

Abstract
In 2008 we published the first set of guidelines for standardizing research in autophagy. Since then, research on this topic has continued to accelerate, and many new scientists have entered the field. Our knowledge base and relevant new technologies have also been expanding. Accordingly, it is important to update these guidelines for monitoring autophagy in different organisms. Various reviews have described the range of assays that have been used for this purpose. Nevertheless, there continues to be confusion regarding acceptable methods to measure autophagy, especially in multicellular eukaryotes. A key point that needs to be emphasized is that there is a difference between measurements that monitor the numbers or volume of autophagic elements (e.g., autophagosomes or autolysosomes) at any stage of the autophagic process vs. those that measure flux through the autophagy pathway (i.e., the complete process); thus, a block in macroautophagy that results in autophagosome accumulation needs to be differentiated from stimuli that result in increased autophagic activity, defined as increased autophagy induction coupled with increased delivery to, and degradation within, lysosomes (in most higher eukaryotes and some protists such as Dictyostelium) or the vacuole (in plants and fungi). In other words, it is especially important that investigators new to the field understand that the appearance of more autophagosomes does not necessarily equate with more autophagy. In fact, in many cases, autophagosomes accumulate because of a block in trafficking to lysosomes without a concomitant change in autophagosome biogenesis, whereas an increase in autolysosomes may reflect a reduction in degradative activity. Here, we present a set of guidelines for the selection and interpretation of methods for use by investigators who aim to examine macroautophagy and related processes, as well as for reviewers who need to provide realistic and reasonable critiques of papers that are focused on these processes. These guidelines are not meant to be a formulaic set of rules, because the appropriate assays depend in part on the question being asked and the system being used. In addition, we emphasize that no individual assay is guaranteed to be the most appropriate one in every situation, and we strongly recommend the use of multiple assays to monitor autophagy. In these guidelines, we consider these various methods of assessing autophagy and what information can, or cannot, be obtained from them. Finally, by discussing the merits and limits of particular autophagy assays, we hope to encourage technical innovation in the field.
Keywords: LC3, autolysosome, autophagosome, flux, lysosome, phagophore, stress, vacuole
Table of Contents
A. Methods for Monitoring Autophagy…462
1. Transmission electron microscopy…462
2. Atg8/LC3 detection and quantification…466
a. Western blotting and ubiquitin-like protein conjugation systems… 466
b. Turnover of LC3-II/Atg8–PE…470
c. GFP-Atg8/LC3 lysosomal delivery and proteolysis…472
d. GFP-Atg8/LC3 fluorescence microscopy…474
e. Tandem mRFP/mCherry-GFP fluorescence microscopy… 479
f. Autophagic flux determination using flow and multispectral imaging cytometry…480
g. Immunohistochemistry…482
3. SQSTM1/p62 and related LC3 binding protein turnover assays…483
4. MTOR, AMPK and Atg1/ULK1…484
5. Additional autophagy-related markers…487
a. ATG9…487
b. ATG12–ATG5…487
c. ATG14…488
d. ATG16L1…488
e. Atg18/WIPI family…488
f. BECN1/Atg6…488
g. DRAM1…489
h. ZFYVE1/DFCP1…489
6. Transcriptional and translational regulation…490
7. Autophagic protein degradation…491
8. Selective types of autophagy…493
a. The Cvt pathway, mitophagy, pexophagy and piecemeal microautophagy of the nucleus in yeast…493
b. Reticulophagy and ribophagy…494
c. Vacuole import and degradation pathway…494
d. Mammalian mitophagy and peroxisome degradation…495
e. Aggrephagy…497
f. Xenophagy…498
g. Lipophagy…499
h. Zymophagy…499
i. Allophagy…499
9. Autophagic sequestration assays…499
10. Turnover of autophagic compartments…500
11. Autophagosome-lysosome colocalization and dequenching assay… 500
12. Tissue fractionation…501
13. Analyses in vivo…502
14. Cell death…504
15. Chaperone-mediated autophagy…504
B. Comments on Additional Methods…507
1. Acidotropic dyes…507
2. Autophagy inhibitors and inducers…508
3. Basal autophagy…511
4. Experimental systems…511
5. Nomenclature…511
C. Methods and Challenges of Specialized Model Systems…512
1. C. elegans…512
2. Chicken B-lymphoid DT40 cells and retina…512
3. Chlamydomonas…512
4. Drosophila…512
5. Filamentous fungi…514
6. Honeybee…514
7. Human…514
8. Hydra…516
9. Large animals…516
10. Lepidoptera…517
11. Neotropical teleosts…517
12. Odontoblasts…517
13. Planarians…518
14. Plants…518
15. Protists…519
16. Rainbow trout …520
17. Sea urchin…520
18. Ticks…520
19. Zebrafish…520
20. Food biotechnology…521
Conclusions and Future Perspectives…522
References…522
Glossary … 543
Index…544
Introduction
Many researchers, especially those new to the field, need to determine which criteria are essential for demonstrating autophagy, either for the purposes of their own research, or in the capacity of a manuscript or grant review.1 This is an important issue, particularly considering that each of us may have his/her own opinion regarding the answer. Unfortunately, the answer is in part a “moving target” as the field evolves,2 and this can be extremely frustrating for researchers who may think they have met those criteria, only to find out that the reviewers of their paper have different ideas. Conversely, as a reviewer, it is tiresome to raise the same objections repeatedly, wondering why researchers have not fulfilled some of the basic requirements for establishing the occurrence of an autophagic process. In addition, drugs that potentially modulate autophagy are increasingly being used in clinical trials, and screens are being performed for new drugs that can modulate autophagy for therapeutic purposes. Clearly it is important to determine whether these drugs are truly affecting autophagy based on a set of accepted criteria. Accordingly, we describe here a basic set of contemporary guidelines that can be used by researchers to plan and interpret their experiments, by clinicians to evaluate the literature with regard to autophagy-modulating therapies, and by both authors and reviewers to justify or criticize an experimental approach.
Several fundamental points must be kept in mind as we establish guidelines for the selection of appropriate methods to monitor autophagy.1 Importantly, there are no absolute criteria for determining autophagic status that are applicable in every biological or experimental context. This is because some assays are inappropriate, problematic or may not work at all in particular cells, tissues or organisms.3-6 In addition, these guidelines are likely to evolve as new methodologies are developed and current assays are superseded. Nonetheless, it is useful to establish guidelines for acceptable assays that can reliably monitor autophagy in many experimental systems. It is important to note that in this set of guidelines the term “autophagy” generally refers to macroautophagy; other autophagy-related processes are specifically designated when appropriate.
For the purposes of this review, the autophagic compartments (Fig. 1) are referred to as the sequestering (pre-autophagosomal) phagophore (previously called the isolation or sequestration membrane7,8),9 the autophagosome,10 the amphisome (generated by the fusion of autophagosomes with endosomes, also referred to as an acidic late autophagosome11),12 the autolysosome (generated by fusion of autophagosomes or amphisomes with a lysosome), and the autophagic body (generated by fusion and release of the internal autophagosomal compartment into the vacuole in fungi and plants; autophagic bodies are not formed within lysosomes/autolysosomes because these lytic organelles are typically smaller than autophagosomes13).8,10 One critical point is that autophagy is a highly dynamic, multi-step process. Like other cellular pathways, it can be modulated at several steps, both positively and negatively. An accumulation of autophagosomes [measured by transmission electron microscopy (TEM) image analysis, as fluorescent GFP-MAP1LC3 (GFP-LC3) dots, or as LC3 lipidation on a western blot], could, for example, reflect induction of autophagy, reduction in autophagosome turnover,14-16 or the inability of turnover to keep pace with increased autophagosome formation (Fig. 1).17 For example, inefficient fusion with endosomes and/or lysosomes, respectively, or perturbation of the transport machinery,18 would inhibit autophagosome maturation to amphisomes or autolysosomes, whereas decreased flux could also be due to inefficient degradation of the cargo once fusion has occurred.19

Figure 1. Schematic model demonstrating the induction of autophagosome formation when turnover is blocked vs. normal autophagic flux, and illustrating the morphological intermediates of macroautophagy. (A) The initiation of autophagy includes the formation of the phagophore, the initial sequestering compartment, which expands into an autophagosome. Completion of the autophagosome is followed by fusion with lysosomes and degradation of the contents, allowing complete flux, or flow, through the entire pathway. This is a different outcome than the situation shown in (B) where induction results in the initiation of autophagy, but a defect in autophagosome turnover due, for example, to a block in fusion with lysosomes or disruption of lysosomal functions will result in an increased number of autophagosomes. In this scenario, autophagy has been induced, but there is no or limited autophagic flux. (C) An autophagosome can fuse with an endosome to generate an amphisome, prior to fusion with the lysosome. (D) Schematic drawing showing the formation of an autophagic body in plants and fungi. The large size of the plant and fungal vacuole relative to autophagosomes allows the release of the single-membrane autophagic body within the vacuole lumen. In cells that lack vacuolar hydrolase activity, or in the presence of inhibitors that block hydrolase activity, intact autophagic bodies accumulate within the vacuole lumen and can be detected by light microscopy. The lysosome of most higher eukaryotes is too small to allow the release of an autophagic body.
Accordingly, the use of autophagy markers such as LC3-II needs to be complemented by assays to estimate overall autophagic flux, or flow, to permit a correct interpretation of the results. That is, autophagic activity includes not just the increased synthesis or lipidation of Atg8/LC3 (LC3 is a mammalian homolog of yeast Atg8), or an increase in the formation of autophagosomes, but, most importantly, flux through the entire system, including lysosomes or the vacuole, and the subsequent release of the breakdown products. Therefore, autophagic substrates need to be monitored dynamically over time to verify that they have reached the lysosome/vacuole, and, when appropriate, are degraded. By responding to perturbations in the extracellular environment, cells tune autophagic flux to meet intracellular metabolic demands. The impact of autophagic flux on cell death and human pathologies therefore demands accurate tools to measure not only the current flux of the system, but also its capacity,20 and its response time, when exposed to a defined insult.21
One approach is to measure the rate of general protein breakdown by autophagy.8,22 Alternatively, it is possible to arrest the autophagic flux at a given point, and then record the time-dependent accumulation of an organelle, an organelle marker, a cargo marker or the entire cargo at the point of blockage; however, the latter assumes there is no feedback of the accumulating structure on its own rate of formation.23 Along the same lines, one can follow the time-dependent decrease of an autophagy-degradable marker (with the caveat that the potential contribution of other proteolytic systems needs to be experimentally addressed). In theory, this can be achieved by blocking autophagic sequestration at specific steps of the pathway (e.g., blocking further induction or nucleation of new phagophores) and by measuring the decrease of markers distal to the block point.14,16,24 The key issue is to differentiate between the often transient accumulation of autophagosomes due to increased induction, from accumulation due to inefficient completion of autophagy, by measuring both the levels of autophagosomes at static time points and by addressing changes in the rates of autophagic degradation of cellular components.19 Both processes have been used to estimate “autophagy,” but unless the experiments can relate changes in autophagosome numbers to a direct or indirect measurement for autophagic flux, they may be difficult to interpret.25 A general caution regarding the use of the term “steady state” is warranted at this point. It should not be assumed that an autophagic system is at steady-state in the strict biochemical meaning of this term, as this implies that the level of autophagosomes does not change with time, and the flux through the system is constant. In these guidelines, we use steady-state to refer to the baseline range of autophagic flux in a system that is not subjected to specific perturbations that increase or decrease that flux.
Autophagic flux refers to the entire process of autophagy including the delivery of cargo to lysosomes (via fusion of the latter with autophagosomes or amphisomes) and its subsequent breakdown and release of the resulting macromolecules back into the cytosol (this may be referred to as productive or complete autophagy). Thus, increases in the level of phosphatidylethanolamine (PE)-modified Atg8/LC3 (Atg8–PE/LC3-II), or even the appearance of autophagosomes are not measures of autophagic flux per se, but can reflect the induction of autophagic sequestration and/or inhibition of autophagosome or amphisome clearance. Also, it is important to realize that while formation of Atg8–PE/LC3-II appears to correlate with the induction of autophagy, we do not know, at present, the actual mechanistic relationship between Atg8–PE/LC3-II formation and the rest of the autophagic process; indeed, it may be possible to execute “self-eating” in the absence of LC3-II.26 As a final note, we also recommend that researchers refrain from the use of the expression “percent autophagy” when describing experimental results, as in “The cells displayed a 25% increase in autophagy.” In contrast, it is appropriate to indicate that the average number of GFP-Atg8/LC3 puncta per cell is increased or a certain percentage of cells display punctate GFP-Atg8/LC3 that exceeds a particular threshold (and this threshold should be clearly defined in the methods), or that there is a particular increase or decrease in the rate of degradation of long-lived proteins, as these are the actual measurements being quantified.
In the previous version of these guidelines,1 the methods were separated into two main sections—steady-state and flux. In some instances, a lack of clear distinction between the actual methodologies and their potential uses made such a separation somewhat artificial. For example, fluorescence microscopy was initially listed as a steady-state method, although this approach can clearly be used to monitor flux as described in this article, especially when considering the increasing availability of new technologies such as microfluidics. Furthermore, the use of multiple time points and/or lysosomal fusion/degradation inhibitors can turn even a typically static method such as TEM into one that monitors flux. Therefore, although we maintain the importance of monitoring autophagic flux and not just induction, this revised set of guidelines does not separate the methods based on this criterion. Readers should be aware that this article is not meant to present protocols, but rather guidelines, including information that is typically not presented in protocol papers. For detailed information on experimental procedures we refer readers to various protocols that have been published elsewhere.27-42
Collectively, we propose the following guidelines for measuring various aspects of selective and nonselective autophagy in eukaryotes.
A. Methods for Monitoring Autophagy
1. Transmission electron microscopy
Autophagy was first detected by TEM in the 1950s (reviewed in ref. 8). The focal degradation of cytoplasmic areas sequestered by the phagophore, which matures into the autophagosome, is the morphological hallmark of autophagy. TEM can be used to monitor both selective and nonselective autophagy. In the case of selective autophagy, the cargo should correspond to the specific substrate being targeted for sequestration—bulk cytoplasm is essentially excluded. In contrast, during nonselective autophagy, the content of the autophagosome is morphologically identical to the cytoplasm, containing similar densities of ribosomes, and intact sequestered organelles are clearly identifiable. Therefore, the use of TEM is a valid and important method both for the qualitative and quantitative analysis of changes in various autophagic structures that sequentially form the phagophore, autophagosome, amphisome, autolysosome and autophagic body (Fig. 1).43 The maturation from the phagophore through the autolysosome is a dynamic and continuous process,44 and, thus, the classification of compartments into discrete morphological subsets can be problematic; however, some basic guidelines can be offered.
Autophagosomes (also referred to as initial autophagic vacuoles, AVi) have a double membrane that is usually at least partly visible as two parallel membrane bilayers separated by an electron-lucent cleft (Fig. 2A).45,46 Autophagosomes contain cytosol and/or organelles that look morphologically intact, i.e., similar to the cytosol and organelles elsewhere in the cell.43,47 Amphisomes48 can sometimes be identified by the presence of small internal vesicles inside the autophagosome/autophagic vacuole (AV).49 These internal vesicles are delivered into the lumen by fusion with multivesicular endosomes. Late/degradative autophagic vacuoles and autolysosomes (AVd) usually have only one limiting membrane, and contain cytoplasmic material and/or organelles at various stages of degradation (Fig. 2A and B).43,47 It should be emphasized that not all vesicles containing electron-dense amorphous material are autolysosomes. The cytoplasmic origin of the contents must still be morphologically identifiable, if morphology is the only criterion that is being used for the identification of autolysosomes. For many biological and pathological situations, examination of both early and late autophagic structures yields valuable data regarding the overall autophagy/lysosomal status in the cells.17 Along these lines, it is possible to use immunocytochemistry to follow particular cytosolic proteins such as CuZn superoxide dismutase (SOD) and carbonic anhydrase to determine the stage of autophagy; the former is much more resistant to lysosomal degradation.50 In some autophagy-inducing conditions it is possible to observe multi-lamellar membrane structures in addition to the conventional double-membrane autophagosomes. The nature of these structures is not fully understood. They may indeed be multiple double layers of phagophores51 and positive for LC3,52 or mere artifacts of fixation.
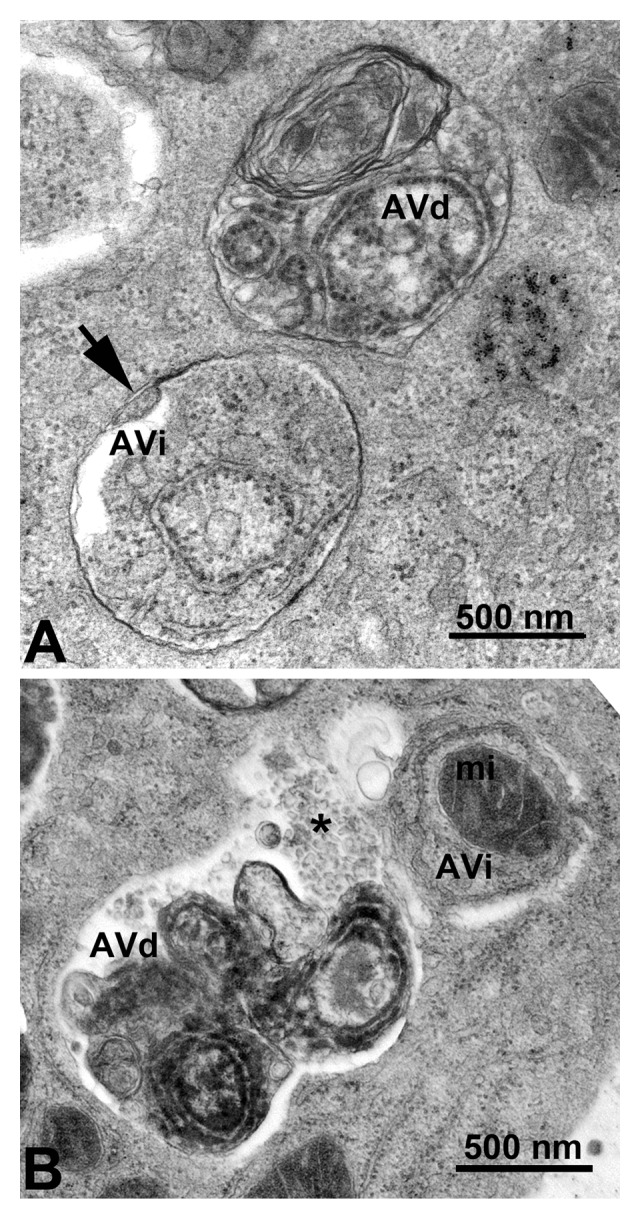
Figure 2. TEM images of autophagic vacuoles in isolated mouse hepatocytes. (A) One autophagosome or early autophagic vacuole (AVi) and one degradative autophagic vacuole (AVd) are shown. The AVi can be identified by its contents (morphologically intact cytoplasm, inlcuding ribosomes, and rough endoplasmic reticulum), and the limiting membrane that is partially visible as two bilayers separated by a narrow electron-lucent cleft, i. e., as a double membrane (arrow). The AVd can be identified by its contents, partially degraded, electron-dense rough endoplasmic reticulum. The vesicle next to the AVd is an endosomal/lysosomal structure containing 5-nm gold particles that were added to the culture medium to trace the endocytic pathway. (B) One AVi, containing rough endoplasmic reticulum and a mitochondrion, and one AVd, containing partially degraded rough endoplasmic reticulum, are shown. Note that the limiting membrane of the AVi is not clearly visible, possibly because it is tangentially sectioned. However, the electron-lucent cleft between the two limiting membranes is visible and helps in the identification of the AVi. The AVd contains a region filled by small internal vesicles (asterisk), indicating that the AVd has fused with a multivesicular endosome. mi, mitochondrion. Image provided by E.-L. Eskelinen.
TEM observations of platinum-carbon replicas obtained by the freeze fracture technique can also supply useful ultrastructural information on the autophagic process. In quickly frozen and fractured cells the fracture runs preferentially along the hydrophobic plane of the membranes, allowing characterization of the limiting membranes of the different types of autophagic vacuoles and visualization of their limited protein intramembrane particles (IMPs, or integral membrane proteins). Several studies have been performed using this technique on yeast,53 as well as on mammalian cells or tissue, first on mouse exocrine pancreas,54 then on mouse and rat liver,55,56 mouse seminal vesicle epithelium24,51 or cancer cell lines (e.g., breast cancer MDA-MB-231)57 to investigate the various phases of autophagosome maturation, and to reveal useful details about the origin and evolution of their limiting membranes.2,8,58-60
The phagophore and the limiting membranes of autophagosomes contain few, or no detectable, IMPs (Fig. 3A and B), when compared with other cellular membranes and to the membranes of lysosomes. In subsequent stages of the autophagic process the fusion of the autophagosome with an endosome and a lysosome results in increased density of IMPs in the membrane of the formed autophagic compartments (amphisomes, autolysosomes; Fig. 3C).8,24,53-56,61,62 Autolysosomes are generally delimited by a single membrane because, in addition to the engulfed material, the inner membrane is degraded by the lytic enzymes. Similarly, the limiting membrane of autophagic bodies in yeast and plants is also quickly broken down under normal conditions. Autophagic bodies can be stabilized, however, by the addition of phenylmethylsulphonylfluoride (PMSF) or genetically by the deletion of the yeast PEP4 gene. Thus, another method to consider for monitoring autophagy in plants and yeast is to count autophagic bodies by TEM using at least two time points. The advantage of this approach is that it can provide accurate information on flux even when the autophagosomes are abnormally small.63,64 Thus, although a high frequency of “abnormal” structures presents a challenge, TEM is still very helpful in analyzing autophagy.

Figure 3. Different autophagic vesicles observed after freeze fracturing in cultured osteosarcoma cells after treatment with the autophagy inducer voacamine.59 (A) Early autophagosome delimited by a double membrane. (B) Inner monolayer of an autophagosome membrane lacking protein particles. (C) Autolysosome delimited by a single membrane rich in protein particles. In the cross-fractured portion (on the right) the profile of the single membrane and the inner digested material are easily visible. Images provided by S. Meschini, M. Condello and A. Giuseppe.
Cautionary notes: Although TEM is one of the most widely used methodologies to monitor autophagy, it is also one of the most problematic due to misinterpretations mostly deriving from methodological artifacts.45,46,65,66 Care in the choice of sample to be analyzed is critical to the success of TEM studies for autophagy. Whereas fixation of in vitro samples is relatively straightforward, fixation of excised tissues requires care to avoid sampling a nonrepresentative or uninformative section of tissue. For instance, if 95% of a tumor is necrotic, TEM analysis of the necrotic core may not be informative, and if the sampling is from the viable rim, this needs to be specified when reported. Ex vivo tissue should be fixed immediately and systematically across samples to avoid changes in autophagy that may occur simply due to elapsed time ex vivo. It is recommended that for tissue samples, perfusion fixation should be used when possible. For yeast, rapid freezing techniques such as high pressure freezing followed by freeze substitution (i.e., dehydration at low temperature) may be particularly useful.
Due to the high potential for sampling artifacts, careful selection of appropriate nonbiased methods of quantification and morphometric/stereological analyses is essential.67-69 Data obtained simply by scoring for the presence or absence of autophagic vacuoles (autophagosomes, autolysosomes) in the section of a cell leads to unreliable results due to variability in cell areas, and autophagic vacuole profiles in the sections. It is more reliable to quantify autophagosome (and/or autolysosome) profiles per total cytoplasmic or cellular area in sections, which still includes an unaccounted variability in the profile size of the autophagic element. The best approach is to estimate the volume occupied by autophagic structures (as percent of cytoplasmic or cellular volume, using volumetric morphometry/stereology) in at least 20 cell profiles per sample (the number needed should be dictated by some form of power analysis, which indicates that the data are significant).20,65,68,70,71 During quantification it is important to make sure that each imaged cell profile is captured and scored at the same magnification, and that every cell profile in the thin section has an equal probability to be included in the counting.
The accurate identification of the autophagosome is a prerequisite for a valid analysis. An additional complication, however, is that maturation of metazoan autophagosomes involves a transition from a double-membrane compartment to single-membrane structures (i.e., amphisomes and autolysosomes).72 In addition, not all double-membrane structures are autophagosomes. Thus, double membranes cannot be relied upon as the sole means for the ultrastructural identification of autophagy-related structures, and it is important to employ expert analysis to avoid misinterpretation of micrographs.46,65,66 In some cases, it may be prudent to employ tomographic reconstructions of the TEM images to confirm that the autophagic compartments are spherical and are not being confused with endomembrane cisternae or damaged mitochondria with similar appearance in thin-sections (e.g., see ref. 73), but this is obviously a time-consuming approach requiring sophisticated equipment. In addition, interpretation of tomographic images can be problematic. For example, starvation-induced autophagosomes should contain cytoplasm (i.e., cytosol and possibly organelles), but autophagosome-related structures involved in specific types of autophagy should show the selective cytoplasmic target, but may be relatively devoid of cytoplasm. Such processes include selective peroxisome or mitochondria degradation (pexophagy or mitophagy, respectively),74,75 targeted degradation of pathogenic microbes (xenophagy),76-81 as well as the yeast biosynthetic cytoplasm-to-vacuole targeting (Cvt) pathway.82 Furthermore, some pathogenic microbes express membrane-disrupting factors during infection (e.g., phospholipases) that disrupt the normal double-membrane architecture of autophagosomes.83 It is not even clear if the sequestering compartments used for specific organelle degradation or xenophagy should be termed autophagosomes or if alternate terms such as pexophagosome,84 mitophagosome and xenophagosome should be used, even though the membrane and mechanisms involved in their formation may be identical to those for starvation-induced autophagosomes; for example, the double-membrane vesicle of the Cvt pathway is referred to as a Cvt vesicle.
It can also be difficult to determine whether material present within a phagosomal structure reflects self-eating, or is from a heterophagic (consumption of components from outside the cell) process. A prominent example is related to apoptosis. Apoptotic bodies from neighboring cells are readily phagocytosed by surviving cells of the same tissue.85,86 Immediately after phagocytic uptake of apoptotic bodies, phagosomes have double limiting membranes. The inner one is from the plasma membrane of the apoptotic body and the outer one is that of the phagocytizing cell. The early heterophagic vacuole formed in this way may appear similar to an autophagosome or, in a later stage, an early autolysosome in that it contains recognizable cytoplasmic material. A major difference, however, is that the surrounding membranes are the thicker (plasma membrane type), rather than the thinner sequestration membrane type (9–10 nm, vs. 7–8 nm, respectively).66 A good feature to distinguish between autophagosomes and double plasma membrane-bound structures is the lack of the distended empty space (characteristic for the sequestration membranes of autophagosomes) between the two membranes of the phagocytic vacuoles. In addition, engulfed apoptotic bodies usually have a larger average size than autophagosomes.87 The problem of heterophagic elements interfering with the identification of autophagic ones is most prominent in cell types with particularly intense heterophagic activity (such as macrophages, and amoeboid or ciliate protists). Special attention has to be paid to this problem in cell cultures or in vivo treatments (e.g., with toxic or chemotherapeutic agents) causing extensive apoptosis.
To decide about the lytic nature of a vacuolar compartment, demonstration of the presence of lysosomal enzymes by traditional (enzyme) cytochemistry or immunocytochemistry is also feasible for identifying post-fusion autophagic compartments. However, when heterophagy and autophagy are going on in parallel, the fusion of secondary lysosomes from both sources happens without distinction. The result will be a mixture of degradative products that may be derived both from heterophagy and autophagy, making it impossible to determine the proportion derived from a single process.
There are numerous structures in cells that resemble, or can be confused with, autophagic vesicles. The most common organelles are mitochondria, and ER, and also (depending on their structure) plastids in plants. Due to the cisternal structure of the ER, double membrane-like structures surrounding mitochondria or other organelles are often observed after sectioning,88 but these can also correspond to cisternae of the ER coming into and out of the section plane.45 If there are ribosomes associated with these membranes they can help distinguish them from the ribosome-free double-membrane of the phagophore and autophagosome. Observation of a mixture of early and late autophagosomes that is modulated by time point of collection and/or brief pulses of bafilomycin A1 (a V-ATPase inhibitor) to trap the cargo in a recognizable early state41 increases the confidence that an autophagic process is being observed. Criteria for distinguishing specialized structures such as melanosomes containing electron dense pigment from autophagosomes should be specified prior to scoring and included in a methods section when reporting the results. Considering that swollen mitochondria can encompass most of the internal structure of small autophagosomes, the possibility exists for scoring errors in every study. To minimize the impact of such errors, exact specification of autophagic elements must be applied. Efforts should be made to clarify the nature of questionable structures by extensive preliminary comparison in many test areas. Elements that still remain questionable should be categorized into special groups and measured separately. Should their later identification become possible, they can be added to the proper category or, if not, kept separate.
Uncertainties of identification and special features of the autophagic process may be clarified by immuno-TEM with gold-labeling,89,90 using antibodies, for example, to cargo proteins of cytoplasmic origin and to LC3 to verify the autophagic nature of the compartment. Although labeling of LC3 can be difficult, good antibodies exist to visualize the GFP moiety of GFP-LC3 reporter constructs.91 Antibodies against an abundant cytosolic protein will result in high background labeling; however, organelle markers work well. Because there are very few characterized proteins that remain associated with the completed structure, the choices for confirmation of its autophagic nature are limited. Furthermore, autophagosome-associated proteins may be cell type-specific. At any rate, the success of this methodology depends on the quality of the antibodies and also on the TEM preparation and fixation procedures utilized. With immuno-TEM, authors should provide controls showing that labeling is specific. This may require a quantification of staining over different cellular compartments.
In addition, statistical information should be provided due to the necessity of showing only a selective number of sections. Again, we note that for quantitative data it is preferable to use proper volumetric analysis rather than just counting numbers of sectioned objects. On the one hand, it must be kept in mind that even volumetric morphometry/stereology only shows either steady-state levels, or a snapshot in a changing dynamic process. Such data by themselves are not informative regarding autophagic flux, unless performed over multiple time points. Alternatively, investigation in the presence and absence of flux inhibitors can reveal the dynamic changes in various stages of the autophagic process.14,20,92,93 For example, if the turnover of autolysosomes is very rapid, a low number/volume will not necessarily be an accurate reflection of low autophagic activity. On the other hand, quantitative analyses indicate that autophagosome volume in many cases does correlate with the rates of protein degradation.94-96 One potential compromise is to perform whole cell quantification of autophagosomes using fluorescence methods, with qualitative verification by TEM,97 to show that the changes in fluorescent puncta reflect increases in autophagic structures.
One additional caveat with TEM, and to some extent with confocal fluorescence microscopy, is that the analysis of a single plane within a cell can be misleading and may make the identification of autophagic structures difficult. Confocal microscopy and fluorescence microscopy with deconvolution software (or with much more work, 3-dimensional TEM) can be used to generate multiple/serial sections of the same cell to reduce this concern; however, in some cases where there is sufficient structural resolution, analysis of a single plane with multiple cells can suffice given practical limitations. Newer EM technologies, including focused ion beam dual-beam EM, should make it much easier to apply three-dimensional analyses. An additional methodology to assess autophagosome accumulation is correlative light and electron microscopy, CLEM, which is helpful in confirming that fluorescent structures are autophagosomes.98,99 Along these lines, the new mini Singlet Oxygen Generator (miniSOG) fluorescent flavoprotein, which is less than half the size of GFP, provides an additional means to genetically tag proteins for CLEM analysis under conditions that are particularly suited to subsequent TEM analysis.100 Combinatorial assays using tandem mRFP-GFP-LC3 (see Tandem mRFP/mCherry-GFP fluorescence microscopy) along with static TEM images should help in the analysis of flux and the visualization of cargo structures.101 Another technique that has proven quite useful for analyzing the complex membrane structures that participate in autophagy is three-dimensional electron tomography,102,103 and cryoelectron microscopy (Fig. 4). Finally, although an indirect measurement, a comparison of the ratio of autophagosomes to autolysosomes by TEM can support alterations in autophagy identified by other procedures.105 In this case it is important to always compare samples to the control of the same cell type and in the same growth phase, as the ratio of autophagosome/autolysosome varies in a cell context-dependent fashion, depending on their clearance activity. It may also be necessary to distinguish autolysosomes from telolysosomes/late secondary lysosomes (the former are actively engaged in degradation, whereas the latter have reached an end point in the breakdown of lumenal contents and are also referred to as residual bodies; see Tissue fractionation) because lysosome numbers generally increase when autophagy is induced.

Figure 4. Cryoelectron microscopy can be used as a three-dimensional approach to monitor the autophagic process. Two computed sections of an electron tomogram of the autophagic vesicle-rich cytoplasm in a hemophagocyte of a semi-thin section after high-pressure freezing preparation. The dashed area is membrane-free (A) but tomography reveals newly formed phagophore-like membranes (B). Image published previously104 and provided by M. Schneider and P. Walter.
With regard to immunohistochemistry using SOD1 as a marker to follow autophagy, it should be noted that a portion of the CuZn superoxide dismutase is also associated with various organelles, including the ER, mitochondria and peroxisomes. In addition, the wild-type SOD1 protein can be oxidized, and this form (SODox) is associated with sporadic cases of amyotrophic lateral sclerosis.106 Thus, the oxidized form of SOD1 might interfere with autophagy, and, if so, it may not be a good choice for a marker to monitor steady-state autophagic flux.
Conclusion: EM can be an extremely informative method for monitoring autophagy; however, it must be performed with extreme caution and rigor to avoid bias, and to ensure correct identification and quantification of autophagic compartments. With TEM, immunogold labeling is strongly recommended as it generally provides the most unequivocal results. Whenever possible, EM should not be the sole method used to monitor autophagy, but rather should be complemented by additional assays as described in this article.
2. Atg8/LC3 detection and quantification
Atg8/LC3 is the most widely monitored autophagy-related protein. In this section we describe multiple assays that utilize this protein, separating the descriptions into several subsections for ease of discussion.
a. Western blotting and ubiquitin-like protein conjugation systems
The Atg8/LC3 protein is a ubiquitin-like protein that can be conjugated to PE (and possibly to phosphatidylserine107). In yeast and several other organisms, the conjugated form is referred to as Atg8–PE. The mammalian homologs of Atg8 constitute a family of proteins subdivided in two subfamilies: LC3 (microtubule-associated protein 1 light chain 3) and GABARAP (GABAA receptor-associated protein). The former is comprised of LC3A, B, B2 and C, whereas the latter family includes GABARAP, GABARAPL1/Atg8L/GEC1 (GABAA receptor-associated protein like 1/Glandular Epithelial Cell 1), GABARAPL2/GATE-16/GEF2 (GABAA receptor-associated protein like 2/Golgi-associated ATPase enhancer of 16 kDa/ganglioside expression factor 2) and GABARAPL3 (GABAA receptor-associated protein like 3).108 The nonlipidated and lipidated forms are usually referred to as LC3-I and LC3-II, or GABARAP and GABARAP–PE, etc. The positions of both Atg8/LC3-I (approximately 18 kDa) and Atg8–PE/LC3-II (approximately 16 kDa) should be indicated on western blots whenever both are detectable.
The mammalian Atg8 homologs share from 29% to 94% sequence identity and have all, apart from GABARAPL3, been demonstrated to be involved in autophagosome biogenesis.109 The LC3 proteins are involved in phagophore formation, with participation of GABARAP subfamily members in later stages of autophagosome formation, in particular phagophore elongation and closure.110 Nevertheless, in most published studies, LC3 has been the primary Atg8 homolog examined in mammalian cells and the one that is typically characterized as an autophagosome marker per se, making this factor the most relevant for this discussion (note that although this protein is referred to as “Atg8” in many other systems, for simplicity we primarily refer to it here as LC3 to distinguish it from the yeast protein). LC3, like the other Atg8 homologs, is initially synthesized in an unprocessed form, proLC3, which is converted into a proteolytically processed form lacking amino acids from the C terminus, LC3-I, and is finally modified into the PE-conjugated form, LC3-II (Fig. 5). Atg8–PE/LC3-II is the only protein marker that is reliably associated with completed autophagosomes, but is also localized to phagophores. In yeast, Atg8 amounts increase at least 10-fold when autophagy is induced.113 In mammalian cells, however, the total levels of LC3 do not necessarily change in a predictable manner, as there may be increases in the conversion of LC3-I to LC3-II, or a decrease in LC3-II relative to LC3-I if degradation of LC3-II via lysosomal turnover is particularly rapid. Both of these events can be seen sequentially in several cell types as a response to total nutrient and serum starvation. In cells of neuronal origin a high ratio of LC3-I to LC3-II is a common finding.114 For instance, SH-SY5Y neuroblastoma cell lines display only a slight increase of LC3-II after nutrient deprivation, whereas LC3-I is clearly reduced. This is likely related to a high basal autophagic flux, as suggested by the higher increase in LC3-II when cells are treated with NH4Cl,115,116 although cell-specific differences in transcriptional regulation of LC3 may also play a role. The pattern of LC3-I to LC3-II conversion seems not only to be cell specific, but also related to the kind of stress to which cells are subjected. For example, the same SH-SY5Y cells display a strong increase of LC3-II when treated with the mitochondrial uncoupler CCCP, a well-known inducer of mitophagy. Thus, neither assessment of LC3-I consumption nor the evaluation of LC3-II levels would necessarily reveal a slight induction of autophagy (e.g., by rapamycin). Also, there is not always a clear precursor/product relationship between LC3-I and LC3-II, because the conversion of the former to the latter is cell type-specific and dependent on the treatment used to induce autophagy. Accumulation of LC3-II can be obtained by interrupting the autophagosome-lysosome fusion step (e.g., by depolymerizing acetylated microtubules with vinblastine, or by raising the lysosomal pH with the lysosomal proton pump inhibitor bafilomycin A1) or by inhibiting lysosome-mediated proteolysis (e.g., with the cysteine protease inhibitor E-64d, the aspartic protease inhibitor pepstatin A, or chloroquine117). Western blotting can be used to monitor changes in LC3 amounts (Fig. 5);25,118 however, even if the total amount of LC3 does increase, the magnitude of the response is generally less than that documented in yeast. It is worth noting that since the conjugated forms of the GABARAP subfamily members are usually undetectable without induction of autophagy in mammalian cells,119 these proteins might be more suitable than LC3 to study and quantify subtle changes in the autophagic flux.

