C KIEs in the glucose series are at the lower end of the range expected for a bimolecular reaction for both the α- and β-series. As the formation of mannosyl and glucosyl triflates from sulfoxides is unaffected by either the anomeric configuration or the configuration at sulfur of the sulfoxides employed,21–24 for preparative reasons the equatorial sulfoxides 5 were employed for the work in the glucose series (Table 2), whereas the axial sulfoxide 3 was used in the mannose series (Table 1).
Table 2.
Experimentally Determined Primary 13C KIE’s for α-and β-Glucopyranosides 7 (Entries 1–4) and 8 (Entries 5–8).
 | ||||
|---|---|---|---|---|
| Entry | Tf2O (equiv) |
Conversion (%) | α:β | KIE (25 °C)*, † |
| α-Glucopyranoside 7 | ||||
| 1 | 1.2 | 31.4 | 1:0.5 | 1.029 |
| 2 | 1.2 | 9.8 | 1:0.7 | 1.025 |
| 3 | 2.5 | 7.1 | 1:1.7 | 1.015 |
| 4 | 1.2 | 12.7 | 1:1 | 1.022 |
| average | 1.023±0.006 | |||
| β-Glucopyranoside 8 | ||||
| 5 | 1.2 | 16.2 | 1:0.5 | 1.017 |
| 6 | 1.2 | 7.1 | 1:0.7 | 1.020 |
| 7 | 2.5 | 12.5 | 1:1.7 | 1.019 |
| 8 | 1.2 | 12.4 | 1:1 | 1.019 |
| average | 1.019±0.001 | |||
Errors quoted in the average values are at 1σ (see Supplementary Information, Tables S1–S4 for details)
To complement the experimental data, and assist in its interpretation, we carried out density functional theory computations of possible transition state structures for glycosylation. We selected the B3LYP/6-311++G(3df,3pd)//B3LYP/6-31G(d,p) level32–34 as a compromise between speed and accuracy as it is sufficient for reproduction of the anomeric effect,16 but fast enough to survey the dozens of possible trajectories for the reactions of both anomers of each glycosyl donor. We also employed a self-consistent reaction field throughout the structure optimizations to capture any bulk medium effects imposed by the solvent used in the experiments above. The four lowest energy transition state structures identified for the formation of α- and β-mannosides and the corresponding glucosides are shown in Figs 1A–D, along with the calculated 13C (and 2H) KIEs (which were indistinguishable whether calculated using the Bigeleisen-Mayer equation or the ratio of rate constants calculated using transition state theory),35 pertinent atomic separations corresponding to forming and breaking bonds and the free energies of activation for each process. Additional views, as well as other relevant structures are provided in the Supplementary Information, computational section.
Figure 1.

Calculated (B3LYP) associative transition states for the reaction of isopropanol with 4,6-O-benzylidene protected manno- and glucopyransyl triflates leading to the formation of the α- and β-glycosides in each case. a) TS for α-mannoside formation, b) TS for β-mannoside formation, c) TS for α-glucoside formation, and d) TS for β-glucoside formation. For each transition state the calculated primary 13C and secondary 2H KIE values, the free energy of activation, the lengths of the partial bonds to the leaving group and to the nucleophile, and the approximate conformation of the pyranose ring are listed.
