

Abstract
A rhodopsin-based homology model of the nucleotide-activated human P2Y2 receptor, including loops, termini, and phospholipids, was optimized with Monte Carlo Multiple Minimum. Docked UTP formed a nucleobase π–π complex with conserved Phe3.32. Selectivity-enhancing 2′-amino-2′-deoxy substitution interacted through π-hydrogen bonding with aromatic Phe6.51 and Tyr3.33. A “sequential ligand composition” approach for docking the flexible dinucleotide agonist Up4U demonstrated a shift of conserved cationic Arg3.29 from the UTP γ position to δ position of Up4U and Up4ribose. Sysnthesized nucleotides were tested as agonists at human P2Y receptors expressed in 1321N1 astrocytoma cells. 2′-Amino and 2-thio modifications synergized to enhance potency and selectivity; compound 8 (8 nM EC50) was 300-fold P2Y2-selective versus P2Y4. 2′-Amine acetylation reduced potency, and trifluoroacetylation produced intermediate potency. 5-Amino nucleobase substitution did not enhance potency through a predicted hydrophilic interaction, possibly because of destabilization of the receptor-favored (N)-ribose conformation. This detailed view of P2Y2 receptor recognition suggests mutations for model validation.
Keywords: G protein–coupled receptor, nucleotides, docking, phospholipase C, pyrimidines, homology modeling
Introduction
P2 nucleotide receptors are activated by a range of naturally occurring extracellular nucleotides and consist of two families: the eight subtypes of P2Y receptors, which are G protein–coupled receptors (GPCRs) (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, and P2Y14), and the seven subtypes of P2X ligand-gated cation channels.1–5 The P2Y2 receptor is activated by endogenous uridine 5′-triphosphate (UTP 1), adenosine 5′-triphosphate (ATP 2), and various dinucleotides (Chart 1). It is preferentially Gq-coupled and stimulates phospholipase Cβ (PLCβ). The P2Y2 receptor, first cloned from mice,6 is expressed in epithelial cells, smooth-muscle cells, endothelial cells, leukocytes, osteoblasts, and cardiomyocytes. In the central nervous system, activation of the receptor is associated with neuronal differentiation.7 The P2Y2 receptor appears to interact with integrins to stimulate chemotaxis and to enhance α-secretase–dependent amyloid precursor protein processing.8,9 Activation of the P2Y2 receptor causes a pronociceptive effect.10 Agonists of the P2Y2 receptor are of interest in the development of therapeutic agents for pulmonary diseases (e.g., cystic fibrosis), salivary gland dysfunction, and ophthalmic diseases.11,12
Chart 1.

Structures of native nucleoside 5′-triphosphate (UTP and ATP) ligands and a dinucleotide (UpnU) ligand of the P2Y2 receptor. EC50 values reported at the human P2Y2 receptor are shown.13,23,24,42
The SAR (structure-activity relationship) of nucleotides in activating the human P2Y2 receptor has been probed.2,3,13 UTP-γ-S 3 and the dinucleoside tetraphosphates INS365 (Up4U) 4a and INS37217 (Up4dC) 4b are potent and relatively stable agonists of the P2Y2 receptor.11,14
The first site-directed mutagenesis and molecular modeling of TMs (transmembrane helical domains) 6 and 7 of the P2Y2 receptor were reported by Erb et al.15 Modeling methods have subsequently undergone significant development. Furthermore, a variety of newly synthesized potent analogues of UTP are available for docking studies. Hence, in the present study, we carried out new molecular modeling studies to predict sites of interaction of the ligands with the P2Y2 receptor and to provide hypotheses for the design of additional analogues. Our recent publications1,13 provided an initial rhodopsin-based homology model, which was refined in the present study. We also have optimized agonist selectivity for the P2Y2 receptor by synthesizing new UTP analogues, which bear modifications at the uracil moiety and ribose ring. Thus, this study integrates the synthesis of a highly selective agonist with modeling studies that use a molecular dynamics (MD) simulation in a phospholipidic and aqueous environment to refine the receptor and a Monte Carlo approach to ligand docking.
Results and Discussion
Structure of the P2Y2 Receptor
A molecular model of the P2Y2 receptor was recently published.1 In contrast to the earlier model constructed by Erb et al15 this model of the P2Y2 receptor is a rhodopsin-based homology model containing not only seven TMs, but also all extracellular and intracellular hydrophilic loops. Here we present a further refined model of the P2Y2 receptor, updated by insertion of a segment of the carboxyl-terminal (CT) domain including the H8 helix (Gly310–Met346) and the amino-terminal (NT) domain. The formal geometry of the model was tested with the ProTable command of Sybyl as well as the Procheck software (Supporting Information).16
Inclusion of the natural environment of the receptor was previously shown to significantly improve the results of MD simulation, providing a more accurate structure of the receptor, especially in its loop and terminal regions.17 Therefore, the model was subjected to 10 ns MD simulation in a hydrated phospholipid bilayer.
The MD trajectory was analyzed, and the root mean square deviations (RMSD) of the P2Y2 receptor atoms, as well as its total energy, were calculated (Supporting Information). The analysis of the variation of the RMSD and the total energy along the trajectory suggested that during MD simulation the receptor structure became stable after 7.5 ns. Thus, we prolonged the simulation up to 10 ns and used the final typical structure for our docking studies. Also, the RMS fluctuations (RMSF) of the P2Y2 receptor residues were calculated from the MD trajectory. As shown in Figure 1, the lowest RMSF values were obtained for amino acid residues located in TM domains, and the highest values corresponded to residues located in the hydrophilic loops and terminal domains. The superimposition of the Cα atoms of the TM domains of the initial structure of the P2Y2 receptor and its typical structure calculated from the final 100 ps of the MD trajectory (Figure 2) demonstrated that the configuration of the loops and terminal domains was most significantly changed.
Figure 1.

a) RMSF of the P2Y2 receptor residues calculated from the 10 ns MD trajectory. b) RMSF of a part of C-terminal region of the P2Y2 receptor. A helical structure of H8 remained stable during the MD simulation.
Figure 2.

Superimposition of the initial structure of the human P2Y2 receptor (white) and its structure obtained after MD simulation (colored by residue position: N-terminus in red, TM1 in orange, TM2 in ochre, TM3 – in yellow, TM4 in green, TM5 in cyan, TM6 in blue, TM7 and C-terminus in purple).
Extracellular Regions
Recent MD simulation of the P2Y6 receptor revealed that EL2 moved toward TM3 and further up into the extracellular space, opening the putative nucleotide binding cavity.18 However, in the case of the P2Y2 receptor, EL2 did not show a significant displacement during the MD simulation. The analysis of the configuration of EL2 in the P2Y2 receptor structure obtained after MD simulation allowed us to propose several key residues that could fix this loop near the TM domain (Table 1). In addition to the disulfide bridge conserved among all GPCRs of the rhodopsin family and formed between two cysteine residues located in TM3 and EL2 (C106 and C183 in the P2Y2 receptor), we found that several charged residues located in EL2 of the P2Y2 receptor can interact with some oppositely charged residues located around the loop (Figure 3A). In particular, three pairs of such residues were observed: R177(EL2)-E20(NT), R180(EL2)-E29(NT), and D185(EL2)-R265(6.55). Throughout this paper we use the GPCR residue indexing system for TM regions, which allows comparison among receptors, as explained in detail elsewhere.17 In addition, E190 can interact with R194; both residues are located at the beginning of EL2. Several other bridges in the extracellular region of the P2Y2 receptor appeared in the model. R24(NT) can interact with D97(EL1) and with D30 (NT). Furthermore, E20(NT) interacted not only with R177(EL2), but also with R26(NT). Finally, C25, located in the NT domain, and C278, located in EL3 formed a disulfide bridge characteristic of all P2Y receptors.
Table 1.
The predicted electrostatic and disulfide bridges observed in the P2Y2 receptor model obtained after MD simulation.
| Electrostatic bridges
| |
|---|---|
| Negatively charged residue | Positively charged residue |
| Glu20 NT | Arg26 NT |
| Glu20 NT | Arg177 EL2 |
| Glu29 NT | Arg180 EL2 |
| Asp30 NT | Arg24 NT |
| Asp97 EL1 | Arg24 NT |
| Asp185 EL2 | Arg 265 6.55 |
| Glu190 EL2 | Arg194 EL2 |
| Asp319 CT | Arg340 CT |
| Asp342 CT | Arg334 CT |
| Asp345 CT | Arg335 CT |
|
| |
| Disulfide bridges | |
|
| |
| Cys25 NT | Cys278 EL3 |
| Cys106 3.25 | Cys183 EL2 |
|
| |
| Putative disulfide bridges | |
|
| |
| Cys132 3.51 | Cys217 5.57 |
| Cys44 1.43 | Cys300 7.47 |
Figure 3.