Figure 5. LC3-I conversion and LC3-II turnover. (A) Expression levels of LC3-I and LC3-II during starvation. Atg5+/+ (wild-type) and Atg5−/− MEFs were cultured in DMEM without amino acids and serum for the indicated times, and then subjected to immunoblot analysis using anti-LC3 antibody and anti-tubulin antibody. E-64d (10 µg/ml) and pepstatin A (10 µg/ml) were added to the medium where indicated. Positions of LC3-I and LC3-II are indicated. The inclusion of lysosomal protease inhibitors reveals that the apparent decrease in LC3-II is due to lysosomal degradation as easily seen by comparing samples with and without inhibitors at the same time points (the overall decrease seen in the presence of inhibitors may reflect decreasing effectiveness of the inhibitors over time). Monitoring autophagy by following steady-state amounts of LC3-II without including inhibitors in the analysis can result in an incorrect interpretation that autophagy is not taking place (due to the apparent absence of LC3-II). Conversely, if there are high levels of LC3-II but there is no change in the presence of inhibitors this may indicate that induction has occurred but that the final steps of autophagy are blocked, resulting in stabilization of this protein. This figure was modified from data previously published in reference 25 and is reproduced by permission of Landes Bioscience, copyright 2007. (B) Lysates of 4 human adipose tissue biopsies were resolved on two 12% polyacrylamide gels, as described previously.111 Proteins were transferred in parallel to either a PVDF or a nitrocellulose membrane, and blotted with anti-LC3 antibody, and then identified by reacting the membranes with an HRP-conjugated anti-rabbit IgG antibody, followed by ECL. The LC3-II/LC3-I ratio was calculated based on densitometry analysis of both bands. *p < 0.05. (C) HEK 293 and HeLa cells were cultured in nutrient-rich medium (DMEM containing10% FCS) or incubated for 4 h in starvation conditions (Krebs-Ringer medium) in the absence (-) or presence (+) of E-64d and pepstatin at 10 µg/ml each (Inhibitors). Cells were then lysed and the proteins resolved by SDS-PAGE. Endogenous LC3 was detected by immunoblotting. Positions of LC3-I and LC3-II are indicated. In the absence of lysosomal protease inhibitors, starvation results in a modest increase (HEK 293 cells) or even a decrease (HeLa cells) in the amount of LC3-II. The use of inhibitors reveals that this apparent decrease is due to lysosome-dependent degradation. This figure was modified from data previously published in reference 112 and is reproduced by permission of Landes Bioscience, copyright 2005. (D) Sequence and schematic representation of the different forms of LC3B. The sequence for the nascent (proLC3) from mouse is shown. The glycine at position 120 indicates the cleavage site for ATG4. After this cleavage, the truncated LC3 is referred to as LC3-I, which is still a soluble form of the protein. Conjugation to PE generates the membrane-associated LC3-II (equivalent to Atg8–PE).
In most organisms, Atg8/LC3 is initially synthesized with a C-terminal extension that is removed by the Atg4 protease. Accordingly, it is possible to use this processing event to monitor Atg4 activity. For example, when GFP is fused at the C terminus of Atg8 (Atg8-GFP), the GFP moiety is removed in the cytosol to generate free Atg8 and GFP. This processing can be easily monitored by western blot.120 It is also possible to use assays with an artificial fluorogenic substrate, or a fusion of LC3B to phospholipase A(2) that allows the release of the active phospholipase for a subsequent fluorogenic assay,121 and there is a FRET-based assay utilizing CFP and YFP tagged versions of LC3B and GABARAPL2 that can be used for high-throughput screening.122 Another method to monitor ATG4 activity in vivo uses the release of Gaussia luciferase from the C terminus of LC3 that is tethered to actin.123 Note that there are four Atg4 homologs in mammals, and they have different activities with regard to the Atg8 subfamilies of proteins.124 ATG4A is able to cleave the GABARAP subfamily, but has very limited activity toward the LC3 subfamily, whereas ATG4B is apparently active against most or all of these proteins. The ATG4C and ATG4D isoforms have minimal activity for any of the Atg8 homologs. In particular because a C-terminal fusion will be cleaved immediately by Atg4, researchers should be careful to correctly specify whether they are using GFP-Atg8/LC3 (an N-terminal fusion, which can be used to monitor various steps of autophagy) or Atg8/LC3-GFP (a C-terminal fusion, which can only be used to monitor Atg4 activity).125
Cautionary notes: There are several important caveats to using Atg8/LC3-II or GABARAP-PE to visualize fluctuations in autophagy. First, changes in LC3-II amounts are tissue- and cell context-dependent.112,126 Indeed, in some cases, autophagosome accumulation detected by TEM does not correlate well with the amount of LC3-II (Tallóczy Z, de Vries RLA, Sulzer D, unpublished results; Eskelinen E-L, unpublished results). This is particularly evident in those cells that show low levels of LC3-II (based on western blotting) because of an intense autophagy flux that consumes this protein,127 or in cell lines having high levels of LC3-II that are tumor-derived, such as MDA-MB-231.112 Conversely, the detectable formation of LC3-II is not sufficient evidence for autophagy, without careful quantification. For example, homozygous deletion of Becn1 does not prevent the formation of LC3-II in embryonic stem cells even though autophagy is substantially reduced, whereas deletion of Atg5 results in the complete absence of LC3-II (see Fig. 5A and Supplemental Data in ref. 128). The same is true for the generation of Atg8–PE in yeast in the absence of ATG6 (see Fig. 7 in ref. 129). Thus, it is important to remember that not all of the autophagy-related proteins are required for Atg8/LC3 processing, including lipidation.129 Vagaries in the detection and amounts of LC3-I vs. LC3-II present technical problems. For example, LC3-I is very abundant in brain tissue, and the intensity of the LC3-I band may obscure detection of LC3-II, unless the polyacrylamide crosslinking density is optimized. Conversely, certain cell lines have much less visible LC3-I compared with LC3-II. In addition, tissues may have asynchronous and heterogeneous cell populations, and this may present challenges when analyzing LC3 by western blotting.
Second, LC3-II also associates with the membranes of nonautophagic structures. For example, some members of the γ-protocadherin family undergo clustering to form intracellular tubules that emanate from lysosomes.130 LC3-II is recruited to these tubules, and appears to promote or stabilize membrane expansion. Furthermore, LC3 can be recruited directly to bacteria-containing phagosome membranes under certain immune activating conditions, for example, TLR-mediated stimulation, in a process designated LC3-associated phagocytosis (LAP)131,132 and also to apoptotic cell-containing phagosome membranes,133,134 macropinosomes,133 and to single-membrane entotic vacuoles.133 TEM analysis of murine macrophage-like RAW 264.7 cells infected with Burkholderia pseudomallei reveals that intracellular bacteria colocalize with GFP-LC3 puncta. However, the TEM analysis further shows that bacteria are either free in the cytosol, or sequestered in single-membrane phagosomes rather than within canonical double-membrane autophagosomes.135 Therefore, in studies of infection of mammalian cells by bacterial pathogens the identity of the LC3-II-labeled compartment as an autophagosome should be confirmed by a second method, such as TEM. It is also worth noting that autophagy induced in response to bacterial infection is not directed solely against the bacteria but can also be a response to remnants of the phagocytic membrane.136 Similar cautions apply with regard to viral infection, as coronaviruses induce the formation of double-membrane vesicles that are coated with LC3-I, and this nonlipidated form of LC3 plays an autophagy-independent role in viral replication.137 Along these lines, with herpes simplex virus infection, an LC3+ autophagosome-like organelle derived from nuclear membranes is observed that contains viral proteins.138
Third, caution must be exercised in general when evaluating LC3 by western blotting, and appropriate standardization controls are necessary. For example, LC3-I may be less sensitive to detection by certain anti-LC3 antibodies. Moreover, LC3-I is more labile than LC3-II, being more sensitive to freezing-thawing and to degradation in SDS sample buffer, so fresh samples should be heated and assessed as soon as possible and should not be subjected to repeated freeze-thaw cycles. A general point to consider when examining transfected cells concerns the efficiency of transfection. A western blot will detect LC3 in the entire cell population, including those that are not transfected. Thus, if transfection efficiency is too low, it may be necessary to use methods, such as fluorescence microscopy, that allow autophagy to be monitored in single cells. The critical point is that the analysis of the gel shift of transfected LC3 or GFP-LC3 can be employed to follow LC3 lipidation only in readily transfected cells.139
When dealing with animal tissues, western blotting of LC3 should be performed on frozen biopsy samples homogenized in the presence of general protease inhibitors (Isidoro C, personal communication; see also Human).140 Caveats regarding detection of LC3 by western blotting have been covered in a review.25 For example, PVDF membranes may result in a stronger LC3-II retention than nitrocellulose membranes, possibly due to a higher affinity for hydrophobic proteins (Fig. 5B; Kovsan J, Rudich A, personal communication), and Triton X-100 may not efficiently solubilize LC3-II in some systems.141 Heating in the presence of 1% SDS, or analysis of membrane fractions,142 may assist in the detection of this protein.
Another important issue concerns the quantification of changes in LC3-II. The previous version of these guidelines specifically stated that the levels of LC3-II should be compared with actin (and here we would modify this to include other appropriate “housekeeping” proteins) and not to that of LC3-I. As a general rule, this still holds true, but there are some exceptions. For example, in some cases actin levels decrease when autophagy is induced, so that it may be more appropriate to determine the ratio of LC3-II to LC3-I. Either method has its potential advantages and disadvantages. For example, if the amount of LC3-I is high relative to LC3-II (as in brain tissues, where the LC3-I signal can be overwhelming), it can be difficult to quantify the change in LC3-II relative to LC3-I. Conversely, by ignoring the level of LC3-I in favor of LC3-II the researcher may miss part of the overall picture of the cellular autophagic response.
Fourth, LC3 is expressed as four isoforms in mammalian cells, LC3A, LC3B, LC3B2 and LC3C,143,144 which exhibit different tissue distributions, and it may be necessary to use different antisera or antibodies that distinguish among these isoforms. A point of caution along these lines is that the increase in LC3A-II vs. LC3B-II levels may not display equivalent changes in all organisms under autophagy-inducing conditions, and it should not be assumed that LC3B is the optimal protein to monitor.145 This supports the important notion that the LC3 isoforms display different functions, but these are yet to be fully elucidated. The commercialized anti-LC3B antibodies also recognize LC3A, but do not recognize LC3C, which shares less sequence homology. It is important to note that LC3C possesses in its primary amino acid sequence the DYKD motif that is recognized with a high affinity by anti-FLAG antibodies. Thus, the standard anti-FLAG M2 antibody can detect and immunoprecipitate overexpressed LC3C, and caution has to be taken in experiments using FLAG-tagged proteins (Biard-Piechaczyk M, Espert L, personal communication).
In addition, it is important to keep in mind the presence of the other subfamily of Atg8 proteins, the GABARAP subfamily (see above).109,146 Certain types of mitophagy induced by BNIP3L/NIX are highly dependent on GABARAP and less dependent on LC3 proteins.147 Furthermore, commercial antibodies for GABARAPL1 also recognize GABARAP,108 which might lead to misinterpretation of experiments, in particular those using immunohistochemical techniques. The problem with cross-reactivity of the anti-GABARAPL1 antibody can be overcome when analyzing these proteins by western blot because they can be resolved during SDS-PAGE using high concentration (15%) gels, with GABARAP migrating faster than GABARAPL1 (Boyer-Guittaut M, personal communication). We therefore advise caution in choosing antibodies for western blotting and immunofluorescence experiments and in interpreting results based on stated affinities of antibodies unless these have been clearly determined. As with any western blot, proper methods of quantification must be used, which are, unfortunately, often not well disseminated, and the readers are referred to an excellent paper on this subject (see ref. 148). Unlike the other members of the GABARAP family, almost no information is available on GABARAPL3, perhaps because it is not yet possible to differentiate between GABARAPL1 and GABARAPL3 proteins, which have 94% identity. As stated by the laboratory that described the cloning of the human GABARAPL1 and GABARAPL3 genes,146 their expression patterns are apparently identical. It is worth noting that GABARAPL3 is the only gene of the GABARAP subfamily that seems to lack an ortholog in mice.146 GABARAPL3 might therefore be considered as a pseudogene without an intron that is derived from GABARAPL1. Hence, until new data are published, GABARAPL3 should not be considered as the fourth member of the GABARAP family.
Fifth, in non-mammalian species, the discrimination of Atg8–PE from the nonlipidated form can be complicated by their nearly identical SDS-PAGE mobilities and the presence of multiple isoforms (e.g., there are 9 in Arabidopsis). In yeast, it is possible to resolve Atg8 (the nonlipidated form) from Atg8–PE by including 6 M urea in the SDS-PAGE separating gel,149 or by using a 15% resolving gel without urea (Reggiori F, personal communication). Similarly, urea combined with prior treatment of the samples with (or without) phospholipase D (that will remove the PE moiety) can often resolve the ATG8 species in plants.150,151 It is also possible to label cells with radioactive ethanolamine, followed by autoradiography to identify Atg8–PE, and a C-terminal peptide can be analyzed by mass spectrometry to identify the lipid modification at the terminal glycine residue. Furthermore, Atg8–PE aberrantly migrates faster than unconjugated Atg8 during SDS-PAGE, even though the former has a larger molecular mass. Special treatments are not needed for the separation of mammalian LC3-I from LC3-II.
Finally, we would like to point out that one general issue with regard to any assay is that it could introduce some type of stress—for example, mechanical stress due to lysis, temperature stress due to heating or cooling a sample, or oxidative stress on a microscope slide, which could lead to potential artifacts including the induction of autophagy.152 This point is not intended to limit the use of any specific methodology, but rather to note that there are no perfect assays; special care should be taken with cells in suspension, however, as the stress resulting from centrifugation can induce autophagy. Therefore, it is important to verify that the positive (e.g., treatment with rapamycin, torin1 or other inducers) and negative (e.g., inhibitor treatment) controls behave as expected in any assays being utilized. Similarly, plasmid transfection or nucleofection can result in the potent induction of autophagy (based on increases in LC3-II or SQSTM1 degradation). In some cell types, the amount of autophagy induced by transfection of a control empty vector may be so high that it is virtually impossible to examine the effect of enforced gene expression on autophagy (Levine B, personal communication). This effect is generally not observed with siRNA transfection; however, it is an issue for plasmid shRNA transfection and for plasmid expression constructs. The use of endotoxin-free DNA reduces, but does not eliminate, this problem. Finally, the precise composition of media components can have profound effects on basal autophagy levels and may need to be modified empirically depending on the cell lines being used.
Conclusion: Atg8/LC3 is often an excellent marker for autophagy; however, it must be kept in mind that there are multiple LC3 isoforms, there is a second family of mammalian Atg8-like proteins (GABARAPs), and antibody affinity (for LC3-I vs. LC3-II) and specificity (for example, for LC3A vs. LC3B) must be considered and/or determined.
b. Turnover of LC3-II/Atg8–PE
Autophagic flux can be measured by inferring LC3-II/Atg8?PE turnover by western blot (Fig. 5C)112 in the presence and absence of lysosomal, or vacuolar, degradation. The relevant parameter in this assay is the difference in the amount of LC3-II in the presence and absence of saturating levels of inhibitors, which can be used to examine the transit of LC3-II through the autophagic pathway; if flux is occurring, the amount of LC3-II will be higher in the presence of the inhibitor.112 Lysosomal degradation can be prevented through the use of protease inhibitors (e.g., pepstatin A and E-64d), compounds such as bafilomycin A1, chloroquine or NH4Cl that neutralize the lysosomal pH,18,114,153,154 or by treatment with agents that block fusion of autophagosomes with lysosomes (note that bafilomycin A1 will ultimately cause a fusion block as well as neutralize the pH155).156 Alternatively, knocking down or knocking out lysosomal-associated membrane protein 2 (LAMP2) represents a genetic approach to block the fusion of autophagosomes and lysosomes [for example, inhibiting LAMP2 in myeloid leukemic cells results in a marked increase of GFP-LC3 dot formation and endogenous LC3-II protein compared with control cells upon autophagy induction during myeloid differentiation (Tschan MP, unpublished data)].157 This approach, however, may be complicated by the compensatory upregulation of macroautophagy that occurs when chaperone-mediated autophagy is blocked,158 unless the LAMP2B isoform is specifically knocked down.159
Generally, an increase in the levels of LC3-II observed with a particular treatment condition in the presence of bafilomycin A1, compared with the treatment alone, is indicative of some degree of flux through the system (i.e., compound/drug treatment plus bafilomycin A1 should result in a higher amount of LC3-II than compound/drug treatment alone); however, a treatment condition increasing LC3-II on its own that has no difference in LC3-II in the presence of bafilomycin A1 compared with treatment alone may suggest a block in autophagy at the terminal stages.6 This procedure has been validated with several autophagy modulators.160 With each of these techniques, it is essential to avoid assay saturation. The duration of the bafilomycin A1 treatment needs to be relatively short (1–2 h) to allow comparisons of the amount of LC3 that is lysosomally degraded over a given time frame under one treatment condition to another treatment condition. Positive control experiments using treatment with known autophagy inducers, along with bafilomycin A1 vs. vehicle, are important to demonstrate the utility of this approach in each experimental context. The same type of assay monitoring the turnover of Atg8–PE can be used to monitor flux in yeast, by comparing the amount of Atg8 present in a wild-type vs. a pep4Δ strain following autophagy induction.161
An additional methodology for monitoring autophagy relies on the observation that a subpopulation of LC3-II exists in a cytosolic form (LC3-IIs) in some cell types.162 The amount of cytosolic LC3-IIs and the ratio between LC3-I and LC3-IIs appears to correlate with changes in autophagy and may provide a more accurate measure of autophagic flux than ratios based on the total level of LC3-II.162 The validity of this method has been demonstrated by comparing autophagic proteolytic flux in rat hepatocytes and hepatoma cells. One advantage of this approach is that it does not require the presence of autophagic or lysosomal inhibitors to block the degradation of LC3-II.
Finally, autophagic flux can be monitored based on the turnover of LC3-II, by utilizing a luminescence-based assay. For example, a reporter assay based on the degradation of Renilla reniformis luciferase (Rluc)-LC3 fusion proteins is well suited for screening compounds affecting autophagic flux.163 In this assay, Rluc is fused N-terminally to either wild-type LC3 (LC3wt) or a lipidation-deficient mutant of LC3 (G120A). Since Rluc-LC3wt, in contrast to Rluc-LC3-G120A, specifically associates with the autophagosomal membranes, Rluc-LC3wt is more sensitive to autophagic degradation. A change in autophagy-dependent LC3-turnover can thus be estimated by monitoring the change in the ratio of luciferase activities between the two cell populations expressing either Rluc-LC3wt or Rluc-LC3-G120A. In its simplest form, the Rluc-LC3-assay can be used to estimate autophagic flux at a single time point by defining the luciferase activities in cell extracts. Moreover, the use of a live cell luciferase substrate makes it possible to monitor changes in autophagic activity in living cells in real time. This method has been successfully used to identify positive and negative regulators of autophagy from cells treated with microRNA, siRNA and small molecule libraries.163-166
Cautionary notes: The main caveat regarding the measurement of LC3-IIs/LC3-I is that this method has only been tested in isolated rat hepatocytes and H4-II-E cells. Thus, it is not yet known whether it is generally applicable to other cell types, and a soluble form of LC3-II (i.e., LC3-IIs) is not observed in many standard cell types including HeLa, HEK 293 and PC12. In addition, the same concerns apply regarding detection of LC3-I by western blotting. It should be noted that the LC3-IIs/LC3-I ratio must be analyzed using the cytosolic fractions rather than the total homogenates. Furthermore, the same caveats mentioned above regarding the use of LC3 for qualitatively monitoring autophagy also apply to the use of this marker for evaluating flux.
The use of a radioactive pulse-chase analysis provides an alternative to lysosomal protease inhibitors,113 although such inhibitors should still be used to verify that degradation is lysosome-dependent. In addition, drugs must be used at concentrations and for time spans that are effective in inhibiting fusion or degradation, but that do not provoke cell death. Thus, these techniques may not be practical in all cell types or in tissues from whole organisms where the use of protease inhibitors is problematic, and where pulse labeling requires artificial short-term culture conditions that may induce autophagy. Another concern when monitoring flux via LC3-II turnover may be seen in the case of a partial autophagy block; in this situation, agents that disrupt autophagy (e.g., bafilomycin A1) will still result in an increase in LC3-II, which may be interpreted as the complete absence of a block resulting from the mutant or compound being tested. Thus, care is needed in interpretation. Furthermore, for characterizing new autophagy modulators, it is ideal to test autophagic flux at early (e.g., 4 h) and late (e.g., 24 h) time-points, since in certain instances, such as with calcium phosphate precipitates, a compound may increase or decrease flux at these two time-points, respectively.167 Finally, many of the chemicals used to inhibit autophagy, such as bafilomycin A1, NH4Cl (see Autophagy inhibitors and inducers below) or chloroquine, also directly inhibit the endocytosis/uncoating of viruses (Smith DR, personal communication), and other endocytic events requiring low pH, as well as exit from the Golgi (Tooze S, personal communication) and as such should be used only with extreme caution in studies investigating autophagy-virus interactions.
One additional consideration is that it may not be absolutely necessary to follow LC3-II turnover if other substrates are being monitored simultaneously. For example, an increase in LC3-II levels in combination with the lysosomal (or ideally autophagy-specific) removal of an autophagic substrate (such as an organelle168,169) that is not a good proteasomal substrate provides an independent assessment of autophagic flux. However, due to the fact that LC3 might be coupled to endosomal membranes and not just autophagosomes, and the levels of well-characterized autophagosome substrates such as SQSTM1 can also be affected by proteasome inhibitors,170 it is probably prudent to monitor both turnover of LC3-II and an autophagosome substrate in parallel.
Another issue relates to the use of protease inhibitors (see Autophagy inhibitors and inducers below). When using lysosomal protease inhibitors, it is of fundamental importance to assess proper conditions of inhibitor concentration and time of pre-incubation to ensure full inhibition of lysosomal cathepsins. In this respect, 1 h of pre-incubation with 10 µg/ml E-64d is sufficient in most cases, since this inhibitor is membrane permeable and rapidly accumulates within lysosomes. On the other hand, pepstatin A is membrane impermeable (ethanol or preferably DMSO must be employed as a vehicle) and requires a prolonged incubation (> 8 h) and a relatively high concentration (> 50 µg/ml) to fully inhibit lysosomal cathepsin D (Fig. 6). An incubation of this duration, however, can be problematic due to indirect effects (see GFP-Atg8/LC3 lysosomal delivery and proteolysis). Also, note that the relative amount of lysosomal cathepsins B and D is cell-specific and changes with culture conditions. In contrast to the protease inhibitors, chloroquine (10 µM) or bafilomycin A1 (1–100 nM) can be added to cells immediately prior to autophagy induction.
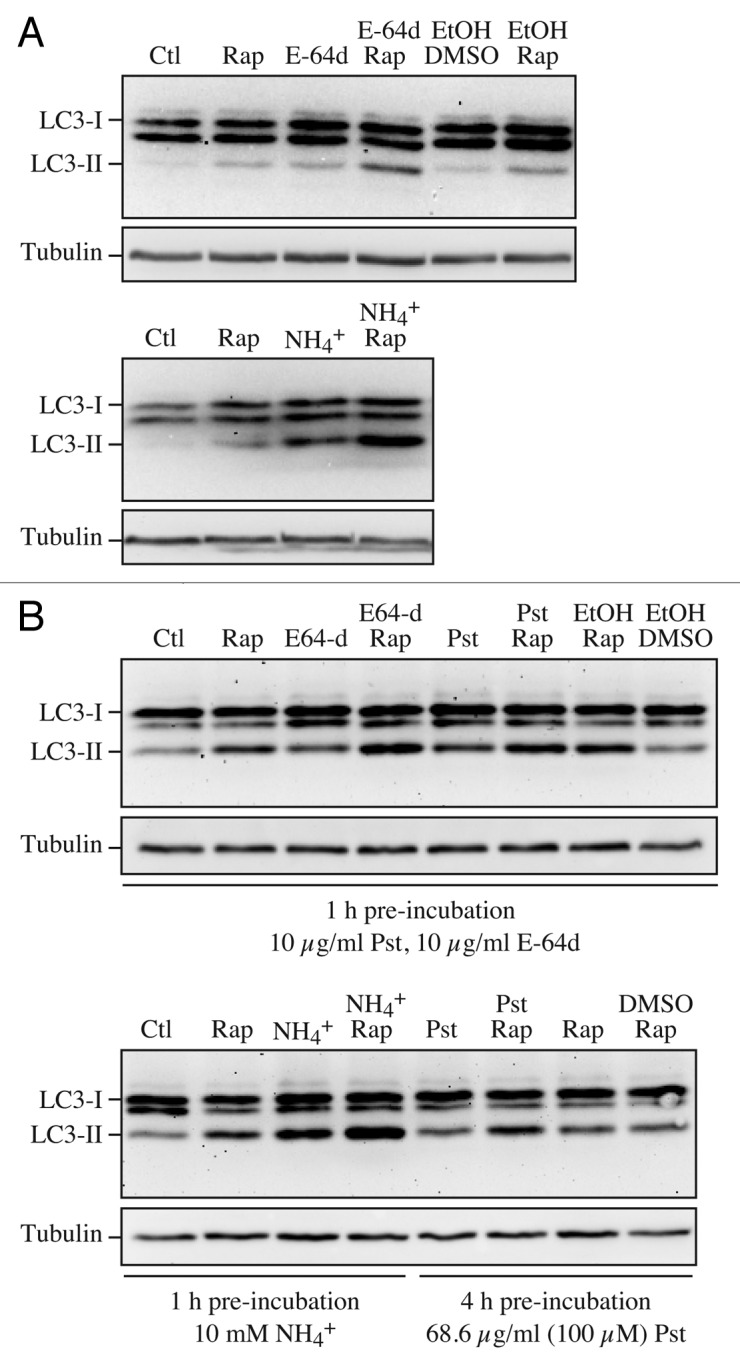
Figure 6. Effect of different inhibitors on LC3-II accumulation. SH-SY5Y human neuroblastoma cells were plated and allowed to adhere for a minimum of 24 h, then treated in fresh medium. Treatments were as follows: rapamycin (Rap), (A) 1 µM, 4 h or (B) 10 µM, 4 h; E-64d, final concentration 10 µg/ml from a 1 mg/ml stock in ethanol (ETOH); NH4Cl (NH4+), final concentration 10 mM from a 1 M stock in water; pepstatin A (Pst), final concentration 10 µg/ml from a 1 mg/ml stock in ethanol, or 68.6 µg/ml from a 6.86 mg/ml stock in DMSO; ethanol or DMSO, final concentration 1%. Pre-incubations in (B) were for 1 or 4 h as indicated. 10 mM NH4Cl (or 30 µM chloroquine, not shown) were the most effective compounds for demonstrating the accumulation of LC3-II. E-64d was also effective in preventing the degradation of LC3-II, with or without a preincubation, but ammomium chloride (or chloroquine) may be more effective. Pepstatin A at 10 µg/ml with a 1 h pre-incubation was not effective at blocking degradation, whereas a 100 µM concentration with 4 h pre-incubation had a partial effect. Thus, alkalinizing compounds are more effective in blocking LC3-II degradation, and pepstatin A must be used at saturating conditions to have any noticeable effect. Images provided by C. Isidoro. Note that the band running just below LC3-I at approximately 17.5 kDa may be a processing intermediate of LC3-I; it is detectable in freshly prepared homogenates, but is less visible after the sample is subjected to a freeze-thaw cycle.
Conclusion: It is important to be aware of the difference between monitoring the steady-state level of Atg8/LC3 and autophagic flux; the latter can be determined by following Atg8/LC3 in the absence and presence of autophagy inhibitors, and/or by examining the autophagy-dependent degradation of appropriate substrates. In particular, if there is any evidence of an increase in LC3-II (or autophagosomes), it is essential to determine whether this represents increased flux, or a block in fusion or degradation through the use of inhibitors such as chloroquine or bafilomycin A1.
c. GFP-Atg8/LC3 lysosomal delivery and proteolysis
GFP-LC3B (hereafter referred to as GFP-LC3) has also been used to follow flux. First, when GFP-Atg8 or GFP-LC3 is delivered to a lysosome/vacuole the Atg8/LC3 part of the chimera is sensitive to degradation, whereas the GFP protein is relatively resistant to hydrolysis (note, however, that GFP fluorescence is quenched by low pH; see GFP-Atg8/LC3 fluorescence microscopy and Tandem mRFP/mCherry-GFP fluorescence microscopy). Therefore, the appearance of free GFP on western blots can be used to monitor lysis of the inner autophagosome membrane and breakdown of the cargo in metazoans (Fig. 7A),161,171,173 or the delivery of autophagosomes to, and the breakdown of autophagic bodies within, the yeast and plant vacuole.151,161,174 Reports on Dictyostelium and mammalian cells highlight the importance of lysosomal pH as a critical factor in the detection of free GFP that results from the degradation of fused proteins. In these cell types, free GFP fragments are only detectable in the presence of nonsaturating levels of lysosomotropic compounds (NH4Cl or chloroquine) or under conditions that attenuate lysosomal acidity; otherwise, the autophagic/degradative machinery appears to be too efficient to allow the accumulation of the proteolytic fragment (Fig. 7B and C).36,172 Hence, a reduction in the intensity of the free GFP band may indicate reduced flux, but it may also be due to efficient turnover. Using a range of concentrations and treatment times of compounds that inhibit autophagy can be useful in distinguishing between these possibilities.175 Since the pH in the yeast vacuole is higher than that in mammalian or Dictyostelium lysosomes, the levels of free GFP fragments are detectable in yeast without the necessity of using lysosomotropic compounds.29 Additionally, in yeast the diffuse fluorescent haze from the released GFP moiety within the vacuole lumen can be observed by fluorescence microscopy.
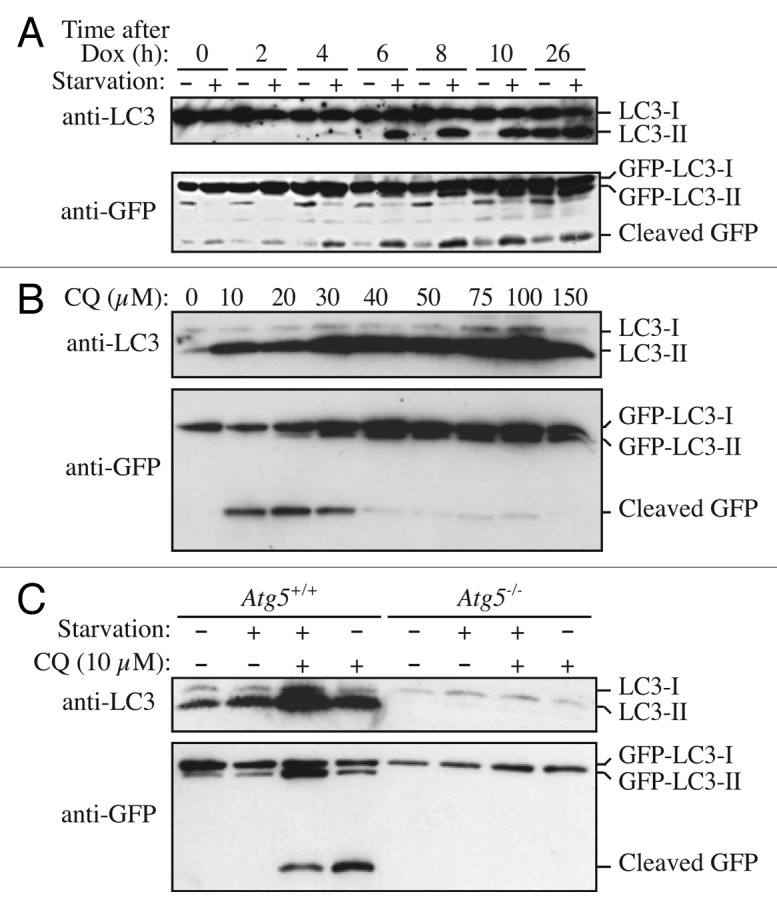
Figure 7. GFP-LC3 processing can be used to monitor delivery of autophagosomal membranes. (A) Atg5−/− MEFs engineered to express Atg5 under the control of the Tet-off promoter were grown in the presence of doxycyline (10 ng/ml) for one week to suppress autophagy. Cells were then cultured in the absence of drug for the indicated times, with or without a final 2 h starvation. Protein lysates were analyzed by western blot using anti-LC3 and anti-GFP antibodies. The positions of untagged and GFP-tagged LC3-I and LC3-II, and free GFP are indicated. This figure was modified from data previously published in reference 171, FEBS Letters, 580, Hosokawa N, Hara Y, Mizushima N, Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size, pp. 2623–2629, copyright 2006, with permission from Elsevier. (B) Differential role of unsaturating and saturating concentrations of lysosomal inhibitors on GFP-LC3 cleavage. HeLa cells stably transfected with GFP-LC3 were treated with various concentrations of chloroquine (CQ) for 6 h. Total lysates were prepared and subjected to immunoblot analysis. (C) CQ-induced free GFP fragments require classical autophagy machinery. Wild-type and Atg5−/− MEFs were first infected with adenovirus GFP-LC3 (100 viral particles per cell) for 24 h. The cells were then either cultured in regular culture medium with or without CQ (10 µM), or subjected to starvation in EBSS buffer in the absence or presence of CQ for 6 h. Total lysates were prepared and subjected to immunoblot analysis. (B and C) are modified from the data previously published in reference 172.
The movement of GFP-LC3 to lysosomes also can be monitored by fluorescence microscopy, although, as mentioned above, the GFP fluorescent signal is more sensitive to acidic pH than other fluorophores (see GFP-Atg8/LC3 fluorescence microscopy). A time-course evaluation of the cell population showing GFP-LC3 puncta can serve to monitor the autophagy flux, since a constant increase in the number of cells accumulating GFP-LC3 puncta is suggestive of defective fusion of autophagosomes with lysosomes, and conversely a decline implies that GFP-LC3 is consumed within newly formed autolysosomes. In either case, it can be problematic to use GFP fluorescence to follow flux, as new GFP-LC3 is continuously being synthesized. A potential solution to this problem for following fluorescence is to use a photoactivatable version of the fluorescent protein,176 which allows this assay to be performed essentially as a pulse/chase analysis. Another alternative is to follow flux using GFP-LC3 fluorescence by adding lysosomal protease or fusion inhibitors to cells expressing GFP-LC3 and monitoring changes in the number of puncta. In this case, the presence of lysosomal inhibitors should increase the number of GFP-LC3-positive structures, and the absence of an effect on the total number of GFP-LC3 puncta or on the percentage of cells displaying numerous puncta is indicative of a defect(s) in autophagic flux.177 The combination of protease inhibitors (to prevent the degradation of GFP) or compounds that modify lysosomal pH such as NH4Cl or chloroquine, or compounds such as bafilomycin A1 or others that block fusion of autophagosomes with lysosomes (e.g., vinblastine) may be most effective in preventing lysosome-dependent decreases in GFP-LC3 puncta. However, because the stability of GFP is affected by lysosomal pH, we advise the use of protease inhibitors whether or not lysosomotropic compounds or fusion inhibitors are included (although lysosomotropic compounds should help stabilize GFP by neutralizing the pH in the lysosome, they do not have an immediate effect on lysosomal hydrolase activity).
Cautionary notes: The GFP-Atg8 processing assay is used routinely to monitor autophagy in yeast. One caveat, however, is that this assay is not always performed in a quantitative manner (for example, western blot exposures need to be in the linear range). Accordingly, an enzymatic assay such as the Pho8Δ60 assay may be preferred (see Autophagic protein degradation),178,179 especially when the differences in autophagic activity need to be precisely determined; however, appropriate caution must be used as with any enzyme assay regarding, for example, substrate concentrations and linearity (note that an equivalent assay has not been developed for higher eukaryotic cells).
The main limitation of the GFP-LC3 processing assay in mammalian cells is that it seems to depend on cell type and culture conditions (Hosokawa N, Mizushima N, unpublished data). Apparently, GFP is more sensitive to mammalian lysosomal hydrolases than to the degradative milieu of the yeast vacuole. Alternatively, the lower pH of lysosomes relative to that of the vacuole may contribute to differences in detecting free GFP. Under certain conditions [such as Earle’s balanced salt solution (EBSS)-induced starvation] in some cell lines, when the lysosomal pH becomes particularly low, free GFP is undetectable because both the LC3-II and free GFP fragments are quickly degraded.172 Therefore, if this method is used it should be accompanied by immunoblotting including controls to address the stability of nonlysosomal GFP such as GFP-LC3-I. It should also be noted that free GFP can be detected when cells are treated with nonsaturating doses of lysosomal inihibitors such as chloroquine, E-64d and bafilomycin A1. The saturating concentrations of these lysosomal inhibitors vary in different cell lines, and it would be better to use a saturating concentration of lysosomal inhibitors when performing an autophagic flux assay.172 Therefore, caution must be exercised in interpreting the data using this assay; it would be helpful to combine an analysis of GFP-LC3 processing with other assays such as the monitoring of endogenous LC3-II by western blot.
Along these lines, a caution concerning the use of the EGFP fluorescent protein for microscopy is that this fluorophore has a relatively neutral pH optimum for fluorescence,180 so that its signal may diminish quickly during live cell imaging due to the acidic environment of the lysosome. It is possible to circumvent this latter problem by imaging paraformaldehyde-fixed cultures that are maintained in a neutral pH buffer, which retains EGFP fluorescence (Kleinman M, Reiners JJ, personal communication). Alternatively, it may be preferable to use a different fluorophore such as monomeric red fluorescent protein (mRFP) or mCherry, which retain fluorescence even at acidic pH.181 On the one hand, a putative advantage of mCherry over mRFP is its enhanced photostability and intensity, which are an order of magnitude higher (and comparable to GFP), enabling acquisition of images at similar exposure settings as are used for GFP, thus minimizing potential bias in interpretation.182 On the other hand, caution is required when evaluating the localization of mCherry fusion proteins during autophagy due to the persistence of the mCherry signal in acidic environments; all tagged proteins are prone to show enrichment in lysosomes during nonspecific autophagy of the cytoplasm, especially at higher expression levels. In addition, red fluorescent proteins (even the monomeric forms) can be toxic due to aggregation.183 DendrA2 is an improved version of the green-to-red photoswitchable fluorescent protein Dendra, which is derived from the octocoral Dendronephthya sp.184 DendrA2 is capable of irreversible photoconversion from a green to a red fluorescent form, but can be used also as a normal GFP or RFP vector. This modified version of the fluorophore has certain properties including a monomeric state, low phototoxic activation and efficient chromophore maturation, which make it suitable for real-time tracking of LC3 and SQSTM1 (Fig. 8; Kaarniranta K, personal communication). Another alternative to mRFP or mCherry is to use the Venus variant of YFP, which is brighter than mRFP and less sensitive to pH than GFP.186
Figure 8. Movement of activated pDendra2-hp62 (orange) from the nucleus (middle) to the aggregate in ARPE-19 cells, revealed by confocal microscopy. Cells were exposed to 5 µM MG132 for 24 h to induce the formation of perinuclear aggregates.185 The cells were then exposed to a UV pulse (the UV-induced area is shown by red lines that are inside of the nucleus) that converts Dendra2 from green to red, and the time shown after the pulse is indicated. SQSTM1/p62 is present in a small nuclear aggregrate, and is shuttled from the nucleus to a perinuclear large protein aggregate (detected as red). Scale bar, 5 µm. Image provided by K. Kaarniranta.
The pH optimum of EGFP is important to consider when using GFP-LC3 constructs, as the original GFP-LC3 marker187 uses the EGFP variant, which may result in a reduced signal upon the formation of amphisomes or autolysosomes. An additional caveat when using the photoactivatable construct PA-GFP180 is that the process of activation by photons may induce DNA damage, which could, in turn, elicit induction of autophagy. Also, GFP is relatively resistant to denaturation, and boiling for 5 min may be needed to prevent the folded protein from being trapped in the stacking gel during SDS-PAGE.
As noted above (see Western blotting and ubiquitin-like protein conjugation systems), Atg4 cleaves the residue(s) that follow the C-terminal glycine of Atg8/LC3 that will be conjugated to PE. Accordingly, it is critical that any chimeras be constructed with the fluorescent tag at the N terminus of Atg8/LC3.
Finally, lysosomal inhibition needs to be carefully controlled. Prolonged inhibition of lysosomal hydrolases (> 6 h) is likely to induce a secondary autophagic response triggered by the accumulated undigested autophagy cargo. This secondary autophagic response can complicate the analysis of the autophagy flux, making it appear more vigorous than it would in the absence of the lysosomal inhibitors.
Conclusion: The GFP-Atg8/LC3 processing assay (the generation of free GFP within the vacuole/lysosome) is a convenient way to monitor autophagy, but it does not work in all cell types, and is not as easy to quantify as enzyme-based assays.
d. GFP-Atg8/LC3 fluorescence microscopy
LC3B, or the protein tagged at its N terminus with a fluorescent protein such as GFP (GFP-LC3), has been used to monitor autophagy through indirect immunofluorescence or direct fluorescence microscopy (Fig. 9), measured as an increase in punctate LC3 or GFP-LC3.187,188 The detection of GFP-LC3/Atg8 is also useful for in vivo studies using transgenic organisms such as Caenorhabditis elegans,189 Dictyostelium discoideum,190 filamentous ascomycetes,191-195 Ciona intestinalis,196 Drosophila melanogaster,197-199 Arabidopsis thaliana,200 Leishmania major202,203 and mice.126 It is also possible to use anti-LC3/Atg8 antibodies for immunocytochemistry or immunohistochemistry,140,204-209 procedures that have the advantages of detecting the endogenous protein, obviating the need for transfection and or the generation of a transgenic organism, as well as avoiding potential artifacts resulting from overexpression (for example, high levels of overexpressed GFP-LC3 can result in its nuclear localization, although the protein can still relocate to the cytosol upon starvation), but we note that it is not always possible to detect endogenous Atg8/LC3. The use of imaging cytometry allows rapid and quantitative measures of the number of LC3 puncta and their relative number in individual or mixed cell types, using computerized assessment, enumeration, and data display (e.g., see refs. 142 and 210). In this respect, the alternative use of an automated counting system may be helpful for obtaining an objective number of puncta per cell. For this purpose, the WatershedCounting3D plug-in for ImageJ may be useful.211,212