The computed transition states in Fig 1 represent what may loosely be termed concerted transition states for nucleophilic substitution reactions in which the departing trifluoromethanesulfonate anion retains some degree of association with the putative glycosyl oxocarbenium ion. While intrinsic reaction coordinate (IRC) calculations on each of these stationary points confirms that the single imaginary vibrational mode corresponds to nucleophile attachment to, and leaving group departure from, the anomeric carbon (see Supplementary Information for IRC data), the differing degrees of C-OTf bond cleavage and C-OiPr bond formation indicate different relative positions along the reaction coordinate. The transition states for the formation of the α-glycosides (Figs 1A and 1C) are highly symmetrical, in that the bond to the incoming alcohol is similar in length to that of the departing triflate, whereas those corresponding to the formation of the β-anomers (Figs 1B and 1D) display nucleophile attachment ahead of leaving group departure. The differences in computed activation energies suggest that formation of the β-mannoside 4 should be very strongly favored over the α-anomer 3, whereas the same comparison for glucose indicates the α-anomer 7 to be considerably favored over the β-isomer 8 – the same preference observed experimentally in these systems on the preparative scale, and to a lesser extent in the present experiments owing to the necessary use of both excess Tf2O and limiting isopropanol; conditions which erode the selectivity due to suppression of the selective ‘associative’ mechanism relative to the competing unselective ‘dissociative’ mechanism.21,22 Importantly, the computed 13C KIEs correspond quite well with the experimental values in these two cases (Figs 1B and 1C), i.e. 1.018 versus 1.023 for 4 and 1.025 versus 1.023 for 7. Furthermore, the calculated secondary 2H KIE of 1.14 for the formation of the β-mannoside 4 is in good agreement with the value of 1.12 some of us measured previously.30
The formation of the α-glucoside 7 through an apparent SN2-like transition state deserves comment as experimentally only the α-glucosyltriflate 6 has been observed;21–23 the implication being that a kinetic scheme is in operation in which the more stable α-glucosyltriflate is in rapid equilibrium with its less stable but more reactive β-isomer, or its functional equivalent a β-contact ion pair. This type of kinetic scheme in which the less stable of two isomers, that are in very rapid equilibrium on the reaction timescale, is the more reactive one leads to a situation in which the product distribution bears no relation to the ratio of starting materials is a manifestation of one of the two boundary conditions of the Curtin-Hammett principle.36 The glaringly inconsistent result is that obtained for formation of the α-mannoside (Fig 1A); the computed 13C KIE of 1.023 differs significantly from the average experimental value of 1.005 (Table 1). The implication is that α- mannoside formation does not proceed through the computed SN2-like transition state of Fig 1A but rather takes place at the other end of the mechanistic spectrum through a dissociative process that at least approaches the intermediacy of a distinct oxocarbenium ion and a triflate anion. The inability of computational methods to locate such transition states,16 without resort to artifact such as the inclusion of a further cation to neutralize the charge on the anion, owing to barrier less internal collapse is widely appreciated. This picture is fully consistent with the fact that the anomeric effect is significantly smaller in the glucopyranosides than in the mannopyranosides,12,37 from which it follows that the Curtin-Hammett scenario involving a minor β-triflate or a closely related contact ion pair is more likely in the gluco- than in the manno-series. The recent demonstration of the existence of β-glycosyltriflates as stable species when the substitution pattern is favorable further supports the possibility of their intervention via a Curtin-Hammet kinetic scheme.38
Overall, a picture emerges (Figure 2) featuring a series of equilibria encompassing at the extremes two covalent glycosy ltriflates, the corresponding contact ion pairs (CIP) and the more loosely associated solvent separated ion pairs (SSIP). For three of the four cases studied the glycosylation reaction has SN2 character but as the KIE is at the lower end of the expected value for such a concerted process the transition states can be considered to be “exploded” and thus possibly in the grey area defined by reaction on a contact ion pair. For this reason the arrows depicting the formation of the two β-products and of the α-glucoside in Figure 2 originate between the covalent triflates and the corresponding CIPs. The exception that proves the rule is the formation of theα-mannoside 3 that is clearly dissociative in character and involves an intermediate that at least approximates a discrete glycosyl oxocarbenium ion. This picture represents what we consider to be the preferred pathways for the formation of the various isomers; it does not exclude the possibility of the existence of other minor pathways such as, for example, the formation of a fraction of the β-mannoside via an oxocarbenium ion-like species.
Figure 2.