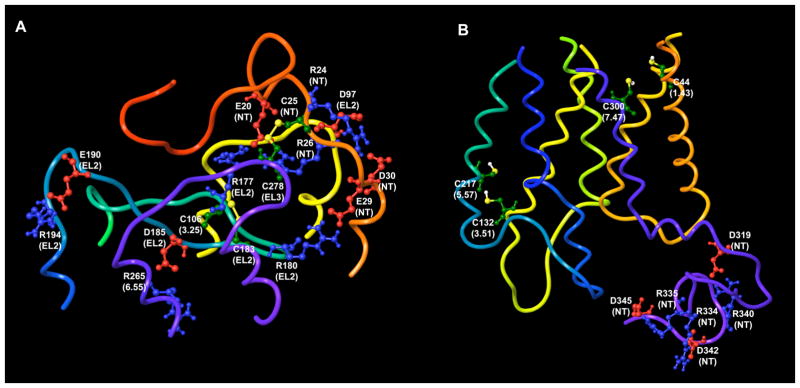
Putative electrostatic and disulfide bridges found in the extracellular (a) and intracellular (b) regions of the model of the P2Y2 receptor
Intracellular Regions
Three possible pairs of charged residues were found in the intracellular region of the P2Y2 receptor: D319-R340, R334-D342, and R335-D345 (Figure 3B). All these residues are located in the CT domain. Our molecular model suggests that ionic interactions between these residues keep the long CT domain in a more compact and configurationally restricted form, giving the entire receptor a more densely packed structure.
The RMSF values obtained for the residues located in the H8 helix (Figure 1B) showed that this part of the receptor remained stable during MD simulation, and H8 was found to be helical in the typical structure of the P2Y2 receptor.
Putative Interhelical Disulfide Bridges
An analysis of the model obtained after MD simulation allowed us to hypothesize that two pairs of cysteine residues, C132(3.51)-C217 (5.57) and C44 (1.43)-C300 (7.47), can be involved in the formation of disulfide bridges (Figure 3B). The distances observed between Cβ atoms of these cysteines (6.2Å and 4.5Å, respectively) are in good agreement with the distances measured experimentally for disulfide-bonded Cys-Cys pairs.19 However, C1.43 and C3.51 are present in the P2Y2 receptor only; in other subtypes of P2Y receptors these residues are F1.43 and Y3.51 (F3.51 in the P2Y13). For this reason, these two proposed disulfide bridges could only exist in the P2Y2 receptor. It has been shown experimentally that in rhodopsin the formation of a disulfide bond between residues located at the 3.51 and 5.57 positions is possible and that a crosslink between Y3.51C and C5.57 inhibits rhodopsin activation.20
Putative Binding Modes of P2Y2 Receptor Agonists
Binding mode of UTP and its analogues
To study the putative binding modes of P2Y2 receptor agonists, we performed molecular docking combined with Monte Carlo Multiple Minimum (MCMM) conformational search analysis on several known nucleotide ligands of this receptor subtype. As described in the Experimental Section, UTP 1 initially was manually docked inside the putative binding site of the P2Y2 receptor. The published data of site-directed mutagenesis combined with computational studies of P2Y receptors were taken into account.1,13,18 Because the Northern (N) conformation of the ribose ring of the ligand was proposed to be important for recognition,21 UTP and all other studied ligands were sketched and initially docked in their (N)-conformation. In addition, the anti-conformation of the ligand base ring was used during the modeling studies. Interestingly, in the original rhodopsin-based model of the P2Y2 receptor,1 the side chain of one of the key cationic residues, namely R3.29, was oriented in the opposite direction from the putative binding pocket, but during the simulation it shifted toward the binding cavity.
After MCMM refinement of the initially obtained docking complex, the α-phosphate group of the UTP triphosphate chain was bonded to R7.39, whereas R6.55 could interact with both α- and γ-phosphate groups and R3.29 interacted with the γ-phosphate group of UTP (Figure 4A). In addition, the γ-phosphate group of the ligand formed H-bonds with H184, located in EL2 directly after the conserved cysteine residue, with the backbone nitrogen atom of D185 and with the hydroxyl group of Y6.59. Another tyrosine residue, Y3.33, was H-bonded to the β-phosphate group.
Figure 4.

The binding modes of various agonists to the human P2Y2 receptor following MCMM calculations. A) UTP 1; B) A 2′-amino-2′-deoxy analogue 8 of UTP. The 2′-amino group is shown in its protonated form, which can be involved in cation-π interactions with F6.51 and can form an additional H-bond with Y3.33; C) 3-Methyl-UTP.13 The 3-methyl group has an unfavorable position between hydroxyl groups of Y1.39 and S7.43, and backbone oxygen atom of P7.40; D) 5-Methyl-UTP 19.13 In the model obtained, the 5-methyl group of 19 is unfavorably located between hydroxyl groups of T182 (EL2) and Y1.39. Also, it is close to the methyl groups of V181 (EL2); E) ATP 2; F) Up4U 4a.
In the model obtained after MCMM calculations, the 2′-hydroxyl group of UTP appeared near the conserved F6.51 (in the P2Y11 and P2Y14 receptors this position is Y6.51). This observation suggests the possibility of OH/π H-bonding between the 2′-hydroxyl group and the aromatic ring of F6.51. Several examples of similar interactions seen in protein crystallographic structures were reviewed by Meyer et al.22 The hypothesis of H-bonding of the 2′-hydroxyl group to the receptor is consistent with the experimental SAR.13 For example, 2′-deoxy-UTP (EC50 = 1.08 μM) is 22-fold less potent than UTP (EC50 = 0.049 μM), whereas the replacement of the 2′-hydroxyl group by a 2′-methoxy group (EC50 = 14.3 μM) reduced the potency further (290-fold weaker than UTP). In the latter case, our model showed that not only was the suggested OH/π H-bond lost, but the 2′-methyl group also interfered sterically with the aromatic ring of F6.51.
Agonist activities of the ligands, taken together with the binding mode obtained, strongly suggest that the hydroxyl group at the 2′-position is important as a donor but not as an acceptor of a H-bond. The binding mode of 2′-deoxy-2′-amino UTP 6 (Table 2, EC50 = 0.062 μM), which was found to maintain potency in activation of the P2Y2 receptor,13 was studied with MCMM calculations. The 2′-amino group (pKa = 6.2 in a related derivative)40,41 was examined in both unprotonated and protonated, positively charged forms. Similarly to the 2′-hydroxyl group of UTP, the 2′-amino group of 6 was found near and oriented toward the aromatic ring of F6.51. Both protonated and unprotonated forms of the ligands interacted with this residue. However, the protonated form of this amino group was involved in a stronger cation/π interaction with F6.51. In addition, the protonated 2′-NH3+ group acted as a H-bond donor to the hydroxyl of Y3.33 in our model (Figure 4B).
Table 2.
In vitro pharmacological data for UTP, 1, and its analogues in the stimulation of PLC at recombinant human P2Y2 and P2Y4 receptors expressed in 1321N1 astrocytoma cells. Unless noted: R1 = OH; X = O; and R2 = H.
 1, 6 – 10, 17 – 19 | |||||
|---|---|---|---|---|---|
| Compound | Modification | Structure | EC50 at hP2Y2 receptor, nMa | EC50 at hP2Y4 receptor, nMa | EC50 at hP2Y6 receptor, nMa |
| 1b | (= UTP) | 49 ± 12 | 73 ± 20 | >10,000e | |
| 2b | (= ATP) | 85 ± 12 | Antagonistd | NEe | |
| 4a | (= Up4U) | 210 ± 30 | 130 ± 10 | 20,000e | |
| 5 | (= Up4ribose) | 1880 ± 30 | ND | ND | |
| 6b | 2′-deoxy-2′-amino | R1 = NH2 | 62 ± 8 | 1200 ± 300 | >100,000 |
| 7b | 2-thio | X = S | 35 ± 4 | 350 ± 10 | ~1500f |
| 8c | 2-thio-2′-deoxy-2′-amino | X = S, R1 = NH2 | 8 ± 2 | 2400 ± 800 | >10,000 |
| 9 | 2-thio-2′-deoxy-2′-trifluoroacetylamino | X = S, R1 = CF3CONH | 470 ± 60 | 8300 ± 1200 | NE |
| 10 | 2-thio-2′-deoxy-2′-acetylamino | X = S, R1 = CH3CONH | 6500 ± 1400 | NE | NE |
| 17 | 5-amino | R2 = NH2 | 5600 ± 1200 | ND | 333 ± 88 |
| 18 | 5-azido | R2 = N3 | 1800 ± 400 | ND | 467 ± 120 |
| 19b | 5-methyl | R2 = CH3 | 480 ± 100 | 3900 ± 1600 | 140 ± 30 |
Agonist potencies reflect stimulation of PLC determined as reported,13,21,30 unless noted, and were calculated using a four-parameter logistic equation and the GraphPad software package (GraphPad, San Diego, CA). EC50 values (mean ± standard error) represent the concentration at which 50% of the maximal effect is achieved. Relative efficacies (%) were determined by comparison with the effect produced by a maximal effective concentration of reference agonist (UTP for P2Y2 and P2Y4, UDP for P2Y6) in the same experiment. The potency of UDP at the P2Y6 receptor was 23 ± 4 nM. If no maximal effect is given, then 100% efficacy was achieved. N = 3.