Figure 9. Changes in the detection and localization of GFP-LC3 upon the induction of autophagy. U87 cells stably expressing GFP-LC3 were treated with PBS, rapamycin (200 nM), or rapamycin in combination with 3-MA (2 mM) for 24 h. Representative fluorescence images of cells counterstained with DAPI (nuclei) are shown. Scale bar, 10 µm. This figure was modified from Figure 6 published in Badr et al. Lanatoside C sensitizes glioblastoma cells to tumor necrosis factor–related apoptosis-inducing ligand and induces an alternative cell death pathway. Neuro-Oncology 2011, 13:1213–24, by permission of Oxford University Press.
Monitoring the endogenous protein, however, obviously depends on the ability to detect it in the system of interest. If the endogenous amount is below the level of detection, the use of an exogenous construct is warranted. In this case, it is important to consider the use of stable transformants vs. transient transfections. On the one hand, stable transformants may have reduced background resulting from the lower gene expression, and there is also the advantage of eliminating artifacts resulting from recent exposure to transfection reagents (see below). Furthermore, with stable transformants more cells can be easily analyzed because nearly 100% of the population will express tagged LC3. On the other hand, one disadvantage of stable transfectants is that the integration sites cannot always be predicted, and expression levels may not be optimal. Therefore, it is worth considering the use of stable episomal plasmids that avoid the problem of unsuitable integration.181 An important advantage of transient transfection is that this approach is better for examining the immediate effects of the transfected protein on autophagy, although it restricts the length of time that the analysis can be performed, and consideration must be given to the induction of autophagy resulting from exposure to the transfection reagents (see below). In addition, a double transfection can be used (e.g., with GFP-LC3 and the protein of interest) to visually tag the cells that express the protein being examined. In conclusion, there is no simple rule for the use of stable vs. transient transfections. When stable transfections are utilized, it is worthwhile screening for clones that give the best signal-to-noise ratio, and when transient transfections are used, it is worthwhile optimizing the GFP-LC3 DNA concentration to give the best signal-to-noise ratio. In clones, the uniformity of expression of GFP-LC3 also makes “thresholding” when scoring puncta-positive cells (see below) much easier. However, there is also a need to beware of the frequent unrepresentative behavior of mammalian cell lines when selecting a single cell clone from a parental pool; therefore, it may be better to use a pool of multiple selected clones to avoid possible artifacts that can arise from the selection and propagation of individual clones from a single transfected cell (although the use of a pool is also problematic as its composition will change over time). Optimization, together with including the appropriate controls, will help overcome the effects of the inherent variability in these analyses.
An additional use of GFP-LC3 is to monitor colocalization with a target during autophagy-related processes such as organelle degradation or the sequestration of pathogenic microbes.213-215 Preincubation of cells stably expressing GFP-LC3 with leupeptin can help stabilize the GFP-LC3 signal during fluorescence microscopy, especially under conditions of induced autophagic flux. Leupeptin is an inhibitor of lysosomal cysteine and serine proteases and will therefore inhibit degradation of membrane-conjugated GFP-LC3 that is present within autolysosomes.
Cautionary notes: Quantification of autophagy by measuring GFP-LC3 puncta (or LC3 by immunofluorescence) can be more tedious, depending on the method used (e.g., high-throughput image analysis can obviously be rapid and efficient), than monitoring LC3-II by western blot; however, the former may be more sensitive and quantitative. Ideally, it is preferable to include both assays and to compare the two sets of results. In addition, if GFP-LC3 is being quantified, it is better to determine the number of puncta corresponding to GFP-LC3 on a per cell basis (or per cell area basis if the cell population is in culture or the cell composition of the tissue is not homogenous, as well as in the case of plant cells, which tend to grow) rather than simply the total number of cells displaying puncta. This latter point is critical because even in nutrient-rich conditions, cells display some basal level of GFP-LC3 puncta, unless they are lacking autophagy-related genes (and even in the latter case it is possible to get puncta of GFP-LC3 depending on the specific conditions). There are, however, practical issues with counting puncta manually and reliably, especially if there are large numbers per cell [although this may be more accurate than relying on a software program, in which case it is important to ensure that only appropriate dots are being counted; applicable programs include ImageJ, Imaris, which may be more accurate (Ktistakis NT, personal communication), and the open-source software CellProfiler216]. Moreover, when autophagosome-lysosome fusion is blocked, larger autophagosomes are detected, possibly due to autophagosome-autophagosome fusion. Although it is possible to detect changes in the size of GFP-Atg8/LC3 puncta by fluorescence microscopy, it is not possible to correlate size with autophagy activity without additional assay methods. Size determinations can be problematic by fluorescence microscopy unless careful standardization is performed,217 and size estimation is not recommended as a method for monitoring autophagy; however, it is possible to quantify the fluorescence intensity of GFP-Atg8/LC3 at specific puncta, which does provide a valid measure of protein recruitment.218
In addition to autophagosome size, the number of puncta visible to the eye will also be influenced by both the level of expression of GFP-LC3 in a given cell and by the exposure time of the microscope, if using widefield microscopy. In many cell types it may be possible to establish a cut-off value for the number of puncta per cell in conditions of “low” and “high” autophagy.219 This can be tested empirically by exposing cells to autophagy-inducing and -blocking agents. Thus, cell populations showing significantly greater proportions of cells with autophagosome numbers higher than the cut-off in perturbation conditions compared with the control cells could provide quantitative evidence of altered autophagy. It is then possible to score the population as the percentage of cells displaying numerous autophagosomes. This approach will only be feasible if the background number of puncta is relatively low. For this method, it is particularly important to count a large number of cells and multiple representative sections of the sample (probably on the order of 50 or more, preferably in at least three different trials, depending on the particular system and experiment, but the critical point is that this determination should be based on statistical power analysis). Accordingly, high-content imaging analysis methods are extremely applicable to provide reliable values. Such methods enable quantification of GFP-LC3 puncta (or overall fluorescence intensity) in thousands of cells per sample (e.g., see refs. 164, 175 and 220). When using automated analysis methods, care must be taken to manually evaluate parameters used to establish background cutoff values for different treatment conditions and cell types. Another note of caution is that treatments affecting cell morphology, leading to the “rounding-up” of cells for example, can result in apparent changes in the number of GFP-LC3 puncta per cell. To avoid misinterpretation of results due to such potential artifacts, manual review of cell images is highly recommended.
To allow comparisons by other researchers attempting to repeat these experiments, it is critical that the authors also specify the baseline number of puncta that are used to define “normal” or “low” autophagy. Furthermore, the cells should also be counted using unbiased procedures (e.g., using a random start point followed by inclusion of all cells at regular intervals), and statistical information should be provided for both baseline and altered conditions, as these assays can be highly variable. One possible method to obtain unbiased counting of GFP-LC3 puncta in a large number of cells is to perform multispectral imaging flow cytometry (see Autophagic flux determination using flow and multispectral imaging cytometry).221 This method can also be used for endogenous LC3, and, therefore, is useful for non-transfected primary cells.222 Multispectral imaging flow cytometry allows characterization of single cells within a population by assessing a combination of morphology and immunofluorescence patterns, thereby providing statistically meaningful data.223 For adherent cell cultures, one caution for flow cytometry is that the techniques necessary to produce single cell suspensions can cause significant injury to the cells, leading to secondary changes in autophagy. Therefore, staining for plasma membrane permeabilization (e.g., cell death) before vs. after isolation is an important control.
An important caveat in the use of GFP-LC3 is that this chimera can associate with aggregates, especially when expressed at high levels in the presence of aggregate-prone proteins, which can lead to a misinterpretation of the results.224 Of note, GFP-LC3 can associate with ubiquitinated protein aggregates;225 however, this does not occur if the GFP-LC3 is expressed at low levels (Rubinsztein DC, unpublished observations). These aggregates have been described in many systems, and are also referred to as Aggresome-Like Induced Structures or ALIS,225-227 dendritic cell ALIS,228 p62 bodies/sequestosomes229 and inclusions. Inhibition of autophagy in vitro and in vivo leads to the accumulation of these aggregates, suggesting a role for autophagy in mediating their clearance.225,226,230-232 One way to control for background levels of puncta is to determine fluorescence from untagged GFP.
The adaptor protein SQSTM1/p62 is required for the formation of ubiquitinated protein aggregates in vitro (see SQSTM1/p62 and related LC3 binding protein turnover assays).229 In this case, the interaction of SQSTM1 with both ubiquitinated proteins and LC3 is thought to mediate delivery of these aggregates to the autophagy system.233,234 Many cellular stresses can induce the formation of aggregates, including transfection reagents,225 or the introduction of foreign DNA (especially if the DNA is not extracted endotoxin free). SQSTM1-positive aggregates are also formed by proteasome inhibition or puromycin treatment. Calcium phosphate transfection of COS7 cells or lipofectamine transfection of MEFs (Pinkas-Kramarski R, personal communication), primary neurons (La Spada AR, personal communication) or neuronal cells (Chu CT, personal communication) transiently increases basal levels of GFP-LC3 puncta and/or the amount of LC3-II. One solution is to examine GFP-LC3 puncta in cells stably expressing GFP-LC3; however, as transfection-induced increases in GFP-LC3 puncta and LC3-II are often transient, another approach is to use cells transfected with GFP, and cells subjected to a mock time-matched transfection as background (negative) controls. A lipidation-defective LC3 mutant where glycine 120 is mutated to alanine is targeted to these aggregates independently of autophagy (likely via its interaction with SQSTM1, see above) and as a result this mutant can serve as another valuable control.225 When carrying out transfections it may be necessary to alter the protocol depending on the level of background fluorescence. For example, changing the medium and waiting 24 to 48 h after the transfection can help to reduce the background level of GFP-LC3 puncta that is due to the transfection reagent (Colombo MI, personal communication). Similarly, when using an mCherry-GFP-SQSTM1 double tag (see Tandem mRFP/mCherry-GFP fluorescence microscopy) in transient transfections it is best to wait 48 h after transfection to reduce the level of aggregate formation and potential inhibition of autophagy (Johansen T, personal communication). Another consideration is that in addition to transfection, viral infection can activate stress pathways in some cells and possibly induce autophagy, again emphasizing the importance of appropriate controls, such as control viruses expressing GFP.235
Ubiquitinated protein aggregate formation and clearance appear to represent a cellular recycling process. Aggregate formation can occur when autophagy is either inhibited or when its capacity for degradation is exceeded by the formation of proteins delivered to the aggregates. In principle, formation of GFP-LC3-positive aggregates represents a component of the autophagy process. However, the formation of GFP-LC3-positive ubiquitinated protein aggregates does not directly reflect either the induction of autophagy (or autophagosome formation), or flux through the system. Indeed, formation of ubiquitinated protein aggregates that are GFP-LC3 positive can occur in autophagy-deficient cells.225 Therefore, it should be remembered that GFP-LC3 puncta likely represent a mix of ubiquitinated protein aggregates in the cytosol, ubiquitinated protein aggregates within autophagosomes and more “conventional” phagophores and autophagosomes bearing other cytoplasmic cargo (this is one example where CLEM could help in resolving this question). In Dictyostelium, inhibition of autophagy leads to huge ubiquitinated protein aggregates containing SQSTM1 and GFP-Atg8, when the latter is co-expressed; the size of the aggregates and the typical presence of only one structure per cell make them easily distinguishable from autophagosomes. Saponin treatment has been used to reduce background fluorescence under conditions where no aggregation of GFP-LC3 is detected in hepatocytes, GFP-LC3 stably-transfected HEK 293235 and human osteosarcoma cells, and in nontransfected cells;236 however, treatment with saponin and other detergents can provoke artifactual GFP-LC3 puncta formation,237 and controls need to be included in such experiments in light of these findings. In general, it is preferable to include additional assays that measure autophagy rather than relying solely on monitoring GFP-LC3. In addition, we recommend that researchers validate their assays by demonstrating the absence or reversal of GFP-LC3 puncta formation in cells treated with pharmacological or RNA interference-based autophagy inhibitors (Table 1). For example, 3-MA is commonly used to inhibit starvation- or rapamycin-induced autophagy,256 but some data indicate that this compound can also have stimulatory effects (see Autophagy inhibitors and inducers).257
Table 1. Genetic and pharmacological regulation of autophagy†.
| Method | Comments |
|---|---|
| 1. 3-methyladenine (3-MA) |
A PtdIns3K inhibitor that effectively blocks an early stage of autophagy by inhibiting the class III PtdIns3K, but is not a specific autophagy inhibitor. 3-MA also inhibits the class I PtdIns3K and can thus, at suboptimal concentrations in long-term experiments, promote autophagy in some systems, as well as affect cell survival through AKT1 and other kinases. |
| 2. 10-NCP |
10-(4′-N-diethylamino)butyl)-2-chlorophenoxazine; an AKT1 inhibitor that induces autophagy in neurons.238 |
| 3. 17-AAG |
An inhibitor of the HSP90-CDC37 chaperone complex, induces autophagy in certain systems (e.g., neurons), but impairs starvation-induced autophagy and mitophagy in others by promoting the turnover of ULK1.239 |
| 4. ATG4C74A |
An active site mutant of ATG4 that is defective for autophagy.240 |
| 5. Bafilomycin A1 (or concanamycin A) |
A V-ATPase inhibitor that causes an increase in lysosomal/vacuolar pH, and ultimately blocks fusion of autophagosomes with the vacuole. |
| 6. Calcium |
An autophagy activator that can be released from ER or lysosomal stores under stress conditions; however, calcium can also inhibit autophagy.241 |
| 7. Chloroquine, NH4Cl |
Lysosomotropic compounds that elevate/neutralize the lysosomal/vacuolar pH. |
| 8. Deletion |
This method provides the most direct evidence for the role of an autophagic component; however, more than one gene involved in autophagy should be targeted to avoid indirect effects. |
| 9. E-64d |
A membrane-permeable cysteine protease inhibitor that can block the activity of a subset of lysosomal hydrolases; should be used in combination with pepstatin A for inhibiting lysosomal protein degradation. |
| 10. Knockdown |
This method provides relatively direct evidence for the role of an autophagic component. However, the efficiency of knockdown varies, as does the stability of the targeted protein. In addition, more than one gene involved in autophagy should be targeted to avoid indirect effects. |
| 11. KU-0063794 |
An MTOR inhibitor that binds the catalytic site and activates autophagy.242,243 |
| 12. Leupeptin |
An inhibitor of cysteine, serine and threonine proteases that can be used in combination with pepstatin A and/or E-64d to block lysosomal protein degradation. Leupeptin is not membrane permeable, so its effect on cathepsins may depend on endocytic activity. |
| 13. microRNA |
Can be used to reduce the levels of target mRNA(s) or block translation. |
| 14. NAADP-AM |
Activates the lysosomal two pore channel and induces autophagy.244 |
| 15. NED-19 |
Inhibits the lysosomal two-pore channel and NAADP-induced autophagy.244 |
| 16. NVP-BEZ235 |
A dual inhibitor of PIK3CD/p110 and the MTOR catalytic site that activates autophagy.245,246 |
| 17. Pathogen-derived |
ICP34.5, vBCL2, vCFLAR/vFLIP, influenza M2, and HIV Nef autophagy inhibitor transfection. |
| 18. Pepstatin A |
An aspartyl protease inhibitor that can be used to partially block lysosomal degradation; should be used in combination with other inhibitors such as E-64d. Pepstatin A is not membrane permeable. |
| 19. Protease inhibitors |
These chemicals inhibit the degradation of autophagic substrates within the lysosome/vacuole lumen. A combination of inhibitors (e.g., leupeptin, pepstatin A and E-64d) is needed for complete blockage of degradation. |
| 20. Rapamycin |
Inhibits MTOR by binding to RPTOR, thus inducing autophagy, but only provides partial inhibition. |
| 21. Resveratrol |
A natural polyphenol that induces autophagy via activation of AMPK.247,248 |
| 22. RNAi |
Can be used to inhibit gene expression. |
| 23. RSVAs |
Synthetic small-molecule analogs of resveratrol that potently activate AMPK and induce autophagy.249 |
| 24. Thapsigargin |
An inhibitor of the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) that inhibits autophagic sequestration through the depletion of intracellular Ca2+ stores;250 however, thapsigargin may also block fusion of autophagosomes with endosomes by interfering with recruitment of RAB7, resulting in autophagosome accumulation.251 Long-term thapsigargin treatment may induce ER stress and a secondary stimulation of autophagy. |
| 25. Torin1 |
A catalytic MTOR inhibitor that induces autophagy and provides more complete inhibition than rapamycin (it inhibits all forms of MTOR).252 |
| 26. Trehalose |
An inducer of autophagy that may be relevant for the treatment of different neurodegenerative diseases.253,254 |
| 27. Tunicamycin |
A glycosylation inhibitor that induces autophagy due to ER stress.255 |
| 28. Vinblastine |
A depolymerizer of both normal and acetylated microbtubles that interferes with autophagosome-lysosome fusion.156 |
| 29. Wortmannin | An inhibitor of PtdIns 3-kinase that blocks autophagy, but is not a specific inhibitor (see 3-MA above). |
†This table is not meant to be complete, as there are many compounds and genetic methods that regulate autophagy, and new ones are being discovered routinely.
Another general limitation of the GFP-LC3 assay is that it requires a system amenable to the introduction of an exogenous gene. Accordingly, the use of GFP-LC3 in primary non-transgenic cells is more challenging. Here again, controls need to be included to verify that the transfection protocol itself does not artifactually induce GFP-LC3 puncta or cause LC3 aggregation. Furthermore, transfection should be performed with low levels of constructs, and the transfected cells should be followed to determine (1) when sufficient expression for detection is achieved, and (2) that during the time frame of the assay, basal GFP-LC3 puncta remain appropriately low. In addition, the demonstration of a reduction in the number of induced GFP-LC3 puncta under conditions of autophagy inhibition is helpful. For some primary cells, delivering GFP-LC3 to precursor cells by infection with recombinant lentivirus, retrovirus or adenovirus,258 and subsequent differentiation into the cell type of interest, is a powerful alternative to transfection of the already differentiated cell type.91
To implement the scoring of autophagy via fluorescence microscopy, one option is to measure pixel intensity. Since the expression of GFP-LC3 may not be the same in all cells—as discussed above—it is possible to use specific imaging software to calculate the standard deviation (SD) of pixel intensity within the fluorescence image and divide this by the mean intensity of the pixels within the area of analysis. This will provide a ratio useful for establishing differences in the degree of autophagy between cells. Cells with increased levels of autophagic activity, and hence a greater number of autophagosomes in their cytosol, are associated with a greater variability in pixel intensity (i.e., a high SD). Conversely, in cells where autophagy is not occurring, GFP-LC3 is uniformly distributed throughout the cytosol and a variation in pixel intensity is not observed (i.e., a low SD) (Campanella M, personal communication).
Although LC3-II is primarily membrane-associated, it is not necessarily associated with autophagosomes as is often assumed; the protein is also found on phagophores, the precursors to autophagosomes, as well as on amphisomes and phagosomes (see Western blotting and ubiquitin-like protein conjugation systems).132,259,260 Along these lines, yeast Atg8 can associate with the vacuole membrane independent of lipidation, so that a punctate pattern does not necessarily correspond to autophagic compartments.261 Thus, the use of additional markers is necessary to specify the identity of an LC3-positive structure; for example, ATG12–ATG5-ATG16L1 would be present on a phagophore, but not an autophagosome. In addition, the site(s) of LC3 conjugation to PE is not definitively known and levels of Atg8–PE/LC3-II can increase even in autophagy mutants that cannot form autophagosomes.262 One method that can be used to examine LC3-II membrane association is differential extraction in Triton X-114, which can be used with mammalian cells,258 or western blot analysis of total membrane fractions following solubilization with Triton X-100, which is helpful in plants.150,151 Another approach is to examine colocalization of LC3 with ATG5 (or other ATG proteins); the ATG12–ATG5 conjugate does not typically remain associated with autophagosomes, meaning that colocalized structures would correspond to phagophores. Importantly, we stress again that numbers of GFP-LC3 puncta, similar to steady-state LC3-II levels, reflect only a snapshot of the numbers of autophagy-related structures (e.g., autophagosomes) in a cell, and not autophagic flux.
Finally, we offer a general note of caution with regard to using GFP. First, the GFP tag is large, in particular relative to the size of LC3; therefore, it is possible that a chimera may behave differently from the native protein in some respects. Second, GFP is not native to most systems, and as such it may be recognized as an aberrant protein and targeted for degradation, which has obvious implications when studying autophagy. Third, some forms of GFP tend to oligomerize, which may interfere with protein function and/or localization. Fourth, EGFP inhibits polyubiquitination263 and may cause defects in other cellular processes. Fifth, not all LC3 puncta represent LC3-II and correspond to autophagosomes.137,264,265 Accordingly it would be prudent to complement any assays that rely on GFP fusions (to Atg8/LC3 or any protein) with additional methods that avoid the use of this fluorophore. Similarly, with the emergence of “super-resolution” microscopy methods such as photoactivated localization microscopy (PALM), new tags are being used (e.g., the EosFP green to red photoconvertible fluorescent protein, or the Dronpa GFP-like protein) that will need to be tested and validated.266
Conclusion: GFP-LC3 provides a marker that is relatively easy to use for monitoring autophagy induction (based on the appearance of puncta), or colocalization; however, it is not a preferred method for determining flux. In addition, it is recommended to use additional assays along with GFP-LC3 fluorescence microscopy to monitor autophagy.
e. Tandem mRFP/mCherry-GFP fluorescence microscopy
A fluorescence assay that is particularly designed to monitor flux relies on the use of a tandem monomeric RFP-GFP-tagged LC3 (tfLC3; Fig. 10).181 The GFP signal is sensitive to the acidic and/or proteolytic conditions of the lysosome lumen, whereas mRFP is more stable. Therefore, colocalization of both GFP and mRFP fluorescence indicates a compartment that has not fused with a lysosome, such as the phagophore or an autophagosome. In contrast, an mRFP signal without GFP corresponds to an amphisome or autolysosome. Other fluorophores such as mCherry are also suitable instead of mRFP,229 and an image-recognition algorithm has been developed to quantify flux of the reporter to acidified compartments.242,267 One of the major advantages of the tandem mRFP/mCherry-GFP reporter method is that it enables simultaneous estimation of both the induction of autophagy and flux through autophagic compartments in essentially native conditions, without requiring any drug treatment. The use of more than one time point allows visualization of increased early autophagosomes followed by increases in late autophagosomes as an additional assurance that flux has been maintained.268 In addition, this method can be used to monitor autophagy in high-throughput drug screening studies.242 The quantification of “yellow only” and “red only” dots in a stable tandem-fluorescent LC3-reporter cell line can be automated by a Cellomics microscope that can be used to assess a huge population of cells (1,000 or more) over a large number of random fields of view. This can give rise to more accurate data than can be achieved by manual assessment of a few selected cells.167,269
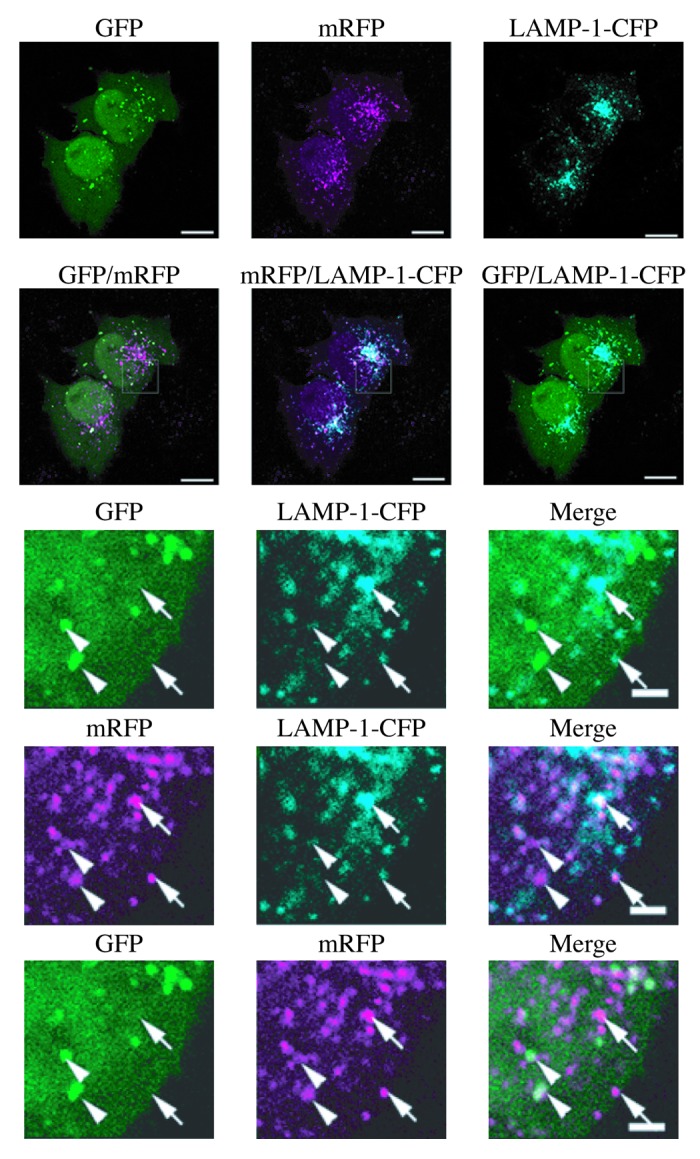
Figure 10. The GFP and mRFP signals of tandem fluorescent LC3 (tfLC3, mRFP-GFP-LC3) show different localization patterns. HeLa cells were cotransfected with plasmids expressing either tfLC3 or LAMP1-CFP. Twenty-four hours after the transfection, the cells were starved in Hanks’ solution for 2 h, fixed and analyzed by microscopy. The lower panels are a higher magnification of the upper panels. Bar, 10 µm in the upper panels and 2 µm in the lower panels. Arrows in the lower panels point to (or mark the location of) typical examples of colocalized signals of mRFP and LAMP1. Arrowheads point to (or mark the location of) typical examples of colocalized particles of GFP and mRFP signals. This figure was previously published in reference 181 and is reproduced by permission of Landes Bioscience, copyright 2007.
An alternative dual fluorescence assay involves the Rosella biosensor. This assay monitors the uptake of material to the lysosome/vacuole and complements the use of the tandem mRFP/mCherry-GFP reporter. The assay is based upon the genetically encoded dual color-emission biosensor Rosella, a fusion between a relatively pH-stable fast-maturing RFP variant, and a pH-sensitive GFP variant. When targeted to specific cellular compartments or fused to an individual protein, the Rosella biosensor provides information about the identity of the cellular component being delivered to the vacuole/lysosome for degradation. Importantly, the pH-sensitive dual color fluorescence emission provides information about the environment of the biosensor during autophagy of various cellular components. In yeast, Rosella has been successfully used to monitor autophagy of cytosol, mitochondria (mitophagy) and the nucleus (nucleophagy).270,271 Furthermore, the Rosella biosensor can be used as a reporter under various conditions including nitrogen depletion-dependent induction of autophagy.270,271 The Rosella biosensor can also be expressed in mammalian cells to follow nonselective autophagy (cytoplasmic turnover), or mitophagy.271
Cautionary notes: The use of tandem mRFP/mCherry-GFP-LC3/Atg8 reporters in live imaging experiments can be complicated by the motion of LC3/Atg8 puncta. As a consequence, conventional confocal microscopy may not allow visualization of colocalized mRFP/mCherry-GFP puncta. In this case, GFP or colocalized puncta represent newly formed autophagic structures whereas mRFP/mCherry-only puncta are ambiguous. Spinning disk confocal microscopy, or rapid acquisition times may be required for imaging tandem mRFP/mCherry-GFP proteins, although these techniques require a brighter fluorescent signal associated with what may be undesirably higher levels of transgene expression. One solution is to use the mTagRFP-mWasabi-LC3 chimera,272a as mTagRFP is brighter than mRFP1 and mCherry, and mWasabi is brighter than EGFP.272b Another possibility is to use fixed cells; however, this presents an additional concern: The use of tandem mRFP/mCherry-GFP relies on the quenching of the GFP signal in the acidic autolysosome; however, fixation solutions are often neutral or weak bases, which will increase the pH of the entire cell. Accordingly, the GFP signal may be restored after fixation (Fig. 11), which would cause an underestimation of the amount of signal that corresponds only to RFP (i.e., in the autolysosome). Thus, the tissue or cell samples must be properly processed to avoid losing the acidic environment of the autolysosomes. In addition, there may be weak fluorescence of EGFP even in an acidic environment (pH between 4 and 5).180,258 Therefore, it may be desirable to choose a monomeric green fluorescent protein that is more acid sensitive than EGFP for assaying autophagic flux.

Figure 11. GFP fluorescence in the autolysosome can be recovered upon neutralization of the pH. (A) GFP-LC3 emits green fluorescence in the autolysosomes of post-mortem processed heart sections. Cryosections of 3.8% paraformaldehyde fixed ventricular myocardium from 3-week old GFP-LC3 transgenic mice at the baseline (control) or starved for 24 h (starved) were processed for immunostaining using a standard protocol (buffered at pH 7.4). Most of the GFP-LC3 puncta are positive for LAMP1, suggesting that the autolysosomes had recovered GFP fluorescence. (B) Colocalization between GFP-LC3 direct fluorescence (green) and indirect immunostaining for GFP (red). Sections processed as in (A) were immunostained for GFP using a red fluorescence-tagged secondary antibody, and the colocalization with GFP fluorescence was examined by confocal microscopy. Almost all of the red puncta emit green fluorescence. Images provided by Xuejun Wang.
Another caution in the interpretation of the tandem fluorescent marker is that colocalization of GFP and mRFP/mCherry might also be seen in the case of impaired proteolytic degradation within autolysosomes, or altered lysosomal pH. Finally, expression of tandem mRFP-GFP-LC3 is toxic to some cancer cell lines relative to GFP-LC3 or RFP-LC3 (Choi KS, personal communication). The cytotoxicity of DsRed and its variants such as mRFP1 is associated with downregulation of BCL2L1/Bcl-xL.273 In contrast to mRFP-GFP-LC3, overexpression of mTagRFP-mWasabi-LC3 does not appear to be toxic to HeLa cells (Lin J, personal communication).
The Rosella assay has not been tested in a wide range of mammalian cell types. Accordingly, the sensitivity and the specificity of the assay must be verified independently until this method has been tested more extensively and used more widely.
Finally, it is ideal to capture the dynamic behavior of autophagy in real time, in order to generate data revealing the rate of formation and clearance of autophagosomes over time, rather than single data points. For example, by acquiring signals from two fluorescent constructs in real time, the rate of change in colocalization signal as a measure of the fusion rate and recycling rate between autophagosomes and lysosomes can be assessed. Importantly, due to the integral dynamic relationship of autophagic flux with the onset of apoptosis and necrosis it is advantageous to monitor cell death and autophagic flux parameters concomitantly over time, which FRET-based reporter constructs make possible.274
Conclusion: The use of tandem fluorescent constructs, which display different emission signals depending on the environment (in particular, GFP fluorescence is particularly sensitive to an acidic pH), provides a convenient way to monitor autophagy flux in many cell types.
f. Autophagic flux determination using flow and multispectral imaging cytometry
Whereas fluorescence microscopy, in combination with novel autophagy probes, has permitted single cell analysis of autophagic flux, automation for allowing medium- to high-throughput analysis has been challenging. A number of methods have been developed that allow the determination of autophagic flux using the fluorescence-activated cell sorter (FACS),154,223,236,275-277 and commercial kits are now available for monitoring autophagy by flow cytometry. These approaches make it possible to capture high-content images of cells in flow (up to 1,000 cells/sec), and are particularly useful for cells that grow in suspension. Optimization of image analysis permits the study of cells with heterogeneous LC3 puncta, thus making it possible to quantify autophagic flux accurately in situations that might perturb normal processes (e.g., microbial infection).277,278 Since EGFP-LC3 is a substrate for autophagic degradation, total fluorescence intensity of EGFP-LC3 can be used to indicate levels of autophagy in living mammalian cells.275 When autophagy is induced, the decrease in total cellular fluorescence can be precisely quantified in large numbers of cells to obtain robust data. In another approach, soluble EGFP-LC3-I can be depleted from the cell by a brief saponin extraction so that the total fluorescence of EGFP-LC3 then represents that of EGFP-LC3-II alone (Fig. 12A).235,236 Since EGFP-LC3 transfection typically results in high relative levels of EGFP-LC3-I, this treatment significantly reduces the background fluorescence due to non-autophagosome-associated reporter protein. By comparing treatments in the presence or absence of lysosomal degradation inhibitors, subtle changes in the flux rate of the GFP-LC3 reporter construct can be detected. If it is not desirable to treat cells with lysosomal inhibitors to determine rates of autophagic flux, a tandem mRFP/mCherry-EGFP-LC3 (or similar) construct can also be used for autophagic flux measurements in FACS experiments (see Tandem mRFP/mCherry-GFP fluorescence microscopy).276

Figure 12. Saponin extraction allows quantification of LC3-II fluorescence by FACS. (A) Schematic diagram of the effects of the saponin wash. Due to the reorganization of the EGFP-LC3 reporter protein, induction of autophagosome formation does not change the total levels of fluorescence in EGFP-LC3-transfected cells. However, extraction of EGFP-LC3-I with saponin results in a higher level of fluorescence in cells with proportionally higher levels of EGFP-LC3-II-containing autophagosomes. This figure was previously published in reference 236 (B) Saponin extraction can also be used to measure flux of endogenous LC3 protein. Human osteosarcoma cells were starved of amino acids and serum by incubation in EBSS, for the indicated times in the presence or absence of a 1 h chloroquine (50 µM) treatment. Cells were then washed with PBS containing 0.05% saponin and processed for FACS analysis for endogenous LC3. These data are provided by K.E. Eng and G.M. McInerney.
These methods, however, require the cells of interest to be transfected with reporter constructs. Since the saponin extraction method can also be combined with intracellular staining for endogenous LC3 protein, subtle changes in autophagic flux can be measured without the need for reporter transfections (Fig. 12B). This enables investigations of autophagic flux in a wide variety of cell types and tissues.
Cautionary notes: Care must be taken when applying flow cytometry measurements to adherent cells, particularly neurons and other cells with interdigitated processes, as the preparation of single cell suspensions entails significant levels of plasma membrane disruption and injury that can secondarily induce autophagy.
Users of the saponin extraction method should carefully titrate saponin concentrations and times of treatment in order to ensure specific extraction of LC3-I in their systems. Also, it has been observed in some cell types that saponin treatment can lead to non-autophagic aggregation of LC3,237 which should be controlled for in these assays (see GFP-Atg8/LC3 fluorescence microscopy).
Cell membrane permeabilization with digitonin and extraction of the nonmembrane-bound form of LC3 allows combined staining of membrane-associated LC3-II protein and any markers for detection of autophagy in relation to other cellular events/processes. Based on this approach a method for monitoring autophagy in different stages of the cell cycle was developed.279 Thus, the presence of basal or starvation-induced autophagy is detected in G1, S and G2/M phases of the cell cycle in MEFs with doxycycline-regulated ATG5 expression. In these experiments cells were gated based on their DNA content, and the relative intensity of GFP-LC3-II and LC3-II expression. This approach might also be used for the detection of autophagic flux in different stages of the cell cycle or subG1 apoptotic cell population by measuring accumulation of LC3-II in the presence or absence of lysosomal inhibitors.
Although GFP-LC3 can be used as a reporter for flow cytometry, it is more stable (which is not necessarily ideal for flux measurements) than GFP-SQSTM1 or GFP-NBR1 (NBR1 is a selective autophagic substrate with structural similarity to SQSTM1280); GFP-SQSTM1 displays the largest magnitude change following the induction of autophagy by amino acid deprivation or rapamycin treatment, and may thus be a better marker for following autophagic flux by this method (confirmed in SH-SY5Y neuronal cell lines stably expressing GFP-SQSTM1; Valente EM, personal communication).281 However, SQSTM1 changes can be cell type and context specific. In some cell types, there is no change in overall SQSTM1 levels despite strong levels of autophagy induction, verified by the tandem mRFP/mCherry-GFP-LC3 reporter as well as Atg7- and lysosome-dependent turnover of cargo proteins (Chu CT, personal observation). In other contexts, a robust loss of SQSTM1 does not correlate with increased autophagic flux as assessed by a luciferase-based measure of flux.166 Thus, appropriate positive and negative controls, and assessment of SQSTM1 mRNA levels, may be needed prior to the use of SQSTM1 as a flux indicator in a particular cellular context.
Conclusion: Medium- to high-throughput analysis of autophagy is possible using flow and multispectral imaging cytometry (Fig. 13). The advantage of this approach is that larger numbers of cells can be analyzed with regard to GFP-LC3 puncta, cell morphology and/or autophagic flux, and concomitant detection of surface markers can be included, potentially providing more robust data than is achieved with other methods.

Figure 13. Assessing autophagy with multispectral imaging cytometry. (A) Bright Detail Intensity (BDI) measures the foreground intensity of bright puncta (that are three pixels or less) within the cell image. For each cell, the local background around the spots is removed before intensity calculation. Thus, autophagic cells with puncta have higher BDI values. (B) Media control (untreated wild type), rapamycin-treated wild-type and Atg5−/− MEFs were gated based on BDI. Representative images of cells with high or low BDI values. Scale bar, 10 µm. Images provided by M.L. Albert.
g. Immunohistochemistry
Immunodetection of ATG proteins (particularly LC3 and BECN1) has been reported as a prognostic factor in various human carcinomas, including lymphoma,140,282 breast carcinoma,283 endometrial adenocarcinoma,284,285 head and neck squamous cell carcinoma,286,287 hepatocellular carcinoma,288,289 gliomas,290 non-small cell lung carcinomas,291 pancreatic292 and colon adenocarcinomas,293-295 as well as in cutaneous and uveal melanomas.296,297 Importantly, this kind of assay should be performed as recommended by the Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK).298 As we identify new drugs for modulating autophagy in clinical applications, this type of information may prove useful in the identification of subgroups of patients for targeted therapy.299-301
In mouse and rat tissues, endogenous LC3, ATG4B, and ATG9A have been detected by immnohistochemical analyses using both paraffin sections and cryosections.208,302-304 When autophagosomes are absent, the localization pattern of LC3 in the cells of various tissues is diffuse and cytosolic. Moreover, intense fibrillary staining of LC3 is detectable along dendrites of intact neurons, whereas granular staining for LC3 appears mainly in the perikarya of neurons in cathepsin D- or cathepsins B- and L-deficient mouse brains.208 In developing retinal tissue in chicken, BECN1 and AMBRA1 are detected by immunofluorescence.305,306 Finally, in non-mammalian vertebrates, BECN1 was detected during follicular atresia in the ovary of three fish species using paraffin sections; a punctate immunostaining for BECN1 was scattered throughout the cytoplasm of the follicular cells when they were in intense phagocytic activity for yolk removal (Rizzo E, unpublished results).
Cautionary notes: One problem with immunohistochemistry for LC3 is that in some tissues this protein can be localized in structures other than autophagosomes. For example, in murine hepatocytes and cardiomyocytes under starved conditions, endogenous LC3 is detected not only in autophagosomes but also on lipid droplets.307 In neurons in ATG7-deficient mice, LC3 is accumulated in ubiquitin- and SQSTM1-positive aggregates.308 Thus, immunodetection of LC3 in cytoplasmic granules is not sufficient to monitor autophagy in vivo.
Conclusion: It has not been clearly demonstrated that immunohistochemistry of ATG proteins in tissues corresponds to autophagy activity, and this area of research needs to be further explored before we can make specific recommendations.
3. SQSTM1/p62 and related LC3 binding protein turnover assays
In addition to LC3, SQSTM1/p62, or other receptors such as NBR1, can also be used as a protein marker, at least in certain settings.25,309 The SQSTM1 protein serves as a link between LC3 and ubiquitinated substrates.98 SQSTM1 and SQSTM1-bound polyubiquitinated proteins become incorporated into the completed autophagosome and are degraded in autolysosomes, thus serving as a readout of autophagic degradation (Fig. 14). Inhibition of autophagy correlates with increased levels of SQSTM1 in mammals and Drosophila, suggesting that steady-state levels of this protein reflect the autophagic status.304,310-315 Similarly, decreased SQSTM1 levels are associated with autophagy activation. The phosphorylation of SQSTM1 at Ser403 appears to regulate its role in the autophagic clearance of ubiquitinated proteins, and anti-phospho-SQSTM1/p62 antibodies can be used to detect the modified form of the protein.234

Figure 14. Regulation of the SQSTM1 protein during autophagy. (A) The level of SQSTM1 during starvation. Atg5+/+ and Atg5−/− MEFs were cultured in DMEM without amino acids and serum for the indicated times, and then subjected to immunoblot analysis using anti-SQSTM1/p62 antibody (Progen Biotechnik). This figure was previously published in reference 25 and is reproduced by permission of Landes Bioscience, copyright 2007. (B) The level of SQSTM1 in the brain of neural cell-specific Atg5 knockout mice. This image was generously provided by Dr. Taichi Hara (Tokyo Medical and Dental University).
Cautionary notes: SQSTM1 contains an LC3 interacting motif as well as a ubiquitin binding domain, and appears to act by linking ubiquitinated substrates with the autophagic machinery. Nonetheless, it would be prudent to keep in mind that SQSTM1 contains domains that interact with several signaling molecules,316 and SQSTM1 may be part of the mechanistic target of rapamycin (MTOR) complex 1 (TORC1).317 Thus, it may have additional functions that need to be considered with regard to its role in autophagy. In the context of autophagy as a stress response, the complexity of using SQSTM1 as an autophagy marker is underscored by its capacity to modulate the NFE2L2/NRF2 anti-oxidant response pathway through a KEAP1 binding domain.318 In fact, SQSTM1 may, itself, be transcriptionally induced by NFE2L2.319 Furthermore, it is necessary to examine endogenous SQSTM1 because overexpression of this protein leads to the formation of protein inclusions. In fact, even endogenous SQSTM1 becomes Triton X-100-insoluble in the presence of protein aggregates and when autophagic degradation is inhibited; thus, results with this protein are often context-dependent. In addition, SQSTM1 participates in proteasomal degradation, and its level may also increase when the proteasome is inhibited.320 Accordingly, the SQSTM1 degradation rate should be analyzed in the presence of an inhibitor such as epoxomicin or lactacystin to determine the contribution from the proteasome (see Autophagy inhibitors and inducers for potential problems with MG132).321 SQSTM1 is also a substrate for CASP6/caspase 6 and CASP8/caspase 8 (as well as CAPN1/calpain 1), which may confound its use in examining cell death and autophagy.322 Another issue is that some phosphatidylinositol 3-kinase (PtdIns3K) inhibitors such as LY294002, and to a lesser extent wortmannin (but apparently not 3-MA)256 can inhibit protein synthesis;323 this might in turn affect the turnover of SQSTM1 and LC3, which could influence conclusions that are drawn from the status of these proteins regarding autophagy flux or aggresome-like induced structures (ALIS) formation. Accordingly, it may be advisable to measure protein synthesis and proteasome activity along with autophagy under inhibitory or activating conditions. With regard to protein synthesis, it is worth noting that this can be monitored through a nonradioactive method.324 Finally, SQSTM1 may be transcriptionally upregulated under certain conditions,227,325 further complicating the interpretation of results. For example, SQSTM1 upregulation, and at least transient increases in the amount of SQSTM1, is seen in some situations where there is an increase in autophagic flux.326-328 Of interest, SQSTM1 hyperexpression at both gene and protein levels can be observed in muscle atrophy induced by cancer, though not by glucocorticoids, suggesting that the stimulus inducing autophagy may also be relevant to the differential regulation of autophagy-related proteins (Penna F, Costelli P, unpublished observations). One solution to problems relating to variations in SQSTM1 synthesis is to use a HaloTag®-SQSTM1/p62 chimera.329 The chimeric protein can be covalently labeled with HaloTag® ligands, and the loss of signal can then be monitored without interference by subsequent changes in protein synthesis. Similarly, a stable cell line expressing EGFP-tagged SQSTM1 under the control of in inducible promoter can be used to assess the rates of SQSTM1 degradation, taking into account the limitations outlined above (see Autophagic flux determination using flow and multispectral imaging cytometry).281 Yet another solution is to employ a radioactive pulse-chase assay to measure the rates of SQSTM1 degradation.330
Western blot analysis using NP40 or Triton X-100 lysis in autophagic conditions typically shows a reduction in SQSTM1 levels. However, this does not necessarily indicate that SQSTM1 is degraded, because SQSTM1 aggregates are insoluble in these detergent lysis conditions.227,331 Moreover, in some instances SQSTM1 levels do not change in the soluble fractions despite autophagic degradation, a finding that might be explained by simultaneous transcriptional induction of the gene encoding SQSTM1, since the soluble fraction accounts only for the diffuse or free form of SQSTM1. Accumulation of SQSTM1 in the Triton X-100-insoluble fraction can be observed when autophagy-mediated degradation is inhibited. Under conditions of higher autophagic flux, accumulation of SQSTM1 in Triton X-100-insoluble fractions may not be observed and SQSTM1 levels may be reduced or maintained. The simplest approach to circumvent many of these problems is by using lysis buffer that allows identification of the entire cellular pool of SQSTM1 (e.g., containing 1% SDS); however, additional assessment of both Triton X-100-soluble and -insoluble fractions will provide further information regarding the extent of SQSTM1 oligomerization.308
To conclusively establish SQSTM1 degradation by autophagy, SQSTM1 levels in both Triton X-100-soluble and -insoluble fractions need to be determined upon treatment with autophagy inducers in combination with autophagy inhibitors, such as those that inhibit the autolysosomal degradation steps (e.g., protease inhibitors, chloroquine or bafilomycin A1, or genetically by knocking out or knocking down LAMP2). Additionally, an alteration in the levels of SQSTM1 may not be immediately evident with changes observed in autophagic flux upon certain chemical perturbations (Sarkar S, personal communication). Whereas LC3 changes may be rapid, clearance of autophagy substrates may require a longer time. Therefore, if LC3 changes are assessed at 6 h or 24 h after a drug treatment, SQSTM1 levels can be tested not only at the same time points, but also at later time points (24 h or 48 h) for determining the maximal impact on substrate clearance. An alternative method is immunostaining for SQSTM1 with and without autophagy inhibitors, which will appear as either a diffuse or punctate pattern. Experiments with autophagy inducers and inhibitors, in combination with western blot and immunostaining analyses, best establish autophagic degradation based on SQSTM1 turnover. A final point, however, is that empirical evidence suggests that the species-specificity of antibodies for detecting SQSTM1 must be taken into account. For example, some commercial antibodies recognize both human and mouse SQSTM1, whereas others detect the human, but not the mouse protein (see the Autophagy Forum at https://www.landesbioscience.com/journals/autophagy/forum/ for information pertaining to anti-SQSTM1/p62 antibodies).332 Another issue with detecting SQSTM1 in the context of human diseases is that it can be mutated (e.g., in Paget disease of bone).333 Thus, care should be taken to ensure that potential mutations are not affecting the epitopes that are recognized by anti-SQSTM1 antibodies when using western blotting to detect this protein.
Conclusion: There is not always a clear correlation between increases in LC3-II and decreases in SQSTM1. Thus, although analysis of SQSTM1 can assist in assessing the impairment of autophagy or autophagy flux, we recommend using SQSTM1 only in combination with other methods such as LC3-II turnover to monitor flux.
4. TOR, AMPK and Atg1/ULK1
Atg1/ULK1 are central components in autophagy that likely act at more than one stage of the process. There are multiple ULK isoforms in mammalian cells including ULK1, ULK2, ULK3, ULK4 and STK36.334 ULK3 is a positive regulator of the Hedgehog signaling pathway,335 and its overexpression induces both autophagy and senescence.336 Along these lines, ectopic ULK3 displays a punctate pattern upon starvation-induced autophagy induction.336 ULK3, ULK4 and STK36, however, lack the domain present on ULK1 and ULK2 that bind ATG13 and RB1CC1/FIP200.337 Thus, ULK3 may play a role that is restricted to senescence, and that is independent of the core autophagy machinery. ULK2 has a higher degree of identity with ULK1 than any of the other homologs, and they may have similar functions that are tissue specific; however, ULK1 may be the predominant isoform involved in autophagy, as knockdown of ULK2 does not affect movement of ATG9.338 The stability and activation of ULK1, but not ULK2, is dependent on its interaction with the HSP90-CDC37 chaperone complex. Pharmacological or genetic inhibition of the chaperone complex increases proteasome-mediated turnover of ULK1, impairing its kinase activity and ability to promote both starvation-induced autophagy and mitophagy.239
AMP-activated protein kinase (AMPK) is a multimeric serine/threonine protein kinase comprising α- (catalytic), β- (scaffold), and γ- (regulatory) subunits. The enzyme activity of AMPK is absolutely dependent on phosphorylation of the α-subunit on Thr172,339,340 and can, therefore, be conveniently monitored by western blotting with a phosphospecific antibody against this site. In some cells, Thr172 is phosphorylated by the Ca2+-activated protein kinase CAMKK2/CaMKKβ; in other cells by the constitutively active kinase STK11/LKB1, regulation of AMPK activity being performed primarily by Thr172-dephosphorylating protein phosphatases such as protein phosphatase 1 and PPP2CB/PP2A.341 Thr172 dephosphorylation is modulated by adenine nucleotides that bind competitively to regulatory sites in the γ-subunit. AMP and ADP inhibit dephosphorylation and promote AMPK activity, whereas Mg2+-ATP has the opposite effect.340 AMPK thus acts as a fine-tuned sensor of the overall cellular energy charge that regulates cellular metabolism to maintain energy homeostasis. Activation of AMPK is also associated with the phosphorylation of downstream enzymes involved in ATP-consuming processes, such as fatty acid (acetyl-CoA carboxylase) and cholesterol (hydroxymethylglutaryl-CoA dehydrogenase) biosynthesis.
The role of AMPK in autophagy is complex, and highly dependent on both cell type and metabolic conditions. In liver cells, AMPK suppresses autophagy at the level of cargo sequestration as indicated by the rapid sequestration-inhibitory effects of a variety of AMPK activators, whereas it appears to stimulate autophagy in many other cell types, including fibroblasts, colon carcinoma cells and skeletal muscle.342-351 Autophagy-promoting effects of AMPK are most evident in cells cultured in a complete medium with serum and amino acids, where cargo sequestration is otherwise largely suppressed.348 Presumably, AMPK antagonizes the autophagy-inhibitory effect of amino acids (at the level of phagophore assembly) by phosphorylating proteins involved in TORC1 signaling, such as TSC2352 and RPTOR/RAPTOR353 as well the TORC1 target ULK1 (see below).354-356
Compound C is an effective and widely used inhibitor of activated (phosphorylated) AMPK.357 However, being a non-specific inhibitor of oxidative phosphorylation,358,359 this drug has been observed to inhibit autophagy under conditions where AMPK is already inactive or knocked out,360 and has even been shown to stimulate autophagy by an AMP-independent mechanism.359,361 Compound C thus cannot be used as a stand-alone indicator of AMPK involvement.
TORC1 is an autophagy-suppressive regulator that integrates growth factor, nutrient and energy signals. In most systems, inhibition of MTOR leads to induction of autophagy, and AMPK activity is generally antagonistic toward MTOR function. TORC1 mediates the autophagy-inhibitory effect of amino acids, which stimulate the MTOR protein kinase through a RRAG/RAG GTPase dimer. Insulin and growth factors activate TORC1 through upstream kinases including AKT1 (protein kinase B), and extracellular signal regulated kinase (MAPK3-MAPK1, or ERK) when the energy supply is sufficient, whereas energy depletion may induce AMPK-mediated TORC1 inhibition and autophagy stimulation, for example, during glucose starvation. Amino acid starvation, on the other hand, can strongly induce autophagy even in cells completely lacking AMPK catalytic activity.362
AMPK and TORC1 regulate autophagy through coordinated phosphorylation of ULK1; under glucose starvation, AMPK promotes autophagy by directly activating ULK1 through phosphorylation, although the exact AMPK-mediated ULK1 phosphorylation site(s) remains unclear.351,354-356 Under conditions of nutrient sufficiency, high TORC1 activity prevents ULK1 activation by phosphorylating alternate ULK1 residues and disrupting the interaction between ULK1 and AMPK. There are commercially available phospho-specific antibodies that recognize different forms of ULK1. For example, phosphorylation at Ser555, an AMPK site, is indicative of increased autophagy in response to nutrient stress, whereas Ser757 is targeted by MTOR to inhibit autophagy. Even the autophagy-suppressive effects of AMPK could, conceivably, be mediated through ULK1 phosphorylation, for example, at the inhibitory site Ser638.363 AMPK inhibits MTOR by phosphorylating and activating TSC2.364 Therefore, AMPK is involved in processes that synergize to activate autophagy, by directly activating ULK1, and indirectly impairing MTOR-dependent inhibition of ULK1. The identification of ULK1 as a direct target of TORC1 and AMPK represents a significant step toward the definition of new tools to monitor the induction of autophagy. However, further studies directed at identifying physiological substrates of ULK1 will be essential to understand how ULK1 activation results in initiation of the autophagy program. Along these lines, ULK1 phosphorylates AMBRA1,365 and the MYLK/MLCK-like protein Sqa,366 as well as ATG13 and RB1CC1/FIP200.367-370 In addition, ULK1 binds to, and phosphorylates, RPTOR, leading to inhibition of TORC1.371 Furthermore, ULK1 itself appears to be able to mediate inhibitory AMPK phosphorylation to generate a negative feedback loop.372
TORC1 activity can be monitored by following the phosphorylation of its substrates, such as EIF4EBP1 (4E-BP1/PHAS-I) and RPS6KB1 (p70S6 kinase) or the latter’s downstream target, the ribosomal protein S6 (RPS6), for which good commercial antibodies are available.373-375 In mammalian cells, the analysis should focus on the phosphorylation of RPS6KB1 at Thr389, and EIF4EBP1 at Thr37 and Thr46, which are directly phosphorylated by TORC1.376 The TORC1-dependent phosphorylation of EIF4EBP1 can be detected as a molecular mass shift by western blot.375 Examining the phosphorylation status of RPS6KB1 and EIF4EBP1 may be a better method for monitoring TORC1 activity than following the phosphorylation of proteins such as RPS6, because the latter is not a direct substrate of TORC1 (although RPS6 phosphorylation is a good readout for RPS6KB1/2 activities, which are directly dependent on MTOR), and it can also be phosphorylated by other kinases such as RPS6KA/RSK. Furthermore, the mechanisms that determine the selectivity as well as the sensitivity of TORC1 for its substrates seem to be dependent on the integrity and configuration of TORC1. For example, rapamycin strongly reduces RPS6KB1 phosphorylation, whereas its effect on EIF4EBP1 is more variable. In the case of rapamycin treatment, EIF4EBP1 can be phosphorylated by TORC1 until rapamycin disrupts TORC1 dimerization and its integrity, whereas RPS6KB1 phosphorylation is quickly reduced when rapamycin simply interacts with MTOR in TORC1 (see Autophagy inhibitors and inducers for information on catalytic MTOR inhibitors such as torin1).376 Since it is likely that other inhibitors, stress, and stimuli may also affect the integrity of TORC1, a decrease or increase in the phosphorylation status of one TORC1 substrate does not necessarily correlate with changes in others, including ULK1. Therefore, reliable antiphospho-ULK1 antibodies should be used to directly examine the phosphorylation state of ULK1, along with additional experimental approaches to analyze the role of the MTOR complex in regulating autophagy.
Activation/assembly of the Atg1 complex in yeast (composed of at least Atg1-Atg13-Atg17-Atg29-Atg31) or the ULK1 complex in mammals (ULK1-RB1CC1-ATG13-C12orf44/ATG101) is one of the first steps of autophagy induction. Therefore, activation of this complex can be assessed to monitor autophagy induction. In yeast, dephosphorylation of Atg13 is associated with activation/assembly of the core complex, which can be followed by immunoprecipitation or western blotting.377-380 In addition, the autophosphorylation of Atg1 at Thr226 is required for its kinase activity and for autophagy induction; this can be detected using phospho-specific antibodies, by immunoprecipitation or western blotting (Fig. 15).381,382 In mammalian cells, the phosphorylation status of ULK1 at the activating sites (Ser317, 467, 555, 637, 777, or Thr574) or dephosphorylation at inactivating sites (Ser638, 757) can be determined using phospho-specific antibodies,355,356 or by western blotting.383 In general, the core complex is stable in mammalian cells, although, as noted above, upstream inhibitors (MTOR) or activators (AMPK) may interact dynamically with it, thereby determining the status of autophagy.

Figure 15.S. cerevisiae cells transformed with a plasmid encoding HA-Atg1 were cultured to mid-log phase and shifted to SD-N (minimal medium lacking nitrogen that induces a starvation response). Immunoblotting was done with anti-HA antibody. The upper band corresponds to autophosphorylation of Atg1. This figure was modified from data previously published in reference 381 and is reproduced by permission of the American Society for Cell Biology, copyright 2011.
One additional topic that bears on ULK1 concerns the process of LC3-associated phagocytosis. LAP is a type of phagocytosis in macrophages that involves the conjugation of LC3 to single-membrane pathogen-containing phagosomes, a process that promotes phagosome acidification and fusion with lysosomes.131 Most of the core autophagy components are required for LAP, but the two processes can be distinguished by the presence or absence, respectively, of a double-membrane sequestering vesicle. ULK1 is not needed for LAP, which provides a more convenient means for distinguishing between the two processes.134
Cautionary notes: A decrease in TORC1 activity is a good measure for autophagy induction; however, TORC1 activity does not necessarily preclude autophagy induction because there are TOR-independent mechanisms that induce autophagy.384-387 Along these lines, whereas in most systems inhibition of MTOR leads to the induction of autophagy, there are instances in commonly used cancer cell lines in which MTOR appears to be a positive effector.388 Furthermore, in adult skeletal muscle, autophagy gene upregulation and autophagosome formation is independent of TORC1 but partially dependent on MTOR complex 2 (TORC2), as shown by the finding that autophagosome formation is increased by knockdown of RICTOR, a component of TORC2, but not TORC1.346 In addition, TORC1 is downstream of AKT1; however, oxidative stress inhibits MTOR, thus allowing autophagy, despite the concomitant activation of AKT1.115 Also, persistent autophagy induction can cause negative feedback that results in the reactivation of MTOR under conditions of ongoing starvation.389 Thus, it is necessary to be cautious in deciding how to monitor the MTOR pathway, and to verify that the pathway being analyzed displays MTOR-dependent inhibition.
One problem in monitoring assembly of the ULK1 complex is the low abundance of endogenous ULK1 in many systems, which makes it difficult to detect phospho-ULK1 by western blot analysis. In addition, Atg1/ULK1 is phosphorylated by multiple kinases, and the amount of phosphorylation at different sites can increase or decrease during autophagy induction. Thus, although there is an increase in phosphorylation at the activating sites upon induction, the overall phosphorylation states of ULK1 and ATG13 are decreased under conditions that lead to induction of autophagy; therefore, monitoring changes in phosphorylation by following molecular mass shifts upon SDS-PAGE may not be informative. In addition, such phosphorylation/dephosphorylation events are expected to occur relatively early (1–2 h) in the signaling cascade of autophagy. Therefore, it is necessary to optimize treatment time conditions. Finally, in Arabidopsis and possibly other eukaryotes, the ATG1 and ATG13 proteins are targets of autophagy, which means that their levels may drop substantially under conditions that induce autophagic turnover.174
At present, the use of Atg1/ULK1 kinase activity as a tool to monitor autophagy is limited because only a few physiological substrates have been identified, and the importance of the Atg1/ULK1-dependent phosphorylation has not been determined. Nonetheless, Atg1/ULK1 kinase activity appears to increase when autophagy is induced, irrespective of the pathway leading to induction. As additional physiological substrates of Atg1/ULK1 are identified it will be possible to follow their phosphorylation in vivo as is done with analyses for MTOR. Nonetheless, it must be kept in mind that monitoring changes in the activity of Atg1/ULK1 is not a direct assay for autophagy, although such changes may correlate with autophagy activity. Furthermore, in some cells ULK1 has functions in addition to autophagy, such as in axonal transport and outgrowth, and its activity state may thus reflect its role in these processes.390-395 Accordingly, other methods as described throughout these guidelines should also be used to follow autophagy directly.
Finally, there is not a complete consensus on the specific residues of ULK1 that are targeted by AMPK or MTOR. Similarly, apparently contradictory data have been published regarding the association of AMPK and MTOR with the ULK1 kinase complex under different conditions. Therefore, caution should be used in monitoring ULK1 phosphorylation or the status of ULK1 association with AMPK until these issues are resolved.
Conclusion: Assays for Atg1/ULK1 can provide detailed insight into the induction of autophagy, but are not a direct measurement of the process. Similarly, analysis of MTOR substrates such as RPS6KB1 and EIF4EBP1 are not recommended readouts for autophagy, and need to be combined with other assays that directly monitor autophagy activity.
5. Additional autophagy-related markers
Although Atg8/LC3 has been the most extensively used protein for monitoring autophagy, other proteins can also be used for this purpose. Here, we discuss some of the more commonly used or better-characterized possibilities.
a. ATG9
ATG9/Atg9 is the only integral membrane ATG protein that is essential for autophagosome formation. ATG9 displays partial colocalization with GFP-LC3.396 Perhaps the most unique feature of ATG9/Atg9, however, is that it localizes to multiple discrete puncta, whereas most Atg proteins are detected primarily in a single punctum or diffusely within the cytosol. Yeast Atg9 may cycle between the phagophore assembly site (PAS) and peripheral reservoirs;397 the latter correspond to tubulovesicular clusters that are precursors to the phagophore.398 Anterograde movement to the PAS is dependent on Atg11, Atg23, Atg27 and actin. Retrograde movement requires Atg1-Atg13, Atg2-Atg18 and the PtdIns3K complex I.399 Mutants such as atg1Δ accumulate Atg9 exclusively at the PAS, and this phenotype forms the basis of the transport of Atg9 after knocking out ATG1 (TAKA) assay.63 In brief, this is an epistasis analysis in which a double-mutant strain is constructed (one of the mutations being atg1Δ) that expresses Atg9-GFP. If the second mutated gene encodes a protein that is needed for Atg9 anterograde transport, the double mutant will display multiple Atg9-GFP puncta. In contrast, if the protein acts along with or after Atg1, all of the Atg9-GFP will be confined to the PAS. Monitoring the localization of ATG9 has not been used extensively in higher eukaryotes, but this protein displays the same type of dependence on Atg1/ULK1 for cycling as seen in yeast,396,399 suggesting that it is possible to follow this protein as an indication of ULK1 and ATG13 function.370
b. ATG12–ATG5
ATG5, ATG12, and ATG16L1, associate with the phagophore and have been detected by fluorescence or immunofluorescence (Fig. 16).400,401 Endogenous ATG5, ATG12 or ATG16L1 puncta formation can be followed to monitor autophagy upregulation. Under physiological conditions, the endogenous proteins are predominantly diffusely distributed throughout the cytoplasm. Upon induction of autophagy, for example during starvation, there is a marked increase in the proportion of cells with punctate ATG5, ATG12 and ATG16L1. Furthermore, inhibitors of autophagosome formation result in a block in this starvation-induced puncta formation, and this assay is very robust in mammalian cells.