Mechanistic picture for the 4,6-O-benzylidene-directed formation of α- and β-gluco- and mannopyranosides
The transition states located for the formation of the two β-glycosides 4 and 8 (Figs 1B and 1D) are further characterized by the presence of a hydrogen bond between the acceptor alcohol and O3 of the donor, which prompts the pyranose ring to adopt a B2,5 conformation in these TS structures. Similarly, the TS structure for the formation of α-glucoside 7 contains a H-bond, but this is now to O2 of the donor in a 4H3 conformation. Whitfield has previously suggested16 that such H-bonds are important in directing glycosylation reactions. Our previous experimental work,39,40 however, indicates a commonality of mechanism for the formation of O- and C-glycosides, with no H-bonding possible in the latter, for benzylidene-directed glucosylation and mannosylation. Other experimental work from our laboratories21,22 confirms the role of C3 substituent in determining stereoselectivity but attributes it to the evolution of the torsional interaction between O2 and O3 as a function of the reaction co-ordinate; clearly further work is necessary to probe the influence of acceptor-donor hydrogen bonding in these reactions.
Conclusion
In conclusion, the agreement between computed and experimentally determined primary 13C KIE values strongly suggests that the 4,6-O-benzylidene directed β-mannosylation, and β- and α-glucosylation reactions proceed through loosely associative transition states in which the incoming nucleophile displaces the leaving group from a loosely bound covalent glycosyl triflate of the opposite configuration. In the case of α-glucoside formation the corollary of this observation is the invocation of a covalent β-glucosyltriflate, or of a β-contact ion pair, and the operation of Curtin-Hammett-type kinetics. The contrast between the computed KIE for the formation of the α-mannoside by a concerted pathway and the experimentally observed value strongly suggests a highly dissociative mechanism for this one example, which approaches the intermediacy of a discrete glycosyl oxocarbenium ion. The demonstration of associative transition states for three of the four reactions studied suggests that the concentrations of both glycosyl acceptor and donor may well impact the stereoselectivity of other glycosylation reactions in solution and points the way to the more rational optimization of reaction conditions. By the same token, the stereoselectivities of concentration dependent glycosylations cannot be expected to translate directly to reactions conducted on polymeric or other supports.
Methods
The 13C NMR spectra for the KIE measurements were recorded at 200 MHz using a 90° pulse, an inter scan delay of 30 s, and 512 scans so as to meet the criterion of a minimum 200/1 signal to noise ratio necessary for the precise integrations required. Calculations were carried out using the Gaussian-09 suite of programs with the results visualized using GaussView 4.0 or 5.0.41 All preparative, spectroscopic, and computational methods are reported in the supplementary information along with full characterization data for all new compounds.
Supplementary Material
Acknowledgments
We thank the Natural Sciences and Engineering Research Council of Canada and the NIH (GM62160) for grant support to DAP and DC, respectively. MH and GEG thank the Ministère de l’Education Nationale de la Recherche et de la Technologie and NSERC, respectively, for scholarships. DAP acknowledges the support of the Canada Research Chairs program.
Footnotes
Author Contributions. LB, DAP and DC designed the project and wrote the manuscript. MH carried out the synthetic work, GEG conducted the computational studies, and NB and MH did the NMR study. MH, NB, LB and DC analyzed the NMR studies, and GEG and DAP analyzed the computational work. All authors discussed the results and commented on the manuscript.
Additional Information. The authors declare no competing financial interests. Correspondence and requests for materials regarding the synthetic work and NMR studies should be addressed to DC, and to DAP for the computational studies.