Agonist potencies from reference 13.
MRS2698.
ATP antagonized the human P2Y4 receptor with a KB of 708 nM.
Agonist potency from reference 26.
NE - no effect at 10 μM.
ND – not determined.
In the receptor-ligand complexes obtained for all studied agonists, the 3′-hydroxyl group of the ligand appeared to be H-bonded with the α-phosphate group of the triphosphate chain. This could be an important intramolecular interaction to stabilize the ribose ring in its (N)-conformation, to facilitate the interaction of the 2′-hydroxyl group with F6.51. This assumption is in agreement with the ligand activities;13,21 both 3′-deoxy-3′-methoxy-UTP and (S)-methanocarba ATP are essentially inactive at the P2Y2 receptor.
Taken together these findings are in good agreement with the SAR data and can explain the experimentally observed differences in activities of UTP and its 3′- and 2′-substituted derivatives: UTP ≈ 2′-NH2-2′-dUTP > 2′-dUTP > 2′-OCH3-2′-dUTP ≫ 3′-OCH3-3′-dUTP.
In our model the oxygen atom at position 4 of the UTP uracil ring was H-bonded to the hydroxyl group of Y1.39, and S7.43 accepted a H-bond from the 3-NH group of the ligand. In contrast, the oxygen atom at position 2 was not involved in H-bonding with the receptor. In addition, F3.32, conserved among all subtypes of P2Y receptors, was found to be involved in π-π interaction with the uracil ring of UTP. Interestingly, in the model obtained for the complex of the P2Y2 receptor with CTP (EC50 = 5.63 μM),13 the hydroxyl group of S7.43 was involved in H-bonding with the nitrogen atom at the 3 position of the nucleobase of CTP, but the NH2-group at the 4 position was unable to form a H-bond with Y1.39. Furthermore, the nucleoside moiety of zebularine-5′-triphosphate (180-fold weaker than UTP)13 docked to the P2Y2 receptor was significantly shifted from the initial position of UTP outwardly toward Y1.39, although the ligand still maintained a H-bond with S7.43. In addition, it was shown that a thiouracil ring can form H-bonded pairs with other nucleobases, which are only slightly less stable than the pairs formed by unmodified uracil.39 This means that 4-thio-UTP could be involved in the same interactions with Y1.39 as UTP. These data argue in favor of the hypothesis that in the case of the P2Y2 receptor, Y1.39 plays a role in the recognition of the functional group at position 4 of the uracil moiety. Available experimental data indicate that 3-methyl-UTP (EC50 = 1.20 μM) is 24-fold weaker than UTP.13 With the aim of explaining the observed differences in activity, we used MCMM calculations to establish a favored binding mode of 3-methyl-UTP, which was compared to the binding mode of UTP. According to this model, the methyl group at position 3 was located between the hydroxyl groups of Y1.39 and S7.43, and the side chain of the serine residue was shifted by 1.5Å away from the ligand. Moreover, the backbone oxygen atom of P7.40 also appeared near the 3-methyl group (Figure 4C).
The binding modes of 5-bromo-(EC50 = 0.75 μM), 5-iodo-(EC50 = 0.83 μM), and 5-methyl UTP (EC50 = 0.48 μM) were determined (Figure 4D), indicating that a hydrophobic group at position 5 of UTP was located unfavorably between the hydrophilic hydroxyl groups of T182 (EL2) and Y1.39. The methyl groups of V181 (EL2) were also located approximately 4Å from the substituent at position 5. Therefore, our model suggested that the introduction of a small hydrophilic group, such as an amino or hydroxyl group, at position 5 could provide additional H-bonds with T182 and Y1.39 and thus favor potency. Also, we predicted that an azido group would interact favorably within the available space between Y40, R180, V181, T182, and P293.
Binding Mode of ATP
The question of whether UTP and ATP bind to the same site in the P2Y2 receptor has already been considered.3 Because ATP 2 (EC50 = 0.085 μM) and UTP 1 (EC50 = 0.049 μM) are nearly equipotent at the P2Y2 receptor, ATP was initially docked (Figure 4E) at the P2Y2 receptor with the ribose in the (N) conformation and the adenine ring in the anti-conformation and subsequently subjected to an MCMM conformational search. Superimposition of the ATP-P2Y2 and the UTP-P2Y2 complexes revealed that the phosphate chains and the ribose rings of these two ligands have identical positions and configurations inside the receptor. The residues interacting with UTP were also found to interact with ATP. In agreement with the binding mode obtained for UTP, F3.32 was involved in a π–π interaction with the adenine ring of ATP. Also, Y1.39 was H-bonded with the N6-amino group, whereas S7.43 was H-bonded with the nitrogen atom at position 1 of the adenine ring of ATP. In addition, Y2.53, conserved among receptors, formed a H-bond with the nitrogen atom at position 1. On this basis, we P2Y1, 2, 4, 6 propose that Y2.53 is likely more important for interactions of the P2Y2 receptor with ATP and its derivatives than with UTP analogues.
Binding mode of Dinucleotide Agonists
The P2Y2 receptor can be activated not only by uracil or adenine triphosphates but also by dinucleotide molecules.23,24 Dinucleotide tetraphosphates, such as Up4U 4a or Up4dC 4b (Chart 1), have the optimal phosphate chain length for activation of the P2Y2 receptor and tend to be more resistant to nucleotidase degradation than the nucleoside triphosphates. In contrast, Up2U, containing only two phosphate groups, is nearly inactive as an agonist, and Up6U is 98-fold weaker than Up4U at the P2Y2 receptor.
To study the binding mode of dinucleotides within the P2Y2 receptor, we docked Up4U to the putative binding site. Docking of molecules that are as complex as Up4U is challenging, because of their large size and great flexibility. In particular, this process is especially difficult in the absence of X-ray structures or even molecular models of the receptor complexed with similar ligands. Several methods for docking of large ligands were described previously.25 In particular, fragmental-guided docking methods decompose a ligand into small fragments and then use favorable binding modes of these fragments to determine the most favorable orientation of the whole ligand.
Here we introduce an alternative “sequential ligand composition” technique, which we applied to the molecular docking of Up4U. With this technique, the structure of UTP docked in the binding site of the P2Y2 receptor and subjected to MCMM calculations was sequentially converted to Up4, Up4-ribose, and Up4U. The MCMM conformational search analysis of the ligand and all receptor residues located within 5Å of the ligand was performed at each stage of the computational conversion of UTP to Up4U to provide an energetically favorable orientation and configuration of the ligand. In contrast to the docking of the entire structure of the ligand (e.g. Up4U) at once, this method can avoid undesirable changes in the configuration of the receptor binding site.
Thus, “sequential ligand composition” provided us with a putative binding mode of Up4U at the P2Y2 receptor (Figure 4F and Supporting Information). In our model, the common uridine triphosphate subset of the Up4U structure had almost the same binding mode as UTP. However, three residues that interacted with the γ-phosphate group of UTP, i.e. R3.29, D185 (EL2), and Y6.59, were found to be bonded to the δ-phosphate group of Up4U. The uracil ring of UTP (uracilI) was involved in a π–π interaction with F3.32, and the uracil ring of the second uridine moiety (uracilII) of Up4U formed π–π interactions with Y3.33, which was also bonded to the β-phosphate group of the ligand. The oxygen atom at the 2 position of the uracilII ring could form H-bonds with Q4.57 and Y3.37. The oxygen atom of the hydroxyl group of Y3.37 also appeared near the 3-NH groupII of the ligand and could be involved in H-bonding with this group. As was observed with UTP, neither of the ribose ring oxygen atoms was H-bonded to the receptor. However, the 3′-hydroxyl groups of both uridineI and uridineII of Up4U were H-bonded to the α and δ phosphate groups, respectively.
The model of receptor-docked Up4U is consistent with a tetraphosphate chain having the optimal length for dinucleotide binding. The phosphate side chain of diuridine diphosphates apparently is too short to allow uridineII to reach its binding pocket. In contrast, ligands with longer phosphate chains, such as Up6U, are too large to be fully accommodated inside the receptor. Also, as discussed by Brunschweiger and Müller2, among various linear dinucleotides only dinucleoside tetraphosphates have the same number of negatively charged oxygen atoms as UTP.
Although H-bonds between the P2Y2 receptor and the ribose ring oxygen of uridineII of Up4U are absent, both 2′- and 3′-hydroxyl groups were found to be H-bonded to the receptor. The 2′-hydroxyl group was found to be H-bonded with the hydroxyl group of Y5.38, and the 3′-hydroxyl group interacted with the backbone NH-group of V4.60. Also, the hydroxyl group of Y6.59 appeared near the 3′-hydroxyl group of the ligand. These observations suggest that Up4-ribose could be active at the P2Y2 receptor, as UDP-glucose is active at the P2Y14 receptor. In the complex obtained for Up4U, both the hydroxyl group of Tyr and the backbone NH-group of Val were found 5.5Å from the ligand.