Figure 16. Confocal microscopy image of HCT116 cells immunostained with human-specific antibody to ATG12. Cells were starved for 8 h or treated with chloroquine (50 µM) for 3 h. Scale bar, 10 µm. Image provided by M. Llanos Valero, M.A. de la Cruz and R. Sanchez-Prieto.
ATG12?ATG5 conjugation has been used in some studies to measure autophagy. In Arabidopsis and some mammalian cells it appears that essentially all of the ATG5 and ATG12 proteins exist in the conjugated form and the expression levels do not change, at least during short-term starvation.150,400-402 Therefore, monitoring ATG12–ATG5 conjugation per se may not be a useful method for following the induction of autophagy. It is worth noting, however, that in some cell lines free ATG5 can be detected,403 suggesting that the amount of free ATG5 may be cell line-dependent. One final parameter that may be considered is that the total amount of the ATG12–ATG5 conjugate may increase following prolonged starvation as has been observed in hepatocytes and both mouse and human fibroblasts (Cuervo AM, personal communication; Sarkar S, personal communication).
c. ATG14
Yeast Atg14 is the autophagy-specific subunit of the Vps34 complex I,404 and a human homolog, named ATG14 (ATG14L/BARKOR), has been identified.405-408 ATG14 localizes primarily to phagophores. The C-terminal fragment of the protein is named the BATS domain (BARKOR/ATG14(L) autophagosome targeting sequence), and is able to direct GFP and BECN1 to autophagosomes in the context of a chimeric protein.409 Currently, a good antibody that can be used to detect the endogenous ATG14 does not exist. ATG14-GFP or BATS-GFP detected by fluorescence microscopy or TEM can be used as a phagophore marker; however, ATG14 is not localized exclusively to phagophores, as it can also be detected on mature autophagosomes as well as the ER.409,410 Accordingly, detection of ATG14 should be performed in combination with other phagophore and autophagosome markers.
d. ATG16L1
ATG16L1 has been used to monitor the movement of plasma membrane as a donor for autophagy, and thus an early step in the process; ATG16L1 is located on phagophores, but not on completed autophagosomes.269,411 ATG16L1 can be detected by immuno-TEM, by immunostaining of Flag epitope-tagged ATG16L1, and/or by the use of GFP-tagged ATG16L1.
e. Atg18/WIPI family
Yeast Atg18412,413 and Atg21262 (or the mammalian WIPI homologs414) are required for both macroautophagy (i.e., nonspecific sequestration of cytoplasm) and autophagy-related processes (e.g., the Cvt pathway,415,416 specific organelle degradation,74 and autophagic elimination of invasive microbes77,78,80,81,417).412 These proteins bind phosphatidylinositol 3-phosphate (PtdIns3P) that is present at the phagophore and autophagosome418,419 and also PtdIns(3,5)P2. Human WIPI1 and WIPI2 function downstream of the phosphatidylinositol 3-kinase class III (PtdIns3KC3) complex I [PIK3C3, BECN1, PIK3R4/VPS15, ATG14] and upstream of both the ATG12 and LC3 ubiquitin-like conjugation systems.418,420,421 Upon the initiation of the autophagic pathway, WIPI1 and WIPI2 bind PtdIns3P and accumulate at limiting membranes, such as those of the endoplasmic reticulum, where they participate in the formation of omegasomes and/or autophagosomes. On the basis of quantitative fluorescence microscopy, this specific WIPI protein localization has been used as an assay to monitor autophagy in human cells.419 Using either endogenous WIPI1 or WIPI2, detected by indirect fluorescence microscopy or EM, or transiently or stably expressed tagged fusions of GFP to WIPI1 or WIPI2, basal autophagy can be detected in cells that display WIPI puncta at autophagosomal membranes. In circumstances of increased autophagic activity, such as nutrient starvation or rapamycin administration, the induction of autophagy is reflected by the elevated number of cells that display WIPI puncta when compared with the control setting. Also, in circumstances of reduced autophagic activity such as wortmannin treatment, the reduced number of WIPI puncta-positive cells reflects the inhibition of autophagy. Basal, induced and inhibited formation of WIPI puncta closely correlate with both the protein level of LC3-II and the formation of GFP-LC3 puncta.419,421 Accordingly, WIPI puncta can be assessed as an alternative to LC3. Automated imaging and analysis of fluorescent WIPI1 (Fig. 17) or WIPI2 puncta represents an efficient and reliable opportunity to combine the detection of WIPI proteins with other parameters. It should be noted that there are two isoforms of WIPI2 (2B and 2D),421 and in C. elegans WIPI4 (EPG-6) has been identified as the WIPI homolog required for autophagy.422 Thus, these proteins, along with the currently uncharacterized WDR45L/WIPI3, provide additional possibilities for monitoring phagophore and autophagosome formation.

Figure 17. Automated WIPI1 puncta image acquisition and analysis monitors the induction and inhibition of autophagy. Stable U2OS clones expressing GFP-WIPI1 were selected using 0.6 μg/ml G418 and then cultured in 96-well plates. Cells were treated for 3 h with nutrient-rich medium (control), nutrient-free medium (EBSS), or with 233 nM wortmannin. Cells were fixed in 3.7% paraformaldehyde and stained with DAPI (5 μg/ml in PBS). An automated imaging and analysis platform was used to determine the number of both GFP-WIPI1 puncta-positive cells and the number of GFP-WIPI1 puncta per individual cell.348 Cells without GFP-WIPI1 puncta are highlighted in red (cell detection) and purple (nuclei detection), whereas GFP-WIPI1 puncta-positive cells are highlighted in yellow (GFP-WIPI1 puncta detection), green (cell detection) and blue (nuclei detection). Bars, 20 µm. These images were provided by S. Pfisterer and T. Proikas-Cezanne.
Cautionary notes: With regard to detection of the WIPI proteins, endogenous WIPI1 puncta cannot be detected in many cell types,418 and the level of transiently expressed GFP-WIPI1 puncta is cell context-dependent;418,419 however, it has been used in human and mouse cell systems348,419 and mCherry-Atg18 also works in transgenic Drosophila (Juhász G, personal communication), although one caution with regard to the latter is that GFP-Atg18 expression can induce autophagy in the fat body of fed larvae (Kiger A, unpublished observation). GFP-WIPI1 and GFP-WIPI2 have been detected on the completed (mature) autophagosome by freeze-fracture analysis,60 but endogenous WIPI2 has not been detected on mRFP-LC3- or LAMP2-positive autophagosomes or autolysosomes using immunolabeling.418 Accordingly, it may be possible to follow the formation and subsequent disappearance of WIPI puncta to monitor autophagy induction and flux using specific techniques. As with GFP-LC3, overexpression of WIPI1 or WIPI2 can lead to the formation of aggregates, which are stable in the presence of PtdIns3K inhibitors.
f. BECN1/Atg6
BECN1/Atg6 and PIK3C3/VPS34 are essential partners in the autophagy interactome that signals the onset of autophagy,404,423,424 and many researchers use BECN1 as a way to monitor autophagy. BECN1 is inhibited by its binding to the anti-apoptotic protein BCL2.425 Autophagy is induced by the release of BECN1 from BCL2 by pro-apoptotic BH3 proteins, phosphorylation of BECN1 by DAPK (at Thr119, located in the BH3 domain),426 or phosphorylation of BCL2 by MAPK8/JNK1 (at Thr69, Ser70 and Ser87).427,428 The relationship between BECN1 and BCL2 is more complex in developing cerebellar neurons as it appears that the cellular levels of BCL2 are, in turn, post-translationally regulated by an autophagic mechanism linked to a switch from immaturity to maturity.429,430 It is important to be aware, however, that certain forms of macroautophagy are induced in a BECN1-independent manner, and are not blocked by PtdIns3K inhibitors.97,431 Interestingly, caspase-mediated cleavage of BECN1 inactivates BECN1-induced autophagy and enhances apoptosis in several cell types,432 emphasizing that the crosstalk between apoptosis and autophagy is complex.
Although a population of BECN1 may localize in proximity to the trans-Golgi network,433 it is also present at the ER and mitochondria.425 In keeping with these observations, in cerebellar organotypic cultures BECN1 co-immunoprecipitates with BCL2 that is primarily localized at the mitochondria and ER, and in a mouse model of neurodegeneration autophagic vacuoles in Purkinje neurons contain partially digested organelles that are immunoreactive for BCL2.430,434 In addition, BECN1 and PIK3C3 can be present in multiple complexes, so caution must be exercised when monitoring localization. On induction of autophagy by various stimuli the presence of BECN1- and PIK3C3-positive macroaggregates can be detected in the region of the Golgi complex by immunofluorescence.115,435 Thus, BECN1-GFP puncta detected by fluorescence microscopy or TEM may serve as an additional marker for autophagy induction;436 however, as with any GFP chimeras there is a concern that the GFP moiety interferes with correct localization of BECN1. To demonstrate that BECN1 or PtdIns3K macroaggregates are an indirect indication of ongoing autophagy, it is mandatory to show their specific association with the process by including appropriate controls with inhibitors (e.g., 3-MA) or autophagy gene silencing. When a BECN1-independent autophagy pathway is induced, such aggregates are not formed regardless of the fact that the cell expresses BECN1 (e.g., as assessed by western blotting) (Isidoro C, personal communication).
g. DRAM1
DRAM1 (damage-regulated autophagic modulator 1) is a gene induced by activated TP53/p53 in response to different types of cellular stress, including DNA damage.438,439 DRAM1 is a small hydrophobic protein with six transmembrane domains. It is detected as a subpopulation in the Golgi and cis-Golgi, colocalizing with GOLGB1/giantin and GOLGA2/GM130, and also in early and late endosomes and lysosomes, colocalizing with EEA1 and LAMP2.439 The elimination of DRAM1 by siRNA blocks autophagy,439,440 as effectively as elimination of BECN1, indicating it is an essential component for this process, although its mechanism of action is not known. The time course of autophagy as a consequence of DRAM1 activation can be monitored following the disappearance of the VRK1 protein, a direct target of this process, by immunoblot.439 Detection of DRAM1 RNA is very easy by qRT-PCR during autophagy;438,439 however, detection of the DRAM1 protein is very difficult because of its small size and hydrophobicity, which makes generation of specific antibodies a complicated process, and in general these have very low sensitivity.
h. ZFYVE1/DFCP1
ZFYVE1/DFCP1 (double FYVE-containing protein 1) binds PtdIns3P that localizes to the ER and Golgi. The ER population of ZFYVE1 is involved in formation of the omegasome.437 Starvation induces the translocation of ZFYVE1 to punctate structures on the ER. ZFYVE1 partially colocalizes with WIPI1 upon nutrient starvation,421 and also with WIPI2.418
Conclusion: Proteins other than Atg8/LC3 can be monitored to follow autophagy, and can be important tools to define specific steps of the process. For example, WIPI puncta formation can be used to monitor autophagy, but, similar to Atg8/LC3, should be examined in the presence and absence of inhibitors. Analysis of WIPI puncta should be combined with other assays because individual members of the WIPI family might also participate in additional, uncharacterized functions apart from their role in autophagy. At present, we caution against the use of changes in BECN1 localization as a marker of autophagy induction.
6. Transcriptional and translational regulation
The induction of autophagy in certain scenarios is accompanied by an increase in the mRNA levels of certain autophagy genes, such as ATG8/Lc3441,442 and Atg12,443 and an autophagy-dedicated microarray was developed as a high-throughput tool to simultaneously monitor the transcriptional regulation of all genes involved in, and related to, autophagy.444 The gene that shows the greatest transcriptional regulation in the liver (in response to starvation and circadian signals) is Ulk1, but others also show more limited changes in mRNA levels including Gabarapl1, Bnip3 and, to a minor extent, Lc3B (Lin JD, personal communication). In several mouse and human cancer cell lines, ER stress and hypoxia increase the transcription of LC3, ATG5 and ATG12 by a mechanism involving the unfolded protein response (UPR). Similarly, a stimulus-dependent increase in LC3B expression is detected in neutrophils undergoing autophagy induction.445 Increased expression of Atg5 in vivo after optic nerve axotomy in mice446 and increased expression of Atg7, Becn1 and Lc3a during neurogenesis at different embryonic stages in the mouse olfactory bulb are also seen.447 LC3 and ATG5 are not required for the initiation of autophagy, but mediate phagophore expansion and autophagosome formation. In this regard, the transcriptional induction of LC3 may be necessary to replenish the LC3 protein that is turned over during extensive ER stress- and hypoxia-induced autophagy.443,448 Thus, assessing the mRNA levels of LC3 and other autophagy-related genes by northern blot or qRT-PCR may provide correlative data relating to the induction of autophagy. It is not clear if these changes are sufficient to induce autophagy, however, and therefore these are not direct measurements.
Of note, large changes in Atg gene transcription just prior to Drosophila salivary gland cell death (that is accompanied by an increase in autophagy) are detected in Atg2, Atg4, Atg5 and Atg7, whereas there is no significant change in Atg8a or Atg8b.449,450 Autophagy is critical for Drosophila midgut cell death, which is accompanied by transcriptional upregulation of all of the Atg genes tested, including Atg8a (Fig. 18).199,451 Similarly, in the silkworm (B. mori) larval midgut the occurrence of autophagy is accompanied by an upregulation of Atg5, Atg6 and Atg8 mRNA levels.452 Transcriptional upregulation of Drosophila Atg8a and Atg8b is also observed in the fat body following induction of autophagy at the end of larval development,453 Atg5, Atg6, Atg8a and Atg18 are upregulated in the ovary of starved flies,454 and an increase in Drosophila Atg8b is observed in cultured Drosophila l(2)mbn cells following starvation (Gorski S, personal communication). An upregulation of plant ATG8 may be needed during the adaptation to reproductive growth; a T-DNA inserted mutation of rice ATG8b blocked the change from vegetative growth to reproductive growth in both homozygous and heterozygous plant lines (Zhang M-Y, unpublished results).
Figure 18. pGFP-Atg8a can be used to monitor autophagy in Drosophila melanogaster. The autophagosome marker pGFP-Atg8a, results in expression of Atg8a fused to GFP from the endogenous Atg8a promoter.199 Live imaging of gastric caeca from Drosophila melanogaster midgut; pGFP-Atg8a puncta (green) and Hoechst 33342 (blue). Midgut from early third instar larvae prior to the onset of cell death (top) and from dying midgut at 2 h after puparium formation (bottom). Bar, 25 µm. Figure provided by D. Denton and S. Kumar.
Similarly, the upregulation of autophagy-related genes (Lc3, Gabarapl1, Bnip3, Atg4b, Atg12l) has been documented at the transcriptional and translational level in several other species (e.g., mouse, rat, trout, Arabidopsis and maize) under conditions of ER stress,443 and diverse types of prolonged (several days) catabolic situations including cancer cachexia, diabetes mellitus, uremia and fasting.151,346,455-457 Along these lines, ATG9 and ATG16L1 are transcriptionally upregulated upon influenza virus infection (Khalil H, personal communication), and in C. elegans, the FOXA transcription factor PHA-4 regulates the expression of several autophagy-related genes.458 Such prolonged induction of the expression of ATG genes has been thought to allow the replenishment of critical proteins (e.g., LC3 and GABARAP) that are destroyed during autophagosome fusion with the lysosome.459 The polyamine spermidine increases life span and induces autophagy in cultured yeast and mammalian cells, as well as in nematodes and flies; in aging yeast, spermidine treatment triggers epigenetic deacetylation of histone H3 through inhibition of histone acetyltransferases, leading to significant upregulation of various autophagy-related transcripts.460
In addition to the ATG genes, transcriptional upregulation of VMP1 (a protein that is involved in autophagy regulation and that remains associated with the completed autophagosome) can be detected in mammalian cells subjected to rapamycin treatment or starvation, and in tissues undergoing disease-induced autophagy.461 VMP1 is an essential autophagy gene that is conserved from Dictyostelium to mammals,232,462 and the VMP1 protein regulates early steps of the autophagic pathway.420 VMP1 is poorly expressed in mammalian cells under nutrient-normal conditions, but is highly upregulated in cells undergoing autophagy, and the expression of VMP1 induces autophagosome formation.
A gene regulatory network, named CLEAR (coordinated lysosomal enhancement and regulation) that controls both lysosome and autophagosome biogenesis was identified using a systems-biology approach.463-465 The basic helix-loop-helix transcription factor EB (TFEB) acts as a master gene of the CLEAR network and positively regulates the expression of both lysosomal and autophagy genes, thus linking the biogenesis of two distinct types of cellular compartments that cooperate in the autophagic pathway. TFEB activity is regulated by starvation,463 and can thus serve as a new tool for monitoring transcriptional regulation connected with autophagy. Along these lines, the erythroid transcription factor GATA1 and its coregulator Friend of GATA1 induce the transcription of multiple genes encoding autophagy components; this developmentally regulated transcriptional response is coupled to increases in autophagosome number as well as the percent of cells that contain autophagosomes.466 Finally, CEBPB/C/EBPβ is a transcription factor that regulates autophagy in response to the circadian cycle.467
Although less work has been done on post-transcriptional regulation, several studies implicate microRNAs in controlling the expression of proteins associated with autophagy.164,468-470
Cautionary notes: Most of the ATG genes do not show significant changes in mRNA levels when autophagy is induced. Even increases in LC3 mRNA can be quite modest and are cell type- and organism-dependent.471 In addition, it is generally better to follow protein levels because that is the ultimate readout that is significant with regard to the initiation and completion of autophagy, although ATG protein amounts do not always change significantly and the extent of increase is again cell type- and tissue-dependent. In some cases (e.g., yeast ATG14), increased transcription is not accompanied by increased protein levels, apparently due to changes in translation efficiency under starvation conditions (Abeliovich H, unpublished data). Finally, changes in autophagy protein levels are not sufficient evidence of autophagy induction, and must be accompanied by additional assays as described herein. Thus, monitoring changes in mRNA levels for either ATG genes or autophagy regulators may provide some evidence for autophagy induction, but should be used along with other methods.
Another general caution pertains to the fact that in any cell culture system different types of cells (for example, those undergoing autophagy or not) exist simultaneously. Therefore, only an average level of protein or mRNA expression can be evaluated with most methods. This means that the results regarding specific changes in autophagic cells could be hidden due to the background of the average data. Along these lines, experiments using single-cell real-time PCR to examine gene expression in individual cardiomyocytes with and without signs of autophagy revealed that the transcription of MTOR markedly and significantly increased in autophagic cells in intact cultures (spontaneously undergoing autophagy) as well as in cultures treated with proteasome inhibitors to induce autophagy (Dosenko V, personal communication). Finally, researchers need to realize that mammalian cell lines may have mutations that alter autophagy signaling or execution; this problem can be avoided by using primary cells.
Conclusion: Although there are changes in ATG gene expression that coincide with, and may be needed for, autophagy, this has not been carefully studied experimentally. Therefore, at the present time we do not recommend the monitoring of ATG gene transcription as a general readout for autophagy unless there is clear documentation that the change(s) correlates with autophagy activity.
7. Autophagic protein degradation
Protein degradation assays represent a well-established methodology for measuring autophagic flux, and they allow good quantification. The general strategy is first to label cellular proteins by incorporation of a radioactive amino acid (e.g., [14C]-leucine, [14C]-valine or [35S]-methionine; although valine may be preferred over leucine due to the strong inhibitory effects of the latter on autophagy), preferably for a long time to achieve sufficient labeling of the long-lived proteins that best represent autophagic substrates, and then to follow this with a long cold-chase so that the assay starts well after labeled short-lived proteins are degraded (which occurs predominantly via the proteasome). Next, the time-dependent release of acid-soluble radioactivity from the labeled protein in intact cells or perfused organs is measured.3,472,473 Note that the inclusion of the appropriate unlabeled amino acid (i.e., valine, leucine or methionine) in the starvation medium at a concentration equivalent to that of other amino acids in the chase medium is necessary; otherwise, the released [14C]-amino acid is effectively re-incorporated into cellular proteins, which results in a significant underestimation of protein degradation. The turnover of specific proteins can also be measured in a pulse-chase regimen using the Tet-ON/OFF system and subsequent western blot analysis.474,475
In this type of assay a considerable fraction of the measured degradation will be non-autophagic, and thus it is important to also measure, in parallel, cell samples treated with autophagy-suppressive concentrations of 3-MA or amino acids, or obtained from mutants missing central ATG components (however, the latter assumes that non-autophagic proteolytic activity remains unchanged, which is unlikely); these values are then subtracted from the total. The complementary approach of using compounds that block other degradative pathways, such as proteasome inhibitors, may cause unexpected results and should generally be avoided due to crosstalk among the degradative systems. For example, blocking proteasome function may activate autophagy.476-479 Thus, when using inhibitors it is critical to know whether the inhibitors being used alter autophagy in the particular cell type and context being examined. In addition, because 3-MA could have some autophagy-independent effects in particular settings it is advisable to verify that the 3-MA-sensitive degradation is also sensitive to general lysosomal inhibitors (such as NH4Cl or leupeptin).
The use of stable isotopes, such as 13C and 15N, in quantitative mass spectrometry-based proteomics allows the recording of degradation rates of thousands of proteins simultaneously. These assays may be applied to autophagy-related questions enabling researchers to investigate differential effects in global protein or even organelle degradation studies.480,481 SILAC (stable isotope labeling with amino acids in cell culture) can also provide comparative information between different treatment conditions, or between a wild type and mutant.
Another assay that could be considered relies on the limited proteolysis of a betaine homocysteine methyltransferase (BHMT) fusion protein. The 44-kDa full-length BHMT protein is cleaved in hepatocyte amphisomes in the presence of leupeptin to generate 32-kDa and 10-kDa fragments.482-485 Accumulation of these fragments is time-dependent and is blocked by treatment with autophagy inhibitors. A modified version of this marker, GST-BHMT, can be expressed in other cell lines where it behaves similar to the wild-type protein.486 Other substrates may be considered for similar types of assays. For example, the neomycin phosphotransferase II-GFP (NeoR-GFP) fusion protein is a target of autophagy.487 Transfection of lymphoblastoid cells with a plasmid encoding NeoR-GFP followed by incubation in the presence of 3-MA leads to an accumulation of the NeoR-GFP protein as measured by flow cytometry.488
A similar western blot assay is based on the degradation of a cytosolic protein fused to GFP. This method has been used in yeast and Dictyostelium cells using GFP-PgkA and GFP-Tkt-1 (phosphoglycerate kinase and transketolase, respectively). In this case the relative amount of the free GFP and the complete fusion protein is the relevant parameter for quantification; although it may not be possible to detect clear changes in the amount of the full-length chimera, especially under conditions of limited flux.29,36 As described above for the marker GFP-Atg8/LC3, nonsaturating levels of lysosomal inhibitors are also needed in Dictyostelium cells to slow down the autophagic degradation, allowing the accumulation and detection of free GFP. It should be noted that this method monitors bulk autophagy since it relies on the passive transit of a cytoplasmic marker to the lysosome. Consequently, it is important to determine that the marker is distributed homogeneously in the cytoplasm.
One of the most useful methods for monitoring autophagy in S. cerevisiae is the Pho8Δ60 assay. PHO8 encodes the vacuolar alkaline phosphatase, which is synthesized as a zymogen before finally being transported to and activated in the vacuole.489 A molecular genetic modification that eliminates the first 60 amino acids prevents the mutant (Pho8Δ60) from entering the endoplasmic reticulum, leaving the zymogen in the cytosol. When autophagy is induced, the mutant zymogen is delivered to the vacuole nonselectively inside autophagosomes along with other cytoplasmic material. The resulting activation of the zymogen can be easily measured by assays for alkaline phosphatase.178 To minimize background activity, it is preferable to have the gene encoding cytosolic alkaline phosphatase (PHO13) additionally deleted (although this is not necessary when assaying certain substrates).
Cautionary notes: Measuring the degradation of long-lived proteins requires prior radiolabeling of the cells, and subsequent separation of acid-soluble from acid-insoluble radioactivity. The labeling can be done with relative ease both in cultured cells, and in live animals.3 In cells, it is also possible to measure the release of an unlabeled amino acid by chromatographic methods, thereby obviating the need for prelabeling;490 however, it is important to keep in mind that amino acid release is also regulated by protein synthesis, which in turn is modulated by many different factors. In either case, one potential problem is that the released amino acid may be further metabolized. For example, branched chain amino acids are good indicators of proteolysis in hepatocytes, but not in muscle cells where they are further oxidized (Meijer AJ, personal communication). In addition, the amino acid can be reincorporated into protein; for this reason, such experiments can be performed in the presence of cycloheximide, but this raises additional concerns (see Turnover of autophagic compartments). In the case of labeled amino acids, a nonlabeled chase is added where the tracer amino acid is present in excess (being cautious to avoid using an amino acid that inhibits autophagy), or by use of single pass perfused organs or superfused cells.491,492 The perfused organ system also allows for testing the reversibility of effects on proteolysis and the use of autophagy-specific inhibitors in the same experimental preparation, which are crucial controls for proper assessment.
If the autophagic protein degradation is low (as it will be in cells in replete medium), it may be difficult to measure it reliably above the relatively high background of non-autophagic degradation. It should also be noted that the usual practice of incubating the cells under “degradation conditions,” that is, in a saline buffer, indicates the potential autophagic capacity (maximal attainable activity) of the cells rather than the autophagic activity that prevails in vivo or under rich culture conditions. Finally, inhibition of a particular degradative pathway is typically accompanied by an increase in a separate pathway as the cell attempts to compensate for the loss of degradative capacity.158,478,493 This compensation might interfere with control measurements under conditions that attempt to inhibit macroautophagy; however, as the latter is the major degradative pathway, the contributions of other types of degradation over the course of this type of experiment are most often negligible.
The Pho8Δ60 assay requires standard positive and negative controls (such as an atg1Δ strain), and care must be taken to ensure the efficiency of cell lysis. Glass beads lysis works well in general, provided that the agitation speed of the instrument is adequate. Instruments designed for liquid mixing with lower speeds should be avoided. We also recommend against holding individual sample tubes on a vortex, as it is difficult to maintain reproducibility; devices or attachments are available to allow multiple tubes to be agitated simultaneously.
Conclusion: Measuring the turnover of long-lived proteins is a good method for determining autophagic flux.
8. Selective types of autophagy
Although autophagy can be nonspecific, in particular during starvation, there are many examples of selective types of autophagy.
a. The Cvt pathway, mitophagy, pexophagy and piecemeal microautophagy of the nucleus in yeast
The precursor form of aminopeptidase I (prApe1) is the major cargo of the Cvt pathway in yeast, a biosynthetic autophagy-related pathway.82 The propeptide of prApe1 is proteolytically cleaved upon vacuolar delivery, and the resulting shift in molecular mass can be monitored by western blot. Under starvation conditions, prApe1 can enter the vacuole through nonspecific autophagy, and thus has been used as a marker for both the Cvt pathway and autophagy. The yeast Cvt pathway is unique in that it is a biosynthetic route that utilizes the autophagy-related protein machinery, whereas other types of selective autophagy are degradative. The latter include pexophagy, mitophagy, reticulophagy, ribophagy and xenophagy, and each process has its own marker proteins, although these are typically variations of other assays used to monitor the Cvt pathway or autophagy. One common type of assay involves the processing of a GFP chimera similar to the GFP-Atg8/LC3 processing assay (see GFP-Atg8/LC3 lysosomal delivery and proteolysis). For example, yeast pexophagy utilizes the processing of Pex14-GFP and Pot1 (thiolase)-GFP,494,495 whereas mitophagy can be monitored by the generation of free GFP from Om45-GFP, Idh1-GFP, Idp1-GFP or mito-DHFR-GFP.496-500 Localization of these mitochondrially-targeted proteins (or specific MitoTracker dyes) or similar organelle markers such as those for the peroxisome [e.g., GFP-SKL with Ser-Lys-Leu at the C terminus that acts as a peroxisomal targeting signal, acyl-CoA oxidase 3 (Aox3-EYFP) that allows simultaneous observation of peroxisome-vacuole dynamics with the single FITC filter set, or GFP-catalase] can also be followed by fluorescence microscopy.412,495,501-503 In addition, yeast mitophagy requires both the Slt2 and Hog1 signaling pathways; the activation and phosphorylation of Slt2 and Hog1 can be monitored with commercially available phospho-specific antibodies (Fig. 19).381 It is also possible to monitor pexophagy in yeasts by the disappearance of activities of specific peroxisome markers such as catalase, alcohol oxidase or amine oxidase in cell-free extracts,504 or permeabilized cell suspensions. Catalase activity, however, is a useful marker only when peroxisomal catalases are the only such enzymes present. In S. cereviseae there are two genes encoding catalase activity, and only one of these gene products is localized in peroxisomes. Plate assays for monitoring the activity of peroxisomal oxidases in yeast colonies are also available.501,505 The decrease in the level of endogenous proteins such as alcohol oxidase or Pot1 can be followed by western blotting,412,506-509 TEM,510 fluorescence microscopy412,511,512 or laser confocal scanning microscopy of GFP-labeled peroxisomes.513,514
Figure 19.S. cerevisiae cells were cultured to mid-log phase and shifted to SD-N for the indicated times. Samples were taken before (+) and at the indicated times after (–) nitrogen starvation. Immunoblotting was done with anti-phospho-Slt2 and anti-phospho-Hog1 antibody. This figure was modified from data previously published in reference 381 and is reproduced by permission of the American Society for Cell Biology, copyright 2011.
In yeast, nonspecific autophagy can be induced by nitrogen starvation conditions, whereas degradative types of selective autophagy generally require a carbon source change or ER stress for efficient induction. For example, to induce a substantial level of mitophagy, cells need to be precultured in a nonfermentable carbon source such as lactate or glycerol to stimulate the proliferation of mitochondria. After sufficient mitochondria proliferation, shifting the cells back to a fermentable carbon source such as glucose will cause the autophagic degradation of superfluous mitochondria.497 It should be noted that in addition to carbon source change, simultaneous nitrogen starvation is also required for efficient mitophagy induction. This is possibly because excessive mitochondria can be segregated into daughter cells by cell division if growth continues.478A similar carbon source change from oleic acid or methanol to ethanol or glucose (with or without nitrogen starvation) can be used to assay for pexophagy.515 In addition, mitophagy can also be induced by culturing the cells in a nonfermentable carbon source to post-log phase. In this case, mitophagy may be induced because the energy demand is lower at post-log phase and the mitochondrial mass exceeds the cell’s needs.75,516,517 It has been suggested by several workers in the field that this type of mitophagy, also known as “stationary phase mitophagy,” reflects a quality-control function that culls defective mitochondria that accumulate in nondividing, respiring cells.518 Similar, pexophagy can be induced by culturing the cells in a peroxisome proliferation medium to post-log phase (Farré J-C, unpublished results). Along these lines, it should also be realized that selective types of autophagy continuously occur at a low level under noninducing conditions. Thus, organelles such as peroxisomes have a finite life span and are turned over at a slow rate by autophagy-related pathways.519
Piecemeal microautophagy of the nucleus (PMN, also micronucleophagy) is another selective autophagic subtype, which targets portions of the nucleus for degradation.520-522 In S. cerevisiae, the nuclear outer membrane, which is continuous with the nuclear ER, forms contact sites with the vacuolar membrane. These nucleus-vacuole junctions (NVJs) are generated by interaction of the outer nuclear membrane protein Nvj1 with the vacuolar protein Vac8.523 Nvj1 further recruits the ER-membrane protein Tsc13, which is involved in the synthesis of very-long-chain fatty acids (VLCFAs) and Osh1, a member of a family of oxysterol-binding proteins. Upon starvation the NVJs bulge into the vacuole and subsequently a PMN-vesicle pinches off into the vacuole. PMN vesicles thus contain nuclear material and are limited by three membranes with the outermost derived from the vacuole, and the two inner ones from the nuclear ER. It is not clear which nuclear components are removed by PMN, but since PMN is not a cell death mechanism per se, most likely superfluous material is recycled. During PMN the NVJs are selectively incorporated into the PMN vesicles and degraded. Accordingly, PMN can be monitored using the proteins that are associated with the NVJs as markers. To quantitatively follow PMN, an assay analogous to the above-described GFP-Atg8/LC3 processing assay has been established using either GFP-Osh1 or Nvj1-GFP. These GFP chimeras are, together with the PMN-vesicles, degraded in the vacuole. Thus, the formation of the relatively proteolysis-resistant GFP detected in western blots correlates with the PMN rate. In fluorescence microscopy, PMN can be visualized with the same constructs, and a chimera of mCherry fused to a nuclear localization signal (NLS-mCherry) can also be used. To assure that the measured PMN rate is indeed due to selective micronucleophagy, appropriate controls such as cells lacking Nvj1 or Vac8 should be included. Detailed protocols for the described assays are provided in reference 524.
b. Reticulophagy and ribophagy
Activation of the UPR in the ER in yeast induces a type of selective macroautophagy of the ER.525-527 This process is termed reticulophagy to be consistent with the terms pexophagy and mitophagy.527 Reticulophagy-associated autophagosomes are lamellar-membraned structures that contain ER proteins. In theory, since reticulophagy is selective, it should be able to sequester parts of the ER that are damaged, and eliminate protein aggregates that cannot be removed in other ways. Reticulophagy may also serve to limit the UPR, by reducing the ER to a normal level after a particular stress condition has ended. Some of the mutated dysferlin protein, LGMB2B/Miyoshi type muscle dystrophy, which accumulates in the ER, is degraded by ER-stress mediated reticulophagy.528 In addition to activation of the UPR, PtdIns3P and its binding proteins could be good markers for reticulophagy.437 Autophagy is also used for the selective removal of ribosomes.529 This process can be monitored by western blot, following the generation of free GFP from Rpl25-GFP or the disappearance of ribosomal subunits such as Rps3. Vacuolar localization of Rpl25-GFP can also be seen by fluorescence microscopy.
Cautionary notes: The Cvt pathway has been demonstrated to occur only in yeast. In addition, the sequestration of prApe1 is specific, even under starvation conditions, as it involves the recognition of the propeptide by a receptor, Atg19, which in turn interacts with the scaffold protein Atg11.530,531 Thus, unless the propeptide is removed, prApe1 is recognized as a selective substrate. Overexpression of prApe1 saturates import by the Cvt pathway, and the precursor form accumulates, but is rapidly matured upon autophagy induction.218 In addition, mutants such as vac8Δ and tlg2Δ accumulate prApe1 under rich conditions, but not during autophagy.380,532Accordingly, it is possible to monitor the processing of prApe1 when overexpressed, or in certain mutant strains to follow autophagy induction. However, even the latter conditions may be misleading, as they do not indicate the size of the autophagosome. The Cvt complex (prApe1 bound to Atg19) is smaller than typical peroxisomes or mitochondrial fragments that are subject to autophagic degradation. Accordingly, particular mutants may display complete maturation of prApe1 under autophagy-inducing conditions, but may still have a defect in other types of selective autophagy, as well as being unable to induce a normal level of nonspecific autophagy.63 For this reason, it is good practice to evaluate autophagosome size and number by TEM. Actually, it is much simpler to monitor autophagic bodies (rather than autophagosomes) in yeast. First, the vacuole is easily identified, making the identification of autophagic bodies much simpler. Second, autophagic bodies can be accumulated within the vacuole, allowing for an increased sample size. It is best to use a strain background that is pep4Δ vps4Δ to prevent the breakdown of the autophagic bodies, and to eliminate confounding vesicles from the multivesicular body pathway. One caveat to the detection of autophagic bodies, however, is that they may coalesce in the vacuole lumen, making it difficult to obtain an accurate quantification.
In general, when working with yeast it is preferable to use strains that have the marker proteins integrated into the chromosome rather than relying on plasmid-based expression, because plasmid numbers can vary from cell to cell. The GFP-Atg8, or similar, processing assay is easy to perform and is suitable for analysis by microscopy as well as western blotting; however, particular care is needed to obtain quantitative data for GFP-Atg8, Pex14-GFP or Om45-GFP, etc. processing assays (see cautionary notes for GFP-Atg8/LC3 lysosomal delivery and proteolysis). An alternative is an organelle targeted Pho8Δ60 assay. For example, mitoPho8Δ60 can be used to quantitatively measure mitophagy.498 In addition, for the GFP-Atg8 processing assay, 2 h of starvation is generally sufficient to detect a significant level of free (i.e., vacuolar) GFP by western blotting as a measure of nonselective autophagy. For selective types of autophagy, the length of induction needed for a clearly detectable free GFP band will vary depending on the rate of cargo delivery/degradation. Usually 6 h of mitophagy induction is needed to be able to detect free GFP (e.g., from Om45-GFP) by western blot under starvation conditions, whereas stationary phase mitophagy typically requires 3 d before a free GFP band is observed.
c. Vacuole import and degradation pathway
In yeast, gluconeogenic enzymes such as fructose-1,6-bisphosphatase (Fbp1, also called FBPase), malate dehydrogenase (Mdh2), isocitrate lyase (Icl1) and phosphoenolpyruvate carboxykinase (Pck1) constitute the cargo of the vacuole import and degradation (Vid) pathway.533 These enzymes are induced when yeast cells are glucose starved (grown in a medium containing 0.5% glucose and potassium acetate). Upon replenishing these cells with fresh glucose (a medium containing 2% glucose), these enzymes are degraded in either the proteasome534-536 or the vacuole533,537 depending on the duration of starvation. Following glucose replenishment after 3 d glucose starvation, the gluconeogenic enzymes are delivered to the vacuole for degradation.538 These enzymes are sequestered in specialized 30- to 50-nm vesicles called Vid vesicles.539 Vid vesicles can be purified by fractionation and gradient centrifugation; western blotting analysis using antibodies against organelle markers and Fbp1, and the subsequent verification of fractions by EM facilitate their identification.539 Furthermore, the amount of marker proteins in the cytosol compared with the Vid vesicles can be examined by differential centrifugation. In this case, yeast cells are lysed and subjected to differential centrifugation. The Vid vesicle-enriched pellet fraction and the cytosolic supernatant fraction are examined with antibodies against Vid24, Vid30, Sec28 and Fbp1.540-542
Vid/endosomes containing their cargo aggregate around endocytic vesicles forming on the plasma membrane and are released into the cytoplasm. The Vid/endosomes can be purified by fractionation and density gradient centrifugation.543 The fractions containing purified Vid/endosomes can be identified by western blot analysis using antibodies against Vid24, Fbp1 and the endosomal marker Pep12. The distribution of Vid vesicles containing cargo destined for endosomes, and finally for the vacuole, can be examined using FM 4–64, a lipophilic dye that stains endocytic compartments and the vacuole limiting membrane.544 In these experiments, starved yeast cells are replenished with fresh glucose and FM 4–64, and cells are collected at appropriate time points for examination by fluorescence microscopy.541 The site of degradation of the cargo in the vacuole can be determined by studying the distribution of Fbp1-GFP, or other Vid cargo markers in wild-type and pep4Δ cells.545 Cells can also be examined for the distribution of Fbp1 at the ultrastructural level by immuno-TEM.543
As actin patch polymerization is required for the delivery of cargo to the vacuole in the Vid pathway, distribution of Vid vesicles containing cargo and actin patches can be examined by actin staining (with phalloidin conjugated to rhodamine) using fluorescence microscopy.543 The distribution of GFP tagged protein and actin is examined by fluorescence microscopy. GFP-Vid24, Vid30-GFP and Sec28-GFP colocalize with actin during prolonged glucose starvation and for up to 30 min following glucose replenishment in wild-type cells; however, colocalization is less obvious by the 60 min time point.540,543
d. Mammalian mitophagy and peroxisome degradation
There is no consensus at the present time with regard to the best method for monitoring mammalian mitophagy. As with any organelle-specific form of autophagy, it is necessary to demonstrate: (1) increased levels of autophagosomes containing mitochondria, (2) maturation of these autophagosomes to culminate with mitochondrial degradation, which can be blocked by specific inhibitors of autophagy or of lysosomal degradation, and (3) whether the changes are due to selective mitophagy or increased mitochondrial degradation during generalized autophagy. Techniques to address each of these points have been reviewed.41
Ultrastructural analysis at early time points can be used to establish selective mitophagy, although a maturation inhibitor may be needed to trap early autophagosomes with recognizable cargo (Fig. 20). Depending on the use of specific imaging techniques, dyes for living cells or antibodies for fixed cells have to be chosen. In any case, transfection of the autophagosomal marker GFP-LC3 and visualization of mitochondria (independent of their mitochondrial membrane potential) makes it possible to determine the association of these two cellular components. This may appear as fluorescence colocalization or as rings of GFP-LC3 surrounding mitochondria in higher resolution images. For live cell imaging microscopy, a method that marks mitochondria independently of mitochondrial membrane potential, such as MitoTracker® Green FM or transfection with a matrix-targeted fluorescent protein, should be used to detect mitochondrial structures. Antibodies that specifically recognize mitochondrial proteins such as VDAC1, TOMM20/TOM20 or complex IV subunit I may be used to visualize mitochondria in immunohistochemical experimental procedures.546,547 Colocalization analyses of mitochondria and autophagosomes provide an indication of the degree of sequestration. TEM can be used to demonstrate the presence of mitochondria within autophagosomes (referred to as mitophagosomes during mitophagy), and this can be coupled with bafilomycin A1 treatment to prevent fusion with the lysosome.41

Figure 20. Autophagosomes with recognizable cargo are rare in cells. (A) To assess relative rates of autophagosome formation, the fusion inhibitor bafilomycin A1 (10 nM) was applied for 2 h prior to fixation with 2% glutaraldehyde in order to trap newly formed autophagosomes. Two different PINK1 shRNA lines exhibit increased AV formation over 2 h compared with the control shRNA line. *p > 0.05 vs. Control. (B) Autophagosomes in bafilomycin A1-treated control cells contain a variety of cytoplasmic structures (left, arrow), while mitochondria comprise a prominent component of autophagosomes in A14 bafilomycin A1-treated (PINK1 shRNA) cells (right, arrow). Scale bar, 500 nm. These data indicate induction of selective mitophagy in PINK1-deficient cells. This figure was modified from Figure 2 published in Chu CT. A pivotal role for PINK1 and autophagy in mitochondrial quality control: implications for Parkinson disease. Hum Mol Genet 2010; 19:R28?37.
The fusion process of mitophagosomes with hydrolase-containing lysosomes represents the next step in the degradation process. To monitor the amount of fused organelles via live cell imaging microscopy, MitoTracker® Green FM, as a mitochondrial marker without significant membrane potential dependence and LysoTracker® Red DND-99 may be used to visualize the fusion process (Fig. 21). Independent of the cell-type specific concentration used for both dyes, we recommend exchanging MitoTracker® Green FM with normal medium after incubation with the dye, whereas it is best to maintain the LysoTracker® Red staining in the incubation medium during the acquisition of images. For immuncytochemical experiments, antibodies specific for mitochondrial proteins and an antibody against the lysosomal-associated membrane protein 1 (LAMP1) can be used. Overlapping signals appear as a merged color and can be used as indicators for successful fusion of autophagosomes that contain mitochondria with lysosomal structures.548 To measure the correlation between two variables by imaging techniques, such as the colocalization of two different stainings, we recommend some form of correlation analysis to assess the value correlating with the strength of the association. This may use, for example, ImageJ software, or other colocalization scores that can be derived from consideration not only of pixel colocalization, but also from a determination that the structures have the appropriate shape. During live-cell imaging, the two structures should move together in more than one frame. Mitophagy can also be quantitatively monitored using a mitochondria-targeted version of the pH-dependent Keima protein.549 The peak of the excitation spectrum of the protein shifts from 440 nm to 586 nm when mitochondria are delivered to acidic lysosomes, which allows easy quantification of mitophagy (Fig. 22). However, it should be noted that long exposure times of the specimen to intense laser light lead to a similar spectral change.

Figure 21. Human fibroblasts showing colocalization of mitochondria with lysosomes. The degree of colocalization of mitochondria with lysosomes in human fibroblasts was measured via live cell imaging microscopy at 37°C and 5% CO2 atmosphere using the ApoTome® technique. LysoTracker® Red DND-99 staining was applied to mark lysosomal structures (red), and MitoTracker® Green FM to visualize mitochondria (green). Hoechst 33342 dye was used to stain nuclei (blue). A positive colocalization is indicated by yellow signals (merge) due to the overlap of LysoTracker® Red and MitoTracker® Green staining (white arrows). Scale bar, 10 µm. Statistical evaluation is performed by calculating the Pearson’s coefficient for colocalizing pixels. Image provided by L. Burbulla and R. Krüger.