References
- 1.Gabius H-J, editor. The Sugar Code. Wiley-VCH: Weinheim; 2009. p. 569. [Google Scholar]
- 2.Martin McGowan ED, Bowman K. Background Paper on Glycosciences and Glycomics in the United States. National Research Council; 2010. [Google Scholar]
- 3.Wu C-Y, Wong C-H. Chemistry and glycobiology. Chem. Commun. 2011;47:6201–6207. doi: 10.1039/c0cc04359a. [DOI] [PubMed] [Google Scholar]
- 4.Seeberger PH. Chemical glycobiology: why now? Nat. Chem. Biol. 2009;5:368–372. doi: 10.1038/nchembio0609-368. [DOI] [PubMed] [Google Scholar]
- 5.Boltje TJ, Buskas T, Boons G-J. Opportunities and challenges in synthetic oligosaccharide and glycoconjugate research. Nature Chemistry. 2009;1:611–622. doi: 10.1038/nchem.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galan MC, Benito-Alifonsoa D, Watt GM. Carbohydrate Chemistry in Drug Discovery. Org. Biomol. Chem. 9:3598–3610. doi: 10.1039/c0ob01017k. [DOI] [PubMed] [Google Scholar]
- 7.Chlubnova I, et al. Natural glycans and glycoconjugates as immunomodulating agents. Natural Products Reports. 2011;28:937–952. doi: 10.1039/c1np00005e. [DOI] [PubMed] [Google Scholar]
- 8.Stallforth P, Lepenies B, Adibekian A, Seeberger PH. Carbohydrates: A Frontier in Medicinal Chemistry. J. Med. Chem. 2009;52:5561–5576. doi: 10.1021/jm900819p. [DOI] [PubMed] [Google Scholar]
- 9.Zhu X, Schmidt RR. New Principles for Glycoside-Bond Formation. Angew. Chem. Int. Ed. 2009;48:1900–1934. doi: 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]
- 10.Demchenko AV, editor. Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance. Wiley-VCH: Weinheim; 2008. p. 501. [Google Scholar]
- 11.Horenstein NA. Mechanism for Nucleophilic Aliphatic Substitution at Glycosides. Advances in Physical Organic Chemistry. 2006;41:275–314. [Google Scholar]
- 12.Sinnott ML. Carbohydrate Chemistry and Biochemistry. RSC Publishing; 2007. [Google Scholar]
- 13.Barresi F, Hindsgaul O. Chemically Synthesized Oligosaccharides, 1994. A Searchable Table of Glycosidic Linkages. J. Carbohydr. Chem. 1995;14:1043–1087. [Google Scholar]
- 14.Bohé L, Crich D. A propos of glycosyl cations and the mechanism of chemical glycosylation. CR Chimie. 2011;14:3–16. doi: 10.1016/j.carres.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith DM, Woerpel KA. Electrostatic interactions in cations and their importance in biology and chemistry. Org. Biomol. Chem. 2006;4:1195–1201. doi: 10.1039/b600056h. [DOI] [PubMed] [Google Scholar]
- 16.Whitfield DM. Computational Studies of the Role of Glycopyranosyl Oxacarbenium Ions in Glycobiology and Glycochemistry. Adv. Carbohydr. Chem. Biochem. 2009;62:83–159. doi: 10.1016/S0065-2318(09)00004-3. [DOI] [PubMed] [Google Scholar]
- 17.Satoh H, Hansen HS, Manabe S, van Gunsteren WF, Hunenberger PH. Theoretical Investigation of Solvent Effects on Glycosylation Reactions: Stereoselectivity Controlled by Preferential Conformations of the Intermediate Oxacarbenium-Counterion Complex. J. Chem. Theor. Comput. 2010;6:1783–1797. doi: 10.1021/ct1001347. [DOI] [PubMed] [Google Scholar]
- 18.Li Z. Computational study of the influence of cyclic protecting groups in stereoselectivity of glycosylation reactions. Carbohydr. Res. 2010;345:1952–1957. doi: 10.1016/j.carres.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Saito K, et al. Indirect Cation-Flow Method: Flash Generation of Alkoxycarbenium Ions and Studies on the Stability of Glycosyl Cations. Angew. Chem. Int. Ed. 2011;50:5153–5156. doi: 10.1002/anie.201100854. [DOI] [PubMed] [Google Scholar]
- 20.Crich D, Sun S. Direct Synthesis of β-Mannopyranosides by the Sulfoxide Method. J. Org. Chem. 1997;62:1198–1199. [Google Scholar]