Selective Recognition for Nucleoside Tri- and Diphosphates at P2Y2 and P2Y6 Receptors
Although UDP is almost inactive at the P2Y2 receptor,2 it is the cognate agonist of the P2Y6 receptor. To explain these differences, we compared residues of the P2Y2 receptor directly involved in ligand interactions with the corresponding residues of the P2Y6 receptor (details are given in Supporting Information). The results obtained allow us to propose that residues R/K6.55, Y/L6.59, and H184/Y178 of the P2Y2/P2Y6 receptors play a critical role in UTP versus UDP recognition. This hypothesis will be tested in future experiments with site-directed mutagenesis.
Exploration of SAR at the P2Y2 Receptor with the Aid of Modeling
To further examine determinants of P2Y2 receptor selectivity, we synthesized several nucleotide derivatives and tested them for activation of the human P2Y2 and P2Y4 receptors expressed in 1321N1 astrocytoma cells (Table 2). The 2′-amino modification of 6, which maintained potency at the P2Y2 receptor,13 was combined with another P2Y2-favorable modification of UTP, i.e. 2-thio, leading to 8. The requirement of a free amine at this position was explored through N-acylation reactions, leading to 9 and 10. Although a 5-methyl modification of the uracil ring of UTP was found to reduce P2Y2 receptor potency,13 we also examined whether introduction of a polar, H-bonding group such as amino at this position would be beneficial. Thus, the 5-amino 17 and 5-azido 18 analogues were evaluated at the receptors.
Chemical Synthesis
2′-Amino-2′-deoxy-2-thiouridine 5′-triphosphate 8 was prepared by standard phosphorylation of the corresponding nucleoside 15. The synthesis of 2′-amino-2′-deoxy-2-thiouridine 15 is depicted in Scheme 1. 2′-Azido-2′-deoxyuridine 12 was obtained via opening of 2,2′-anhydrouridine with NaN3 in DMF.27 After selective mesylation of the 5′-hydroxyl group of 12, the resulting 5′-methanesulfonate ester 13 was converted to 2′-azido-2′-deoxy-2-O-ethyluridine (14) according to the method of Manoharan and coworkers.28 In this reaction, 2,5′-anhydro-2′-azido-2′-deoxyuridine was generated in situ and subsequently opened with ethoxide. Treatment of 14 with H2S in anhydrous pyridine allowed simultaneous 2-thionation and reduction of the 2′-azido group, resulting in 2′-amino-2′-deoxy-2-thiouridine (15). The 2-thionation was confirmed by the 13C NMR resonance signal of C-2, which shifts to a low magnetic field (177.66 ppm) compared to uridine (150.66 ppm).
Scheme 1.

Preparation of the synthetic intermediate 15, a 2′-amino-2′-deoxy-2-thio nucleoside. Reagents and conditions. (a) NaN3, DMF, 150° C, 15 h; (b) MsCl, pyridine, 0° C, 61%; (c) NaHCO3/EtOH, reflux, 36 h, 57%; (d) H2S, pyridine, 50°C, 250 psi, 24 h, 77%.
Attempts to directly phosphorylate compound 15 by a standard method failed to yield the desired 5′-triphosphate 8. Therefore, the 2′-amino group was protected by trifluoroacetylation. The phosphorylation of the protected nucleoside 16 was performed as shown in Scheme 2.29 During purification by ion exchange chromatography, partial hydrolysis yielded a mixture of 8 and 9, which was readily separable by HPLC. The acetylation of the 2′-amino group of 8 to obtain 10 was performed as shown in Scheme 3. Up4ribose 5 was prepared as shown in Scheme 4.
Scheme 2.
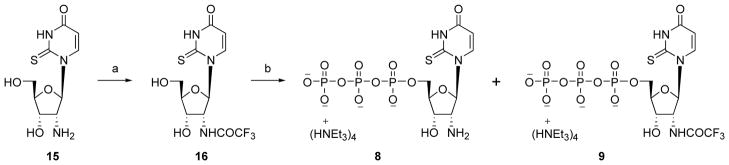
Synthesis of 2′-amino-2′-deoxy-2-thioUTP 8 and its N-trifluoroacetyl derivative 9. Reagents and conditions. (a) DIEA, ethyl trifluoroacetate, DMF, room temperature, 13 h, 88%; (b) (i) POCl3, PO(OCH3)3, Proton Sponge, 0° C, 2 h, (ii) (Bu3NH+)2P2O7H2, Bu3N, DMF, 10 min, (iii) 0.2 M triethylammonium bicarbonate solution, room temperature, 1 h, 12% (8), 7% (9).
Scheme 3.
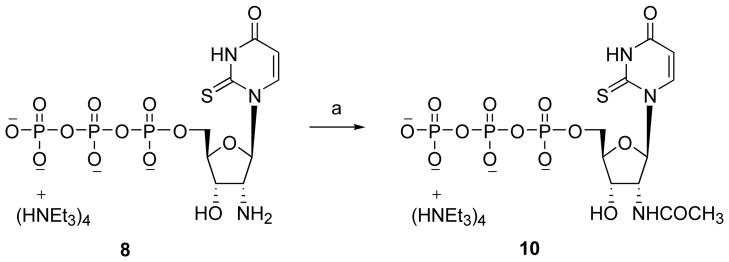
Synthesis of 2′-acetylamino-2′-deoxy-2-thioUTP 10. Reagents and conditions. (a) acetic anhydride, H2O, room temperature, 6 h, 57%.
Scheme 4.
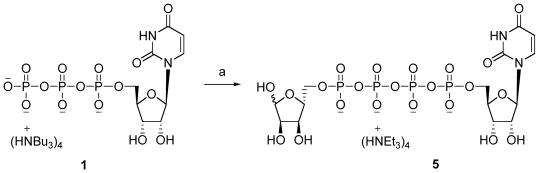
Synthesis of Up4-5′-ribose 5. Reagents and conditions. (a) (i) DCC, DMF, room temperature, 1h, (ii) D-ribose-5-phosphate, DMF, room temperature, 48 h, 18%.
Pharmacological Activity
Activation of PLC by a range of concentrations of each nucleotide derivatives was studied in 1321N1 astrocytoma cells stably expressing the human P2Y2, P2Y4 and P2Y6 receptors (Table 2), by reported methods.13,21,30,31 Tritiated inositol phosphates produced from a radiolabeled myo-inositol precursor were measured using a standard ion exchange method.
An intermediate structure related to the modeling by “sequential ligand composition”, Up4-[5′]-ribose 5, activated the P2Y2 receptor with an EC50 of 1.88 μM, i.e. 9-fold less potent than Up4U
The 5′-triphosphate 8, which combined potency-enhancing features derived from our previous studies, was 300-fold selective in activation of the P2Y2 receptor in comparison with the P2Y4 receptor (Figure 5). Compound 8 and other 2′-deoxy-2′-amino analogues, 6, 9, and 10, were nearly inactive at the P2Y6 receptor. The binding mode of this potent and selective analogue was studied with MCMM calculations, and the results were similar to those for 6, in which the 2′-amino group in its protonated form was involved in a cation-π interaction with F6.51 and as a H-bond donor to Y3.33 (Figure 4B). The potency of 8 was greatly reduced upon acetylation (10) and reduced to a lesser degree upon trifluoroacetylation (9). These observations are consistent with the binding modes obtained for compound 10. In the model of the P2Y2 receptor complex with compound 10 (Supporting Information), the H-bond between F6.51 and the NH-group of the acetamide moiety was not observed. Moreover, the methyl group of this moiety undesirably appeared near the OH-group of Y3.33 and the NH2-group of R6.55. The oxygen atom of the acetyl group was not involved in interactions with the receptor. We speculate that in the case of compound 9, the CF3-group, which is more electronegative than CH3, provides some favorable interactions with positively charged R6.55, improving the potency of compound 10.
Figure 5.
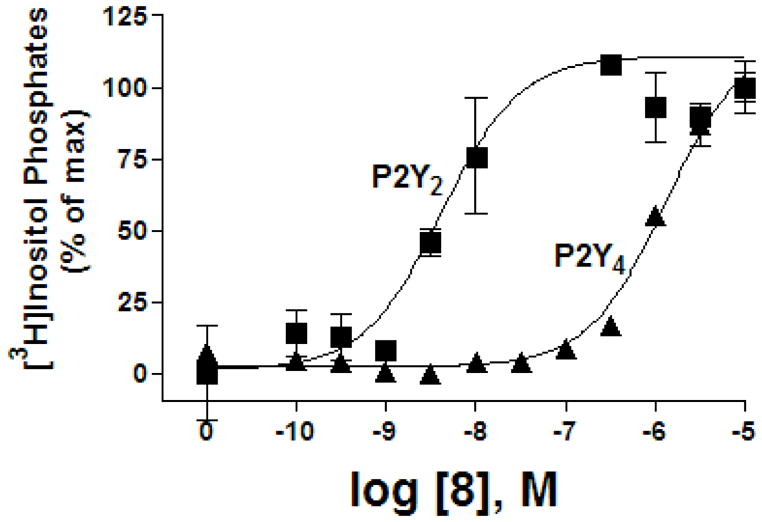
Activation by compound 8 of PLC in 1321N1 astrocytoma cells expressing the human P2Y2 receptor or P2Y4 receptor.