Figure 22. Detection of mitophagy in primary cortical neurons using mitochondria-targeted Keima. Neurons transfected with mito-Keima were visualized using 458 nm (green, mitochondria at neutral pH) and 561 nm (red, mitochondria in acidic pH) laser lines and 575 nm band pass filter. Compared with the control (A) wild-type PINK1 overexpression (B) increases the number of the mitochondria exposed to acidic conditions. Scale bar, 2 µm. (C) Quantification of red dots suggests increased mitophagy in wild-type PINK1 but not in the kinase dead PINK1K219M-overexpressing neurons. Figure provided by V. Choubey and A. Kaasik.
The third and last step of the degradation process is the monitoring of the amount of remaining mitochondria by analyzing the mitochondrial mass. This final step provides the opportunity to determine the efficiency of degradation of dysfunctional, aged or impaired mitochondria. Mitochondrial mass can either be measured by a FACS technique using MitoTracker® Green FM or, on a single cell basis, by either live cell imaging or immuncytochemistry with use of MitoTracker® Green FM or MitoTracker® Red FM for the former, and antibodies specifically raised against different mitochondrial proteins for the latter. Immunoblot analysis of the levels of mitochondrial proteins from different mitochondrial subcompartments is valuable for validating the data from FACS or microscopy studies. EM can also be used to verify loss of entire mitochondria, and PCR (or fluorescence microscopy) to quantify mitochondrial DNA.
In addition to monitoring the steady-state levels of different steps of mitophagy, investigation of the mitophagic flux is needed to decipher which steps within this process potentially fail to result in efficient mitochondrial degradation. Therefore, appropriate treatment may be applied to prevent mitochondrial degradation at distinct steps of the process.
Certain cellular models require stress conditions to measure the mitochondrial degradation capacity, as basal levels are too low to reliably assess organelle clearance. Hence, it may be useful to pretreat the cells with uncoupling agents, such as CCCP that stimulate mitochondrial degradation and allow measurements of mitophagic activity; however, it should be kept in mind that, although helpful to stimulate mitochondrial degradation, this treatment is not physiological. Another method to induce mitophagy is by expressing and activating a mitochondrially-localized fluorescent protein photosensitizer such as Killer Red.550 The excitation of Killer Red results in an acute increase of superoxide that causes mitochondrial damage resulting in mitophagy (Kim PK, unpublished results). The advantage of using a genetically encoded photosensitizer is that it allows for both spatial and temporal control in inducing mitophagy.
Relatively little work has been done in the area of mammalian peroxisome degradation by autophagy, and at present it is not known if this is a selective process. Typically, peroxisomes are induced by treatment with hypolipidemic drugs such as clofibrate or dioctyl phthalate, and degradation is induced by drug withdrawal, although starvation without prior proliferation can also be used. Loss of peroxisomes can be followed enzymatically or by immunoblot, monitoring enzymes such as fatty acyl-CoA oxidase (note that this enzyme is sometimes abbreviated “AOX,” but should not be confused with the enzyme alcohol oxidase that is frequently used in assays for yeast pexophagy) or catalase, and also by EM.551,552 Finally, a HaloTag®-PTS1 marker that is targeted to peroxisomes has been used to fluorescently label the organelle.553
Cautionary notes: There are many assays that can be used to monitor specific types of autophagy, but caution must be used in choosing an appropriate marker(s). It is best to monitor more than one protein, and to include an inner membrane or matrix component in the analysis. In particular, it is not sufficient to follow a single mitochondrial outer membrane protein because these can be degraded independently of mitophagy. Although the localization of PARK2/PARKIN to mitochondria as monitored by fluorescence microscopy is associated with the early stages of protonophore uncoupler (CCCP)-driven mitochondria degradation,169 this by itself cannot be used as a marker for mitophagy, as these events can be dissociated.554 PARK2 translocates to damaged mitochondria and ubiquitinates a wide range of outer membrane proteins including VDAC1, MFN1/2 and TOMM20.547,555-557 This results in the preferential degradation of mitochondrial outer membrane proteins by the proteasome, while inner membrane proteins and mitochondrial DNA558 remain intact. Monitoring loss of a single protein such as TOMM20 by western blot or fluorescence microscopy to follow mitophagy may thus be misleading.555 MitoTracker dyes are widely used to stain mitochondria and, when colocalized with GFP-LC3, they can function as a marker for mitophagy. However, staining with certain MitoTracker dyes depends on mitochondrial activity and membrane potential, so that damaged, or sequestered nonfunctional mitochondria may not be stained.
Although it is widely assumed that macroautophagy is the major mechanism for degradation of entire organelles, there are multiple mechanisms that may account for the disappearance of mitochondrial markers. These include proteasomal degradation of outer membrane proteins and/or proteins that fail to correctly translocate into the mitochondria, degradation due to proteases within the mitochondria, and reduced biosynthesis or import of proteins. In addition to mitophagy, mitochondria can be eliminated by extrusion from the cell (mitoptosis).559 Thus, it is advisable to use a variety of complementary methods to monitor mitochondria loss including TEM, single cell analysis of LC3 fluorescent puncta, and western blot, in conjunction with flux inhibitors and specific inhibitors of autophagy induction compared with inhibitors of the other major degradation systems (see cautions in Autophagy inhibitors and inducers).
Likewise, although the mechanism(s) of peroxisome degradation in mammals awaits further elucidation, it can occur by both autophagic and proteasome-dependent mechanisms.560 Thus, controls are needed to determine the extent of degradation that is due to the proteasome. Moreover, two additional degradation mechanisms have been suggested: the action of the peroxisome-specific Lon protease and the membrane disruption effect of 15-lipoxygenase.561
e. Aggrephagy
Aggrephagy is the selective removal of aggregates by macroautophagy.562 This process can be followed in vitro (in cell culture) and in vivo (in mice) by monitoring the levels of an aggregate-prone protein such as an expanded polyglutamine (polyQ)-containing protein or mutant SNCA (α-synuclein). Levels are quantified by immunofluorescence or traditional immunoblot. Similarly, fluorescently tagged aggregated proteins such as polyQ80-CFP can be monitored via immunoblot and immunofluorescence. A polyQ80-luciferase reporter, which forms aggregates, can also be used to follow aggrephagy.563 A nonaggregating polyQ19-luciferase or untagged full-length luciferase serves as a control. The ratio of luciferase activity from these two constructs can be calculated to determine autophagic flux.
Autophagic degradation of endogenous aggregates such as lipofuscin can in some cell types be monitored by fluorescence microscopy, utilizing the autofluorescence of lipofuscin particles. The amount of lipofuscin in primary human adipocytes can be reduced by activation of autophagy, and the amount of lipofuscin is dramatically reduced in adipocytes from patients with type 2 diabetes and chronically enhanced autophagy.209
Cautionary notes: Caution must be used when performing immunoblots of aggregated proteins, as many protein aggregates fail to enter the resolving gel and are retained in the stacking gel. In addition, the polyQ80-luciferase in the aggregated state lacks luciferase activity whereas soluble polyQ80-luciferase retains activity. Therefore, caution must be used when interpreting results with these vectors, as treatments that increase aggrephagy or enhance protein aggregation can lead to a decrease in luciferase activity.564 Finally, soluble polyQ reporters can be degraded by the proteasome; thus, changes in the ratio of polyQ19-luciferase:polyQ80-luciferase may also reflect proteasomal effects and not just changes in autophagic flux.
f. Xenophagy
The macroautophagy pathway has emerged as an important cellular factor in both innate and adaptive immunity. Many in vitro and in vivo studies have demonstrated that genes encoding macroautophagy components are required for host defense against infection by bacteria, parasites and viruses. Macroautophagy may be defective in human diseases such as inflammatory bowel disease, since genes encoding essential macroautophagy components have been linked to the disease. Xenophagy is often used as a term to describe autophagy of microbial pathogens, mediating their capture and delivery to lysosomes for degradation. Since xenophagy presents an immune defense, it is not surprising that microbial pathogens have evolved strategies to overcome it. The interactions of such pathogens with the autophagy system of host cells are complex and have been the subject of several excellent reviews.76-81,565-570 Here we will make note of a few key considerations when studying interactions of microbial pathogens with the autophagy system.
LC3 is commonly used as a marker of macroautophagy. However, studies have established that LC3 can promote phagosome maturation independently of macroautophagy through LC3-associated phagocytosis (see cautionary notes in Atg8/LC3 detection and quantification). Other studies show that macroautophagy of Salmonella Typhimurium is dependent on ATG9, an essential macroautophagy protein, whereas LC3 recruitment to bacteria does not require ATG9.571 In contrast, macroautophagy of these bacteria requires ubiquitination of target proteins (not yet identified) and recruitment of three ubiquitin-binding adaptor proteins, SQSTM1,572 CALCOCO2/NDP52573 and OPTN.574 Therefore, the currently available criteria to differentiate LAP from macroautophagy include: (1) LAP involves LC3 recruitment to bacteria in a manner that requires reactive oxygen species production by an NADPH oxidase. It should be noted that most cells express at least one member of the NADPH oxidase (also known as NOX) family. Targeting expression of the common CYBA/p22phox subunit is an effective way to disrupt the NOX NADPH oxidases. Scavenging of reactive oxygen species by antioxidants such as resveratrol and α-tocopherol is also an effective way to inhibit LAP. In contrast, N-acetylcysteine, which raises cellular glutathione levels, does not inhibit LAP.575 (2) Macroautophagy of bacteria requires ATG9, while LAP apparently does not.571 (3) LAP involves single-membrane structures. For LAP, CLEM (with LC3 as a marker) is expected to show single-membrane structures that are LC3+ LAP.131 In contrast, macroautophagy is expected to generate double-membrane structures surrounding cargo (which may include single membrane phagosomes, giving rise to triple-membrane structures571). (4) Macroautophagy of bacteria requires protein ubiquitination and ubiquitin-binding adaptors (SQSTM1, CALCOCO2, OPTN, and possibly others), whereas LAP does not. It is anticipated that more specific markers of LAP will be identified as these phagosomes are further characterized.
Nonmotile Listeria monocytogenes can be targeted to double-membrane autophagosomes upon antibiotic treatment,576 which indicates that macroautophagy serves as a cellular defense against microbes in the cytosol. However, subsequent studies have revealed that macroautophagy can also target pathogens within phagosomes, damaged phagosomes or the cytosol. Therefore, when studying microbial interactions by EM, many structures can be visualized, with any number of membranes encompassing microbes, all of which may be LC3+.577 As discussed above, single-membrane structures that are LC3+ may arise through LAP, and we cannot rule out the possibility that both LAP and macroautophagy may operate at the same time to target the same phagosome.
Viruses can also be targeted by autophagy, and in turn can act to inhibit autophagy. For example, infection of a cell by influenza and dengue viruses578 or enforced expression of the hepatitis B virus C protein579 have profound consequences for autophagy, as viral proteins such as NS4A stimulate autophagy and protect the infected cell against apoptosis, thus extending the time in which the virus can replicate. Conversely, the herpes simplex virus ICP34.5 protein inhibits autophagy by targeting BECN1.580 While the impact of ICP34.5′s targeting of BECN1 on virus replication in cultured permissive cells is minimal, it has a significant impact upon pathogenesis in vivo, most likely through interfering with activation of CD4+ T cells.581,582 Care must be taken in determining the role of autophagy in virus replication, as some viruses such as vaccinia use double-membrane structures that form independently of the autophagy machinery.583 Similarly, dengue virus replication, which appears to involve a double-membrane compartment, requires the ER rather than autophagosomes,584 whereas coronaviruses use a nonlipidated version of LC3 (see Atg8/LC3 detection and quantification).137 Yet another type of variation is seen with hepatitis C virus, which requires BECN1, ATG4B, ATG5 and ATG12 for initiating replication, but does not require these proteins once an infection is established.585
Finally, it is important to realize that there may be other macroautophagy-like pathways that have yet to be characterized. For example, in response to cytotoxic stress (treatment with etoposide), autophagosomes are formed in an ATG5- and ATG7-independent manner.26 While this does not rule out involvement of other macroautophagy regulators/components in the formation of these autophagosomes, it does establish that the canonical macroautophagy pathway involving LC3 conjugation is not involved. In contrast, RAB9 is required for this alternative pathway, potentially providing a useful marker for analysis of these structures. Returning to xenophagy, Mycobacterium tuberculosis can be targeted to autophagosomes in an ATG5-independent manner.586 Furthermore, up to 25% of intracellular Salmonella typhimurium are observed in multi-lamellar membrane structures resembling autophagosomes in Atg5−/− MEFs.572 These findings indicate that an alternate macroautophagy pathway is relevant to host-pathogen interactions. Moreover, differences are observed that depend on the cell type being studied. Yersinia pseudotuberculosis is targeted to autophagosomes where they can replicate in bone marrow-derived macrophages,587 whereas in RAW264.7 and J774 cells, bacteria are targeted both to autophagosomes, and LC3-negative, single-membrane vacuoles (Lafont F, personal communication).
g. Lipophagy
The specific macroautophagic degradation of lipid droplets represents another type of selective autophagy.588 Lipophagy requires the core autophagic machinery and can be monitored by following triglyceride content, or total lipid levels using BODIPY 493/503 or HCS LipidTOX neutral lipid stains with fluorescence microscopy, cell staining with Oil Red O, or ideally label-free techniques such as CARS or SRS microscopy. TEM can also be used to monitor lipid droplet size and number.
Cautionary notes: With regard to changes in the cellular neutral lipid content, the presence and potential activation of cytoplasmic lipases that are unrelated to lysosomal degradation must be considered.
h. Zymophagy
Zymophagy is a protective mechanism induced during pancreatitis that results in the selective degradation of activated zymogen granules, which are deleterious for the pancreatic acinar cells.589 This process can be monitored by TEM, identifying autophagosomes containing secretory granules, by following SQSTM1 degradation by western blot, and by examining the subcellular localization of VMP1-EGFP, which relocates to granular areas of the cell upon zymophagy induction. Colocalization of trypsinogen (which is packaged within zymogen granules) and LC3, or of GFP-ubiquitin (which is recruited to the activated granules) with RFP-LC3 can also be observed by indirect or direct immunofluorescence microscopy, respectively.
i. Allophagy
In metazoans, mitochondria, and hence mitochondrial DNA, from the sperm is eliminated by an autophagic process. This process of allogeneic (nonself) organelle autophagy is termed “allophagy.”590,591 During allophagy in C. elegans, both paternal mitochondria and membranous organelles (a sperm-specific membrane compartment) are eliminated by the 16-cell stage (100–120 min post-fertilization).592,593 The degradation process can be monitored in living embryos with GFP::ubiquitin, which appears in the vicinity of the sperm chromatin (labeled for example with mCherry-histone H2B) on the membranous organelles within 3 min after fertilization. GFP fusions and antibodies specific for LGG-1 and LGG-2 (Atg8/LC3 homologs), which appear next to the sperm DNA, membranous organelles and mitochondria (labeled with CMXRos or mitochondria-targeted GFP) within 15 to 30 min post-fertilization, can be used to verify the autophagic nature of the degradation. TEM can also be utilized to demonstrate the presence of mitochondria within autophagosomes in the early embryo.
Conclusion: There are many assays that can be used to monitor specific types of autophagy, but caution must be used in choosing an appropriate marker(s). The potential role of other degradative pathways for any individual organelle or cargo marker should be considered, and it is advisable to use more than one marker or technique.
9. Autophagic sequestration assays
Autophagic activity can also be monitored by the sequestration of autophagic cargo, using either an (electro)injected, inert cytosolic marker such as [3H]raffinose,594 or an endogenous cytosolic protein such as lactate dehydrogenase (LDH),595 in the latter case along with treatment with a protease inhibitor (e.g., leupeptin) to prevent intralysosomal degradation of the protein marker. The assay simply measures the transfer of cargo from the soluble (cytosol) to the insoluble (sedimentable) cell fraction (which includes autophagic compartments), with no need for a sophisticated subcellular fractionation (a filtration assay would presumably work just as well as centrifugation, although it would be necessary to verify that the filtration membrane does not destroy the integrity of the postnuclear supernatant compartments). The cargo marker can be quantified by an enzymatic assay, or by western blotting. In principle, any intracellular component can be used as a cargo marker, but cytosolic enzymes having low sedimentable backgrounds are preferable. Membrane-associated markers are less suitable, and proteins such as LC3, which are part of the sequestering system itself, will have a much more complex relationship to the autophagic flux than a pure cargo marker such as LDH.
In yeast, sequestration assays are typically done by monitoring protease protection of an autophagosome marker or a cargo protein. For example, prApe1, and GFP-Atg8 have been used to follow completion of the autophagosome.596 The relative resistance or sensitivity to an exogenous protease in the absence of detergent is an indication of whether the autophagosome (or other sequestering vesicle) is complete or incomplete, respectively. Thus, this method also distinguishes between a block in autophagosome formation vs. fusion with the vacuole. The critical issues to keep in mind involve the use of appropriate control strains and/or proteins, and deciding on the correct reporter protein. In addition to protease protection assays, sequestration can be monitored by fluorescence microscopy during pexophagy of methanol-induced peroxisomes, using GFP-Atg8 as a pexophagosome marker, and BFP-SKL to label the peroxisomes. The vacuolar sequestration process during micropexophagy can also be monitored by formation of the vacuolar sequestration membrane (VSM) stained with FM 4–64.501,507
Sequestration assays can be designed to measure flux through individual steps of the autophagy pathway. For example, intralysosomally degraded sequestration probes such as [14C]-lactate or LDH will mark prelysosomal compartments in the absence of degradation inhibitors. Hence, their accumulation in such compartments can be observed when fusion with lysosomes is suppressed, for example, by a microtubule inhibitor such as vinblastine.597 Furthermore, lactate hydrolysis can be used to monitor the overall autophagic pathway (autophagic lactolysis).598 One caveat, however, is that inhibitors may affect sequestration indirectly, for example, by modifying the uptake and metabolism (including protein synthesis) of autophagy-suppressive amino acids (see Autophagy inhibitors and inducers).
A variation of this approach applicable to mammalian cells includes live cell imaging. Autophagy induction is monitored as the movement of cargo, such as mitochondria, to GFP-LC3-colocalizing compartments, and then fusion/flux is measured by delivery of cargo to lysosomal compartments.258,599 In addition, sequestration of fluorescently tagged cytosolic proteins into membranous compartments can be measured, as fluorescent puncta become resistant to the detergent digitonin.600 Use of multiple time points and monitoring colocalization of a particular cargo with GFP-LC3 and lysosomes can also be used to assess sequestration of cargo with autophagosomes as well as delivery to lysosomes.601
Cautionary notes: The electro-injection of radiolabeled probes is technically demanding, but the use of an endogenous cytosolic protein probe is very simple and requires no pretreatment of the cells other than with a protease inhibitor. Another concern with electro-injection is that it can affect cellular physiology, so it is necessary to verify that the cells behave properly under control situations such as amino acid deprivation. An alternate approach for incorporating exogenous proteins into mammalian cell cytosol is to use “scrape-loading,” a method that works for cells that are adherent to tissue culture plates.602 Finally, these assays work well with hepatocytes but may be problematic with other cell types, and it can be difficult to load the cell while retaining the integrity of the compartments in the post-nuclear supernatant (Tooze S, unpublished results). General points of caution to be addressed with regard to live cell imaging relate to photobleaching of the fluorophore, cell injury due to repetitive imaging, autofluorescence in tissues containing lipofuscin, and the pH sensitivity of the fluorophore.
There are several issues to keep in mind when monitoring sequestration by the protease protection assay in yeast.596 First, as discussed in Selective types of autophagy, prApe1 is not an accurate marker for nonspecific autophagy; import of prApe1 utilizes a receptor (Atg19) and a scaffold (Atg11) that make the process specific. In addition, vesicles that are substantially smaller than autophagosomes can effectively sequester the Cvt complex. Another problem is that prApe1 cannot be used as an autophagy reporter for mutants that are not defective in the Cvt pathway, although this can be bypassed by using a vac8Δ background.603 At present, the prApe1 assay cannot be used in any system other than yeast. The GFP-Atg8 protease protection assay avoids these problems, but the signal-to-noise ratio is typically substantially higher. In theory, it should be possible to use this assay in other cell types, but at present no publications report its use other than in yeast.
Conclusion: Sequestration assays present another valid method for monitoring autophagy, and in particular for discriminating between conditions where the autophagosome is complete (but not fused with the lysosome/vacuole) or open (i.e., a phagophore). These assays can also be modified to measure autophagic flux.
10. Turnover of autophagic compartments
Inhibitors of autophagic sequestration (e.g., amino acids, 3-MA or wortmannin) can be used to monitor the disappearance of autophagic elements (phagophores, autophagosomes, autolysosomes) to estimate their half-life by TEM morphometry/stereology. The turnover of the autophagosome or the autolysosome will be differentially affected if fusion or intralysosomal degradation is inhibited.14,16,24,604 The duration of such experiments is usually only a few hours; therefore, long-term side effects or declining effectiveness of the inhibitors can be avoided. It should be noted that fluorescence microscopy has also been used to monitor the half-life of autophagosomes, monitoring GFP-LC3 in the presence and absence of bafilomycin A1 or following GFP-LC3 after starvation and recovery in amino acid-rich medium (see Atg8/LC3 detection and quantification).18,605
Cautionary notes: The inhibitory effect must be strong and the efficiency of the inhibitor needs to be tested under the experimental conditions to be employed. Cycloheximide is sometimes used as an autophagy inhibitor, but this is problematic because of the many potential indirect effects. Cycloheximide inhibits translational elongation, and therefore protein synthesis. In addition, it decreases the efficiency of protein degradation in several cell types (Cuervo AM, personal communication) including hematopoietic cells (Edinger A, personal communication). Treatment with cycloheximide causes a potent increase in TORC1 activity, which can decrease autophagy, in part as a result of the increase in the amino acid pool resulting from suppressed protein synthesis (Shen H-M, personal communication).606,607 In addition, at high concentrations (in the millimolar range) cycloheximide inhibits complex I of the mitochondrial respiratory chain,608,609 but this is not a problem, at least in hepatocytes, at low concentrations (10 -20 µM) that are sufficient to prevent protein synthesis (Meijer AJ, personal communication).
Conclusion: The turnover of autophagic compartments is a valid, but less preferred method, for monitoring flux, and cycloheximide must be used in these experiments with caution.
11. Autophagosome-lysosome colocalization and dequenching assay
Another method to demonstrate the convergence of the autophagic pathway with a functional degradative compartment is to incubate cells with the bovine serum albumin derivative dequenched (DQ)-BSA that has been labeled with the red-fluorescent BODIPY TR-X dye; this conjugate will accumulate in lysosomes. The labeling of DQ-BSA is so extensive that the fluorophore is self-quenched. Proteolysis of this compound results in dequenching and the release of brightly fluorescent fragments. Thus, DQ-BSA is useful for detecting intracellular proteolytic activity as a measure of a functional lysosome.610
Furthermore, DQ-BSA labeling can be combined with GFP-LC3 to monitor colocalization, and thus visualize the convergence, of amphisomes with a functional degradative compartment (DQ-BSA is internalized by endocytosis). This method can also be used to visualize fusion events in real-time experiments by confocal microscopy (live cell imaging). Along similar lines, other approaches for monitoring convergence are to follow the colocalization of RFP-LC3 and LysoSensor Green (Bains M, Heidenreich KA, personal communication), mCherry-LC3 and LysoSensor Blue,259 or tagged versions of LC3 and LAMP1 (Macleod K, personal communication) or CD63258 as a measure of the fusion of autophagosomes with lysosomes. It is also possible to trace autophagic events by visualizing the pH-dependent excitation changes of the coral protein Keima.549 This quantitative technique is capable of monitoring the fusion of autophagosomes with lysosomes, that is, the formation of an autolysosome, and the assay does not depend on the analysis of LC3.
Cautionary notes: Some experiments require the use of inhibitors (e.g., 3-MA or wortmannin) or overexpression of proteins (e.g., RAB7 dominant negative mutants) that may also affect the endocytic pathway or the delivery of DQ-BSA to lysosomes (e.g., wortmannin causes the swelling of late endosomes611). In this case, the lysosomal compartment can be labeled with DQ-BSA overnight before treating the cells with the drugs, or prior to the transfection.
Conclusion: DQ-BSA provides a relatively convenient means for monitoring lysosomal protease function, and to follow the fusion of amphisomes with the lysosome. Colocalization of autophagosomes (fluorescently tagged LC3) with lysosomal proteins or dyes can also be monitored.
12. Tissue fractionation
The study of autophagy in the organs of larger animals, in large numbers of organisms with very similar characteristics, or in tissue culture cells provides an opportunity to use tissue fractionation techniques as has been possible with autophagy in rat liver.34,48,612-617 Because of their sizes [smaller than nuclei but larger than membrane fragments (microsomes)], differential centrifugation can be used to obtain a subcellular fraction enriched in mitochondria and organelles of the autophagic-lysosomal system, which can then be subjected to density gradient centrifugation to enrich autophagosomes, amphisomes, autolysosomes and lysosomes.34,48,617-621 Any part of such a fraction can be considered to be a representative sample of tissue constituents and used in quantitative biochemical, centrifugational and morphological studies of autophagic particle populations.
The simplest studies of the autophagic process take advantage of sequestered marker enzymes, changes in location of these enzymes, differences in particle/compartment size and differential sensitivity of particles of different sizes to mechanical and osmotic stress (for example, acid hydrolases are found primarily in membrane-bound compartments and their latent activities cannot be measured unless these membranes are lysed). Such a change in enzyme accessibility can be used to follow the time course of an exogenously induced, or naturally occurring, autophagic process.612,614,616
Quantitative localization of enzymatic activity (or any other marker) to specific cytoplasmic particle populations and changes in the location of such markers during autophagy can be performed using rate sedimentation ultracentrifugation (see Autophagic sequestration assays).618 Similar results can be obtained with isopycnic centrifugation where particles enter a density gradient (sometimes made with sucrose but iso-osmotic media such as iodixanol, metrizamide and Nycodenz may be preferred as discussed below under Cautionary notes) and are centrifuged until they reach locations in the gradient where their densities are equal to those of the gradient.618
The fractionation of organelles can also be evaluated by protein-correlation-profiling (PCP), a quantitative mass spectrometry-based proteomics approach. Similar to the biochemical assays described above, gradient profiles of marker proteins can be recorded and compared with proteins of interest.278 Compared with classical biochemical approaches, PCP allows the proteome-wide recording of protein gradient profiles.
Particle populations in subcellular fractions evaluated with quantitative biochemical and centrifugational approaches can also be studied with quantitative morphological methods. Detailed morphological study of the particle populations involved in the autophagic process usually requires the use of EM. The thin sections required for such studies pose major sampling problems in both intact cells622 and subcellular fractions.618 With the latter, 2,000,000 sections can be obtained from each 0.1 ml of pellet volume, so any practical sample size is an infinitesimally small subsample of the total sample.618 However, through homogenization and resuspension, complex and heterogeneous components of subcellular fractions become randomly distributed throughout the fraction volume. Therefore, any aliquot of that volume can be considered a random sample of the whole volume. What is necessary is to conserve this property of subcellular fractions in the generation of a specimen that can be examined with the electron microscope. This can be done with the use of a pressure filtration procedure.618,623 Because of the thinness of the sections, multiple sections of individual particles are possible so morphometric/stereological methods622 must be used to determine the volume occupied by a given class of particles, as well as the size distribution and average size of the particle class. From this information the number of particles in a specific particle class can be calculated.624 Examination of individual profiles gives information on the contents of different types of particles and their degree of degradation, as well as their enclosing membranes.612,614
Cautionary notes: When isolating organelles from tissues and cells in culture it is essential to use disruption methods that do not alter the membrane of lysosomes and autophagosomes, compartments that are particularly sensitive to some of those procedures. For example teflon/glass motor homogenization is suitable for tissues with abundant connective tissue, such as liver, but for circulating cells or cells in culture, disruption by nitrogen cavitation is a good method to preserve lysosomal membrane stability;625 however, this method is not suitable for small samples and may not be readily available. Other methods, including “Balch” or “Dounce” homogenizers also work well.626,627 During the isolation procedure it is essential to always use iso-osmotic solutions to avoid hypotonic or hypertonic disruption of the organelles. In that respect, because lysosomes are able to take up sucrose if it is present at high concentrations, the use of sucrose gradients for the isolation of intact lysosome-related organelles is strongly discouraged.
As with the isolation of any other intracellular organelle, it is essential to assess the purity of each preparation, as there is often considerable variability from experiment to experiment due to the many steps involved in the process. Correction for purity can be done through calculation of recovery (percentage of the total activity present in the homogenate) and enrichment (dividing by the specific activity in the homogenate) of enzymes or protein markers for those compartments (e.g., β-hexosaminidase is routinely used to assess lysosomal purity, but enzymes such as CTSB/cathepsin B may also be used and may provide more accurate readouts).625 Because of the time-consuming nature of quantitative morphological studies, such studies should not be performed until simpler biochemical procedures have established the circumstances most likely to give meaningful morphometric/stereological results.
Finally, it is worthwhile noting that not all lysosomes are alike. For example, there are differences among primary lysosomes, autolysosomes and telolysosomes. Furthermore, what we refer to as “lysosomes” are actually a very heterogeneous pool of organelles that simply fulfill five classical criteria, having a pH < 5.6, mature cathepsins, the presence of LAMP proteins, a single membrane, and the absence of endosomal and recycling compartment markers (e.g., the mannose-6-phosphate receptor or RAB5). But even applying those criteria we can separate lysosomes with clear differences in their proteome and other properties, and these distinct populations of lysosomes are likely to participate in different functions in the cell (see Chaperone-mediated autophagy).628
Conclusion: Considering the limited methods available for in vivo analysis of autophagy, tissue fractionation is a valid, although relatively laborious, method for monitoring this process. Care must be taken to ensure that sample analysis is representative.
13. Analyses in vivo
Monitoring autophagic flux in vivo or in organs is one of the least developed areas at present, and ideal methods relative to the techniques possible with cell culture may not exist. Importantly, the level of basal autophagy, time course of autophagic induction, and the bioavailabilty of autophagy-stimulating and -inhibiting drugs is likely tissue specific. Moreover, basal autophagy or sensitivity to autophagic induction may vary with animal age, sex or strain background. Therefore methods may need to be optimized for the tissue of interest. One method for in vivo studies is the analysis of GFP-LC3/Atg8 (see GFP-Atg8/LC3 fluorescence microscopy above). Autophagy can be monitored in tissue (e.g., skeletal muscle, liver and retina) in vivo in transgenic mice systemically expressing GFP-LC3,126,446 or in other models by transfection with GFP-LC3 plasmids.346 In addition, tissue-specific GFP-LC3 mice have been generated for monitoring cardiac myocytes.629,630 In these settings, GFP fluorescent puncta are indicative of autophagic structures. In addition, cleavage of GFP-LC3 to generate free GFP can be evaluated. This has been successfully performed in mouse liver,172,631 suggesting the GFP-LC3 cleavage assay may also be applied to in vivo studies. Alternatively, confocal laser scanning microscopy, which makes it possible to obtain numerous sections and substantial data about spatial localization features, can be a suitable system for studying autophagic structures (especially for whole mount embryo in vivo analysis).632 In addition, this method can be used to obtain quantitative data through densitometric analysis of fluorescent signals.633
Another possibility is immunohistochemical staining, an important procedure that may be applicable to human studies as well, considering the role of autophagy in neurodegeneration, myopathies and cardiac disease where samples may be limited to biopsy/autopsy tissue. Immunodetection of LC3 as definite puncta is possible in paraffin-embedded tissue sections and fresh frozen tissue, by either immunohistochemistry or immunofluorescence;140,634-637 however, this methodology has not received extensive evaluation, and does not lend itself well to dynamic assays. Other autophagic substrates can be evaluated via immunohistochemistry and include SQSTM1, NBR1, ubiquitinated inclusions and protein aggregates. Similarly, autophagy can be evaluated by measuring levels of these autophagic substrates via traditional immunoblot; however, their presence or absence needs to be cautiously interpreted as some of these substrates can accumulate with either an increase or a decrease in autophagic flux (see SQSTM1/p62 and related LC3 binding protein turnover assays). Bone marrow transfer has been used to document in vivo the role of autophagy in the reverse cholesterol transport pathway from peripheral tissues or cells (e.g., macrophages) to the liver for secretion in bile and for excretion,638 and a study shows that transglutaminase type 2 protein levels decrease in mouse liver in vivo upon starvation in an autophagy-dependent manner (and in human cell lines in vitro in response to various stimuli; Piacentini M, personal communication), presenting additional possible methods for following autophagy activity.
It is also possible to analyze tissues ex vivo, and these studies can be particularly helpful in assessing autophagic flux as they avoid the risks of toxicity and bioavailabilty of compounds such as bafilomycin A1 or other autophagy inhibitors. Along these lines, autophagic flux can be determined by western blot in retinas placed in culture for 4 h with protease inhibitors. This method could be used in tissues that can remain “alive” for several hours in culture such as the retina (Boya P, unpublished observations) and brain slices (Desai S, unpublished observations).
Several studies have demonstrated the feasibility of monitoring autophagic flux in vivo in skeletal muscle. Starvation is one of the easiest and most rapid methods for stimulating the autophagic machinery in skeletal muscles. Twelve h of fasting in mice may be sufficient to trigger autophagy in muscle,639,640 but the appropriate time should be determined empirically. Data about the autophagic flux can be obtained by treating mice with, for example, chloroquine,640 leupeptin641 or colchicine153 and then monitoring the change in accumulation of LC3 (see cautionary notes). This type of analysis can also be done with liver, by comparing the LC3-II level in untreated liver (obtained by a partial hepatectomy) to that following subsequent exposure to chloroquine (Skop V, Papackova Z, Cahová M, personal communication). Additional reporter assays to monitor autophagy flux in vivo need to be developed, including tandem fluorescent-LC3 transgenic mice, or viral vectors to express this construct in vivo in localized areas.
Some biochemical assays may be used to at least provide indirect correlative data relating to autophagy, in particular when examining the role of autophagy in cell death. For example, cellular viability is related to high CTSB activity and low CTSD/cathepsin D activities.642 Therefore, the appearance of the opposite levels of activities may be one indication of the initiation of autophagy (lysosome)-dependent cell death. The question of “high” vs. “low” activities can be determined by comparison to the same tissue under control conditions, or to a different tissue in the same organism, depending on the specific question.
With regard to living mammals, a minimally invasive method that may be used even in humans is to measure the arterio-venous amino acid exchange rate in the peripheral tissues as a measure of postabsorptive protein catabolism. In humans, the insulin- and amino acid-sensitive postabsorptive (autophagic) net protein catabolism in the peripheral (mostly skeletal muscle) tissue can be measured by determining the amino acid exchange rate across the lower extremities, as defined by the difference between the plasma amino acid concentrations in the femoral artery and femoral vein multiplied by the blood flow.643-645 Amino acid exchange studies show that the peripheral tissues take up amino acids during the post-prandial (fed) state and release amino acids in the postabsorptive (fasted) state, i.e., in a state with relatively low plasma insulin and amino acid levels. This post-absorptive release of amino acids is strongly inhibited by infusion of insulin or by exogenous supply of amino acids, suggesting that it is mainly mediated by a lysosomal/autophagic mechanism of protein catabolism.643-650 However, the relative contribution of autophagy to the post-absorptive release of amino acids may be changed in disease states (see cautionary notes).
Cautionary notes: The major hurdle with in vivo analyses is the identification of autophagy-specific substrates and the ability to “block” autophagosome degradation with a compound such as bafilomycin A1. Regardless, it is still essential to adapt the same rigors for measuring autophagic flux in vitro to measurements made with in vivo systems. Moreover, as with cell culture, in order to substantiate a change in autophagic flux it is not adequate to rely solely on the analysis of static levels or changes in LC3-II protein levels on western blot using tissue samples. To truly measure in vivo autophagic flux using LC3-II as a biomarker, it is necessary to block lysosomal degradation of the protein. Several studies have successfully done this in select tissues in vivo. Certain general principles need to be kept in mind: (a) Any autophagic blocker, whether leupeptin, bafilomycin A1, chloroquine or microtubule depolarizing agents such as colchicine or vinblastine must significantly increase basal LC3-II levels. The turnover of LC3-II or rate of basal autophagic flux is not known for tissues in vivo, and therefore short treatments (e.g., 4 h) may not be as effective as blocking for longer times (e.g., 12 to 24 h). (b) The toxicity of the blocking agent needs to be considered (e.g., treating animals with bafilomycin A1 for 2 h can be quite toxic) and food intake must be monitored. If long-term treatment is needed to see a change in LC3-II levels, then confirmation that the animals have not lost weight may be needed. Mice may lose a substantial portion of their body weight when deprived of food for 24 h, and starvation is a potent stimulus for the activation of autophagy. (c) The bioavailability of the agent needs to be considered. For example, many inhibitors such as bafilomycin A1 or chloroquine have relatively poor bioavailability to the central nervous system. To overcome this problem, intracerebroventricular injection can be performed.
When analyzing autophagic flux in vivo, one major limitation is the variability between animals. Different animals do not always activate autophagy at the same time. To improve the statistical relevance and avoid unclear results, these experiments should be repeated more than once and each experiment should include several animals. Induction of autophagy in a time-dependent manner by fasting mice for different times requires appropriate caution. Mice are nocturnal animals, so they preferentially move and eat during the night, while they mostly rest during daylight. Therefore, in such experiments it is better to start food deprivation early in the morning, in order to avoid the possibility that the animals have already been fasting for several hours. The use of chloroquine is technically easier, since it only needs one intraperitoneal injection per day, but the main concern is that chloroquine has some toxicity. Chloroquine suppresses the immunological response in a manner that is not due to its pH-dependent lysosomotropic accumulation (chloroquine interferes with lipopolysaccharide-induced TNF/TNFα gene expression by a nonlysosmotropic mechanism),651 as well as through its pH-dependent inhibition of antigen presentation.652 Therefore, chloroquine treatment should be used for short times and at doses that do not induce severe collateral effects, which may invalidate the measurement of the autophagic flux, and care must be exercised in using chloroquine for studies on autophagy that involve immunological aspects. It is also important to have time-matched controls for in vivo analyses. That is, having only a zero hour time point control is not sufficient because there may be substantial diurnal changes in basal autophagy.467 For example, variations in basal flux in the liver associated with circadian rhythm may be several fold, which can equal or exceed the changes due to starvation. Along these lines, in order to allow comparisons of a single time-point it is important to specify what time of day the measurement is taken, and the lighting conditions under which the animals are housed. It is also important that the replicate experiments are conducted at the same time of day. Controlling for circadian effects can greatly reduce the mouse-to-mouse variability in autophagy markers and flux (Haspel JA, Choi AMK, personal communication).
The amino acid exchange rate, which has been suggested as a minimal invasive marker for measuring autophagic flux in vivo should be used with special caution. Postprandial suppression of amino acid release by peripheral tissues is strongly mediated by insulin and amino acids and is therefore thought to be mediated by autophagy (see above). However, the ubiquitin-proteasome system likely confounds this investigation, as this pathway can also be inhibited by insulin and amino acids.653 In addition, multiple disease states have been associated with an altered activity of the ubiquitin-proteasome system, further complicating the use of the amino acid exchange rate as a marker of autophagy.654
When analyzing basal autophagic level in vivo using GFP-LC3 transgenic mice,126 one pitfall is that GFP-LC3 expression is driven by the cytomegalovirus enhancer and β-actin (CAG) promoter, so that the intensity of the GFP signal may not always represent the actual autophagic activity, but rather the CAG promoter activity in individual cells. For example, GFP-LC3 transgenic mice exhibit prominent fluorescence in podocytes, but rarely in tubular epithelial cells in the kidney,126 but a similar GFP pattern is observed in transgenic mice carrying CAG promoter-driven non-tagged GFP.655 Furthermore, proximal tubule-specific ATG5-deficient mice656 display a degeneration phenotype earlier than podocyte-specific ATG5-deficient mice,657 suggesting that autophagy, and hence LC3 levels, might actually be more prominent in the former.
One caution in using approaches that monitor ubiquitinated aggregates is that the accumulation of ubiquitin may indicate a block in autophagy, inhibition of proteasomal degradation, or may correspond to structural changes in the substrate proteins that hinder their degradation. In addition, only cytosolic and not nuclear ubiquitin is subject to autophagic degeneration. It is helpful to analyze aggregate degradation in an autophagy-deficient control strain, such as an autophagy mutant mouse, whenever possible to determine whether an aggregate is being degraded by an autophagic mechanism. This type of control will be impractical for some tissues such as those of the central nervous system because the absence of autophagy leads to rapid degeneration. Accordingly, the use of Atg16l1 hypomorphs or Becn1+/- heterozygotes may help circumvent this problem.
Conclusion: Although the techniques for analyzing autophagy in vivo are not as advanced as those for cell culture, it is still possible to follow this process (including flux) by monitoring, for example, GFP-LC3 by fluorescence microscopy, and SQSTM1 and NBR1 by immunohistochemistry and/or western blotting.
14. Cell death
In a limited number of cases, autophagy has been established as the cause of cell death;97,199,658-664 although opposite results have been reported using analogous experimental settings.665 Furthermore, many of the papers claiming a causative role of autophagy in cell death fail to provide adequate evidence.666 Other papers suffer from ambiguous use of the term “autophagic cell death,” which was coined in the 1970s667 in a purely morphological context to refer to cell death with autophagic features (especially the presence of numerous secondary lysosomes); this was sometimes taken to suggest a role of autophagy in the cell death mechanism, but death-mediation was not part of the definition.668 Unfortunately, the term “autophagic cell death” is now used in at least three different ways: (a) autophagy-associated cell death (the original meaning); (b) autophagy-mediated cell death (which could involve a standard mechanism of cell death such as apoptosis, but triggered by autophagy); (c) a distinct mechanism of cell death, independent of apoptosis or necrosis. Clearly claim (b) is stronger than claim (a), and needs to be justified by proof that inhibiting autophagy, through either genetic or chemical means, prevents cell death.669 Claim (c) is still stronger, because, even if the cell death is blocked by autophagy inhibition, proof needs to be provided that the cell death mechanism is not apoptosis or necrosis.670 In view of the current confusion it may be preferable to replace the term “autophagic cell death” by other terms such as “autophagy-associated cell death” or “autophagy-mediated cell death,” unless the criteria in claim (c) above have been satisfied. Along these lines, it is preferable to use the term “autophagy-dependent cell death” instead of “autophagy-mediated cell death” when it is proven that autophagy is a pre-requisite for the occurrence of cell death, but it is not proven that autophagy mechanistically mediates the switch to cell death. A special caution should also be taken when describing developmental programmed cell death in plants, which is in most cases executed by the growing lytic vacuoles and therefore is referred to as “vacuolar cell death.”671 Although the morphology of vacuolar cell death resembles a combination of macro- and microautophagy, there is no genetic evidence yet that this death requires the core autophagic machinery. Finally, the relationship between autophagy and cell death may significantly differ as a function of the model system being studied. For example, upon induction by starvation of multicellular development in the protist Dictyostelium, autophagy (or at least Atg1) is required to protect against starvation-induced cell death, allowing vacuolar developmental cell death to take place instead.672,673 Autophagy may be involved not only in allowing this death to occur, but also in the vacuolization process itself.674
Cautionary notes: In brief, rigorous criteria must be met in order to establish a death-mediating role of autophagy, as this process typically promotes cell survival. These include a clear demonstration of autophagic flux as described in this article, as well as verification that inhibition of autophagy prevents cell death [claim (b) above; if using a knockdown approach, at least two ATG genes should be targeted], and that other mechanisms of cell death are not responsible [claim (c) above]. As part of this analysis, it is necessary to examine the effect of the specific treatment, conditions or mutation on cell viability using several methods.675 In the case of postmitotic cells such as neurons or retinal cells, cell death—and cell rescue by autophagy inhibition—can usually be established in vivo by morphological analysis,676 and in culture by cell counts and/or measurement of the release of an enzyme such as LDH into the medium at early and late time points; however, a substantial amount of neuronal cell death occurs during neurogenesis, making it problematic to carry out a correct analysis in vivo or ex vivo.677,678 In populations of rapidly dividing cells, the problems may be greater. A commonly used method is the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, or a related assay using a similar, or a water-soluble, tetrazolium salt. The main concern with the MTT assay is that it measures mitochondrial activity, but does not allow a precise determination of cellular viability or cell death, whereas methods that show cell death directly (e.g., trypan blue exclusion) fail to establish the viability of the remaining cell population.679 Accordingly, a preferred alternative is to accurately quantify cell death by appropriate cytofluorometric or microscopy assays.675 Moreover, long-term clonogenic assays should be employed when possible to measure the effective functional survival of cells.
Conclusion: In most systems, ascribing death to autophagy based solely on morphological criteria is insufficient; autophagic cell death can only be demonstrated as death that is suppressed by the inhibition of autophagy, through either genetic or chemical means.669 In addition, more than one assay should be used to measure cell death.
15. Chaperone-mediated autophagy
The primary characteristic that makes chaperone-mediated autophagy (CMA) different from the other autophagic variants described in these guidelines is that it does not require formation of intermediate vesicular compartments (autophagosomes or microvesicles) for the import of cargo into lysosomes.680,681 Instead, the CMA substrates are translocated across the lysosomal membrane through the action of HSPA8/HSC70 located in the cytosol and lysosome lumen, and the lysosome membrane protein LAMP2A. To date, CMA has only been identified in mammalian cells, and accordingly this section refers only to studies in mammals.
The following methods are commonly utilized to determine if a protein is a CMA substrate (see ref. 682 for experimental details): (a) Analysis of the amino acid sequence of the protein to identify the presence of a KFERQ-related motif that is an absolute requirement for all CMA substrates.683 (b) Colocalization studies with lysosomal markers (typically LAMP2A and/or LysoTracker) to identify a fraction of the protein associated with lysosomes. The increase in association of the putative substrate under conditions that upregulate CMA (such as prolonged starvation) or upon blockage of lysosomal proteases (to prevent the degradation of the protein) helps support the hypothesis that the protein of interest is a CMA substrate. However, association with lysosomes is necessary but not sufficient to consider a protein an authentic CMA substrate, because proteins delivered by other pathways to lysosomes will also behave in a similar manner. A higher degree of confidence can be attained if the association is preferentially with the subset of lysosomes active for CMA (i.e., those containing HSPA8 in their lumen), which can be separated from other lysosomes following published procedures.628 (c) Co-immunoprecipitation of the protein of interest with cytosolic HSPA8. Due to the large number of proteins that interact with this chaperone, it is usually better to perform affinity isolation with the protein of interest and then analyze the isolated proteins for the presence of HSPA8 rather than vice versa. (d) Co-immunoprecipitation of the protein of interest with LAMP2A.684 Due to the fact that the only antibodies specific for the LAMP2A variant (the only one of the three LAMP2 variants involved in CMA105,685) are generated against the cytosolic tail of LAMP2A, where the substrate also binds, it is necessary to affinity isolate the protein of interest and then analyze for the presence of LAMP2A. Immunoblot for LAMP2A in the precipitate can only be done with the antibodies specific for LAMP2A and not just those that recognize the lumenal portion of the protein that is identical in the other LAMP2 variants. If the protein of interest is abundant inside cells, co-immunoprecipitations with LAMP2A can be done in total cellular lysates, but for low abundance cellular proteins, preparation of a membrane fraction (enriched in lysosomes) by differential centrifugation may facilitate the detection of the population of the protein bound to LAMP2A. (e) Selective upregulation and blockage of CMA to demonstrate that degradation of the protein of interest changes with these manipulations. Selective chemical inhibitors for CMA are not currently available. Note that general inhibitors of lysosomal proteases (e.g., bafilomycin A1, NH4Cl, leupeptin) also block the degradation of proteins delivered to lysosomes by other autophagic and endosomal pathways. The most selective way to block CMA is by knockdown of LAMP2A, which causes this protein to become a limiting factor.105 The other components involved in CMA, including HSPA8, HSP90, GFAP, and EEF1A1/eF1α are all multifunctional cellular proteins, making it difficult to interpret the effects of knockdowns. Overexpression of LAMP2A684 is also a better approach to upregulate CMA than the use of chemical modulators. The two compounds demonstrated to affect degradation of long-lived proteins in lysosomes,686 6-aminonicotinamide and geldanamycin, lack selectivity, as they affect many other cellular processes. In addition, in the case of geldanamycin, the effect on CMA can be the opposite (inhibition rather than stimulation) depending on the cell type (this is due to the fact that the observed stimulation of CMA is actually a compensatory response to the blockage of HSP90 in lysosomes, and different cells activate different compensatory responses).687 (f) The most conclusive way to prove that a protein is a CMA substrate is by reconstituting its direct translocation into lysosomes using a cell-free system.682 This method is only possible when the protein of interest can be purified, and it requires the isolation of the population of lysosomes active for CMA. Internalization of the protein of interest inside lysosomes upon incubation with the isolated organelle can be monitored using protease protection assays (in which addition of an exogenous protease removes the protein bound to the cytosolic side of lysosomes, whereas it is inaccessible to the protein that has reached the lysosomal lumen; note that pre-incubation of lysosomes with lysosomal protease inhibitors before adding the substrate is required to prevent the degradation of the translocated substrate inside lysosomes).688 The use of exogenous protease requires numerous controls (see ref. 682) to guarantee that the amount of protease is sufficient to remove all the substrate outside lysosomes, but will not penetrate inside the lysosomal lumen upon breaking the lysosomal membrane. The difficulties in the adjustment of the amount of protease, has led to the development of a second method that is more suitable for laboratories that have no previous experience with these procedures. In this case, the substrate is incubated with lysosomes untreated or previously incubated with inhibitors of lysosomal proteases, and uptake is determined as the difference of protein associated with lysosomes not incubated with inhibitors (in which the only remaining protein will be the one associated with the cytosolic side of the lysosomal membrane) and those incubated with the protease inhibitors (which contain both the protein bound to the membrane and that translocated into the lumen).689 Confidence that the lysosomal internalization is by CMA increases if the uptake of the substrate can be competed with proteins previously identified as substrates for CMA (e.g., GAPDH/glyceraldehyde-3-phosphate dehydrogenase or ribonuclease A, both commercially available as purified proteins), but is not affected by the presence of similar amounts of nonsubstrate proteins (such as ovalbumin or PPIA/cyclophilin A). Blockage of uptake by pre-incubation of the lysosomes with antibodies against the cytosolic tail of LAMP2A also reinforces the hypothesis that the protein is a CMA substrate.
In other instances, rather than determining if a particular protein is a CMA substrate, the interest may be to analyze possible changes in CMA activity under different conditions or in response to different modifications. We enumerate here the methods, from lower to higher complexity that can be utilized to measure CMA in cultured cells and in tissues (see ref. 682 for detailed experimental procedures). (a) Measurement of changes in the intracellular rates of degradation of long-lived proteins, when combined with inhibitors of other autophagic pathways, can provide a first demonstration in support of changes that are due to CMA. For example, CMA is defined as lysosomal degradation upregulated in response to serum removal but insensitive to PtdIns3K inhibitors. (b) Measurement of levels of CMA components is insufficient to conclude changes in CMA because this does not provide functional information, and changes in CMA components can also occur under other conditions. However, analysis of the levels of LAMP2A can be used to support changes in CMA detected by other procedures. Cytosolic levels of HSPA8 remain constant and are not limiting for CMA, thus providing no information about this pathway. Likewise, changes in total cellular levels of LAMP2A do not have an impact on this pathway unless they also affect their lysosomal levels (i.e., conditions in which LAMP2A is massively overexpressed lead to its targeting to the plasma membrane where it cannot function in CMA). It is advisable that changes in the levels of these two CMA components are confirmed to occur in lysosomes, either by colocalization with lysosomal markers when using image-based procedures or by performing immunoblot of a lysosomal enriched fraction (purification of this fraction does not require the large amounts of cells/tissue necessary for the isolation of the subset of lysosomes active for CMA). (c) Tracking changes in the subset of lysosomes active for CMA. This group of lysosomes is defined as those containing HSPA8 in their lumen (note that LAMP2A is present in both lysosomes that are active and inactive for CMA, and it is the presence of HSPA8 that confers CMA capability). Immunogold or immunofluorescence against these two proteins (LAMP2A and HSPA8) makes it possible to quantify changes in the levels of these lysosomes present at a given time, which correlates well with CMA activity.628 (d) Analysis of lysosomal association of fluorescent artificial CMA substrates. Two different fluorescent probes have been generated to track changes in CMA activity in cultured cells using immunofluorescence or FACS analysis.628 These probes contain the KFERQ and context sequences in frame with photoswitchable or photoactivated fluorescent proteins. Activation of CMA results in the mobilization of a fraction of the cytosolic probe to lysosomes and the subsequent change from a diffuse to a punctate pattern. CMA activity can be quantified as the number of fluorescent puncta per cell or as the decay in fluorescence activity over time because of degradation of the artificial substrate. Because the assay does not allow measuring accumulation of the substrate (which must unfold for translocation), it is advisable to perform a time-course analysis to determine gradual changes in CMA activity. Antibodies against the fluorescent protein in combination with inhibitors of lysosomal proteases can be used to monitor accumulation of the probe in lysosomes over a period of time, but both the photoswitchable and the unmodified probe will be detected by this procedure.690 As for any other fluorescence probe based on analysis of intracellular “puncta” it is essential to include controls to confirm that the puncta are indeed lysosomes (colocalization with LysoTracker or LAMPs and lack of colocalization with markers of cytosolic aggregation such as ubiquitin), and do not reach the lysosomes through other autophagic pathways (insensitivity to PtdIns3K inhibitors and sensitivity to LAMP2A knockdown are good controls in this respect). (e) Direct measurement of CMA using in vitro cell free assays. Although the introduction of the fluorescent probes should facilitate measurement of CMA in many instances, they are not applicable for tissue samples. In addition, because the probes measure binding of substrate to lysosomal membranes it is important to confirm that enhanced binding does not result from defective translocation. Lastly, the in vitro uptake assays are also the most efficient way to determine primary changes in CMA independently of changes in other proteolytic systems in the cells. These in vitro assays are the same ones described in the previous section on the identification of proteins as substrates of CMA, but are performed in this case with purified proteins previously characterized to be substrates for CMA. In this case the substrate protein is always the same and what changes is the source of lysosomes (from the different tissues or cells that are to be compared). As described in the previous section, binding and uptake can be analyzed separately using lysosomes previously treated or not with protease inhibitors. The analysis of the purity of the lysosomal fractions prior to performing functional analysis is essential to conclude that changes in the efficiency to take up the substrates results from changes in CMA rather than from different levels of lysosomes in the isolated fractions. Control of the integrity of the lysosomal membrane and sufficiency of the proteases are also essential to discard the possibility that degradation is occurring outside lysosomes because of leakage, or that accumulation of substrates inside lysosomes is due to enhanced uptake rather than to decreased degradation.
Cautionary notes: The discovery of a new selective form of protein degradation in mammals named endosomal-microautophagy (e-MI)691 has made it necessary to reconsider some of the criteria that applied in the past for the definition of a protein as a CMA substrate. The KFERQ-like motif, previously considered to be exclusive for CMA, is also used to mediate selective targeting of cytosolic proteins to the surface of late endosomes. Once there, substrates can be internalized in microvesicules that form from the surface of these organelles in an ESCRT-dependent manner. HSPA8 has been identified as the chaperone that binds this subset of substrates and directly interacts with lipids in the late endosomal membrane, acting thus as a receptor for cytosolic substrates in this compartment. At a practical level, to determine if a KFERQ-containing protein is being degraded by CMA or e-MI the following criteria can be applied: (a) Inhibition of lysosomal proteolysis (for example with NH4Cl and leupeptin) blocks degradation by both pathways. (b) Knockdown of LAMP2A inhibits CMA but not e-MI. (c) Knockdown of components of ESCRTI and II (e.g., VPS4 and TSG101) inhibits e-MI but not CMA. (d) Interfering with the capability to unfold the substrate protein blocks its degradation by CMA, but does not affect e-MI of the protein. In this respect soluble proteins, oligomers and protein aggregates can undergo e-MI, but only soluble proteins can be CMA substrates. (e) In vitro uptake of e-MI substrates can be reconstituted using isolated late endosomes whereas in vitro uptake of CMA substrates can only be reconstituted using lysosomes.
Another pathway that needs to be considered relative to CMA is chaperone-assisted selective autophagy (CASA).692 CASA is dependent on HSPA8 and LAMP2 (although it is not yet known if it is dependent solely on the LAMP2A isoform). Thus, a requirement for these two proteins is not sufficient to conclude that a protein is degraded by CMA.
Conclusion: One of the key issues with the analysis of CMA is verifying that the protein of interest is an authentic susbstrate. Methods for monitoring CMA that utilize fluorescent probes are available that eliminate the need for the isolation of CMA-competent lysosomes, one of the most difficult aspects of assaying this process.
B. Comments on Additional Methods
1. Acidotropic dyes
Among the older methods for following autophagy is staining with acidotropic dyes such as monodansylcadaverine (MDC),693 acridine orange,694 Neutral Red,632 LysoSensor Blue695 and LysoTracker Red.198,696
Cautionary notes: Although MDC was first described as a specific marker of autophagic vacuoles697 subsequent studies have suggested that this, and other acidotropic dyes, are not specific markers for early autophagosomes,258 but rather label later stages in the degradation process. For example, autophagosomes are not acidic, and MDC staining can be seen in autophagy-defective mutants401 and in the absence of autophagy activation.698 MDC may also show confounding levels of background labeling unless narrow bandpass filters are used. However, in the presence of vinblastine, which blocks fusion with lysosomes, MDC labeling increases, suggesting that under these conditions MDC can label late-stage autophagosomes.699 Along these lines, cells that overexpress a dominant negative version of RAB7 (the T22N mutant) show colocalization of this protein with MDC; in this case fusion with lysosomes is also blocked700 indicating that MDC does not just label lysosomes. Finally, MDC labeling could be considered to be an indicator of autophagy when the increased labeling of cellular compartments by this dye is prevented by treatment with autophagy inhibitors such as wortmannin or 3-MA.
Overall, staining with MDC or its derivative monodansylamylamine (MDH)693 is not, by itself, a sufficient method for monitoring autophagy. Similarly, LysoTracker Red, Neutral Red and acridine orange are not ideal markers for autophagy because they primarily detect lysosomes. These markers are, however, useful for monitoring selective autophagy when used in conjunction with protein markers or other dyes. For example, increased colocalization of mitochondria with both GFP-LC3 and LysoTracker can be used as evidence of autophagic cargo delivery to lysosomes. Moreover, LysoTracker Red has been used to provide correlative data on autophagy in Drosophila melanogaster fat body cells (Fig. 23).197,198 However, additional assays, such as GFP-Atg8/LC3 fluorescence or EM, should be used to substantiate results obtained with acidotropic dyes whenever possible. One important caution when co-imaging with LysoTracker Red and a green-fluorescing marker (e.g., GFP-LC3 or MitoTracker Green) is that it is necessary to control for rapid red-to-green photoconversion of the LysoTracker, which can otherwise result in an incorrect interpretation of colocalization.701
Figure 23. LysoTracker Red stains lysosomes and can be used to monitor autophagy in Drosophila. Live fat body tissues from Drosophila were stained with LysoTracker Red (red) and Hoechst 33342 (blue) to stain the nucleus. Tissues were isolated from fed (left) or 3 h starved (right) animals. Bar, 25 µm. This figure was modified from data presented in reference 198, Scott RC, Schuldiner O, Neufeld TP, Role and regulation of starvation-induced autophagy in the Drosophila fat body, Dev Cell 2004; 7:167–78, copyright 2004, with permission from Elsevier.
Some of the confusion regarding the interpretation of results with these dyes stems in part from the nomenclature in this field. Indeed, the discussion of acidotropic dyes points out why it is advisable to differentiate between the terms “autophagosome” and “autophagic vacuole,” although they are occasionally, and incorrectly, used interchangeably. The autophagosome is the sequestering compartment generated by the phagophore. The fusion of an autophagosome with an endosome or a lysosome generates an amphisome or an autolysosome, respectively. The early autophagosome is not an acidic compartment, whereas amphisomes and autolysosomes are acidic. Earlier names for these compartments are “initial autophagic vacuole (AVi),” “intermediate autophagic vacuole (AVi/d)” and “degradative autophagic vacuole (AVd),” respectively. Thus, acidotropic dyes can stain late autophagic vacuoles (in particular autolysosomes), but not the initial autophagic vacuole, the early autophagosome. With the above caveats in mind, the combined use of early and late markers of autophagy is highly encouraged, and when quantifying mammalian lysosomes, it is important to keep in mind that increases in both lysosome size and number are frequently observed. Finally, in order to avoid confusion with the plant and fungal vacuole, the equivalent organelle to the lysosome, we recommend the use of the term “autophagosome” instead of “autophagic vacuole,” and the use of “autophagic compartment” when the specific nature of the structure is not known.
Conclusion: Given the development of better techniques that are indicators of autophagy, the use of acidotropic dyes to study this process is discouraged, and relying entirely on such dyes is not acceptable.
2. Autophagy inhibitors and inducers
In many situations it is important to demonstrate an effect resulting from inhibition or stimulation of autophagy (see ref. 702 for a partial listing of regulatory compounds), and a few words of caution are worthwhile in this regard. Most chemical inhibitors of autophagy are not entirely specific, and it is important to consider possible dose- and time-dependent effects. Accordingly, it is generally preferable to analyze specific loss-of-function Atg mutants. However, it must be kept in mind that some apparently specific Atg gene products may have autophagy-independent roles (e.g., ATG5 in cell death, and the PIK3C3-containing complexes—including BECN1—in apoptosis, endosomal function and protein trafficking).403,432,703-706 Therefore, the experimental conditions of inhibitor application and their side effects must be carefully considered. In addition, it must be emphasized once again that autophagy, as a multistep process, can be inhibited at different stages. Sequestration inhibitors, including 3-MA, LY294002 and wortmannin, inhibit class I as well as class III PtdIns3Ks.134,257,707 The class I enzymes generate products [PtdIns(3,4,5)P3] that inhibit autophagic sequestration, whereas the class III product (PtdIns3P) generally stimulates autophagic sequestration. The overall effect of these inhibitors is typically to block autophagy because the class III enzymes that are required to activate autophagy act downstream of the negative regulatory class I enzymes, although cell death may ensue in cell types that are dependent upon high levels of AKT1 for survival. The effect of 3-MA (but not that of wortmannin) is further complicated by the fact that it has different temporal patterns of inhibition, causing a long-term suppression of the class I PtdIns3K, but only a transient inhibition of the class III enzyme. In cells incubated in a complete medium for extended periods of time, 3-MA may, therefore (particularly at suboptimal concentrations), promote autophagy by inhibition of the class I enzyme.257 Thus, wortmannin may be considered as an alternative to 3-MA for autophagy inhibition.257 However, wortmannin can induce the formation of vacuoles that may have the appearance of autophagosomes, although they are swollen late endocytic compartments.611 Furthermore, studies have demonstrated that inhibition of autophagy with 3-MA or wortmannin can have effects on cytokine transcription, processing and secretion, particularly IL1 family members,708-710 but 3-MA also inhibits the secretion of some cytokines (e.g., TNF, IL6) in an autophagy-independent manner (Harris J, unpublished observations). Thus, in studies where the effect of autophagy inhibition on specific cellular processes is being investigated, is is important to confirm results using other methods, such as RNA silencing. Due to these issues, it is of great interest that inhibitors with specificity for the class III PtdIns3Ks, and their consequent effects on autophagy, have been described.165
Cycloheximide, a commonly used protein synthesis inhibitor in mammals, is also an inhibitor of sequestration in vivo,14-16,92,604,711-715 and in various cell types in vitro,344,716 and it has been utilized to investigate the dynamic nature of the regression of various autophagic elements.14-16,24,92,712,713 The mechanism of action of cycloheximide in short-term experiments is not clear, but it has no direct relation to the inhibition of protein synthesis.344 This latter activity, however, may complicate certain types of analysis when using this drug.
A significant challenge for a more detailed analysis of the dynamic role of autophagy in physiological and pathophysiological processes, for instance with regard to cancer and cancer therapy, is to find more specific inhibitors of autophagy signaling which do not affect other signaling cascades. For example, in the context of cellular radiation responses it is well known that PtdIns3Ks (e.g., ATM, PRKDC/DNA-PKcs), in addition to signaling through the PtdIns3K-AKT1 pathway, have a major role in the regulation of DNA-damage repair.717 However, 3-MA, which is a nonspecific inhibitor of class 3 PtdIns3Ks, can alter the function of other classes of this enzyme, which are involved in the DNA-damage repair response. This is of particular importance for investigations into the role of radiation-induced autophagy in cellular radiation sensitivity or resistance.718,719
Most other inhibitory drugs act at post-sequestration steps. These types of agents have been used in many experiments to both inhibit endogenous protein degradation and to increase the number of autophagic compartments. They cause the accumulation of sequestered material in either autophagosomes or autolysosomes, or both, because they allow autophagic sequestration to proceed. The main categories of these types of inhibitors include the vinca alkaloids (e.g., vinblastine) and other microtubule poisons that inhibit fusion, inhibitors of lysosomal enzymes (e.g., leupeptin, pepstatin A and E-64d), and compounds that elevate lysosomal pH (e.g., inhibitors of vacuolar-type ATPases such as bafilomycin A1 and concanamycin A (another V-ATPase inhibitor), and weak base amines including methyl- or propylamine, chloroquine, and Neutral Red, some of which slow down fusion). Ammonia is a very useful agent for the elevation of lysosomal pH in short-term experiments, but has been reported to cause a stimulation of autophagy during long-term incubation of cells in a full medium,720 under which conditions a good alternative might be methylamine or propylamine.721 Along these lines, it should be noted that the half-life of glutamine in cell culture media is approximately two weeks due to chemical decomposition, which results in media with lowered glutamine and elevated ammonia concentrations that can affect the autophagic flux (either inhibiting or stimulating autophagy, depending on the concentration722). Thus, the use of freshly prepared cell culture media with glutamine is advised, to help reduce experimental variation. A special note of caution is also warranted in regard to chloroquine. Although this chemical is commonly used as an autophagy inhibitor, chloroquine may initially stimulate autophagy (Dorsey FC, personal communication; Franco R, personal communication).
Some data suggest that some nanomaterials may also be novel inhibitors of autophagy, by as yet unidentified mechanisms.723 It is worth noting that lysosomal proteases fall into three general groups, cysteine, aspartic acid and serine proteases. Therefore, the fact that leupeptin, a serine and cysteine protease inhibitor, has little or no effect does not necessarily indicate that lysosomal degradation is not taking place; a combination of leupeptin, pepstatin A and E-64d may be a more effective treatment. However, it should also be pointed out that these protease inhibitors can exert inhibitory effects not only on lysosomal proteases, but also on cytosolic proteases; that is, degradation of proteins might be blocked through inhibition of cytosolic instead of lysosomal proteases. Conversely, it should be noted that MG132 (Z-leu-leu-leu-al) and its related peptide aldehydes are commonly used as proteasomal inhibitors, but they can also inhibit certain lysosomal hydrolases such as cathepsins and calpains.724 Thus, any positive results using MG132 do not rule out the possibility of involvement of the autophagy-lysosome system. Therefore, even if MG132 is effective, it is important to confirm the result using more specific proteasomal inhibitors such as lactacystin or epoxomicin. Finally, there are significant differences in cell permeability among protease inhibitors. For example, E-64d is membrane permeable, whereas leupeptin and pepstatin A are not (although there are derivatives that display greater permeability such as pepstatin A methyl ester).725 Thus, when analyzing whether a protein is an autophagy substrate, caution should be taken in utilizing these protease inhibitors to block autophagy.
As with the PtdIns3K inhibitors, many autophagy-suppressive compounds are not specific. For example, okadaic acid726 is a powerful general inhibitor of both type 1 (protein phosphatase 1) and type 2A (PPP2CB/PP2A) protein phosphatases.727 Bafilomycin A1 and other compounds that raise the lysosomal pH may have indirect effects on any acidified compartments. Moreover, treatment with bafilomycin A1 for extended periods (18 h) can cause significant disruption of the mitochondrial network in cultured cells (Gegg ME, personal communication), and either bafilomycin A1 or concanamycin A cause swelling of the Golgi in plants,728 and increase cell death by apoptosis in cancer cells (Rao VA, personal communication). Bafilomycin A1 is often used at a final concentration of 100 nM, but much lower concentrations such as 1 nM may be sufficient to inhibit autophagic-lysosomal degradation and are less likely to cause indirect effects;154,729,730 however, appropriate inhibitory concentrations should be empirically determined for each cell type.6 One final caution with regard to bafilomycin A1 is that activation of lysosomally localized TORC1 depends on an active V-ATPase.364 This means that treatment with bafilomycin A1 could elevate the autophagic flux through MTOR inhibition, while simultaneously interfering with flux by inhibiting fusion. Accordingly, other compounds may be preferred for flux measurements. Thus, although these various agents can inhibit different steps of the autophagic pathway, their potential side effects must be considered in interpretation of the secondary consequences of autophagy inhibition, especially in long-term studies. For example, lysosomotropic compounds can increase the rate of autophagosome formation by inhibiting TORC1.731 Along these lines, chloroquine treatment may cause an apparent increase in the formation of autophagosomes possibly by blocking fusion with the lysosome (Dorsey FC, Cleveland JL, personal communication). This conclusion is supported by the finding that chloroquine reduces the colocalization of LC3 and LysoTracker despite the presence of autophagosomes and lysosomes (Simon AK, personal communication). Concanamycin A blocks sorting of vacuolar proteins in plant cells in addition to inhibiting vacuolar acidification.732 Furthermore, in addition to causing the accumulation of autophagic compartments, many of these drugs seem to be stimulators of sequestration in many cell types, especially in vivo.93,235,604,712,716,733-737 Although it is clear why these drugs cause the accumulation of autophagic compartments, it is not known why they stimulate sequestration. One possibility, at least for hepatocytes, is that the inhibition of protein degradation reduces the intracellular amino acid pool, which in turn upregulates sequestration. A time-course study of the changes in both the intra- and extracellular fractions may provide accurate information regarding amino acid metabolism. For these various reasons, it is important to include appropriate controls; along these lines, MTOR inhibitors such as rapamycin or amino acid deprivation can be utilized as positive controls for inducing autophagy. In many cell types, however, the induction of autophagy by rapamycin is relatively slow, or transient, allowing more time for indirect effects; thus, rapamycin may fail to activate autophagy in cultured primary neurons, despite its potent stimulation of autophagy in some cancer cell lines,238,738,739 and, similarly, it does not induce autophagy in human neuroblastoma SH-SY5Y cells, which can differentiate into neuron-like cells (Diaz-Nido J, personal communication). Thus, glucose depletion may be much more efficient at inducing autophagy than rapamycin or amino acid starvation in neurons in culture (Germain M, Slack R, personal communication); although a number of compounds can also be quite efficient autophagy inducers in neurons including the calpain inhibitor calpeptin.238,740,741 Several small molecule inhibitors, including torin1, PP242, KU-0063794, PtdIns-103 and NVP-BEZ235, have been developed that target the catalytic domain of MTOR in an ATP-competitive manner.154,252,742-745 In comparison to rapamycin, these catalytic MTOR inhibitors are more potent, and hence are stronger autophagy agonists in most cell lines.242,252,746 The use of these second-generation MTOR inhibitors may reveal that some reports of mTOR-independent autophagy may actually reflect the use of the relatively weak inhibitor rapamycin. Furthermore, the use of these compounds has revealed a role for TORC1 and TORC2 as independent regulators of autophagy.747 Finally, a specialized class of compounds with α,β-unsaturated ketone structure tends to induce autophagic cell death, accompanied by changes in mitochondrial morphology; since the cytotoxic action of these compounds is efficiently blocked by N-acetyl-l-cysteine, the β-position in the structure may interact with an SH group of the targeted molecules.748 Due to the potential pleiotropic effects of various drug treatments, it is incumbent upon the researcher to demonstrate that autophagy is indeed inhibited, by using the methodologies described herein. Accordingly, it is critical to verify the effect of a particular biochemical treatment with regard to its effects on autophagy induction or inhibition when using a cell line that was previously uncharacterized for the chemical being used. Similarly, cytotoxicity of the relevant chemical should be assessed.
The use of gene deletions/inactivations (e.g., in primary or immortalized Atg−/− MEFs,401 plant T-DNA or transposon insertion mutants,200,749 or in vivo using transgenic knockout models750,751 including Cre-lox based “conditional” knockouts230,231) or functional knockdowns (e.g., with RNAi against ATG genes) is the preferred approach when possible because these methods allow a more direct assessment of the resulting phenotype; however, different floxed genes are deleted with varying efficiency, and the proportion deleted must be carefully quantified (Hwang S, Virgin IV HW, personal communication). Studies also suggest that microRNAs may be used for blocking gene expression.164,468,469,752 In certain contexts, it is advisable when using a knockout or knockdown approach to examine multiple autophagy-related genes to exclude the possibility that the phenotype observed is due to effects on a non-autophagic function(s) of the corresponding protein, especially when examining the possibility of autophagic cell death (in contrast, if examining whether perturbation induces clearance of a substrate via autophagy, a single ATG gene knockout is probably sufficient). This is particularly the case in evaluating BECN1, which interacts with anti-apoptotic BCL2 family proteins,425 or when low levels of a target protein are sufficient for maintaining autophagy as is the case with ATG5.171 With regard to ATG5, a better approach may be to use a dominant negative (K130R) version.706,753,754 Along these lines, and as stated above for the use of inhibitors, when employing a knockout or especially a knockdown approach, it is again incumbent upon the researcher to demonstrate that autophagy is actually inhibited, by using the methodologies described herein. Finally, we note that the long-term secondary consequences of gene knockouts or knockdowns are likely much more complex than the immediate effects of the actual autophagy inhibition. To overcome this concern, inducible knockout systems might be useful.171,315 One additional caveat to knockdown experiments is that pathogen-associated molecular pattern (PAMP) recognition pathways can be triggered by double-stranded RNAs (dsRNA), like siRNA probes, or the viral vector systems that deliver shRNA.755 Some of these, like TLR-mediated RNA recognition,756 can influence autophagy by either masking any inhibitory effect or compromising autophagy independent of the knockdown probe. Therefore, nontargeting (scrambled) siRNA or shRNA controls should be used with the respective transfection or transduction methods in the experiments that employ ATG knockdown. Another strategy to specifically interfere with autophagy is to use dominant negative inhibitors. Delivery of these agents by transient transfection, adenovirus, or TAT-mediated protein transduction offers the possibility of their use in cell culture or in vivo.753 However, since autophagy is an essential metabolic process for many cell types and tissues, loss of viability due to autophagy inhibition always has to be a concern when analyzing cell death-unrelated questions. In this respect it is noteworthy that some cell-types of the immune system such as dendritic cells260 seem to tolerate loss of autophagy fairly well, whereas others such as T and B cells are compromised in their development and function after autophagy inhibition.757,758
In addition to pharmacological inhibition, RNA silencing, gene knockout and dominant negative RAB and ATG protein expression, pathogen-derived autophagy inhibitors can also be considered to manipulate autophagy. Along these lines ICP34.5, viral BCL2 homologs and viral CFLAR/FLIP of herpesviruses block autophagosome formation,425,580,759 whereas M2 of influenza virus and HIV Nef block autophagosome degradation.278,760 However, as with other tools discussed in this section, transfection or transduction of viral autophagy inhibitors should be used in parallel with other means of autophagy manipulation, because these proteins are used for the regulation of usually more than one cellular pathway by the respective pathogens.
There are fewer compounds that act as inducers of autophagy, but the initial characterization of this process was due in large part to the inducing effects of glucagon, which appears to act through indirect inhibition of MTOR via the activation of STK11/LKB1-AMPK.615,616,761 Currently, the most commonly used inducer of autophagy is rapamycin, an allosteric inhibitor of TORC1, although one caution is that MTOR is a major regulatory protein that is part of the insulin signaling pathway, and it controls processes other than autophagy, so that rapamycin will ultimately affect many metabolic pathways.379,762-764 In particular, the strong effects of MTOR on protein synthesis may be a confounding factor when analyzing the effects of rapamycin. MTOR-independent regulation can be achieved through lithium, sodium valproate and carbamazepine, compounds that lower the myo-inositol-1,4,5-triphosphate levels.765 In vivo treatment of embryos with cadmium results in an increase in autophagy, probably to counter the stress, allowing cell survival through the elimination/recycling of damaged structures.632 Autophagy may also be regulated by the release of calcium from the endoplasmic reticulum under stress conditions;212,726,766 however, additional calcium signals from other stores such as the mitochondria and lysosomes could also play an important role in autophagy induction. The activation of the lysosomal two pore channel (TPC), by nicotinic acid adenine dinucleotide phosphate (NAADP) induces autophagy, which can selectively be inhibited by the TPC blocker NED-19, or by pre-incubation with BAPTA, showing that lysosomal calcium also modulates autophagy.244
Another way to induce autophagy, both in cultured cells and in vivo, is through transcriptional control. For example, this can be achieved either through overexpression or post-translational activation of the gene encoding TFEB (see Transcriptional and translational regulation), a transcriptional regulator of the biogenesis of both lysosomes and autophagosomes.463,464 Similarly, adenoviral-mediated expression of the transcription factor CEBPB/C/EBPβ induces autophagy in hepatocytes.467
Relatively little is known about direct regulation via the ATG proteins, but there is some indication that tamoxifen acts to induce autophagy by increasing the expression of BECN1 in MCF7 cells.767 However, in U87MG cells treated with tamoxifen BECN1 does not appear to be upregulated, whereas the levels of LC3-II and SQSTM1 are increased, while LAMP2B is downregulated and CTSD and CTSL1 activities are almost completely blocked (Choi KS, personal communication). Thus, the effect of tamoxifen may differ depending on the cell type. Other data suggest that tamoxifen acts by blocking cholesterol biosynthesis, and that the sterol balance may determine whether autophagy acts in a protective vs. cytotoxic manner.768,769 Finally, screens have identified small molecules that induce autophagy independently of rapamycin, and allow the removal of misfolded or aggregate-prone proteins,770,771 suggesting that they may prove useful in therapeutic applications. However, caution should be taken because of the crosstalk between autophagy and the proteasomal system. For example, trehalose, an MTOR-independent autophagy inducer,253 can compromise proteasomal activity in cultured primary neurons.739
Conclusion: Rapamycin is much less effective at inhibiting MTOR and inducing autophagy than catalytic inhibitors, and the latter should therefore be considered for use instead of rapamycin; however, it must be kept in mind that catalytic inhibitors also affect MTORC2. The main concern with pharmacological manipulations is pleiotropic effects of the compound being used. Accordingly, genetic confirmation is preferred whenever possible.
3. Basal autophagy
Basal levels of LC3-II or GFP-LC3 puncta may change according to the time after addition of fresh medium to cells, and this can lead to misinterpretations of what basal autophagy means. This is particularly important when comparing the levels of basal autophagy between different cell populations (such as knockout vs. wild-type clones). If cells are very sensitive to nutrient supply and display a high variability of basal autophagy, the best experimental condition is to monitor the levels of basal autophagy at different times after the addition of fresh medium. One example is the chicken lymphoma DT40 cells (see Chicken B-lymphoid DT40 cells below) and their knockout variant for all three inositol 1,4,5-trisphosphate receptor isoforms.241,772,773 In these cells, no differences in basal levels of LC3-II can be observed up to 4 h after addition of fresh medium, but differences can be observed after longer times (Vicencio JM, Szabadkai G, personal communication). This concept should also be applied to experiments in which the effect of a drug upon autophagy is the subject of study. If the drugs are added after a time in which basal autophagy is already high, then the effects of the drug can be masked by the cell’s basal autophagy, and wrong conclusions may be drawn. To avoid this, fresh medium should be added first in order to reduce and equilibrate basal autophagy in cells under all conditions, and then the drugs can be added. The basal autophagy levels of the cell under study must be identified beforehand in order to know the time needed to reduce basal autophagy.
A similar caution must be exercised with regard to cell culture density and hypoxia. When cells are grown in normoxic conditions at high cell density, HIF1A/HIF-1α is stabilized at levels similar to that obtained with low-density cultures under hypoxic conditions.774 This results in the induction of BNIP3 and BNIP3L/NIX and “hypoxia”-induced autophagy, even though the conditions are theoretically normoxic.775 Therefore, researchers need to be careful about cell density in order to avoid accidental induction of autophagy.
It should be realized that also in yeast species, medium changes can trigger a higher “basal” level of autophagy in the cells. In the methylotrophic yeast species Pichia pastoris and Hansenula polymorpha a shift of cells grown in batch from glucose to methanol results in stimulation of autophagy.776,777 A shift to a new medium can be considered a stress situation. Thus, it appears to be essential to cultivate the yeast cells for a number of hours to stabilize the level of basal autophagy, before performing experiments intended to study levels of (selective) autophagy (e.g., pexophagy). Finally, plant root tips cultured in nutrient-sufficient medium display constitutive autophagic flux (i.e., a basal level), which is enhanced in nutrient-deprived medium.696,778,779
Conclusion: The levels of basal autophagy can vary substantially and can mask the effects of the experimental parameters being tested. Changes in media and growth conditions need to be examined empirically to determine affects on basal autophagy and the appropriate times for subsequent manipulations.
4. Experimental systems
Throughout these guidelines we have noted that it is not possible to state explicit rules that can be applied to all experimental systems. For example, some techniques may not work in particular cell types or organisms. In each case, the efficacy of autophagy promotors, inhibitors and measurement technique must be empirically determined, which is why it is important to include appropriate controls. Differences may also be seen between in vivo or perfused organ studies and cell culture analyses. For example, insulin has no effect on proteolysis in suspended rat hepatocytes, in contrast to the result with perfused rat liver. The insulin effect reappears, however, when isolated hepatocytes are incubated in stationary dishes780,781 or are allowed to settle down on the matrix (Häussinger D, personal communication). The reason for this might be that autophagy regulation by insulin and some amino acids requires volume sensing via integrin-matrix interactions and also intact microtubules.782-784 Along these lines, the use of whole embryos makes it possible to investigate autophagy in multipotent cells, which interact among themselves in their natural environment, bypassing the disadvantages of isolated cells that are deprived of their normal network of interactions.632 In general, it is important to keep in mind that results from one particular system may not be generally applicable to others.
Conclusion: Although autophagy is conserved from yeast to human, there may be tremendous differences in the specific details among systems. Thus, results based on one system should not be assumed to be applicable to another.
5. Nomenclature
In order to minimize confusion regarding nomenclature, we make the following notes: In general, we follow the conventions established by the nomenclature committees for each model organism whenever appropriate guidelines are available, and briefly summarize the information here using “ATG1” as an example for yeast and mammals. The standard nomenclature of autophagy-related genes, mutants and proteins for yeast is ATG1, atg1 (or atg1Δ in the case of deletions) and Atg1, respectively, according to the guidelines adopted by the Saccharomyces Genome Database (http://www.yeastgenome.org/gene_guidelines.shtml). For mammals we follow the recommendations of the International Committee on Standardized Genetic Nomenclature for Mice (www.informatics.jax.org/mgihome/nomen/), which dictates the gene and protein designations Atg1 and ATG1 (for all rodents), respectively, and the guidelines for human genes established by the HUGO Nomenclature Committee (http://www.genenames.org/guidelines.html), which states that human gene symbols are in the form ULK1, and recommends that proteins use the same designation without italics, as with ULK1.
C. Methods and Challenges of Specialized Model Systems
There are now a large number of model systems being used to study autophagy. These guidelines cannot cover every detail, and this article is not meant to provide detailed protocols. Nonetheless, we think it is useful to briefly discuss what techniques can be used in these systems, and to highlight some of the specific concerns and/or challenges. We also refer readers to the three volumes of Methods in Enzymology that provide additional information for “nonstandard” model systems.38-40
1. C. elegans
C. elegans has a single ortholog of most yeast Atg proteins; however, two nematode homologs exist for Atg4 and Atg8.785 Multiple studies have established C. elegans as a useful multicellular genetic model to delineate the autophagy pathway and associated functions (see for example refs. 189, 462, 592, 593 and 786). The LGG-1/Atg8/LC3 reporter is the most commonly used tool to detect autophagy in C. elegans. Similar to Atg8, which is incorporated into the double membrane of autophagic vesicles during autophagy,113,187,442 the C. elegans LGG-1 localizes into cytoplasmic puncta under conditions known to induce autophagy. Fluorescent reporter fusions of LGG-1/Atg8 with GFP, DsRED or mCherry have been used to monitor autophagosome formation in vivo, in the nematode. These reporters can be expressed either in specific cells and tissues or throughout the animal.189,593,787,788 LGG-2 is the second LC3 homolog and is also a convenient marker for autophagy either fused to GFP,789 especially when expressed from an integrated transgene to prevent its germline silencing,592 or using specific antibodies.592 The exact function of LGG-1 vs. LGG-2 remains to be addressed.
For observing autophagy by GFP-LC3 fluorescence in C. elegans, it is best to use integrated versions of GFP-LC3592,593,790 (GFP::LGG-1 and GFP::LGG-2; Fig. 24) rather than extrachromosomal transgenic strains189,789 because the latter show variable expression among different animals or mosaic expression (Kang C, personal communication; Galy V, personal communication). It is also possible to carry out indirect immunofluorescence microscopy using antibodies against endogenous LGG-1,462,593 or LGG-2.592 In addition, with the integrated version, or with antibodies directed against endogenous LGG-1, it is possible to perform a western blot analysis for lipidation, at least in embryos,790 and in the whole animal,593 respectively. Finally, we point out the increasing availability of instruments that are capable of “super-resolution” fluorescence microscopy, which will further enhance the value and possibilities afforded by this technology.791,792