- 21.Crich D. Mechanism of a Chemical Glycosylation. Acc. Chem. Res. 2010;43:1144–1153. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- 22.Aubry S, Sasaki K, Sharma I, Crich D. Influence of Protecting Groups on the Reactivity and Selectivity of Glycosylation: Chemistry of the 4,6-O-Benzylidene Protected Mannopyranosyl Donors and Related Species. Top. Curr. Chem. 2011;301:141–188. doi: 10.1007/128_2010_102. [DOI] [PubMed] [Google Scholar]
- 23.Crich D, Sun S. Are Glycosyl Triflates Intermediates in the Sulfoxide Glycosylation Method? A Chemical and 1H, 13C, and 19F-NMR Spectroscopic Investigation. J. Am. Chem. Soc. 1997;119:11217–11223. [Google Scholar]
- 24.Crich D. Chemistry of Glycosyl Triflates: Synthesis of β-Mannopyranosides. J. Carbohydr. Chem. 2002;21:663–686. [Google Scholar]
- 25.Berti PJ, Tanaka KSE. Transition State Analysis Using Multiple Kinetic Isotope Effects: Mechanisms of Enzymatic and Non-enzymatic Glycoside Hydrolysis and Transfer. Advances in Physical Organic Chemistry. 2002;37:239–313. [Google Scholar]
- 26.Chan J, Lewis AR, Gilbert M, Karwaski M-F, Bennet AJ. A direct NMR method for the measurement of competitive kinetic isotope effects. Nature Chem. Biol. 2010;6:405–407. doi: 10.1038/nchembio.352. [DOI] [PubMed] [Google Scholar]
- 27.Melander L, Saunders WH. Reaction Rates of Isotopic Molecules. Wiley-Interscience; 1980. [Google Scholar]
- 28.Singleton DA, Szymanski MJ. Simultaneous Determination of Intermolecular and Intramolecular 13C and 2H Kinetic Isotope Effects at Natural Abundance. J. Am. Chem. Soc. 1999;121:9455–9456. [Google Scholar]
- 29.Lee JK, Bain AD, Berti PJ. Probing the transition states of four glucoside hydrolyses with 13C kinetic isotope effects measured at natural abundance by NMR spectroscopy. J. Am. Chem. Soc. 2004;126:3769–3776. doi: 10.1021/ja0394028. [DOI] [PubMed] [Google Scholar]
- 30.Crich D, Chandrasekera NS. Mechanism of 4,6-O-Benzylidene Directed β-Mannosylation as Determined by α-Deuterium Kinetic Isotope Effects. Angew. Chem. Int. Ed. 2004;43:5386–5389. doi: 10.1002/anie.200453688. [DOI] [PubMed] [Google Scholar]
- 31.Crich D, Smith M, Yao Q, Picione J. 2,4,6-Tri-tert-butylpyrimidine (TTBP): A Cost Effective, Readily Available Alternative to the Hindered Base 2,6-Di-tert-butylpyridine and its 4-Substituted Derivatives in Glycosylation and Other Reactions. Synthesis. 2001:323–326. [Google Scholar]
- 32.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 33.Becke AD. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A. 1988:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 34.Becke AD. Density Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 35.Bigeleisen J, Mayer MG. J. Chem. Phys. 1947;15:261–267. [Google Scholar]
- 36.Seeman JI. Effect of Conformational Change on Reactivity in Organic Chemistry. Chem. Rev. 1983;83:83–134. [Google Scholar]
- 37.Lemieux RU. Rearrangements and Isomerizations in Carbohydrate Chemistry. In: De Mayo P, editor. Molecular Rearrangements. Part 2. Interscience Publishers; 1964. pp. 709–769. [Google Scholar]
- 38.Walvoort MTC, Lodder G, Mazurek J, Overkleeft HS, Codée JDC, van der Marel GA. Equatorial Anomeric Triflates from Mannuronic Acid Esters. J. Am. Chem. Soc. 2009;131:12080–12081. doi: 10.1021/ja905008p. [DOI] [PubMed] [Google Scholar]
- 39.Crich D, Sharma I. Is Donor-Acceptor Hydrogen Bonding Necessary for 4,6-O-Benzylidene-directed β-Mannopyranosylation? Stereoselective Synthesis of β-C-Mannopyranosides and α-C-Glucopyranosides. Org. Lett. 2008;10:4731–4734. doi: 10.1021/ol8017038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crich D, Sharma I. Influence of the O3 Protecting Group on Stereoselectivity in the Preparation of C-Mannopyranosides with 4,6-O-Benzylidene Protected Donors. J. Org. Chem. 2010;75:8383–8391. doi: 10.1021/jo101453y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frisch M, et al. Gaussian 09. Gaussian, Inc. Wallingford, CT: 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.