The 5-position was predicted to accommodate an azido group as well as a small, polar, H-bonding group, such as an amino group. Building on these observations, we measured the activities of 5-amino 17 and 5-azido 18 UTP derivatives at the human P2Y2 receptor (Table 2). However, the results indicated that both structures are less active than 5-methyl-UTP 19 at this receptor.13 Similar to certain other uracil-substituted uridine 5′-triphosphates,26 compounds 17 – 19 also potently activated the human P2Y6 receptor.
Introduction of an amino or azido group at position 5 of UTP apparently influences the conformation of the ribose ring. It was shown previously,32 through NMR analysis combined with semiempirical calculations, that electron-donating groups at position 5 of uracil nucleotides can destabilize the receptor-preferred (N) conformation of the ribose ring.
Conclusions
This detailed view of molecular recognition at the P2Y2 receptor suggests mutations for further model validation. In this study we report a highly potent and selective agonist, the 2′-amino-2-thio derivative of UTP, compound 8, which should prove very useful as a pharmacological probe for studying P2Y2 receptor action. Modeling provided an explanation for the general stabilizing effect of the 2′-amino modification of UTP in P2Y2 receptor recognition. A “sequential ligand composition” approach was adopted for docking flexible dinucleotide agonists such as Up4U. This modeling approach, in which ligands are docked to the receptor, including loops and terminal regions; embedded in a phospholipid bilayer; and optimized with MCMM, promises to be helpful in the design of additional potent and selective P2Y2 receptor agonists.
Experimental Section
Chemical Synthesis
All reagents were from standard commercial sources and of analytic grade. Compounds 17 and 18 were purchased from ALT, Inc. (Lexington, KY). 2,2′-Anhydrouridine (11) was obtained from Wako Chemicals GmbH (Neuss, Germany). Precoated Merck silica gel F254 plates were used for TLC, and spots were examined under ultraviolet light at 254 nm and further visualized by sulphuric acid-anisaldehyde spray. Column chromatography was performed on ICN silica gel (63–200 μm, 60Å, ICN Biochemicals, Eschwege, Germany). For compounds 11–15, NMR spectra were obtained with a Varian Mercury 300 MHz spectrometer. Chemical shifts are given in ppm (δ) relative to the residual solvent signal, in the case of DMSO-d6 2.54 ppm for 1H and 40.5 ppm for 13C. Structural assignment was confirmed with COSY and DEPT experiments. All signals assigned to hydroxyl groups were exchangeable with D2O. Exact mass measurements were performed on a quadruple/orthogonal–acceleration time-of-flight (Q/oaTOF) tandem mass spectrometer (qToF 2, Micromass, Manchester, UK) equipped with a standard electrospray ionization (ESI) interface. Samples were infused in a 2-propanol/water (1:1) mixture at 3 μL/min. For compounds 16 and 8–10, 1H NMR spectra were obtained with a Varian Gemini 300 spectrometer (Varian, Inc., Palo Alto, CA). 31P NMR spectra were recorded at room temperature with a Varian XL 300 spectrometer (121.42 MHz); orthophosphoric acid (85%) was used as an external standard. Purity of compounds was checked with a Hewlett–Packard 1100 HPLC equipped with a Zorbax Eclipse 5 μm XDB-C18 analytical column (250 ×4.6 mm; Agilent Technologies Inc., Palo Alto, CA). Purity was measured with two different solvent systems. (System A: 5 mM TBAP (tetrabutylammonium dihydrogenphosphate)-CH3CN from 80:20 to 40:60 in 20 min; flow rate 1 mL/min. System B: 10 mM TEAA (triethylammonium acetate)-CH3CN from 80:20 to 60:40 in 20 min; flow rate 1 mL/min. System C: 10 mM TEAA - CH3CN from 100:0 to 80:20 in 20 min; flow rate 1 mL/min.) Purifications by HPLC were performed under the following conditions: Luna 5 μ RP-C18 (2) semipreparative column, 250 ×10.0 mm, Phenomenex, Torrance, CA; flow rate 2 mL/min. System D: 10 mM TEAA-CH3CN from 100:0 to 90:10 in 20 min. System E: 10 mM TEAA-CH3CN from 100:0 to 95:5 in 30 min; High-resolution mass measurements were performed on a Micromass/Waters LCT Premier Electrospray Time of Flight mass spectrometer coupled with a Waters HPLC system (Waters Corp., Milford, MA).
2′-Azido-2′-deoxy-5′-O-methanesulfonyluridine (13)
Methanesulfonyl chloride (210 μL, 2.7 mmol) was added to a solution of 12 (610 mg, 2.3 mmol) in 10 mL pyridine at −78°C. The reaction was stirred for 1 h at 0°C, quenched with aqueous saturated NaHCO3, extracted with CH2Cl2, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (CH2Cl2:MeOH 96:4) to obtain compound 13 as a white solid (472 mg, 61%). 1H NMR (DMSO-d6):δ 3.23 (s, 3H, CH3), 4.06 (m, 1H, H-4′), 4.25–4.48 (m, 4H, H-2′, H-3′, H-5′A, and H-5′B), 5.68 (dd, J = 7.9, 2.1 Hz, 1H, H-6), 5.82 (d, J = 5.3 Hz, 1H, 3′-OH), 6.2 (d, J = 5.6 Hz, 1H, H-1′), 7.62 (d, J = 8.2 Hz, 1H, H-5), 11.47 (s, 1H, NH); HRMS (ESI-MS) for C10H14N5O7S: [M+H]+: found, 348.0618; calcd., 348.0613.
2′-Azido-2′-deoxy-2-O-ethyluridine (14)
Compound 13 (465 mg, 1.3 mmol) was refluxed in absolute ethanol (60 mL) in the presence of anhydrous sodium bicarbonate (281 mg, 3.3 mmol) under a nitrogen atmosphere for 36 h. After cooling to room temperature, the reaction was diluted with ethyl acetate and the precipitated sodium salt was removed by filtration. The filtrate was concentrated in vacuo and purified on a silica gel column (CH2Cl2:MeOH 95:5) to obtain compound 14 (222 mg, 57%) as a white solid. 1H NMR (DMSO-d6):δ 1.33 (t, J = 6.9 Hz, 3H, CH3), 3.56–3.63 (m, 1H, H-5′A), 3.67–3.74 (m, 1H, H-5′B), 3.92 (m, 1H, H-4′), 4.12 (app t, J = 4.8 Hz, 1H, H-3′), 4.27–4.39 (m, 3H, CH2 and H-2′), 5.23 (t, J = 4.8 Hz, 1H, 5′-OH), 5.85 (m, 2H, H-6 and 3′-OH), 6.01 (d, J = 5.4 Hz, 1H, H-1′), 7.98 (d, J = 7.8 Hz, 1H, H-5); HRMS (ESI-MS) for C11H15N5O5Na: [M+Na]+: found, 320.0974; calcd., 320.0971.
2′-Amino-2′-deoxy-2-thiouridine (15)
In a Parr apparatus compound 14 (210 mg, 0.71 mmol) was dissolved in pyridine (40 mL), cooled to −50°C and saturated with H2S. The mixture was heated at 50°C for 24 h, resulting in a pressure of 250 psi. After cooling to room temperature, the remaining H2S was released, the solvent was evaporated, and the residue was purified on a silica gel column (CH2Cl2:MeOH 90:10), yielding compound 15 (141 mg, 77%), after subsequent crystallization from MeOH. 1H NMR (DMSO-d6): δ 3.32 (dd, J = 6.1, 5.0 Hz, 1H, H-2′), 3.56–3.67 (m, 2H, H-5′A and H-5′B), 3.94 (m, 2H, H-3′ and H-4′), 5.19 (t, J = 4.7 Hz, 5′-OH), 5.41 (br s, 1H, 3′-OH), 6.01 (d, J = 8.2 Hz, 1H, H-6), 6.53 (d, J = 6.0 Hz, 1H, H-1′), 8.09 (d, J = 8.2 Hz, 1H, H-5); 13C NMR (DMSO-d6): δ 59.74 (C-2′), 61.43 (C-5′), 70.91 (C-3′), 86.50 (C-4′), 93.49 (C-1′), 107.32 (C-5), 141.66 (C-6), 160.24 (C-4), 177.66 (C-2), HRMS (ESI-MS) for C9H14N3O4S1: [M+H]+: found, 260.0692; calcd., 260.0704.