Figure 24. GFP::LGG-1 and GFP::LGG-2 are autophagy markers in C. elegans. (A–F) Animals were generated that carry an integrated transgene expressing a GFP-tagged version of lgg-1, the C. elegans ortholog of mammalian MAP1LC3. Representative green fluorescence images in the pharyngeal muscles of (A) control RNAi animals without starvation, (B) control RNAi animals after 9 d of starvation, (C) atg-7 RNAi animals after 9 d of starvation, (D) starvation-hypersensitive gpb-2 mutants without leucine after 3 d of starvation, and (E) gpb-2 mutants with leucine after 3 d of starvation. The arrows show representative GFP::LGG-1-positive punctate areas that label pre-autophagosomal and autophagosomal structures. (F) The relative levels of PE-conjugated and unconjugated GFP::LGG-1 were determined by western blotting. These figures were modified from data previously published in Kang C, You YJ, Avery L, Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev 2007, 21:2161–71, Copyright © 2007, Genes & Development by Cold Spring Harbor Laboratory Press and Kang C, Avery L, Systemic regulation of starvation response in Caenorhabditis elegans. Genes Dev 2009, 23:12–7, Copyright © 2011, Genes & Development by Cold Spring Harbor Laboratory Press, www.genesdev.org. (G and H) GFP:LGG-2 serves as a marker for autophagosomes in early C. elegans embryos. (G) GFP::LGG-2 expressed in the germline from an integrated transgene reveals the formation of autophagosomes (green) around sperm-inherited membranous organelles (red). DNA of the two pronuclei is stained (blue). (H) Later during development, GFP::LGG-2-positive structures are present in all cells of the embryo. Scale bar, 10 µm. Images provided by V. Galy.
2. Chicken B-lymphoid DT40 cells and retina
The chicken B-lymphoid DT40 cell line represents a suitable tool for the analysis of autophagic processes in a nonmammalian vertebrate system. In DT40 cells, foreign DNA integrates with a very high frequency by homologous recombination compared with random integration. This makes the cell line a very valuable tool for the generation of cellular gene knockouts. Generally, the complete knockout of autophagy-regulatory proteins is preferable compared with RNAi-mediated knockdown, since in some cases these proteins function normally when expressed at reduced levels.171 Different Atg-deficient DT40 cell lines already exist, including atg13−/−, ulk1−/−, ulk2−/−, ulk1/2−/−,793 becn1−/− and rb1cc1/fip200−/− (Stork B, personal communication). Many additional non-autophagy-related gene knockout DT40 cell lines have been generated and are commercially available.794
DT40 cells are highly proliferative (the generation time is approximately 10 h) and knockout cells can be easily reconstituted with cDNAs by retroviral gene transfer for the mutational analysis of signaling pathways. DT40 cells mount an autophagic response upon starvation in EBSS,793 and autophagy can be analyzed by a variety of assays in this cell line. Steady-state methods that can be used include TEM, LC3 western blotting and fluorescence microscopy; flux measurements include monitoring LC3-II turnover and tandem mRFP/mCherry-GFP-LC3 fluorescence microscopy. Using atg13−/− and ulk1/2−/− DT40 cells, it was shown that ATG13 and its binding capacity for RB1CC1 are mandatory for both basal and starvation-induced autophagy in this cell line, whereas ULK1/2 and in vitro-mapped ULK1-dependent phosphorylation sites of ATG13 appear to be dispensable for these processes.793
Another useful system is chick retina, which can be used for monitoring autophagy at different stages of development. For example, lipidation of LC3 is observed during starvation, and can be blocked with a short-term incubation with 3-MA.305,306 LEP-100 antibody is commercially available for the detection of this lysosomal protein.
Cautionary notes: Since the DT40 cell line derives from a chicken bursal lymphoma, not all ATG proteins and autophagy-regulatory proteins are detected by the commercially available antibodies produced against their mammalian orthologs. The chicken genome is almost completely assembled, which facilitates the design of targeting constructs. However, in the May 2006 chicken (Gallus gallus) v2.1 assembly, 5% of the sequence has not been anchored to specific chromosomes, and this might also include autophagy regulatory genes. It is possible that there is some divergence within the signaling pathways between mammalian and nonmammalian model systems. One example might be the role of ULK1/2 in starvation-induced autophagy described above. Additionally, neither rapamycin nor torin1 seem to be potent inducers of autophagy in DT40 cells, although MTOR activity is completely repressed as detected by phosphorylated RPS6KB western blotting.793 Finally, DT40 cells represent a transformed cell line, being derived from an avian leukosis virus (ALV)-induced bursal lymphoma. Thus, DT40 cells release ALV into the medium, and the 3′-long-terminal repeat has integrated upstream of the c-myc gene, leading to an increased c-myc expression.795 Both circumstances might influence basal and starvation-induced autophagy.
3. Chlamydomonas
It is possible to detect Atg8 modification as well as an increase in the amount of the protein by western blotting in response to autophagy activation.207 Detection of Atg8 by immunofluorescence microscopy assays is also a reliable method to study autophagy, although it is recommended that this be combined with western blot analysis.
4. Drosophila
Drosophila provides an excellent system for in vivo analysis of autophagy. In this case, the problem of animal-to-animal variability can be circumvented by the use of clonal mutant cell analysis, a major advantage of this model system. In this scenario, somatic clones of cells are induced that either overexpress the gene of interest, or silence the gene through expression of a transgenic RNA interference construct, or homozygous mutant cells are generated. These gain- or loss-of-function clones are surrounded by wild-type cells, which serve as an internal control for autophagy induction. In such an analysis, autophagy in these genetically distinct cells is always compared with neighboring cells of the same tissue, thus eliminating most of the variability and also ruling out potential non-cell-autonomous effects that may arise in mutant animals. Along these lines, clonal analysis should be an integral part of in vivo Drosophila studies when possible.
LC3-II western blotting using antibodies against mammalian proteins does not work in Drosophila (Baehrecke E, Denton D, Kumar S, Neufeld T, unpublished results). Western blotting has been used successfully in Drosophila by monitoring flies expressing human GFP-LC3101,197 or using an antibody directed against the endogenous Atg8 protein.454,796 In addition, cultured Drosophila (S2) cells can be stably transfected with GFP fused to Drosophila Atg8a, which generates easily resolvable GFP-Atg8a and GFP-Atg8a–PE forms that respond to autophagic stimuli (Wilkinson S, personal communication). Similarly, cultured Drosophila cells (l(2)mbn or S2) stably transfected with EGFP-humanLC3B respond to autophagy stimuli (nutrient deprivation) and inhibitors (3-MA, bafilomycin A1) as expected, and can be used to quantify GFP-LC3 puncta, which works best using fixed cells with the aid of an anti-GFP antibody.797a However, in the Drosophila eye, overexpression of GFP-Atg8 results in a significant increase in Atg8–PE by western blot, and this occurs even in control flies in which punctate GFP-Atg8 is not detected by immunofluorescence (Fanto M, unpublished results), and in transfected Drosophila Kc167 cells, uninducible but persistent GFP-Atg8 puncta are detected (Kiger A, unpublished results). In contrast, expression of GFP-LC3 under the control of the rh1 promoter in wild-type flies did not result in the formation of LC3-II detectable by western blot, nor the formation of punctate staining; however, increased GFP-LC3 puncta by immunofluorescence or LC3-II by western blot were observed upon activation of autophagy.797b Autophagy can also be monitored with mCherry-Atg18, which is displayed in punctate patterns that are very similar to mCherry-Atg8a (Juhász G, personal communication). Tandem fluorescence reporters have been established in Drosophila in vivo, where GFP-mCherry-Atg8a is expressed in the nurse cells of the developing egg chamber.662 A Drosophila transgenic line (Ref(2)P-GFP) and a specific antibody against Ref(2)P, the Drosophila SQSTM1 homolog, are available to follow SQSTM1 expression and localization.312,368
5. Filamentous fungi
As in yeast, autophagy is involved in nutrient recycling during starvation.193,194,798-801 In addition, macroautophagy seems to be involved in many normal developmental processes such as sexual and asexual reproduction, where there is a need for reallocation of nutrients from one part of the mycelium to another to supply the developing spores and spore-bearing structures.194,798,799,801,802 Similarly, autophagy also affects conidial germination under nitrogen-limiting conditions.194 In Podospora anserina, autophagy has been studied in relation to incompatibility reactions between mating strains where it seems to play a prosurvival role.192,802 Of special interest to many researchers of autophagy in filamentous fungi has been the possible involvement of autophagy in plant pathogen infection and growth inside the host.193,798,799,803-805 Autophagy also appears to be necessary for the development of aerial hyphae,194,798,802,804 and for appresorium function in Magnaporthe oryzae and Colletotrichum orbiculare.193,803,804 Some of these effects could be caused by the absence of autophagic processing of storage lipids (lipophagy) to generate glycerol for increasing turgor.798,804,805
Methods for functional analysis of autophagy have been covered in a review article.806 Most studies on autophagy in filamentous fungi have involved deleting some of the key genes necessary for autophagy, followed by an investigation of what effects this has on the biology of the fungus. Most commonly, ATG1 and/or ATG8 has been deleted.193,798,799,801,802,804 To confirm that the deletion(s) affects autophagy, the formation of autophagic bodies in the wild type and the mutant can be compared. In filamentous fungi the presence of autophagic bodies can be detected using MDC staining,193,798 TEM193,799 or fluorescence microscopy to monitor Atg8 tagged with a fluorescent protein.194,801,802 This type of analysis is most effective after increasing the number of autophagic bodies by starvation, in combination with decreasing the degradation of the autophagic bodies through the use of the protease inhibitor PMSF,193,799,801,802 or alternatively by adding the autophagy-inducing drug rapamycin.194,798 In filamentous fungi it might also be possible to detect the accumulation of autophagic bodies in the vacuoles using differential interference contrast (DIC) microscopy.801,802 Additional information regarding the timing of autophagy induction can be gained by monitoring transcript accumulation of ATG1 and/or ATG8 using qRT-PCR.799
6. Honeybee
The reproductive system of bees, or insects with meroistic polytrophic ovaries, with regard to the ovary developmental cycle can be a very useful tool to analyze and monitor physiological autophagy. Both queen and worker ovaries of Africanized A. mellifera display time-regulated features of cell death that are, however, linked to external stimuli.807 Features of apoptosis and autophagy are frequently associated with the degeneration process in bee organs, but only more recently has the role of autophagy been highlighted in degenerating bee tissues. TEM is the primary method currently being used to monitor autophagy by following the formation of autophagosomes and autolysosomes. This can be combined with cytochemical and immunohistochemical detection of acid phosphatase as a marker for autolysosomes.808,809 Acidotropic dyes can also be used to follow autophagy in bee organs, as long as the cautions noted in this article are followed. The honeybee genome has been sequenced, and differential gene expression has been used to monitor Atg18 in bees parasitized by Varroa destructor.810
7. Human
Considering that much of the research conducted today is directed at understanding the functioning of the human body, in both normal and disease states, it is pertinent to include humans and primary human tissues and cells as important models for the investigation of autophagy. Although clinical studies are not readily amenable to these types of analyses, it should be kept in mind that the TORC1 inhibitor rapamycin is available as a clinically approved drug (e.g., sirolimus). Furthermore, fresh biopsies of some human tissues are possible to obtain. Blood, in particular, as well as samples of adipose and muscle tissues, can be obtained from needle biopsies or from elective surgery. For example, in a large study, adipocytes were isolated from pieces of adipose tissue (obtained during surgery) and examined for insulin signaling and autophagy. It was demonstrated that autophagy was strongly upregulated (based on LC3 flux, EM, and lipofuscin degradation) in adipocytes obtained from obese patients with type 2 diabetes compared with nondiabetic subjects.209
The major caveat of the work concerning autophagy on human tissue is the problem of postmortem times and fixation. Postmortem times are typically longer in autopsy material than when surgical biopsies are obtained. For tumors, careful sampling to avoid necrosis, hemorrhagic areas and non-neoplastic tissue is required. The problem of fixation is that it can diminish the antibody binding capability; in addition, especially in autopsies, material is not obtained immediately after death.811,812 The possibilities of postmortem autolysis and fixation artifacts must always be taken into consideration when interpreting changes attributed to autophagy.813 Analyses of these types of samples require not only special antigen retrieval techniques, but also histopathological experience to interpret autophagy studies by immunohistochemistry, immunofluorescence or TEM.
The situation is even worse with TEM, where postmortem delays can cause vacuolization. Researchers experienced in the analysis of TEM images corresponding to autophagy should be able to identify these potential artifacts because autophagic vacuoles should contain cytoplasm. While brain biopsies may be usable for high quality TEM (Figs. 25 and 26), this depends upon proper handling at the intraoperative consultation stage, and such biopsies are performed infrequently except for brain tumor diagnostic studies. An analysis that examined liver and skeletal muscle from critically ill patients utilized tissue biopsies that were taken within 30 ± 20 min after death and were flash-frozen in liquid nitrogen followed by storage at -80°C.815 Samples could subsequently be used for EM and western blot analysis.
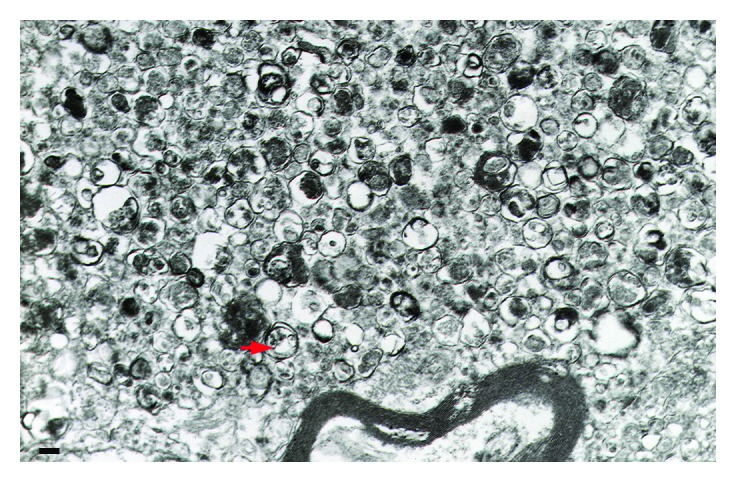
Figure 25. A large dystrophic neurite from a brain biopsy of a patient with Gerstmann-Sträussler-Scheinker (GSS) disease not unlike those reported for Alzheimer disease.814 This structure is filled with innumerable autophagic vacuoles, some of which are covered by a double membrane. Electron dense lysosomal-like structures are also visible. The red arrow points to a double-membrane autophagic compartment. Scale bar, 200 nm. Image provided by P. Liberski.