2′-Trifluoroacetylamino-2′-deoxy-2-thiouridine (16)
N,N-Diisopropylethylamine (0.006 mL, 0.035 mmol) and ethyl trifluoroacetate (0.004 mL, 0.035 mmol) were added sequentially to a solution of 15 (6 mg, 0.023 mmol) in DMF (1 mL). The reaction mixture was stirred for 13 h at room temperature. The solvent was removed in vacuo, and the residue was purified by preparative thin-layer chromatography (CH2Cl2-MeOH, 90:10) to afford 16 (7.2 mg, 88%) as a white solid. 1H NMR (CD3OD) δ 3.84 (m, 2H, H-5′A and H-5′B), 4.14 (dd, J = 5.4, 2.7 Hz, 1H, H-4′), 4.34 (dd, J = 5.7, 3.0 Hz, 1H, H-3′), 4.58 (dd, J = 6.6, 6.3 Hz, 1H, H-2′), 6.01 (d, J = 8.1 Hz, 1H, H-6), 7.12 (d, J = 6.9 Hz, 1H, H-1′), 8.25 (d, J = 8.1 Hz, 1H, H-5); HRMS m/z found 356.0520 (M + H+)−. C11H13N3O5F3S requires 356.0528.
2′-Amino-2′-deoxy-2-thiouridine 5′-triphosphate triethylammonium salt (8) and 2′-Trifluoroacetylamino-2′-deoxy-2-thiouridine 5′-triphosphate triethylammonium salt (9)
Phosphorous oxychloride (0.004 mL, 0.04 mmol) was added to a solution of 16 (7.2 mg, 0.020 mmol) and Proton Sponge (4 mg, 0.033 mmol) in trimethyl phosphate (1 mL) at 0°C. The reaction mixture was stirred for 2 h at 0°C. Then a mixture of tributylamine (0.02 mL, 0.08 mmol) and tributylammonium pyrophosphate (1.6 moles C12H27N per mole H4PO7, 62 mg, 0.131 mmol) in DMF (0.3 mL) was added at once. After 10 min, 0.2 M triethylammonium bicarbonate solution (2 mL) was added, and the clear solution was stirred at room temperature for 1 h. The latter was lyophilized overnight and the resulting residue was purified by ion-exchange column chromatography with a Sephadex-DEAE A-25 resin with a linear gradient (0.01–0.5 M) of 0.5 M ammonium bicarbonate as the mobile phase to get a mixture of 8 and 9 as the ammonium salts. The mixture was purified by HPLC (System D) to obtain 8 (2.2 mg, 12 %) and 9 (1.4 mg, 7 %) as the triethylammonium salts. Compound 8: 1H NMR (D2O) δ 1.28 (t, J = 7.5 Hz, 36H, N(CH2CH3)3), 3.20 (q, J = 7.5 Hz, 24H, N(CH2CH3)3), 4.15 (dd, J = 6.9, 5.7 Hz, 1H, H-2′), 4.26 (m, 1H, H-5′A), 4.37 (m, 1H, H-5′B), 4.48 (m, 1H, H-4′), 4.86 (m, 1H, H-3′), 6.31 (d, J = 8.1 Hz, 1H, H-6), 7.22 (d, J = 7.2 Hz, 1H, H-1′), 8.12 (d, J = 8.1 Hz, 1H, H-5); 31P NMR (D2O) δ −22.64 (t, J = 20.2 Hz), −11.22 (d, J = 20.2 Hz), −9.63 (d, J = 19.6 Hz); HRMS m/z found 497.9529 (M − H+)−. C9H15N3O13P3S requires 497.9538; purity > 98% by HPLC. (System A: 15.2 min; System B: 6.6 min). Compound 9: 1H NMR (D2O) δ 1.28 (t, J = 7.5 Hz, 36H, N(CH2CH3)3), 3.20 (q, J = 7.5 Hz, 24H, N(CH2CH3)3), 4.32 (m, 2H, H-5′A and H-5′B), 4.58 (m, 1H, H-4′), 4.68 (m, 1H, H-2′), 4.80 (hidden by the water peak, 1H, H-3′), 6.26 (d, J = 8.1 Hz, 1H, H-6), 6.92 (d, J = 5.4 Hz, 1H, H-1′), 8.13 (d, J = 8.4 Hz, 1H, H-5); 31P NMR (D2O) δ −22.28 (t, J = 19.6 Hz), −11.04 (d, J = 19.6 Hz), −8.54 (m); HRMS m/z found 593.9364 (M − H+)−. C11H14N3O14P3F3S requires 593.9361; purity > 98% by HPLC. (System A: 18.3 min, System B: 7.7 min).
2′-Acetylamino-2′-deoxy-2-thiouridine 5′-triphosphate triethylammonium salt (10)
Acetic anhydride (0.07 mL, 0.74 mmol) was added to a solution of 8 (0.5 mg, 0.55 μmol) in H2O (0.5 mL) at room temperature. After the reaction mixture was stirred for 6 h, the solvent was removed in vacuo. The residue was purified by HPLC (System D) to obtain 10 (0.3 mg, 57 %) as the triethylammonium salts. 1H NMR (D2O) δ 1.28 (t, J = 7.5 Hz, 36H, N(CH2CH3)3), 3.20 (q, J = 7.5 Hz, 24H, N(CH2CH3)3), 4.29 (m, 2H, H-5′A and H-5′B), 4.36 (m, 1H, H-4′), 4.53 (m, 2H, H-2′ and H-3′), 6.27 (d, J = 8.1 Hz, 1H, H-6), 6.98 (d, J = 5.6 Hz, 1H, H-1′), 8.08 (d, J = 8.1 Hz, 1H, H-5); 31P NMR (D2O) δ −21.31 (t, J = 20.5 Hz), −10.84 (d, J = 20.8 Hz), −7.62 (m); HRMS m/z found 539.9660 (M − H+)−. C11H17N3O14P3S requires 539.9644; purity > 98% by HPLC. (System A: 17.2 min; System B: 6.7 min).
Diuridine 5′,5′-tetraphosphate ammonium salt (4a)
Compound 4a was synthesized by following the procedures of ref. 23. HRMS m/z found 788.9842 (M − H+)−. C18H25N4O23P4 requires 788.9860; purity > 98% by HPLC. (System A: 20.5 min; System C: 7.7 min).
Uridine-5′-tetraphosphate-5′-ribose triethylammonium salt (5)
Uridine 5′-triphosphate trisodium salt (25 mg, 0.05 mmol) and D-ribose-5-monophosphate disodium salt (59 mg, 0.19 mmol) were converted to the tributylammonium salts by treatment with the ion-exchange resin (DOWEX 50WX2-200 (H)) and tributylamine. After removal of the solvent, the resulting residue was dried under high-vacuum overnight. To a solution of uridine 5′-triphosphate tributylammonium salt (0.05 mmol) in DMF (2 mL) was added N,N′-dicyclohexylcarbodiimide (22 mg, 0.11 mmol) and the mixture stirred for 1 h at room temperature. A solution of D-ribose-5-monophosphate tributylammonium salt (0.19 mmol) in DMF (2 mL) was added to the reaction mixture with stirring continued at room temperature for 48 h. After removal of the solvent, the residue was purified by ion-exchange column chromatography with Sephadex-DEAE A-25 resin using a linear gradient (0.01–0.5 M) of 0.5 M ammonium bicarbonate as the mobile phase. Compound 5 (7.0 mg, 18 %) was additionally purified by HPLC (System E). 1H NMR (D2O) δ 1.28 (t, J = 7.5 Hz, 36H, N(CH2CH3)3), 3.20 (q, J = 7.5 Hz, 24H, N(CH2CH3)3), 4.11 (m, 3H, H-ribose), 4.28 (m, 4H, H-4′, H-5′, 1H-ribose), 4.42 (m, 3H, H-2′, H-3′, 1H-ribose), 5.22 (d, J = 2.4 Hz, 3/5H, H-1′ribose), 5.41 (d, J = 3.6 Hz, 2/5H, H-1′ribose), 5.99 (d, J = 8.3 Hz, 1H, H-5), 6.02 (d, J = 5.4 Hz, 1H, H-1′), 7.99 (d, J = 8.3 Hz, 1H, H-6); 31P NMR (D2O) δ −24.55 (m), −12.83 (m), −12.43 (m); HRMS m/z found 694.9720 (M − H+)−. C14H23N2O22P4 requires 694.9693; purity > 99% by HPLC. (System A: 18.6 min; System C: 7.1 min).
Molecular Modeling
The published molecular model of the human P2Y2 receptor1 was updated by insertion of a part of the C-terminal (CT) domain including the H8 helix and the N-terminal (NT) domain. The P2Y2 CT domain (residues Gly310–Met346) was modeled by homology to bovine rhodopsin. We used the published sequence alignment,1 to superimpose the backbone atoms of the TM domain of the P2Y2 receptor on the corresponding atoms of bovine rhodopsin (PDB ID = 1U19)33 with Sybyl 7.1.34 The rhodopsin TM domains and its hydrophilic loops (Ala26–Met309) as well as Gly310 of the P2Y2 receptor (last residue in the published model) subsequently were removed from the receptor structures. The remaining rhodopsin NT (Met1–Glu25) and CT (residues starting from Asn310) domains were connected to Gly22 and Ala309 of the P2Y2 receptor, respectively. The residues of the CT domain were replaced with the corresponding residues of the P2Y2 receptor (residues Gly310–Met346) with Sybyl 7.1.
An attempt to apply the same technique for modeling of the NT domain failed because of overlap of the rhodopsin NT domain and the extracellular loops (ELs) of the P2Y2 receptor. For this reason, the configuration of the P2Y2 receptor NT domain (Met1–Gly22) was predicted with the Loopy program from the Jackal package,35 and 1000 initial conformations were generated. The option of energy minimization of the generated candidates was used.
The P2Y2 receptor model was minimized with Sybyl 7.1 in the Amber7 FF99 force field. The energy minimization of side chains of the terminal domains initially was performed with constrained positions of all other atoms of the P2Y2 receptor until an energy gradient lower than 0.05 kcal•mol−1•Å−1 was reached. The minimization was continued without any constraints in the terminal domains, but with constraint of the atoms of the TM helices and hydrophilic loops. The structure was then minimized, fixing only the backbone atoms of the TM α-helices until the energy gradient lower than 0.05 kcal•mol−1•Å−1 was reached. Finally, an unconstrained energy minimization of the whole P2Y2 receptor model was performed until the energy gradient was lower than 0.05 kcal•mol−1•Å−1. The formal geometry of the model was tested with the ProTable command of Sybyl 7.1 as well as the Procheck software.18
Molecular Dynamics Simulation of the P2Y2 Receptor
The molecular dynamics (MD) simulation of the P2Y2 receptor was performed on the NIH Biowulf Cluster (Bethesda, MD) with CHARMM 32a2 software.36 The protocol used for the system construction and MD simulation was described previously.18,37 The constructed system included the P2Y2 receptor surrounded by 100 DOPC lipids, 5964 TIP3 water molecules, and 107 Cl− and K+ ions, for a total of 37,242 atoms. The MD simulation was performed for 10 ns under conditions of CPT (constant pressure and temperature), as previously described.18 A hexagonal unit cell (69.3_69.3_85Å) and hexagonal periodic boundary conditions in all directions were used.
For the first 4.5 ns of MD simulation, nuclear Overhauser effect (NOE) restraints were applied within TM7 to the distances between each backbone carbonyl oxygen atom of residue n and the backbone NH-group of the residue n+4. The restraints were applied to all residues of TM7 with the exception of prolines and residues before and after prolines. MD simulation performed without such restraints led to a disordered secondary structure of TM7. Similar findings were described previously for the P2Y6 receptor.18 After 4.5 ns the MD simulation was continued without any restraints, and the helical structure of the TM7 remained stable until the end of the simulation. The typical structure of the P2Y2 receptor calculated from the last 100 ps of the MD trajectory was used as final model.
Manual Molecular Docking
The structure of UTP containing all hydrogen atoms initially was sketched with Sybyl 7.1. The ligand geometry was optimized in the Tripos force field with Gasteiger-Hückel atomic charges.34 Available site-directed mutagenesis data and previously published results of molecular modeling1,13,18 were used, with the DOCK command of the Sybyl 7.1 package,34 to manually preposition UTP, with an anti-conformation of the uracil ring and an (N)-conformation of the ribose ring, inside the putative binding site of the P2Y2 receptor. The tri-phosphate chain of the ligand was placed between the R3.29, R6.55, and R7.39. The nucleobase ring was located near Y1.39, Y2.53, and S7.43.
The manual molecular docking procedure was performed in several stages. During the first stage of the molecular docking process, only the bonds of the ligand were flexible. When the most energetically favorable location and conformation of the ligand was found, the molecular docking procedure was repeated with flexible bonds of the receptor. During each iteration of the docking process, the minimization of the binding site with the ligand inside was performed until an RMS of 0.01 kcal•mol−1•Å−1 was reached. Finally, the energy of the entire obtained protein-ligand complex was minimized until an energy gradient lower than 0.01 kcal•mol−1•Å−1 was reached.
Conformational Analysis of UTP, ATP, and Their Derivatives
The conformational analysis of the studied mononucleotides located in the putative binding site of the P2Y2 receptor was performed with the MCMM and the mixed torsional/low-mode sampling methods implemented in MacroModel 9.0 software.38 MCMM calculations initially were performed for UTP and all residues located within 5Å of the ligand, with a shell of constrained atoms with a radius of 2Å. The following parameters were used: MMFFs force field, water as an implicit solvent, maximum of 1000 iterations of the Polak-Ribier Conjugate Gradient (PRCG) minimization method with a convergence threshold of 0.05 kJ•mol−1•Å−1, number of conformational search steps = 100, energy window for saving structures = 1000 kJ•mol−1. The ligand-receptor complex obtained after MCMM calculations was subjected to an additional 100 steps of the mixed torsional/low-mode conformational search. The structure of UTP docked in the receptor subsequently was transformed to other studied UTP analogues by substitution of functionalities, and MCMM calculations were performed for each analogue, setting the number of steps to 100 and the energy window for saving structures to 100 kJ•mol−1.
Molecular Docking of Up4U
The “subsequent ligand composition” technique was introduced to dock the dinucleotide Up4U to the P2Y2 receptor. The UTP binding mode obtained after MCMM calculations was used as a starting point. Initially, the δ-phosphate group was added to UTP located inside the receptor. The resulting uridine 5′-tetraphosphate and all residues located within 5Å of the ligand were subjected to MCMM calculations with a shell of constrained atoms within a radius of 2Å. The following parameters were used: MMFFs force field, water as an implicit solvent, maximum of 1000 iterations of the (PRCG) minimization method with a convergence threshold of 0.05 kJ•mol−1•Å−1, number of conformational search steps = 100, energy window for saving structures = 100 kJ•mol−1. A ribose moiety in its (N)-conformation was then attached to the δ-phosphate group of the ligand. The obtained molecule and all residues located within 5Å were also subjected to MCMM calculations with the same parameters. The uracil ring in its anti-conformation then was attached to the ligand. The resulting Up4U and all receptor residues located within 5Å from the ligand were subjected to MCMM calculations with the specified parameters. Finally, Up4U and all receptor residues located within 5Å of the ligand were subjected to an additional 100 steps of the mixed torsional/low-mode conformational search.
Supplementary Material
Acknowledgments
Mass spectral measurements were carried out by Dr. John Lloyd and NMR measurements by Wesley White (NIDDK). This research was supported in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases. This work was supported by National Institutes of Health grants GM38213 and HL34322 to T.K. Harden. We thank C. Hoffmann (Univ. of Würzburg, Germany) and C. Müller and A. El-Tayeb (Univ. of Bonn, Germany) for helpful discussion.