Figure 26. A high-power electron micrograph from a brain biopsy showing autophagic vacuoles in a case of ganglioglioma. Scale bar, 200 nm. Image provided by P. Liberski.
A major limitation of studying patient biopsies is that only static measurements can be performed. This limitation does not apply, however, for dynamic experiments on tissue biopsies or cells derived from biopsies, as described above.209 Multiple measurements over time, especially when deep (vital) organs are involved, are impossible and ethically not justifiable. Hence, quantitative flux measurements are virtually impossible in patients. To overcome these problems to the extent possible and to gain a more robust picture of the autophagic status, observational studies need to include two different aspects. First, a static marker for phagophore or autophagosome formation needs to be measured. This can be done by assessing ultrastructural changes with TEM and/or on the molecular level by measuring LC3-II protein levels. Second, accumulation of autophagy substrates, such as SQSTM1 and (poly)ubiquitinated proteins can provide information on the overall efficacy of the pathway and can be a surrogate marker of the consequences of altered autophagic flux, especially when autophagy is insufficient, although these changes can also be affected by the ubiquitin-proteasome system as mentioned above. In addition, and even more so when problems with specific pathways are suspected (e.g., mitophagy), specific substrates of these pathways should be determined. Again, none of these measurements on its own provides enough information on (the efficacy of) autophagy, because other processes may confound every single parameter. However, the combination of multiple analyses should be informative. Although still in its infancy with regard to autophagy, it is worth pointing out that mathematical modeling has the power to bridge whole body in vivo data with in vitro data from tissues and cells. The usefulness of so-called hierarchical or multilevel modeling has thus been demonstrated when examining the relevance of insulin signaling to glucose uptake in primary human adipocytes compared with whole-body glucose homeostasis.816
The amino acid exchange rate, which has been suggested as a minimal invasive marker for measuring autophagic flux in vivo should be used with special caution, as its utility is debatable in disease models (see Analyses in vivo).
Finally, a stepwise process can be proposed for linking changes in the autophagic pathway to changes in disease outcome. First, in an observational study, the changes in the autophagic pathway (see above) should be quantified and linked to changes in disease outcome. To prove causality, a subsequent autophagy-modifying intervention should be tested in a randomized study. Before an intervention study is performed in human patients, the phenotype of (in)active autophagy contributing to poor outcome should be established in a validated animal model of the disease. For the validation of the hypothesis in an animal model, a similar two-step process is suggested, with the assessment of the phenotype in a first stage, followed by a proof-of-concept intervention study (see Large animals).
8. Hydra
Hydra is a freshwater cnidarian animal that provides a unique model system to test autophagy either in the context of nutrient deprivation, as these animals easily survive several weeks of starvation,817,818 or in the context of regeneration, because in the absence of protease inhibitors, bisection of the animals leads to an uncontrolled wave of autophagy; in the latter case, an excess of autophagy in the regenerating tip immediately after amputation is deleterious.819-821 Most components of the autophagy and MTOR pathways are evolutionarily conserved in Hydra.818 For steady-state measurements, autophagy can be monitored by western blot for ATG8/LC3, by immunofluorescence (using antibodies to ATG8/LC3, LBPA or RSK), or with dyes such as MitoFluor Red 589 and LysoTracker Red. Flux measurements can be made by following ATG8/LC3 turnover using lysosomal protease inhibitors (leupeptin and pepstatin A), or in vivo labeling using LysoTracker Red. It is also possible to monitor MTOR activity with phosphospecific antibodies to RPS6KB kinase and EIF4EBP1, or to examine gene expression by semiquantitative RT-PCR, using primers that are designed for Hydra. Autophagy can be induced by RNAi-mediated knockdown of Kazal1,819,820 or with rapamycin treatment, and can be inhibited with wortmannin or bafilomycin A1.817,818
9. Large animals
Assessment of autophagy (and, in particular, autophagic flux) in clinically relevant large animal models is critical in establishing its (patho)physiological role in multiple disease states. For example, evidence obtained in swine suggests that upregulation of autophagy may protect the heart against damage caused by acute myocardial infarction or “heart attack.”822 Autophagy also plays an important role in the development and remodeling of the bovine mammary gland. In vitro studies with the use of a three-dimensional culture model of bovine mammary epithelial cells (MECs) have shown that this process is involved in the formation of fully developed alveoli-like structures.823 Earlier studies show that intensified autophagy is observed in bovine MECs at the end of lactation and during the dry period, when there is a decrease in the levels of lactogenic hormones, increased expression of auto/paracrine apoptogenic peptides, increased influence of sex steroids and enhanced competition between the intensively developing fetus and the mother organism for nutritional and bioactive compounds.824,825 These studies were based on some of the methods described elsewhere in these guidelines, including GFP-Atg8/LC3 fluorescence microscopy, TEM, and western blotting of LC3 and BECN1. Creation of a specific GFP-LC3 construct by insertion of cDNA encoding bovine LC3 into the pEGFP-C1 vector makes it possible to observe induction of autophagy in bovine MECs in a more specific manner than can be achieved by immunofluoresce techniques, in which the antibodies do not show specific reactivity to bovine cells and tissues.823,825 However, it is important to remember that definitive confirmation of cause-and-effect is challenging for studies on large animals, given the lack or poor availability of specific antibodies and other molecular tools, the frequent inability to utilize genetic approaches, and the often prohibitive costs of administering pharmacological inhibitors in these translational preparations.
In contrast with cell culture experiments, precise monitoring of autophagic flux is practically impossible in large animals. Theoretically, repetitive analyses of small tissue biopsies should be performed to study ultrastructural and molecular alterations over time in the presence or absence of an autophagy inhibitor (e.g., chloroquine). However, several practical problems impede applicability of this approach. First, repetitive sampling of small needle biopsies in the same animal (a major challenge by itself) could be assumed to induce artifacts following repetitive tissue destruction, especially when deep (vital) organs are involved. In addition, chemical inhibitors of autophagy have considerable side effects and toxicity, hampering their usage. Also, the general physical condition of an animal may confound results obtained with administration of a certain compound, for instance altered uptake of the compound when perfusion is worse.
Therefore, in contrast to cells, where it is more practical to accurately document autophagic flux, we suggest the use of a stepwise approach in animal models to provide a proof-of-concept with an initial evaluation of sequellae of (in)active autophagy and the relation to the outcome of interest.
First, prior to an intervention, the static ultrastructural and molecular changes in the autophagic pathway should be documented and linked to the outcome of interest (organ function, muscle mass or strength, survival, etc.). These changes can be evaluated by light microscopy, EM and/or by molecular markers such as LC3-II. In addition, the cellular content of specific substrates normally cleared by autophagy should be quantified, as such measurement, despite the static nature, could provide a clue about the results of altered autophagic flux in vivo. These autophagic substrates can include SQSTM1 and (poly)ubiquitinated substrates or aggregates, but also specific substrates such as damaged mitochondria. As noted above, measurement of these autophagic substrates is mainly informative when autophagic flux is prohibited/insufficient, and, individually, all have specific limitations for interpretation. As mentioned several times in these guidelines, no single measurement provides enough information on its own to reliably assess autophagy, and all measurements should be interpreted in view of the whole picture. In every case, both static measurements reflecting the number of autophagosomes (ultrastructural and/or molecular) and measurements of autophagic substrates as surrogate markers of autophagic flux need to be combined. Depending on the study hypothesis, essential molecular markers can further be studied to pinpoint at which stage of the process autophagy may be disrupted.
After having identified a potential role of autophagy in mediating an outcome in a clinically relevant animal model, an autophagy-modifying intervention should be tested. For this purpose, an adequately designed, randomized controlled study of sufficient size on the effect of a certain intervention on the phenotype and outcome can be performed in a large animal model. Alternatively, the effect of a genetic intervention can be studied in a small animal model with clinical relevance to the studied disease.
As mentioned above, exact assessment of autophagic flux requires multiple time points, which cannot be done in the same animal. Alternatively, different animals can be studied for different periods of time. Due to the high variability between animals, however, it is important to include a sufficiently high number of animals per time point. This thus limits feasibility and the number of time points that can be investigated. The right approach to studying autophagy in large animals likely differs depending on the question that is being addressed. Several shortcomings regarding the methodology, inherent to working with large animals, can be overcome by an adequate study design. As for every study question, the use of an appropriate control group with a sufficient number of animals is crucial in this regard.
10. Lepidoptera
Some of the earliest work in the autophagy field was performed in the area of insect metamorphosis.667 Microscopy and biochemical research revealed autophagy during the metamorphosis of American silkmoths and the tobacco hornworm, Manduca sexta, and included studies of the intersegmental muscles, but they did not include molecular analysis of autophagy. Overall, these tissues cannot be easily maintained in culture, and antibodies against mammalian proteins do not often work. Accordingly, these studies were confined to biochemical measurements and electron micrographs. During metamorphosis, the bulk of the larval tissue is removed by autophagy and other forms of proteolysis.826 Bombyx mori is now used as a representative model among Lepidoptera, for studying not only the regulation of autophagy in a developmental setting, but also the relations between autophagy and apoptosis. The advantages of this model are the large amount of information gathered on its developmental biology, physiology and endocrinology, the availability of numerous genetic and molecular biology tools, and a completely sequenced genome.827 The basic studies of B. mori autophagy have been performed in four main larval systems: the silk gland, the fat body, the midgut and the ovary. The methods used for these studies are comparatively similar, starting from EM, which is the most widely used method to follow the changes of various autophagic structures and other features of the cytosol and organelles that are degraded during autophagy.452,828-831 Immuno-TEM also can be used, when specific antibodies for autophagic markers are available. Acidotropic dyes such as MDC and LysoTracker Red staining have been used as markers for autophagy in silk moth egg chambers combined always with additional assays.828,829 Acidic phosphatase also can be used as a marker for autolysosomal participation in these tissues.452,830,832 Systematic cloning and analysis revealed that homologs of most of the Atg genes identified in other insect species such as Drosophila are present in B. mori, and 11 Atg genes have now been identified in the silkworm genome, as well as other genes involved in the MTOR signal transduction pathway.833,834 Variations in the expression of several of these genes have been monitored not only in silkworm larval organs, where autophagy is associated with development,452,833-835 but also in the fat body of larvae undergoing starvation.833
In the IPLB-LdFB cell line, derived from the fat body of the caterpillar of the gypsy moth Lymantria dispar, indirect immunofluorescence experiments have demonstrated an increased number of Atg8-positive dots in cells with increased autophagic activity; however, western blotting did not reveal the conversion of Atg8 into Atg8–PE. Instead, a single band with an approximate molecular mass of 42 kDa was observed that was independent of the percentage of cells displaying punctate Atg8 (Malagoli D, unpublished results). In contrast, with B. mori midgut, the use of an antibody specific for BmAtg8 makes it possible to monitor BmAtg8 processing to BmAtg8–PE by western blotting.452 Thus, the utility of monitoring Atg8 in insects may depend on the particular organism and antibody.
11. Neotropical teleosts
In tropical environments, fish have developed different reproductive strategies, and many species have the potential for use as biological model in cell and molecular biology, especially for studying the mechanisms that regulate gametogenesis and embryo development. In these fish, the ovary is a suitable experimental model system for studying autophagy and its interplay with cell death programs due to the presence of postovulatory follicles (POFs) and atretic follicles (AFs), which follow different routes during ovarian remodeling after spawning.836 In the fish reproductive biology, POFs are excellent morphological indicators of spawning, whereas AFs are relevant biomarkers of environmental stress. In addition, many freshwater teleosts of commercial value do not spawn spontaneously in captivity, providing a suitable model for studying the mechanisms of follicular atresia under controlled conditions.837 When these species are subjected to induced spawning, the final oocyte maturation (resumption of meiosis) occurs, and POFs are formed and quickly reabsorbed in ovaries after spawning.838 Assessment of autophagy in fish has been primarily made using TEM at different times of ovarian regression.839 Due to the difficulty of obtaining antibodies specific for each fish species, immunodetection of ATG-proteins (mainly LC3 and BECN1) by immunohistochemistry associated with analyses by western blotting can be performed using antibodies that are commercially available for other vertebrates. Such studies suggest dual roles for autophagy in follicular cells;836 however, evaluation of the autophagic flux in different conditions is critical for establishing its physiological role during follicular regression and ovarian remodeling after spawning. Given the ease of obtaining samples and monitoring them during development, embryos of these fish are also suitable models for studying autophagy that is activated in response to different environmental stressors, particularly in studies in vivo.
12. Odontoblasts
Odontoblasts are long-lived dentin-forming postmitotic cells, which evolved from neural crest cells early during vertebrate evolution. These cells are aligned at the periphery of the dental pulp and are maintained during the entire healthy life of a tooth. As opposed to other permanent postmitotic cells such as cardiac myocytes or central nervous system neurons, odontoblasts are significantly less protected from environmental insult, such as dental caries and trauma. Mature odontoblasts develop a well-characterized autophagic-lysosomal system, including a conspicuous autophagic vacuole that ensures turnover and degradation of cell components. Immunocytochemical and TEM studies make it possible to monitor age-related changes in autophagic activity in human odontoblasts.840
13. Planarians
Planarians are one of the favorite model systems in which to study regeneration and stem cell biology, and represent a unique model where it is possible to investigate autophagy in the context of regeneration, stem cells and growth. Currently the method used to detect autophagy is TEM. A detailed protocol adapted to planarians has been described.841,842 However, complementary methods to detect autophagy are also needed, since TEM cannot easily distinguish between activation and blockage of autophagy, which would both be observed as an accumulation of autophagosomes. Detection of autophagy by other methods are being developed (González-Estévez C, personal communication), including immunohistochemistry and western blotting approaches for the planarian homolog of LC3. Several commercial antibodies against human LC3 have been tried for cross-reactivity without success (see the Autophagy Forum for details) and three planarian-specific antibodies have been generated. Some preliminary results show that LysoTracker Red can be a useful technique on whole-mount planarians. Interestingly, most of the components of the autophagy and MTOR signaling machinery are evolutionarily conserved in planarians, and an RNA interference screen is being performed. Whether autophagy genes vary at the mRNA level during starvation and after depletion of MTOR signaling components is also currently being investigated (González-Estévez C, personal communication).
14. Plants
The fluorophore of the red fluorescent protein shows a relatively high stability under acidic pH conditions. Thus, chimeric RFP fusion proteins that are sequestered within autophagosomes and delivered to the plant vacuole can be easily detected by fluorescence microscopy. Furthermore, fusion proteins with some versions of RFP tend to form intracellular aggregates, allowing the development of a visible autophagic assay for plant cells.843 For example, fusion of cytochrome b5 and the original (tetrameric) RFP generate an aggregated cargo protein that displays cytosolic puncta of red fluorescence and, following vacuolar delivery, diffuse staining throughout the vacuolar lumen. This system allows autophagy to be monitored through fluorescence microscopy with minimum damage to intact plant cells. In addition, the size difference between the intact and processed cargo protein allows the quantification of autophagic degradation through the detection of RFP after separation of total protein by gel electrophoresis, similar to the GFP-Atg8/LC3 processing assay described for yeast and mammals.
Arabidopsis cells can be stably transfected with GFP fused to plant ATG8, and the lipidated and nonlipidated forms can be separated by SDS-PAGE.150 Furthermore, the GFP-ATG8 processing assay is particularly robust in Arabidopsis and can be observed not only by western blotting but also by a diffuse fluorescent haze that appears in vacuoles together with punctate GFP fluorescence, which is presumed to represent free soluble GFP and GFP-ATG8–PE still bound to autophagic bodies, respectively.151,174 Thus, as with other systems, autophagosome formation in plants can be monitored through the use of fluorescent protein fusions to ATG8, and by TEM (Fig. 27). A tandem fluorescence reporter system is also available in Arabidopsis.844