Footnotes
Supporting Information Available: Figures illustrating the change in energy and RMSD of the P2Y2 receptor during the MD simulation, UDP and Up4U docked inside the P2Y2 receptor, and UTP docked in a P2Y2/P2Y6 chimeric receptor. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Costanzi S, Mamedova L, Gao ZG, Jacobson KA. Architecture of P2Y nucleotide receptors: Structural comparison based on sequence analysis, mutagenesis, and homology modeling. J Med Chem. 2004;47:5393–5404. doi: 10.1021/jm049914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brunschweiger A, Müller CE. P2 receptors activated by uracil nucleotides--an update. Curr Med Chem. 2006;13:289–312. doi: 10.2174/092986706775476052. [DOI] [PubMed] [Google Scholar]
- 3.Jacobson KA, Jarvis MF, Williams M. Purine and pyrimidine (P2) receptors as drug targets. J Med Chem. 2002;45:4057–4093. doi: 10.1021/jm020046y. [DOI] [PubMed] [Google Scholar]
- 4.Jacobson KA, Costanzi S, Ohno M, Joshi BV, Besada P, Xu B, Tchilibon S. Molecular recognition at purine and pyrimidine nucleotide (P2) receptors. Curr Top Med Chem. 2004;4:805–819. doi: 10.2174/1568026043450961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobson KA, Mamedova L, Joshi BV, Besada P, Costanzi S. Molecular recognition at adenine nucleotide (P2) receptors in platelets. Semin Thromb Hemost. 2005;31:205–216. doi: 10.1055/s-2005-869526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lustig KD, Shiau AK, Brake AJ, Julius D. Expression cloning of an ATP receptor from mouse neuroblasoma cells. Proc Natl Acad Sci USA. 1993;90:5113–5117. doi: 10.1073/pnas.90.11.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arthur DB, Georgi S, Akassoglou K, Insel PA. Inhibition of apoptosis by P2Y2 receptor activation: novel pathways for neuronal survival. J Neurosci. 2006;26:3798–3804. doi: 10.1523/JNEUROSCI.5338-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bagchi S, Liao Z, Gonzalez FA, Chorna NE, Seye CI, Weisman GA, Erb L. The P2Y2 nucleotide receptor requires interaction with αv integrins to communicate with Gostimulate chemotaxis. J Biol Chem. 2005;280:39058–39066. [Google Scholar]
- 9.Camden JM, Schrader AM, Camden RE, González FA, Erb L, Seye CI, Weisman GA. P2Y2 nucleotide receptors enhance α-secretase-dependent amyloid precursor protein processing. J Biol Chem. 2005;280:18696–18702. doi: 10.1074/jbc.M500219200. [DOI] [PubMed] [Google Scholar]
- 10.Davis BM, Malin SA, Koerber HR, Albers KM, Koller BH, Molliver DC. Mice lacking the P2Y2 receptor have deficits in noxious thermal sensation and neuronal responses to capsaicin. Soc. For Neuroscience, 2005 National Meeting; p. Abstract 393.16. [Google Scholar]
- 11.Yerxa BR, Sabater JR, Davis CW, Stutts MJ, Lang-Furr M, Picher M, Jones AC, Cowlen M, Dougherty R, Boyer J, Abraham WM, Boucher RC. Pharmacology of INS37217 [P(1)-(uridine 5′)-P(4)-(2′-deoxycytidine 5′)tetraphosphate, tetrasodium salt] a next-generation P2Y2 receptor agonist for the treatment of cystic fibrosis. J Pharmacol Exp Ther. 2002;302:871–880. doi: 10.1124/jpet.102.035485. [DOI] [PubMed] [Google Scholar]
- 12.Kellerman D, Evans R, Mathews D, Shaffer C. Inhaled P2Y2 receptor agonists as a treatment for patients with cystic fibrosis lung disease. Adv Drug Deliv Rev. 2002;54:1463–1474. doi: 10.1016/s0169-409x(02)00154-0. [DOI] [PubMed] [Google Scholar]
- 13.Jacobson KA, Costanzi S, Ivanov AA, Tchilibon S, Besada P, Gao ZG, Maddileti S, Harden TK. Structure activity and molecular modeling analyses of ribose- and base-modified uridine 5′-triphosphate analogues at the human P2Y2 and P2Y4 receptors. Biochem Pharmacol. 2006;71:540–549. doi: 10.1016/j.bcp.2005.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malmsjö M, Adner M, Harden TK, Pendergast W, Edvinsson L, Erlinge D. The stable pyrimidines UDPβS and UTPγS discriminate between the P2 receptors that mediate vascular contraction and relaxation of the rat mesenteric artery. Br J Pharmacol. 2000;131:51–56. doi: 10.1038/sj.bjp.0703536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erb L, Garrad R, Wang Y, Quinn T, Turner JT, Weisman GA. Site-directed mutagenesis of P2U purinoceptors. J Biol Chem. 1995;270:4185–4188. doi: 10.1074/jbc.270.9.4185. [DOI] [PubMed] [Google Scholar]
- 16.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 17.Mehler EL, Periole X, Hassan SA, Weinstein H. Key issues in the computational simulation of GPCR function: representation of loop domains. J Comput Aided Mol Des. 2002;16:841–853. doi: 10.1023/a:1023845015343. [DOI] [PubMed] [Google Scholar]
- 18.Costanzi S, Joshi BV, Maddileti S, Mamedova L, Gonzalez-Moa MJ, Marquez VE, Harden TK, Jacobson KA. Human P2Y6 receptor: Molecular modeling leads to the rational design of a novel agonist based on a unique conformational preference. J Med Chem. 2005;48:8108–8111. doi: 10.1021/jm050911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ballesteros JA, Shi L, Javitch A. Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol Pharmacol. 2001;60:1–19. [PubMed] [Google Scholar]
- 20.Yu H, Oprian DD. Tertiary interactions between transmembrane segments 3 and 5 near the cytoplasmic side of rhodopsin. Biochemistry. 1999;38:12033–12040. doi: 10.1021/bi9909492. [DOI] [PubMed] [Google Scholar]
- 21.Kim HS, Ravi RG, Marquez VE, Maddileti S, Wihlborg A-K, Erlinge D, Malmsjö M, Boyer JL, Harden TK, Jacobson KA. Methanocarba modification of uracil and adenine nucleotides: High potency of Northen ring conformation at P2Y1, P2Y2, P2Y4, P2Y11, but not P2Y6 receptors. J Med Chem. 2002;45:208–218. doi: 10.1021/jm010369e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer EA, Castellano RK, Diederich F. Interactions with aromatic rings in chemical and biological recognition. Angew Chem Int Ed. 2003;42:1210–1250. doi: 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- 23.Shaver SR, Rideout JL, Pendergast W, Douglass JG, Brown EG, Boyer JL, Patel RI, Redick CC, Jones AC, Picher M, Yerxa BR. Structure-activity relationships of dinucleotides: potent and selective agonists of P2Y receptors. Purinergic Signalling. 2005;1:183–191. doi: 10.1007/s11302-005-0648-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pendergast W, Yerxa BR, Douglass JG, Shaver SR, Dougherty RW, Redick CC, Sims IF, Rideout JL. Synthesis and P2Y receptor activity of a series of uridine dinucleotides 5′-polyphosphates. Bioorg Med Chem Lett. 2001;11:157–160. doi: 10.1016/s0960-894x(00)00612-0. [DOI] [PubMed] [Google Scholar]
- 25.Fradera X, Mestres J. Guided docking approaches to structure-based design and screening. Curr Top Med Chem. 2004;4:687–700. doi: 10.2174/1568026043451104. [DOI] [PubMed] [Google Scholar]
- 26.El-Tayeb A, Qi A, Müller CE. Synthesis and structure-activity relationships of uracil nucleotide derivatives and analogues as agonists at the human P2Y2, P2Y4, and P2Y6 receptors. J Med Chem. 2006 doi: 10.1021/jm060848j. in press. [DOI] [PubMed] [Google Scholar]
- 27.Verheyden JPH, Wagner D, Moffatt JG. Synthesis of some pyrimidine 2′-amino-2′-deoxynucleosides. J Org Chem. 1971;36:250–254. doi: 10.1021/jo00801a002. [DOI] [PubMed] [Google Scholar]
- 28.Rajeev KG, Prakash TP, Manoharan M. 2′-Modified-2-thiothymidine oligonucleotides. Org Lett. 2003;5:3005–3008. doi: 10.1021/ol0348607. [DOI] [PubMed] [Google Scholar]
- 29.Halbfinger E, Major DT, Ritzmann M, Ubl J, Reiser G, Boyer JL, Harden KT, Fischer B. Molecular recognition of modified adenine nucleotides by the P2Y1-receptor. 1. A synthetic, biochemical, and NMR approach. J Med Chem. 1999;42:5325– 5337. doi: 10.1021/jm990156d. [DOI] [PubMed] [Google Scholar]
- 30.Nicholas RA, Lazarowski ER, Watt WC, Li Q, Harden TK. Uridine nucleotide selectivity of three phospholipase C-activating P2 receptors: Identification of a UDP-selective, a UTP-selective, and an ATP- and UTP-specific receptor. Mol Pharmacol. 1996;50:224–229. [PubMed] [Google Scholar]
- 31.Brown HA, Lazarowski ER, Boucher RC, Harden TK. Evidence that UTP and ATP regulate phospholipase C through a common extracellular 5′-nucleotide receptor in human airway epithelial cells. Mol Pharmacol. 1991;40:648–655. [PubMed] [Google Scholar]
- 32.Poznanski J, Felczak K, Kulikowski T, Remin M. 1H NMR Conformational study of antiherpetic C5-substituted 2′-deoxyuridines: insight into the nature of structure-activity relationships. Biochem Biophys Res Comm. 2000;272:64–74. doi: 10.1006/bbrc.2000.2725. [DOI] [PubMed] [Google Scholar]
- 33.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 Å crystal structure. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 34.SYBYL 7.1, Tripos Inc., 1699 South Hanley Road, St. Louis, Missouri, 63144, USA
- 35.http://honiglab.cpmc.columbia.edu/programs/jackal/intro.html
- 36.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM - a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 37.Woolf TB, Roux B. Structure, energetics, and dynamics of lipid-protein interactions: A molecular dynamics study of the gramicidin A channel in a DMPC bilayer. Proteins. 1996;24:92–114. doi: 10.1002/(SICI)1097-0134(199601)24:1<92::AID-PROT7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 38.Mohamadi FN, Richards GJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. MacroModel - an integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J Comput Chem. 1990;11:440–467. [Google Scholar]
- 39._poner J, Leszczynski J, Hobza P. Thioguanine and thiouracil: hydrogen-bonding and stacking properties. J Phys Chem A. 1997;101:9489–9495. [Google Scholar]
- 40.Verheyden JPH, Wagner D, Moffatt JG. Synthesis of some pyrimidine 2′-amino-2′-deoxynucleosides. J Org Chem. 1971;36:250–245. doi: 10.1021/jo00801a002. [DOI] [PubMed] [Google Scholar]
- 41.Aurup H, Tuschl T, Benseler F, Ludwig J, Eckstein F. Oligonucleotide duplexes containing 2′-amino-2′-deoxycytidines: thermal stability and chemical reactivity. Nucl Acid Res. 1994;22:20–24. doi: 10.1093/nar/22.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lazarowski ER, Watt WC, Stutts MJ, Brown HA, Boucher RC, Harden TK. Enzymatic synthesis of UTP gamma S, a potent hydrolysis resistant agonist of the P2U-purinoceptors. Br J Pharmacol. 1996;117:203–209. doi: 10.1111/j.1476-5381.1996.tb15175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.