Figure 27. Detection of macroautophagy in tobacco BY-2 cells. (A) Induction of autophagosomes in tobacco BY-2 cells expressing YFP-NtAtg8 (shown in green for ease of visualization) under conditions of nitrogen limitation (Induced). Arrowheads indicate autophagosomes that can be seen as a bright green dot. No such structure was found in cells grown in normal culture medium (Control). Bar, 10 µm. N, nucleus; V, vacuole. (B) Ultrastructure of an autophagosome in a tobacco BY-2 cell cultured for 24 h without a nitrogen source. Bar, 200 µm. AP, autophagosome; P, plastid; CW, cell wall. This image was provided by K. Toyooka (RIKEN Plant Science Center).
Furthermore, it is possible to use plant homologs of SQSTM1 and NBR1 (JOKA2 in tobacco845 and NBR1 in Arabidopsis844) as markers for autophagy when constructed as fluorescent chimeras. It is worth noting that in some plant models including Arabidopsis200,402 and somatic embryos of Picea abies (Norway spruce) (Minina A, Bozhkov PV, personal communication), the high turnover rate of Atg8-decorated autophagosomes precludes their detection in untreated cells using fluorescent markers (GFP/RFP-Atg8, MDC or LysoTracker) and necessitates pretreatment with concanamycin A. However, the latter treatment will increase vacuolar pH, which may subsequently interfere with its detection by MDC and LysoTracker.
In some systems, including fungi and plants, the size of the vacuole is sufficiently large such that fusion of the autophagosome results in the release of the inner vesicle into the organelle lumen; the resulting single-membrane vesicle is termed an autophagic body (Fig. 1).200,846 The accumulation of autophagic bodies can be detected by light microscopy in cells that lack vacuolar hydrolase activity (e.g., the pep4Δ yeast mutant) or in the presence of inhibitors that interfere with hydrolase activity (e.g., PMSF or concanamycin A). Using Nomarski optics (differential interference contrast) it is easy to distinguish and quantify yeast vacuoles that lack autophagic bodies from those that have accumulated them, and the same is true for plants.
Other methods described throughout these guidelines can also be used in plants. For example, in tobacco cells cultured in sucrose starvation medium, the net degradation of cellular proteins can be measured by a standard protein assay; this degradation is inhibited by 3-MA and E-64c (an analog of E-64d), and is thus presumed to be due to autophagy.847 In addition, the cytoplasm in tobacco cells that is present in transvacuolar strands that cross the large central vacuole can be seen by TEM to disappear during starvation-induced autophagy.847
Cautionary notes: Although the detection of vacuolar RFP can be applied to both plant cell lines and to intact plants, it is not practical to measure RFP fluorescence in intact plant leaves, due to the very high red fluorescence of chloroplasts. Furthermore, different autophagic induction conditions cause differences in protein synthesis rates; thus, special care should be taken to monitor the efficiency of autophagy by quantifying the intact and processed cargo proteins. With regard to autophagic body accumulation, it is difficult to quantify their number and/or volume, although their presence or absence can be examined by light microscopy or TEM. In addition, the accumulation of autophagic bodies requires the inhibition of vacuolar hydrolase activity. Therefore, to demonstrate turnover, the assay must be performed either in the absence and presence of appropriate inhibitors, or in both a wild-type strain and a strain with a mutation/deletion in a gene encoding a vacuolar hydrolase(s) (see Selective types of autophagy). Otherwise, accumulation of autophagic bodies could instead indicate a defect in the lysis/degradation step of autophagy. Finally, this method is not well suited for systems other than plants or fungi because lysosomes are too small for detection by standard (i.e., nonfluorescence) light microscopy, and fusion with autophagosomes does not generate autophagic bodies (Fig. 1).
15. Protists
An essential role of autophagy during the differentiation of parasitic protists (formerly called protozoa) is clearly emerging. Only a few of the known ATG genes are present in these organisms, which raises the question about the minimal system that is necessary for the normal functioning of autophagy. The reduced complexity of the autophagic machinery in many protists provides a simplified model to investigate the core mechanisms of autophagosome formation necessary for selective proteolysis, and will open a completely new area in autophagy research. Some of the standard techniques used in other systems can be applied to protists including indirect immunofluorescence using antibodies generated against ATG8, the generation of stable lines expressing mCherry- or GFP-fused ATG8 for live microscopy, and immuno-TEM analyses. Extrachromosomal constructs of GFP-ATG8 also work well with lower eukaryotes,201-203 as do other fluorescently-tagged ATG proteins including ATG5 and ATG12.
The unicellular amoeba Dictyostelium discoideum provides another useful system for monitoring autophagy.848 The primary advantage of Dictyostelium is that it has a unique life cycle that involves a transition from a unicellular to a multicellular form. Upon starvation, up to 100,000 single cells aggregate by chemotaxis and form a multicellular structure that undergoes morphogenesis and cell-type differentiation. Development proceeds via the mound stage, the tipped aggregate and a motile slug and culminates with the formation of a fruiting body that is composed of a ball of spores supported by a thin, long stalk made of vacuolized dead cells. Development is dependent on autophagy and, at present, all of the generated mutants in Dictyostelium autophagy genes display developmental phenotypes of varying severity.848,849 D. discoideum is also a versatile model to study infection with human pathogens and the role of autophagy in the infection process. The susceptibility of D. discoideum to microbial infection and its strategies to counteract pathogens are similar to those in higher eukaryotes.850 Along these lines, Dictyostelium utilizes some of the proteins involved in autophagy that are not present in S. cerevisiae including C12orf44/Atg101, Uvrag and Vmp1, in addition to the core Atg proteins. Autophagy can be monitored in Dictyostelium by fluorescence microscopy of GFP-Atg8 or GFP-Atg18, by TEM and by western blot to detect free GFP or the loss of SQSTM1.36,152,848
One cautionary note with regard to the use of GFP-ATG8 in protists is that these organisms display some “nonclassical” variations in their ATG proteins. For example, Leishmania contains many apparent ATG8-like proteins (the number varying per species; e.g., up to 25 in L. major) grouped in four families, but only one labels true autophagosomes even though the others form puncta,202 and ATG12 requires truncation to provide the C-terminal glycine before it functions in the canonical way. Unusual variants in protein structures also exist in other protists, including the malaria parasite Plasmodium falciparum, which expresses ATG8 with a terminal glycine not requiring cleavage to become functional in autophagy.851 Thus, in each case care needs to be applied and the use of the protein to monitor autophagy validated. In addition, due to possible divergence in the upstream signaling kinases, classical inhibitors such as 3-MA, or inducers like rapamycin, which are not as potent for trypanosomes852 or apicomplexan parasites as in mammalian cells or yeast, must be used with caution (Coppens I, personal communication);201 however, RNAi knockdown of TORC1 (e.g., TOR1 or RPTOR) is effective in inducing autophagy.
The scuticociliate Philasterides dicentrarchi has proven to be a good experimental organism for identifying autophagy-inducing drugs or for autophagy initiation by starvation-like conditions, since this process can be easily induced and visualized in this ciliate.853 In scuticociliates, the presence of autophagic vacuoles can be detected by TEM, fluorescence microscopy or confocal laser scanning microscopy by using dyes such as MitoTracker Deep Red FM© and MDC.
Finally, a novel autophagy event has been found in Tetrahymena thermophila, which is a free-living ciliated protist. A remarkable, virtually unique feature of the ciliates is that they maintain spatially differentiated germline and somatic nuclear genomes within a single cell. The germline genome is housed in the micronucleus, while the somatic genome is housed in the macronucleus. These nuclei are produced during sexual reproduction (conjugation), which involves not only meiosis and mitosis of the micronucleus and its products, but also degradation of some of these nuclei as well as the parental old macronucleus. Hence, there should be a mechanism governing the degradation of these nuclei. The inhibition of PtdIns3Ks with wortmannin or LY294002 results in the accumulation of additional nuclei during conjugation.854 During degradation of the parental old macronucleus, the envelope of the nucleus becomes MDC- and LysoTracker Red-stainable without sequestration of the nucleus by a double membrane and with the exposure of certain sugars and phosphatidylserine on the envelope.855 Subsequently, lysosomes fuse only to the old parental macronucleus, but other co-existing nuclei such as developing new macro- and micronuclei are unaffected.855 This evidence suggests that selective autophagy may be involved in the regulation of the degradation of nuclei during conjugation, but its mechanism apparently differs from the classical pathway. Indeed, no ATG genes homologous to those in yeast are characterized in this organism to date.
16. Rainbow trout
Salmonids (e.g., salmon, rainbow trout) experience long periods of fasting often associated with seasonal reductions in water temperature and prey availability or spawning migrations. As such, they represent an interesting model system for studying and monitoring the long-term induction of autophagy. Moreover, the rainbow trout (Oncorhynchus mykiss) displays unusual metabolic features that may allow us to gain a better understanding of the nutritional regulation of this degradative system (i.e., a high dietary protein requirement, an important use of amino acids as energy sources, and an apparent inability to metabolize dietary carbohydrates). It is also probably one of the most deeply studied fish species with a long history of research performed in physiology, nutrition, ecology, genetics, pathology, carcinogenesis and toxicology.856 Its relatively large size compared with model fish such as zebrafish or medaka, makes rainbow trout a particularly well-suited alternative model to carry out biochemical and molecular studies on specific tissues or cells that are impossible to decipher in small fish models. The genomic resources in rainbow trout are now being extensively developed; a high-throughput DNA sequencing program of EST has been initiated associated with numerous transcriptomics studies,857-860 and the full genome sequence is now available.
Most components of the autophagy and associated signaling pathways (AKT1, TOR, AMPK, FOXO) are evolutionarily conserved in rainbow trout;457,861-863 however, not all ATG proteins and autophagy-regulatory proteins are detected by the commercially available antibodies produced against their mammalian orthologs. Nonetheless, the expressed sequence transcript databases facilitate the design of targeting constructs. For steady-state measurement, autophagy can be monitored by western blot or by immunofluorescence using antibodies to ATG8/LC3.863 Flux measurements can be made in a trout cell culture model (e.g., in primary culture of trout myocytes) by following ATG8/LC3 turnover in the absence and presence of bafilomycin A1. It is also possible to monitor the mRNA levels of ATG genes by real-time PCR using primer sequences chosen from trout sequences available in the above-mentioned expressed sequence transcript database. A major challenge in the near future will be to develop for this model the use of RNAi-mediated gene silencing in order to analyze the role of some signaling proteins in the control of autophagy, and also the function of autophagy-related genes in this species.
17. Sea urchin
Sea urchin embryo is an appropriate model system for studying and monitoring autophagy and other defense mechanisms activated during physiological development and in response to stress.632 This experimental model offers the possibility of detecting LC3 through both protein gel blot and immunofluorescence in situ analysis. Furthermore, in vivo staining of autolysosomes with acidotropic dyes can also be performed. Studies on whole embryos make it possible to obtain qualitative and quantitative data for autophagy and also to get information about spatial localization aspects in cells that interact among themselves in their natural environment. Cautionary notes include the standard recommendation that it is always preferable to combine molecular and morphological parameters to support the validity of the data.
18. Ticks
In the hard tick Haemaphysalis longicornis, endogenous autophagy-related proteins (Atg6 and Atg12) can be detected by western blotting and/or by immunohistochemical analysis of midgut sections.864,865 It is also possible to detect endogenous Atg3 and Atg8 by western blotting using antibodies produced against the H. longicornis proteins (Umemiya-Shirafuji R, unpublished results). Commercial antibodies against mammalian ATG orthologs (ATG3, ATG5, and BECN1) can also be used for western blotting; however, when the tick samples include blood of a host animal, the animal species immunized with autophagy-related proteins should be checked before use to avoid nonspecific background cross-reactivity. In addition to these methods, TEM is recommended to detect autophagosomes and autolysosomes. Although acidotropic dyes can be useful as a marker for autolysosomes in some animals, careful attention should be taken when using the dyes in ticks. Since the midgut epithelial cells contain acidic organelles (e.g., lysosomes) that are related to blood digestion during blood feeding, this method may cause confusion. It is difficult to distinguish between autophagy (autolysosomes) and blood digestion (lysosomes) with acidotropic dyes. Another available monitoring method is to assess the mRNA levels of tick ATG genes by real-time PCR.866 However, this method should be used along with other approaches such as western blotting, immunostaining, and TEM as described in this article. Unlike model insects, such as Drosophila, powerful genetic tools to assess autophagy are still not established in ticks. However, RNAi-mediated gene silencing is currently being developed to analyze the function of autophagy-related genes in ticks during nonfeeding periods (Umemiya-Shirafuji R, unpublished results).
19. Zebrafish
Zebrafish have many characteristics that make them a valuable vertebrate model organism for the analysis of autophagy. For example, taking advantage of the transparency of embryos, autophagosome formation can be visualized in vivo during development using transgenic GFP-Lc3 and GFP-Gabarap fish. The addition of 1-phenyl-2-thiourea (PTU) to media inhibits melanogenesis allowing visualization of later-stage embryos. Lysosomes can also be readily detected in vivo by the addition of LysoTracker Red to fish media prior to visualization. Additionally, protocols have been developed to monitor Lc3 protein levels and conjugation to PE by western blot analysis using commercially available Lc3 antibodies.35,879
Because of their translucent character and external fertilization and development, this vertebrate has proven to be an exceptional choice for developmental research. In situ hydridization of whole embryos can be performed to determine expression patterns. Knockdown of gene function is performed by treatment with morpholinos; the core autophagy machinery protein Gabarap,880 and regulatory proteins such as the phosphoinositide phosphatase Mtmr14,881 Rptor and Mtor,882 have all been successfully knocked down by morpholino treatment. However, research is currently somewhat hampered by the deficiency of efficient methods for targeted gene mutations and deletions.
Zebrafish are ideal organisms for in vivo drug discovery and/or verification because of their relatively small size and aqueous habitat, and several chemicals have been identified that modulate zebrafish autophagy activity.879 Many chemicals can be added to the media and are absorbed directly through the skin. Because of simple drug delivery and the onset of neurodegenerative disease phenotypes at the larval stage, zebrafish are a promising organism for the study of autophagy’s role in neurodegenative disease. Along these lines, a zebrafish model of Huntington disease has been developed.740
20. Food biotechnology
Required for yeast cell survival under a variety of stress conditions, autophagy has the potential to contribute to the outcome of many food fermentation processes. For example, autophagy induction is observed during the primary fermentation of synthetic grape must,867 and during sparkling wine production (secondary fermentation).868 A number of genome-wide studies have identified vacuolar functions and autophagy as relevant processes during primary wine fermentation or for ethanol tolerance, based on gene expression data or cell viability of knockout yeast strains;867,869-873 however, understanding the actual relevance of autophagy in yeast-driven food fermentation processes would require addressing this issue experimentally using some of the methods available for S. cerevisiae as described in these guidelines.
Autophagy is a target for some widespread food preservatives used to prevent yeast-dependent spoilage. For example, the effect of benzoic acid is exacerbated when concurrent with nitrogen starvation.874 This opened the way to devise strategies to improve the usefulness of sorbic and benzoic acid, taking advantage of their combination with stress conditions that would require functional autophagy for yeast cell survival.875 Practical application of these findings would also require extending this research to other relevant food spoilage yeast species, which would be of obvious practical interest.
In the food/health interface, the effect of some food bioactive compounds on autophagy in different human cell types has already attracted some attention.876,877 Interpreting the results of this type of research, however, warrants two cautionary notes.878 First, the relationship between health status and autophagic activity is obviously far from being direct. Second, experimental design in this field must take into account the actual levels of these molecules in the target organs after ingestion, as well as exposure time and their transformations in the human body. In addition, attention must be paid to the fact that several mechanisms might contribute to the observed biological effects. Thus, relevant conclusions about the actual involvement of autophagy on the health-related effect of food bioactive compounds would only be possible by assaying the correct molecules in the correct concentrations.
Conclusions and Future Perspectives
There is no question that research on the topic of autophagy has expanded dramatically since the publication of the first set of guidelines.1 To help keep track of the field we have published a glossary of autophagy-related molecules and processes,883,884 and there are now databases that are specifically dedicated to autophagy including the Human Autophagy-dedicated Database (HADb; www.autophagy.lu) and the Autophagy Database (http://tp-apg.genes.nig.ac.jp/autophagy/).
With this continued influx of new researchers we think it is critical to try to define standards for the field. Accordingly, we have highlighted the uses and caveats of an expanding set of recommended methods for monitoring macroautophagy in a wide range of systems (Table 2). Importantly, investigators need to determine whether they are evaluating levels of early or late autophagic compartments, or autophagic flux. If the question being asked is whether a particular condition changes autophagic flux (i.e., the rate of delivery of autophagy substrates to lysosomes or the vacuole, followed by degradation), then assessment of steady-state levels of autophagosomes (e.g., by counting GFP-LC3 puncta, monitoring the amount of LC3-II without examining turnover, or by single time point electron micrographs) is not sufficient as an isolated approach. In this case it is also necessary to directly measure the flux of autophagosomes and/or autophagy cargo (e.g., in wild-type cells compared with autophagy-deficient cells, the latter generated by treatment with an autophagy inhibitor or resulting from ATG gene knockdowns). Collectively, we strongly recommend the use of multiple assays whenever possible, rather than relying on the results from a single method.
Table 2. Recommended methods for monitoring autophagy.
| Method | Description |
|---|---|
| 1. Electron microscopy |
Quantitative electron microscopy, immuno-TEM; monitor autophagosome number, volume |
| 2. Atg8/LC3 western blotting |
Western blot. The analysis is performed in the absence and presence of lysosomal protease or fusion inhibitors to monitor flux; an increase in the LC3-II amount in the presence of the inhibitor is usually indicative of flux |
| 3. GFP-Atg8/LC3 lysosomal delivery and proteolysis |
Western blot ± lysosomal fusion or degradation inhibitors; the generation of free GFP indicates lysosomal/vacuolar delivery |
| 4. GFP-Atg8/LC3 fluorescence microscopy |
Fluorescence microscopy, FACS to monitor vacuolar/lysosomal localization. Also, increase in punctate GFP-Atg8/LC3 or Atg18/WIPI |
| 5. Tandem mRFP/mCherry-GFP fluorescence microscopy, Rosella |
Flux can be monitored as a decrease in green/red (yellow) fluorescence (phagophores, autophagosomes) and an increase in red fluorescence (autolysosomes) |
| 6. SQSTM1/p62 and related LC3 binding protein turnover |
The amount of SQSTM1 increases when autophagy is inhibited and decreases when autophagy is induced |
| 7. MTOR, AMPK and Atg1/ULK1 kinase activity |
Western blot, immunoprecipitation or kinase assays |
| 8. WIPI fluorescence microscopy |
Quantitative fluorescence analysis using endogenous WIPI proteins, or GFP- or Myc-tagged versions. Suitable for high-throughput imaging procedures. |
| 9. Transcriptional and translational regulation |
Northern blot, or qRT-PCR, autophagy-dedicated microarray |
| 10. Autophagic protein degradation |
Turnover of long-lived proteins to monitor flux |
| 11. Pex14-GFP, GFP-Atg8, Om45-GFP, mitoPho8Δ60 |
A range of assays can be used to monitor selective types of autophagy. These typically involve proteolytic maturation of a resident enzyme or degradation of a chimera, which can be followed enzymatically or by western blot |
| 12. Autophagic sequestration assays |
Lysosomal accumulation by biochemical or multilabel fluorescence techniques, and TEM with a maturation inhibitor |
| 13. Turnover of autophagic compartments |
Electron microscopy with morphometry/stereology |
| 14. Autophagosome-lysosome colocalization and dequenching assay |
Fluorescence microscopy |
| 15. Sequestration and processing assays in plants |
Chimeric RFP fluorescence and processing, and light and electron microscopy |
| 16. Tissue fractionation |
Centrifugation, western blot and electron microscopy |
| 17. Degradation of endogenous lipofuscin | Fluorescence microscopy |
As a final reminder, we stated at the beginning of this article that this set of guidelines is not meant to be a formulaic compilation of rules, because the appropriate assays depend in part on the question being asked and the system being used. Rather, these guidelines are presented primarily to emphasize key issues that need to be addressed such as the difference between measuring autophagy components, and flux or substrate clearance; they are not meant to constrain imaginative approaches to monitoring autophagy. Indeed, it is hoped that new methods for monitoring autophagy will continue to be developed, and new findings may alter our view of the current assays. Similar to the process of autophagy, this is a dynamic field, and we need to remain flexible in the standards we apply.
Glossary
For a more complete listing of autophagy-related terms and definitions we refer readers to Klionsky DJ, Baehrecke EH, Brumell JH, Chu CT, Codogno P, Cuervo AM, etal. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 2011; 7:1273–94.
Amphisome: Intermediate compartment formed by the fusion of an autophagosome with an endosome; this compartment has not yet fused with a lysosome (also referred to as an acidic late autophagosome).
Autolysosome: A degradative compartment formed by the fusion of an autophagosome or amphisome with a primary lysosome or telolysosome. Upon completion of degradation, or when degradation has reached an end point, this compartment (again) becomes a telolysosome (also referred to as a residual body).
Autophagosome: A cytosolic membrane-bound compartment typically denoted by a limiting double membrane. The early autophagosome in particular contains cytoplasmic components and organelles that are morphologically unchanged because the compartment has not fused with a lysosome and lacks proteolytic enzymes.
Autophagy: A collection of processes typically involving degradative delivery of a portion of the cytoplasm to lysosomes or the plant or fungal vacuole that does not involve direct transport through the endocytic or vacuolar protein sorting, Vps, pathways.
Chaperone-mediated autophagy (CMA): Import and degradation of soluble cytosolic proteins by chaperone-dependent, direct translocation across the lysosomal membrane.
Cytoplasm-to-vacuole targeting (Cvt): A biosynthetic pathway in fungi that transports resident hydrolases to the vacuole through a selective autophagy-related process.
Lysosome: A degradative organelle in higher eukaryotes that compartmentalizes a range of hydrolytic enzymes and maintains a highly acidic pH. A primary lysosome is a relatively small compartment that has not yet participated in a degradation process, whereas secondary lysosomes are sites of present or past digestive activity. The secondary lysosomes include autolysosomes and telolysosomes. Autolysosomes/early secondary lysosomes are larger compartments actively engaged in digestion, whereas telolysosomes/late secondary lysosomes do not have significant digestive activity and contain residues of previous digestions. Both may contain material of either autophagic or heterophagic origin.
Macroautophagy: The largely nonspecific autophagic sequestration of cytoplasm into a double- or multiple-membrane-delimited compartment (an autophagosome) of nonlysosomal/vacuolar origin and its subsequent degradation by the lysosomal system. Note that certain proteins and organelles may be selectively degraded via a macroautophagy-related process, and conversely, some cytosolic components such as cytoskeletal elements are selectively excluded.
Microautophagy: Uptake and degradation of cytoplasm by protrusion, invagination or septation of the lysosome or vacuole membrane.
Mitophagy: The selective autophagic sequestration and degradation of mitochondria.
Nucleophagy: The selective autophagic degradation of the nucleus.
Pexophagy: A selective type of autophagy involving the sequestration and degradation of peroxisomes; can occur by a micro- or macroautophagic process.
Phagophore: Membrane cisterna that has been implicated in an initial event during formation of the autophagosome. Previously referred to as the “isolation membrane.”
Phagophore assembly site (PAS): A perivacuolar compartment or location that is involved in the formation of Cvt vesicles and autophagosomes in yeast. The PAS may supply membranes during the formation process or may be an organizing center where most of the autophagic machinery resides, at least transiently.
Phosphatidylinositol 3-kinase (PtdIns3K): A family of enzymes that add a phosphate group to the 3′ hydroxyls on the inositol ring of phosphoinositides. The class III phosphatidylinositol 3-kinases are stimulatory for autophagy, whereas class I enzymes are inhibitory.
Reticulophagy: A type of selective macroautophagy of the ER. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. Activation of the UPR in yeast induces reticulophagy.
Ribophagy: The selective autophagic sequestration and degradation of ribosomes.
Vacuole: The fungal and plant counterpart of the lysosome; this organelle also carries out storage and osmoregulatory functions. The plants’ bona fide equivalent of the lysosome is the lytic vacuole.
Xenophagy: The selective degradation of microbes (e.g., bacteria, fungi, parasites and/or viruses) through an autophagy-related mechanism.
Index
A
Acridine Orange 63
Atg8–PE conjugation 18, 23, 24, 26, 27, 33, 69, 70, 73, 74
Atg12–Atg5 conjugation 43
Atg12–Atg5, Atg16 fluorescence 33, 43
ATG16L1 43, 44, 46, 60
Atg18 43, 44, 46, 70
Autophagic body accumulation 75
Autophagosome characteristics 16–23, 25–29, 32–36, 38, 39, 42–44, 47, 48, 50–52, 54–57, 59, 60, 63–66, 69–71, 73–78
B
BHMT 48
Bovine mammary epithelial cells 72
C
C. elegans 44, 46, 55, 68, 69
Calcium 27, 32, 34, 66
Cell death 16, 27, 31, 32, 36, 39, 46, 49, 58, 60, 64–66, 70, 73
Chicken DT40 cells 67, 68
Chlamydomonas 68
Chloroquine 23, 26–30, 34, 38, 40, 43, 58, 59, 64, 65, 72
Correlative light and electron microscopy (CLEM) 21, 33, 54
D
Degradation assays 47
Dictyostelium 28, 30, 33, 46, 48, 60, 75
DQ-BSA 56, 57
E
Electron microscopy 16, 18, 21, 22, 78
Endosomal-microautophagy 62
EPG proteins 44
F
Flow cytometry 32, 36, 37, 48
Fluorescence microscopy 18, 21, 25, 28–33, 35, 36, 43, 44, 49–53, 55, 56, 58, 60, 68, 70, 72, 74, 75, 78
G
GFP-Atg8/LC3 processing 30, 49, 74
GFP-LC3 fluorescence microscopy 35
H
HIV Nef 34, 66
Honeybee 70
Hydra 72
I
ICP34.5 34, 54, 66
Influenza M2 34
Inhibitors 34, 36, 37, 39, 40–42, 44, 45, 47, 48, 51, 53, 55, 56, 58, 59, 61–69, 72, 75, 78
K
Keima 52, 56
L
Large animals 72, 73
LC3-I and LC3-II western blot 69, 70
LC3-IIs 27
LC3-associated phagocytosis (LAP) 25
Lipofuscin 53, 56, 70, 78
Long-lived protein degradation 18, 48, 61, 78
LysoTracker Red 61–63, 65, 72–74, 76, 77
M
3-methyladenine 34
Mitophagy 20, 23, 25, 34, 35, 40, 48–53, 71
Monodansylcadaverine, MDC 63
MTOR activity 68, 72
P
p62 western blot 16, 22–33, 40–42, 45, 47, 48, 50, 51, 53, 55, 58–60, 68–73
Pexophagy 20, 48–50, 53, 55, 67
Planarians 74
Plants 16, 17, 19, 21, 26, 33, 60, 65, 74, 75, 78
PolyQ protein turnover 53
Protists 21, 75
R
Rainbow trout 76
Reticulophagy 49, 50
RFP chimera processing 78
Rosella 35, 36, 78
S
Saponin 33, 36, 38
Sea urchin 76
Sequestration assays 55–57, 78
SQSTM1 western blot 16, 22–33, 39–42, 45, 47, 48, 50, 51, 53–55, 58–60, 68–73
T
TAKA assay 43
Tandem mRFP/mCherry-GFP-LC3 21, 35, 36, 68
Trehalose 34, 66
V
Viral Bcl-2 66
Viral CFLAR/FLIP 66
W
WIPI1 44, 45
WIPI2 44, 45
WIPI4 44
Wortmannin 34, 40, 44, 45, 56, 57, 63, 64, 72, 76
X
Xenophagy 20, 49, 54
Z
Zebrafish 76, 77
Zymophagy 55
Acknowledgments
In a rapidly expanding and highly dynamic field such as autophagy, it is possible that some authors who should have been included on this manuscript have been missed. D.J.K. extends his apologies to researchers in the field of autophagy who, due to oversight or any other reason, could not be included. This work was supported by National Institutes of Health Public Health Service grant GM53396 to D.J.K. Due to space and other limitations, it is not possible to include all other sources of financial support.
Glossary
Abbreviations:
- 3-MA
3-methyladenine
- AMPK
AMP-activated protein kinase
- Ape1
aminopeptidase I
- Atg
autophagy-related
- AV
autophagic vacuole
- CLEAR
coordinated lysosomal enhancement and regulation
- CLEM
correlative light and electron microscopy
- CMA
chaperone-mediated autophagy
- Cvt
cytoplasm-to-vacuole targeting
- DQ-BSA
dequenched bovine serum albumin
- e-MI
endosomal microautophagy
- EBSS
Earle’s balanced salt solution
- FACS
fluorescence-activated cell sorter
- GFP
green fluorescent protein
- IMP
intramembrane particle
- LAMP2
lysosome-associated membrane protein 2
- LAP
LC3-associated phagocytosis
- LC3
microtubule-associated protein 1 light chain 3 (MAP1LC3)
- MDC
monodansylcadaverine
- mRFP
monomeric red fluorescent protein
- MTOR
mechanistic target of rapamycin
- NVJ
nucleus-vacuole junction
- RPS6KB
ribosomal protein S6 kinase, 70kDa, polypeptide 2
- PAS
phagophore assembly site
- PE
phosphatidylethanolamine
- PMN
piecemeal microautophagy of the nucleus
- PtdIns3K
phosphatidylinositol 3-kinase
- PtdIns3KC3
PtdIns3K class III
- Rluc
Renilla reniformis luciferase
- SD
standard deviation
- SOD
superoxide dismutase
- TEM
transmission electron microscopy
- tfLC3
tandem fluorescent LC3
- TORC1
TOR complex I
- TR-FRET
time-resolved fluorescence resonance energy transfer
- UPR
unfolded protein response
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/19496
References
- 1.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–7. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 3.Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3:181–206. doi: 10.4161/auto.3678. [DOI] [PubMed] [Google Scholar]
- 4.Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491–502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–26. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S, et al. In search of an “autophagomometer”. Autophagy. 2009;5:585–9. doi: 10.4161/auto.5.5.8823. [DOI] [PubMed] [Google Scholar]
- 7.Klionsky DJ. The autophagosome is overrated! Autophagy. 2011;7:353–4. doi: 10.4161/auto.7.4.14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eskelinen E-L, Reggiori F, Baba M, Kovács AL, Seglen PO. Seeing is believing: the impact of electron microscopy on autophagy research. Autophagy. 2011;7:935–56. doi: 10.4161/auto.7.9.15760. [DOI] [PubMed] [Google Scholar]
- 9.Seglen PO. Regulation of autophagic protein degradation in isolated liver cells. In: Glaumann H and Ballard FJ, eds. Lysosomes: Their Role in Protein Breakdown. London: Academic Press, 1987:369-414. [Google Scholar]
- 10.de Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–92. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 11.Dunn WA., Jr. Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol. 1994;4:139–43. doi: 10.1016/0962-8924(94)90069-8. [DOI] [PubMed] [Google Scholar]
- 12.Gordon PB, Seglen PO. Prelysosomal convergence of autophagic and endocytic pathways. Biochem Biophys Res Commun. 1988;151:40–7. doi: 10.1016/0006-291X(88)90556-6. [DOI] [PubMed] [Google Scholar]
- 13.Dice JF, Klionsky DJ. Artophagy: the art of autophagy--macroautophagy. Autophagy. 2010;6:320–1. doi: 10.4161/auto.6.3.11263. [DOI] [PubMed] [Google Scholar]
- 14.Kovács J, Fellinger E, Kárpáti AP, Kovács AL, László L, Réz G. Morphometric evaluation of the turnover of autophagic vacuoles after treatment with Triton X-100 and vinblastine in murine pancreatic acinar and seminal vesicle epithelial cells. Virchows Arch B Cell Pathol Incl Mol Pathol. 1987;53:183–90. doi: 10.1007/BF02890242. [DOI] [PubMed] [Google Scholar]
- 15.Kovács J, Fellinger E, Kárpáti PA, Kovács AL, László L. The turnover of autophagic vacuoles: evaluation by quantitative electron microscopy. Biomed Biochim Acta. 1986;45:1543–7. [PubMed] [Google Scholar]
- 16.Kovács J, László L, Kovács AL. Regression of autophagic vacuoles in pancreatic acinar, seminal vesicle epithelial, and liver parenchymal cells: a comparative morphometric study of the effect of vinblastine and leupeptin followed by cycloheximide treatment. Exp Cell Res. 1988;174:244–51. doi: 10.1016/0014-4827(88)90158-9. [DOI] [PubMed] [Google Scholar]
- 17.Chu CT. Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol. 2006;65:423–32. doi: 10.1097/01.jnen.0000229233.75253.be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem. 2006;281:36303–16. doi: 10.1074/jbc.M607031200. [DOI] [PubMed] [Google Scholar]
- 19.Kovács AL, Reith A, Seglen PO. Accumulation of autophagosomes after inhibition of hepatocytic protein degradation by vinblastine, leupeptin or a lysosomotropic amine. Exp Cell Res. 1982;137:191–201. doi: 10.1016/0014-4827(82)90020-9. [DOI] [PubMed] [Google Scholar]
- 20.Tóth S, Nagy K, Pálfia Z, Réz G. Cellular autophagic capacity changes during azaserine-induced tumour progression in the rat pancreas. Up-regulation in all premalignant stages and down-regulation with loss of cycloheximide sensitivity of segregation along with malignant transformation. Cell Tissue Res. 2002;309:409–16. doi: 10.1007/s00441-001-0506-7. [DOI] [PubMed] [Google Scholar]
- 21.Loos B, Engelbrecht AM. Cell death: a dynamic response concept. Autophagy. 2009;5:590–603. doi: 10.4161/auto.5.5.8479. [DOI] [PubMed] [Google Scholar]
- 22.Seglen PO, Gordon PB, Grinde B, Solheim A, Kovács AL, Poli A. Inhibitors and pathways of hepatocytic protein degradation. Acta Biol Med Ger. 1981;40:1587–98. [PubMed] [Google Scholar]
- 23.Ktistakis NT, Andrews S, Long J. What is the advantage of a transient precursor in autophagosome biogenesis? Autophagy. 2011;7:118–22. doi: 10.4161/auto.7.1.13697. [DOI] [PubMed] [Google Scholar]
- 24.Kovács AL, Réz G, Pálfia Z, Kovács J. Autophagy in the epithelial cells of murine seminal vesicle in vitro. Formation of large sheets of nascent isolation membranes, sequestration of the nucleus and inhibition by wortmannin and 3-ethyladenine. Cell Tissue Res. 2000;302:253–61. doi: 10.1007/s004410000275. [DOI] [PubMed] [Google Scholar]
- 25.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 26.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–8. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 27.Kanki T, Kang D, Klionsky DJ. Monitoring mitophagy in yeast: the Om45-GFP processing assay. Autophagy. 2009;5:1186–9. doi: 10.4161/auto.5.8.9854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grandér D, Kharaziha P, Laane E, Pokrovskaja K, Panaretakis T. Autophagy as the main means of cytotoxicity by glucocorticoids in hematological malignancies. Autophagy. 2009;5:1198–200. doi: 10.4161/auto.5.8.10122. [DOI] [PubMed] [Google Scholar]
- 29.Welter E, Thumm M, Krick R. Quantification of nonselective bulk autophagy in S. cerevisiae using Pgk1-GFP. Autophagy. 2010;6:794–7. doi: 10.4161/auto.6.6.12348. [DOI] [PubMed] [Google Scholar]
- 30.Raju D, Jones NL. Methods to monitor autophagy in H. pylori vacuolating cytotoxin A (VacA)-treated cells. Autophagy. 2010;6:138–43. doi: 10.4161/auto.6.1.10222. [DOI] [PubMed] [Google Scholar]
- 31.Geng J, Klionsky DJ. Determining Atg protein stoichiometry at the phagophore assembly site by fluorescence microscopy. Autophagy. 2010;6:144–7. doi: 10.4161/auto.6.1.10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swanlund JM, Kregel KC, Oberley TD. Investigating autophagy: quantitative morphometric analysis using electron microscopy. Autophagy. 2010;6:270–7. doi: 10.4161/auto.6.2.10439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Ney PA. Reticulocyte mitophagy: monitoring mitochondrial clearance in a mammalian model. Autophagy. 2010;6:405–8. doi: 10.4161/auto.6.3.11245. [DOI] [PubMed] [Google Scholar]
- 34.Seglen PO, Brinchmann MF. Purification of autophagosomes from rat hepatocytes. Autophagy. 2010;6:542–7. doi: 10.4161/auto.6.4.11272. [DOI] [PubMed] [Google Scholar]
- 35.He C, Klionsky DJ. Analyzing autophagy in zebrafish. Autophagy. 2010;6:642–4. doi: 10.4161/auto.6.5.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calvo-Garrido J, Carilla-Latorre S, Mesquita A, Escalante R. A proteolytic cleavage assay to monitor autophagy in Dictyostelium discoideum. Autophagy. 2011;7:1063–8. doi: 10.4161/auto.7.9.16629. [DOI] [PubMed] [Google Scholar]
- 37.Xu F, Liu XH, Zhuang FL, Zhu J, Lin FC. Analyzing autophagy in Magnaporthe oryzae. Autophagy. 2011;7:525–30. doi: 10.4161/auto.7.5.15020. [DOI] [PubMed] [Google Scholar]
- 38.Klionsky DJ. Autophagy: Lower Eukaryotes and Non-Mammalian Systems, Part A. Amsterdam: Academic Press/Elsevier, 2008. [Google Scholar]
- 39.Klionsky DJ. Autophagy in Mammalian Systems, Part B. Amsterdam: Academic Press/Elsevier, 2008. [Google Scholar]
- 40.Klionsky DJ. Autophagy in Disease and Clinical Applications, Part C. Amsterdam: Academic Press/Elsevier, 2008. [Google Scholar]
- 41.Zhu J, Dagda RK, Chu CT. Monitoring mitophagy in neuronal cell cultures. Methods Mol Biol. 2011;793:325–39. doi: 10.1007/978-1-61779-328-8_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. http://www.landesbioscience.com/journals/autophagy/protocols/
- 43.Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen E-L. Monitoring autophagy by electron microscopy in Mammalian cells. Methods Enzymol. 2009;452:143–64. doi: 10.1016/S0076-6879(08)03610-0. [DOI] [PubMed] [Google Scholar]
- 44.Eskelinen E-L. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- 45.Eskelinen E-L. To be or not to be? Examples of incorrect identification of autophagic compartments in conventional transmission electron microscopy of mammalian cells. Autophagy. 2008;4:257–60. doi: 10.4161/auto.5179. [DOI] [PubMed] [Google Scholar]
- 46.Eskelinen E-L, Kovács AL. Double membranes vs. lipid bilayers, and their significance for correct identification of macroautophagic structures. Autophagy. 2011;7:931–2. doi: 10.4161/auto.7.9.16679. [DOI] [PubMed] [Google Scholar]
- 47.Eskelinen E-L. Fine structure of the autophagosome. In: Deretic V, ed. Autophagosome and Phagosome. Totowa, NJ: Humana Press, 2008:11-28. [DOI] [PubMed] [Google Scholar]
- 48.Berg TO, Fengsrud M, Strømhaug PE, Berg T, Seglen PO. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem. 1998;273:21883–92. doi: 10.1074/jbc.273.34.21883. [DOI] [PubMed] [Google Scholar]
- 49.Eskelinen E-L. Macroautophagy in mammalian cells. In: Saftig P, ed. Lysosomes. Georgetown, TX: LandesBioscience/Eurekah.com, 2005. [Google Scholar]
- 50.Rabouille C, Strous GJ, Crapo JD, Geuze HJ, Slot JW. The differential degradation of two cytosolic proteins as a tool to monitor autophagy in hepatocytes by immunocytochemistry. J Cell Biol. 1993;120:897–908. doi: 10.1083/jcb.120.4.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kovács AL, Pálfia Z, Réz G, Vellai T, Kovács J. Sequestration revisited: integrating traditional electron microscopy, de novo assembly and new results. Autophagy. 2007;3:655–62. doi: 10.4161/auto.4590. [DOI] [PubMed] [Google Scholar]
- 52.Gao W, Kang JH, Liao Y, Ding WX, Gambotto AA, Watkins SC, et al. Biochemical isolation and characterization of the tubulovesicular LC3-positive autophagosomal compartment. J Biol Chem. 2010;285:1371–83. doi: 10.1074/jbc.M109.054197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baba M, Osumi M, Ohsumi Y. Analysis of the membrane structures involved in autophagy in yeast by freeze-replica method. Cell Struct Funct. 1995;20:465–71. doi: 10.1247/csf.20.465. [DOI] [PubMed] [Google Scholar]
- 54.Réz G, Meldolesi J. Freeze-fracture of drug-induced autophagocytosis in the mouse exocrine pancreas. Lab Invest. 1980;43:269–77. [PubMed] [Google Scholar]
- 55.Punnonen E-L, Pihakaski K, Mattila K, Lounatmaa K, Hirsimäki P. Intramembrane particles and filipin labelling on the membranes of autophagic vacuoles and lysosomes in mouse liver. Cell Tissue Res. 1989;258:269–76. doi: 10.1007/BF00239447. [DOI] [PubMed] [Google Scholar]
- 56.Fengsrud M, Erichsen ES, Berg TO, Raiborg C, Seglen PO. Ultrastructural characterization of the delimiting membranes of isolated autophagosomes and amphisomes by freeze-fracture electron microscopy. Eur J Cell Biol. 2000;79:871–82. doi: 10.1078/0171-9335-00125. [DOI] [PubMed] [Google Scholar]
- 57.Rao VA, Klein SR, Bonar SJ, Zielonka J, Mizuno N, Dickey JS, et al. The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. J Biol Chem. 2010;285:34447–59. doi: 10.1074/jbc.M110.133579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krick R, Mühe Y, Prick T, Bredschneider M, Bremer S, Wenzel D, et al. Piecemeal microautophagy of the nucleus: genetic and morphological traits. Autophagy. 2009;5:270–2. doi: 10.4161/auto.5.2.7639. [DOI] [PubMed] [Google Scholar]
- 59.Meschini S, Condello M, Calcabrini A, Marra M, Formisano G, Lista P, et al. The plant alkaloid voacamine induces apoptosis-independent autophagic cell death on both sensitive and multidrug resistant human osteosarcoma cells. Autophagy. 2008;4:1020–33. doi: 10.4161/auto.6952. [DOI] [PubMed] [Google Scholar]
- 60.Proikas-Cezanne T, Robenek H. Freeze-fracture replica immunolabelling reveals human WIPI-1 and WIPI-2 as membrane proteins of autophagosomes. J Cell Mol Med. 2011;15:2007–10. doi: 10.1111/j.1582-4934.2011.01339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kovács J, Réz G, Kovács AL, Csák J, Zboray G. Autophagocytosis: freeze-fracture morphology, effects of vinblastine and influence of transcriptional and translational inhibitors. Acta Biol Med Ger. 1982;41:131–5. [PubMed] [Google Scholar]
- 62.Hirsimäki Y, Hirsimäki P, Lounatmaa K. Vinblastine-induced autophagic vacuoles in mouse liver and Ehrlich ascites tumor cells as assessed by freeze-fracture electron microscopy. Eur J Cell Biol. 1982;27:298–301. [PubMed] [Google Scholar]
- 63.Cheong H, Yorimitsu T, Reggiori F, Legakis JE, Wang C-W, Klionsky DJ. Atg17 regulates the magnitude of the autophagic response. Mol Biol Cell. 2005;16:3438–53. doi: 10.1091/mbc.E04-10-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xie Z, Nair U, Klionsky DJ. Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell. 2008;19:3290–8. doi: 10.1091/mbc.E07-12-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sigmond T, Fehér J, Baksa A, Pásti G, Pálfia Z, Takács-Vellai K, et al. Qualitative and quantitative characterization of autophagy in Caenorhabditis elegans by electron microscopy. Methods Enzymol. 2008;451:467–91. doi: 10.1016/S0076-6879(08)03228-X. [DOI] [PubMed] [Google Scholar]
- 66.Kovács AL, Vellai T, Müller F. Autophagy in Caenorhabditis elegans In: Klionsky DJ, ed. Autophagy. Georgetown, Texas: Landes Bioscience, 2004:217-23. [Google Scholar]
- 67.Weibel ER. Practical Methods for Biological Morphometry. Academic Press, New York, 1979. [Google Scholar]
- 68.Williams MA. Quantitative methods in biology: Practical methods in electron microscopy. Amsterdam, New York, Oxford: North-Holland Publishing Company, 1977. [Google Scholar]
- 69.Howard V, Reed MG. Unbiased stereology; three dimensional measurement in microscopy. U Bios Scientific Publishers, 1998. [Google Scholar]
- 70.Punnonen EL, Reunanen H. Effects of vinblastine, leucine, and histidine, and 3-methyladenine on autophagy in Ehrlich ascites cells. Exp Mol Pathol. 1990;52:87–97. doi: 10.1016/0014-4800(90)90061-H. [DOI] [PubMed] [Google Scholar]
- 71.Kovács AL, László L, Fellinger E, Jakab A, Orosz A, Réz G, et al. Combined effects of fasting and vinblastine treatment on serum insulin level, the size of autophagic-lysosomal compartment, protein content and lysosomal enzyme activities of liver and exocrine pancreatic cells of the mouse. Comp Biochem Physiol B. 1989;94:505–10. doi: 10.1016/0305-0491(89)90189-2. [DOI] [PubMed] [Google Scholar]
- 72.Shelburne JD, Arstila AU, Trump BF. Studies on cellular autophagocytosis. The relationship of autophagocytosis to protein synthesis and to energy metabolism in rat liver and flounder kidney tubules in vitro. Am J Pathol. 1973;73:641–70. [PMC free article] [PubMed] [Google Scholar]
- 73.Reyes FC, Chung T, Holding D, Jung R, Vierstra R, Otegui MS. Delivery of prolamins to the protein storage vacuole in maize aleurone cells. Plant Cell. 2011;23:769–84. doi: 10.1105/tpc.110.082156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dunn WA, Jr., Cregg JM, Kiel JAKW, van der Klei IJ, Oku M, Sakai Y, et al. Pexophagy: the selective autophagy of peroxisomes. Autophagy. 2005;1:75–83. doi: 10.4161/auto.1.2.1737. [DOI] [PubMed] [Google Scholar]
- 75.Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy. 2011;7:297–300. doi: 10.4161/auto.7.3.14502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bélanger M, Rodrigues PH, Dunn WA, Jr., Progulske-Fox A. Autophagy: a highway for Porphyromonas gingivalis in endothelial cells. Autophagy. 2006;2:165–70. doi: 10.4161/auto.2828. [DOI] [PubMed] [Google Scholar]
- 77.Birmingham CL, Brumell JH. Autophagy recognizes intracellular Salmonella enterica serovar Typhimurium in damaged vacuoles. Autophagy. 2006;2:156–8. doi: 10.4161/auto.2825. [DOI] [PubMed] [Google Scholar]
- 78.Colombo MI, Gutierrez MG, Romano PS. The two faces of autophagy: Coxiella and Mycobacterium. Autophagy. 2006;2:162–4. doi: 10.4161/auto.2827. [DOI] [PubMed] [Google Scholar]
- 79.Ogawa M, Sasakawa C. Shigella and autophagy. Autophagy. 2006;2:171–4. doi: 10.4161/auto.2829. [DOI] [PubMed] [Google Scholar]
- 80.Vergne I, Singh S, Roberts E, Kyei G, Master S, Harris J, et al. Autophagy in immune defense against Mycobacterium tuberculosis. Autophagy. 2006;2:175–8. doi: 10.4161/auto.2830. [DOI] [PubMed] [Google Scholar]
- 81.Yoshimori T. Autophagy vs. Group A Streptococcus. Autophagy. 2006;2:154–5. doi: 10.4161/auto.2822. [DOI] [PubMed] [Google Scholar]
- 82.Lynch-Day MA, Klionsky DJ. The Cvt pathway as a model for selective autophagy. FEBS Lett. 2010;584:1359–66. doi: 10.1016/j.febslet.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Birmingham CL, Canadien V, Gouin E, Troy EB, Yoshimori T, Cossart P, et al. Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy. 2007;3:442–51. doi: 10.4161/auto.4450. [DOI] [PubMed] [Google Scholar]
- 84.Klionsky DJ. Protein transport from the cytoplasm into the vacuole. J Membr Biol. 1997;157:105–15. doi: 10.1007/s002329900220. [DOI] [PubMed] [Google Scholar]
- 85.Dini L, Pagliara P, Carlà EC. Phagocytosis of apoptotic cells by liver: a morphological study. Microsc Res Tech. 2002;57:530–40. doi: 10.1002/jemt.10107. [DOI] [PubMed] [Google Scholar]
- 86.Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, et al. Nomenclature Committee on Cell Death Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005;12:1463–7. doi: 10.1038/sj.cdd.4401724. [DOI] [PubMed] [Google Scholar]
- 87.Réz G, Pálfia Z, Fellinger E. Occurrence and inhibition by cycloheximide of apoptosis in vinblastine-treated murine pancreas. A role for autophagy? Acta Biol Hung. 1991;42:133–40. [PubMed] [Google Scholar]
- 88.Giammarioli AM, Gambardella L, Barbati C, Pietraforte D, Tinari A, Alberton M, et al. Differential effects of the glycolysis inhibitor 2-deoxy-D-glucose on the activity of pro-apoptotic agents in metastatic melanoma cells, and induction of a cytoprotective autophagic response. Int J Cancer. 2012 doi: 10.1002/ijc.26420. In press. [DOI] [PubMed] [Google Scholar]
- 89.Mayhew TM. Quantitative immunoelectron microscopy: alternative ways of assessing subcellular patterns of gold labeling. Methods Mol Biol. 2007;369:309–29. doi: 10.1007/978-1-59745-294-6_15. [DOI] [PubMed] [Google Scholar]
- 90.Mayhew TM, Lucocq JM, Griffiths G. Relative labelling index: a novel stereological approach to test for non-random immunogold labelling of organelles and membranes on transmission electron microscopy thin sections. J Microsc. 2002;205:153–64. doi: 10.1046/j.0022-2720.2001.00977.x. [DOI] [PubMed] [Google Scholar]
- 91.Schmid D, Pypaert M, Münz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kovács J. Regression of autophagic vacuoles in seminal vesicle cells following cycloheximide treatment. Exp Cell Res. 1983;144:231–4. doi: 10.1016/0014-4827(83)90460-3. [DOI] [PubMed] [Google Scholar]
- 93.Réz G, Csák J, Fellinger E, László L, Kovács AL, Oliva O, et al. Time course of vinblastine-induced autophagocytosis and changes in the endoplasmic reticulum in murine pancreatic acinar cells: a morphometric and biochemical study. Eur J Cell Biol. 1996;71:341–50. [PubMed] [Google Scholar]
- 94.Kovács AL, Grinde B, Seglen PO. Inhibition of autophagic vacuole formation and protein degradation by amino acids in isolated hepatocytes. Exp Cell Res. 1981;133:431–6. doi: 10.1016/0014-4827(81)90336-0. [DOI] [PubMed] [Google Scholar]
- 95.Mortimore GE, Hutson NJ, Surmacz CA. Quantitative correlation between proteolysis and macro- and microautophagy in mouse hepatocytes during starvation and refeeding. Proc Natl Acad Sci U S A. 1983;80:2179–83. doi: 10.1073/pnas.80.8.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mortimore GE, Lardeux BR, Adams CE. Regulation of microautophagy and basal protein turnover in rat liver. Effects of short-term starvation. J Biol Chem. 1988;263:2506–12. [PubMed] [Google Scholar]
- 97.Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol. 2007;170:75–86. doi: 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Øvervatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–14. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Razi M, Tooze SA. Correlative light and electron microscopy. Methods Enzymol. 2009;452:261–75. doi: 10.1016/S0076-6879(08)03617-3. [DOI] [PubMed] [Google Scholar]
- 100.Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, et al. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 2011;9:e1001041. doi: 10.1371/journal.pbio.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Castillo K, Rojas-Rivera D, Lisbona F, Caballero B, Nassif M, Court FA, et al. BAX inhibitor-1 regulates autophagy by controlling the IRE1α branch of the unfolded protein response. EMBO J. 2011;30:4465–78. doi: 10.1038/emboj.2011.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen E-L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5:1180–5. doi: 10.4161/auto.5.8.10274. [DOI] [PubMed] [Google Scholar]
- 103.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. Electron tomography reveals the endoplasmic reticulum as a membrane source for autophagosome formation. Autophagy. 2010;6:301–3. doi: 10.4161/auto.6.2.11134. [DOI] [PubMed] [Google Scholar]
- 104.Schneider EM, Lorezn M, Walther P. Autophagy as a hallmark of hemophagocytic diseases In: Gorbunov N, ed. Autophagy: Principles, regulation and roles in disease: Nova Science Publishers, 2012. [Google Scholar]
- 105.Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2006;103:5805–10. doi: 10.1073/pnas.0507436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13:1396–403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sou YS, Tanida I, Komatsu M, Ueno T, Kominami E. Phosphatidylserine in addition to phosphatidylethanolamine is an in vitro target of the mammalian Atg8 modifiers, LC3, GABARAP, and GATE-16. J Biol Chem. 2006;281:3017–24. doi: 10.1074/jbc.M505888200. [DOI] [PubMed] [Google Scholar]
- 108.Le Grand JN, Chakrama FZ, Seguin-Py S, Fraichard A, Delage-Mourroux R, Jouvenot M, et al. GABARAPL1 (GEC1): original or copycat? Autophagy. 2011;7:1098–107. doi: 10.4161/auto.7.10.15904. [DOI] [PubMed] [Google Scholar]
- 109.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–12. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 110.Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29:1792–802. doi: 10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kovsan J, Blüher M, Tarnovscki T, Klöting N, Kirshtein B, Madar L, et al. Altered autophagy in human adipose tissues in obesity. J Clin Endocrinol Metab. 2011;96:E268–77. doi: 10.1210/jc.2010-1681. [DOI] [PubMed] [Google Scholar]
- 112.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 113.Huang W-P, Scott SV, Kim J, Klionsky DJ. The itinerary of a vesicle component, Aut7p/Cvt5p, terminates in the yeast vacuole via the autophagy/Cvt pathways. J Biol Chem. 2000;275:5845–51. doi: 10.1074/jbc.275.8.5845. [DOI] [PubMed] [Google Scholar]
- 114.Cai Q, Lu L, Tian J-H, Zhu Y-B, Qiao H, Sheng Z-H. Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron. 2010;68:73–86. doi: 10.1016/j.neuron.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Castino R, Fiorentino I, Cagnin M, Giovia A, Isidoro C. Chelation of lysosomal iron protects dopaminergic SH-SY5Y neuroblastoma cells from hydrogen peroxide toxicity by precluding autophagy and Akt dephosphorylation. Toxicol Sci. 2011;123:523–41. doi: 10.1093/toxsci/kfr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Michiorri S, Gelmetti V, Giarda E, Lombardi F, Romano F, Marongiu R, et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 2010;17:962–74. doi: 10.1038/cdd.2009.200. [DOI] [PubMed] [Google Scholar]
- 117.Ahlberg J, Berkenstam A, Henell F, Glaumann H. Degradation of short and long lived proteins in isolated rat liver lysosomes. Effects of pH, temperature, and proteolytic inhibitors. J Biol Chem. 1985;260:5847–54. [PubMed] [Google Scholar]
- 118.McLeland CB, Rodriguez J, Stern ST. Autophagy monitoring assay: qualitative analysis of MAP LC3-I to II conversion by immunoblot. Methods Mol Biol. 2011;697:199–206. doi: 10.1007/978-1-60327-198-1_21. [DOI] [PubMed] [Google Scholar]
- 119.Chakrama FZ, Seguin-Py S, Le Grand JN, Fraichard A, Delage-Mourroux R, Despouy G, et al. GABARAPL1 (GEC1) associates with autophagic vesicles. Autophagy. 2010;6:495–505. doi: 10.4161/auto.6.4.11819. [DOI] [PubMed] [Google Scholar]
- 120.Kim J, Huang W-P, Klionsky DJ. Membrane recruitment of Aut7p in the autophagy and cytoplasm to vacuole targeting pathways requires Aut1p, Aut2p, and the autophagy conjugation complex. J Cell Biol. 2001;152:51–64. doi: 10.1083/jcb.152.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shu CW, Drag M, Bekes M, Zhai D, Salvesen GS, Reed JC. Synthetic substrates for measuring activity of autophagy proteases: autophagins (Atg4) Autophagy. 2010;6:936–47. doi: 10.4161/auto.6.7.13075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li M, Chen X, Ye Q-Z, Vogt A, Yin X-M. A high-throughput FRET-based assay for determination of Atg4 activity. Autophagy. 2012;8:401–12. doi: 10.4161/auto.18777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ketteler R, Seed B. Quantitation of autophagy by luciferase release assay. Autophagy. 2008;4:801–6. doi: 10.4161/auto.6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li M, Hou Y, Wang J, Chen X, Shao ZM, Yin X-M. Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J Biol Chem. 2011;286:7327–38. doi: 10.1074/jbc.M110.199059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Klionsky DJ. For the last time, it is GFP-Atg8, not Atg8-GFP (and the same goes for LC3) Autophagy. 2011;7:1093–4. doi: 10.4161/auto.7.10.15492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Castino R, Lazzeri G, Lenzi P, Bellio N, Follo C, Ferrucci M, et al. Suppression of autophagy precipitates neuronal cell death following low doses of methamphetamine. J Neurochem. 2008;106:1426–39. doi: 10.1111/j.1471-4159.2008.05488.x. [DOI] [PubMed] [Google Scholar]
- 128.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–22. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 129.Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001;20:5971–81. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hanson HH, Kang S, Fern´ndez-Monreal M, Oung T, Yildirim M, Lee R, et al. LC3-dependent intracellular membrane tubules induced by gamma-protocadherins A3 and B2: a role for intraluminal interactions. J Biol Chem. 2010;285:20982–92. doi: 10.1074/jbc.M109.092031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–7. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 132.Sanjuan MA, Milasta S, Green DR. Toll-like receptor signaling in the lysosomal pathways. Immunol Rev. 2009;227:203–20. doi: 10.1111/j.1600-065X.2008.00732.x. [DOI] [PubMed] [Google Scholar]
- 133.Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol. 2011;13:1335–43. doi: 10.1038/ncb2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108:17396–401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 135.Gong L, Cullinane M, Treerat P, Ramm G, Prescott M, Adler B, et al. The Burkholderia pseudomallei type III secretion system and BopA are required for evasion of LC3-associated phagocytosis. PLoS One. 2011;6:e17852. doi: 10.1371/journal.pone.0017852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dupont N, Lacas-Gervais S, Bertout J, Paz I, Freche B, Van Nhieu GT, et al. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe. 2009;6:137–49. doi: 10.1016/j.chom.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 137.Reggiori F, Monastyrska I, Verheije MH, Calì T, Ulasli M, Bianchi S, et al. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe. 2010;7:500–8. doi: 10.1016/j.chom.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.English L, Chemali M, Duron J, Rondeau C, Laplante A, Gingras D, et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat Immunol. 2009;10:480–7. doi: 10.1038/ni.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Plowey ED, Cherra SJ, III, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008;105:1048–56. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nicotra G, Mercalli F, Peracchio C, Castino R, Follo C, Valente G, et al. Autophagy-active beclin-1 correlates with favourable clinical outcome in non-Hodgkin lymphomas. Mod Pathol. 2010;23:937–50. doi: 10.1038/modpathol.2010.80. [DOI] [PubMed] [Google Scholar]
- 141.Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- 142.Chu CT, Plowey ED, Dagda RK, Hickey RW, Cherra SJ, III, Clark RS. Autophagy in neurite injury and neurodegeneration: in vitro and in vivo models. Methods Enzymol. 2009;453:217–49. doi: 10.1016/S0076-6879(08)04011-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.He H, Dang Y, Dai F, Guo Z, Wu J, She X, et al. Post-translational modifications of three members of the human MAP1LC3 family and detection of a novel type of modification for MAP1LC3B. J Biol Chem. 2003;278:29278–87. doi: 10.1074/jbc.M303800200. [DOI] [PubMed] [Google Scholar]
- 144.Shpilka T, Weidberg H, Pietrokovski S, Elazar Z. Atg8: an autophagy-related ubiquitin-like protein family. Genome Biol. 2011;12:226. doi: 10.1186/gb-2011-12-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zois CE, Koukourakis MI. Radiation-induced autophagy in normal and cancer cells: towards novel cytoprotection and radio-sensitization policies? Autophagy. 2009;5:442–50. doi: 10.4161/auto.5.4.7667. [DOI] [PubMed] [Google Scholar]
- 146.Xin Y, Yu L, Chen Z, Zheng L, Fu Q, Jiang J, et al. Cloning, expression patterns, and chromosome localization of three human and two mouse homologues of GABAA receptor-associated protein. Genomics. 2001;74:408–13. doi: 10.1006/geno.2001.6555. [DOI] [PubMed] [Google Scholar]
- 147.Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gassmann M, Grenacher B, Rohde B, Vogel J. Quantifying Western blots: pitfalls of densitometry. Electrophoresis. 2009;30:1845–55. doi: 10.1002/elps.200800720. [DOI] [PubMed] [Google Scholar]
- 149.Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, et al. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol. 2000;151:263–76. doi: 10.1083/jcb.151.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chung T, Phillips AR, Vierstra RD. ATG8 lipidation and ATG8-mediated autophagy in Arabidopsis require ATG12 expressed from the differentially controlled ATG12A AND ATG12B loci. Plant J. 2010;62:483–93. doi: 10.1111/j.1365-313X.2010.04166.x. [DOI] [PubMed] [Google Scholar]
- 151.Chung T, Suttangkakul A, Vierstra RD. The ATG autophagic conjugation system in maize: ATG transcripts and abundance of the ATG8-lipid adduct are regulated by development and nutrient availability. Plant Physiol. 2009;149:220–34. doi: 10.1104/pp.108.126714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.King JS, Veltman DM, Insall RH. The induction of autophagy by mechanical stress. Autophagy. 2011;7:1490–9. doi: 10.4161/auto.7.12.17924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ju JS, Varadhachary AS, Miller SE, Weihl CC. Quantitation of “autophagic flux” in mature skeletal muscle. Autophagy. 2010;6:929–35. doi: 10.4161/auto.6.7.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Degtyarev M, De Mazière A, Orr C, Lin J, Lee BB, Tien JY, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol. 2008;183:101–16. doi: 10.1083/jcb.200801099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy. 2008;4:849–50. doi: 10.4161/auto.6845. [DOI] [PubMed] [Google Scholar]
- 156.Xie R, Nguyen S, McKeehan WL, Liu L. Acetylated microtubules are required for fusion of autophagosomes with lysosomes. BMC Cell Biol. 2010;11:89. doi: 10.1186/1471-2121-11-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.González-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquère S, et al. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118:3091–102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- 158.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–5. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 159.Trincheri NF, Follo C, Nicotra G, Peracchio C, Castino R, Isidoro C. Resveratrol-induced apoptosis depends on the lipid kinase activity of Vps34 and on the formation of autophagolysosomes. Carcinogenesis. 2008;29:381–9. doi: 10.1093/carcin/bgm271. [DOI] [PubMed] [Google Scholar]
- 160.Sarkar S, Ravikumar B, Rubinsztein DC. Autophagic clearance of aggregate-prone proteins associated with neurodegeneration. Methods Enzymol. 2009;453:83–110. doi: 10.1016/S0076-6879(08)04005-6. [DOI] [PubMed] [Google Scholar]
- 161.Shintani T, Klionsky DJ. Cargo proteins facilitate the formation of transport vesicles in the cytoplasm to vacuole targeting pathway. J Biol Chem. 2004;279:29889–94. doi: 10.1074/jbc.M404399200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Karim MR, Kanazawa T, Daigaku Y, Fujimura S, Miotto G, Kadowaki M. Cytosolic LC3 ratio as a sensitive index of macroautophagy in isolated rat hepatocytes and H4-II-E cells. Autophagy. 2007;3:553–60. doi: 10.4161/auto.4615. [DOI] [PubMed] [Google Scholar]
- 163.Farkas T, Høyer-Hansen M, Jäättelä M. Identification of novel autophagy regulators by a luciferase-based assay for the kinetics of autophagic flux. Autophagy. 2009;5:1018–25. doi: 10.4161/auto.5.7.9443. [DOI] [PubMed] [Google Scholar]
- 164.Frankel LB, Wen J, Lees M, Høyer-Hansen M, Farkas T, Krogh A, et al. microRNA-101 is a potent inhibitor of autophagy. EMBO J. 2011;30:4628–41. doi: 10.1038/emboj.2011.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Farkas T, Daugaard M, Jäättelä M. Identification of small molecule inhibitors of phosphatidylinositol 3-kinase and autophagy. J Biol Chem. 2011;286:38904–12. doi: 10.1074/jbc.M111.269134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Szyniarowski P, Corcelle-Termeau E, Farkas T, Høyer-Hansen M, Nylandsted J, Kallunki T, et al. A comprehensive siRNA screen for kinases that suppress macroautophagy in optimal growth conditions. Autophagy. 2011;7:892–903. doi: 10.4161/auto.7.8.15770. [DOI] [PubMed] [Google Scholar]
- 167.Sarkar S, Korolchuk V, Renna M, Winslow A, Rubinsztein DC. Methodological considerations for assessing autophagy modulators: a study with calcium phosphate precipitates. Autophagy. 2009;5:307–13. doi: 10.4161/auto.5.3.7664. [DOI] [PubMed] [Google Scholar]
- 168.Iwata J, Ezaki J, Komatsu M, Yokota S, Ueno T, Tanida I, et al. Excess peroxisomes are degraded by autophagic machinery in mammals. J Biol Chem. 2006;281:4035–41. doi: 10.1074/jbc.M512283200. [DOI] [PubMed] [Google Scholar]
- 169.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nogalska A, Terracciano C, D’Agostino C, King Engel W, Askanas V. p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol. 2009;118:407–13. doi: 10.1007/s00401-009-0564-6. [DOI] [PubMed] [Google Scholar]
- 171.Hosokawa N, Hara Y, Mizushima N. Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett. 2006;580:2623–9. doi: 10.1016/j.febslet.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 172.Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, et al. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy. 2011;7:188–204. doi: 10.4161/auto.7.2.14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Gutierrez MG, Saka HA, Chinen I, Zoppino FC, Yoshimori T, Bocco JL, et al. Protective role of autophagy against Vibrio cholerae cytolysin, a pore-forming toxin from V. cholerae. Proc Natl Acad Sci U S A. 2007;104:1829–34. doi: 10.1073/pnas.0601437104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Suttangkakul A, Li F, Chung T, Vierstra RD. The ATG1/ATG13 protein kinase complex is both a regulator and a target of autophagic recycling in Arabidopsis. Plant Cell. 2011;23:3761–79. doi: 10.1105/tpc.111.090993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Balgi AD, Fonseca BD, Donohue E, Tsang TC, Lajoie P, Proud CG, et al. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One. 2009;4:e7124. doi: 10.1371/journal.pone.0007124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Patterson GH, Lippincott-Schwartz J. Selective photolabeling of proteins using photoactivatable GFP. Methods. 2004;32:445–50. doi: 10.1016/j.ymeth.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 177.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–87. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 178.Noda T, Klionsky DJ. The quantitative Pho8Δ60 assay of nonspecific autophagy. Methods Enzymol. 2008;451:33–42. doi: 10.1016/S0076-6879(08)03203-5. [DOI] [PubMed] [Google Scholar]
- 179.Klionsky DJ. Monitoring autophagy in yeast: the Pho8Delta60 assay. Methods Mol Biol. 2007;390:363–71. doi: 10.1007/978-1-59745-466-7_24. [DOI] [PubMed] [Google Scholar]
- 180.Patterson GH, Knobel SM, Sharif WD, Kain SR, Piston DW. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys J. 1997;73:2782–90. doi: 10.1016/S0006-3495(97)78307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–60. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 182.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–72. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 183.Strack RL, Keenan RJ, Glick BS. Noncytotoxic DsRed derivatives for whole-cell labeling. Methods Mol Biol. 2011;699:355–70. doi: 10.1007/978-1-61737-950-5_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gurskaya NG, Verkhusha VV, Shcheglov AS, Staroverov DB, Chepurnykh TV, Fradkov AF, et al. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol. 2006;24:461–5. doi: 10.1038/nbt1191. [DOI] [PubMed] [Google Scholar]
- 185.Ryhänen T, Hyttinen JM, Kopitz J, Rilla K, Kuusisto E, Mannermaa E, et al. Crosstalk between Hsp70 molecular chaperone, lysosomes and proteasomes in autophagy-mediated proteolysis in human retinal pigment epithelial cells. J Cell Mol Med. 2009;13:3616–31. doi: 10.1111/j.1582-4934.2008.00577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Rekas A, Alattia JR, Nagai T, Miyawaki A, Ikura M. Crystal structure of venus, a yellow fluorescent protein with improved maturation and reduced environmental sensitivity. J Biol Chem. 2002;277:50573–8. doi: 10.1074/jbc.M209524200. [DOI] [PubMed] [Google Scholar]
- 187.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Badr CE, Wurdinger T, Nilsson J, Niers JM, Whalen M, Degterev A, et al. Lanatoside C sensitizes glioblastoma cells to tumor necrosis factor-related apoptosis-inducing ligand and induces an alternative cell death pathway. Neuro Oncol. 2011;13:1213–24. doi: 10.1093/neuonc/nor067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Meléndez A, Tallóczy Z, Seaman M, Eskelinen E-L, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301:1387–91. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- 190.Otto GP, Wu MY, Kazgan N, Anderson OR, Kessin RH. Macroautophagy is required for multicellular development of the social amoeba Dictyostelium discoideum. J Biol Chem. 2003;278:17636–45. doi: 10.1074/jbc.M212467200. [DOI] [PubMed] [Google Scholar]
- 191.Liu XH, Liu TB, Lin FC. Monitoring autophagy in Magnaporthe oryzae. Methods Enzymol. 2008;451:271–94. doi: 10.1016/S0076-6879(08)03219-9. [DOI] [PubMed] [Google Scholar]
- 192.Pinan-Lucarré B, Paoletti M, Dementhon K, Coulary-Salin B, Clavé C. Autophagy is induced during cell death by incompatibility and is essential for differentiation in the filamentous fungus Podospora anserina. Mol Microbiol. 2003;47:321–33. doi: 10.1046/j.1365-2958.2003.03208.x. [DOI] [PubMed] [Google Scholar]
- 193.Veneault-Fourrey C, Barooah M, Egan M, Wakley G, Talbot NJ. Autophagic fungal cell death is necessary for infection by the rice blast fungus. Science. 2006;312:580–3. doi: 10.1126/science.1124550. [DOI] [PubMed] [Google Scholar]
- 194.Kikuma T, Ohneda M, Arioka M, Kitamoto K. Functional analysis of the ATG8 homologue Aoatg8 and role of autophagy in differentiation and germination in Aspergillus oryzae. Eukaryot Cell. 2006;5:1328–36. doi: 10.1128/EC.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Nolting N, Bernhards Y, Pöggeler S. SmATG7 is required for viability in the homothallic ascomycete Sordaria macrospora. Fungal Genet Biol. 2009;46:531–42. doi: 10.1016/j.fgb.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 196.Baghdiguian S, Martinand-Mari C, Mangeat P. Using Ciona to study developmental programmed cell death. Semin Cancer Biol. 2007;17:147–53. doi: 10.1016/j.semcancer.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 197.Rusten TE, Lindmo K, Juhász G, Sass M, Seglen PO, Brech A, et al. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev Cell. 2004;7:179–92. doi: 10.1016/j.devcel.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 198.Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004;7:167–78. doi: 10.1016/j.devcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 199.Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, et al. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19:1741–6. doi: 10.1016/j.cub.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Yoshimoto K, Hanaoka H, Sato S, Kato T, Tabata S, Noda T, et al. Processing of ATG8s, ubiquitin-like proteins, and their deconjugation by ATG4s are essential for plant autophagy. Plant Cell. 2004;16:2967–83. doi: 10.1105/tpc.104.025395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Besteiro S, Brooks CF, Striepen B, Dubremetz J-F. Autophagy protein Atg3 is essential for maintaining mitochondrial integrity and for normal intracellular development of Toxoplasma gondii tachyzoites. PLoS Pathog. 2011;7:e1002416. doi: 10.1371/journal.ppat.1002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Williams RA, Tetley L, Mottram JC, Coombs GH. Cysteine peptidases CPA and CPB are vital for autophagy and differentiation in Leishmania mexicana. Mol Microbiol. 2006;61:655–74. doi: 10.1111/j.1365-2958.2006.05274.x. [DOI] [PubMed] [Google Scholar]
- 203.Williams RA, Woods KL, Juliano L, Mottram JC, Coombs GH. Characterization of unusual families of ATG8-like proteins and ATG12 in the protozoan parasite Leishmania major. Autophagy. 2009;5:159–72. doi: 10.4161/auto.5.2.7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Elsässer A, Vogt AM, Nef H, Kostin S, Möllmann H, Skwara W, et al. Human hibernating myocardium is jeopardized by apoptotic and autophagic cell death. J Am Coll Cardiol. 2004;43:2191–9. doi: 10.1016/j.jacc.2004.02.053. [DOI] [PubMed] [Google Scholar]
- 205.Knaapen MW, Davies MJ, De Bie M, Haven AJ, Martinet W, Kockx MM. Apoptotic versus autophagic cell death in heart failure. Cardiovasc Res. 2001;51:304–12. doi: 10.1016/S0008-6363(01)00290-5. [DOI] [PubMed] [Google Scholar]
- 206.Kostin S, Pool L, Elsässer A, Hein S, Drexler HC, Arnon E, et al. Myocytes die by multiple mechanisms in failing human hearts. Circ Res. 2003;92:715–24. doi: 10.1161/01.RES.0000067471.95890.5C. [DOI] [PubMed] [Google Scholar]
- 207.Pérez-Pérez ME, Florencio FJ, Crespo JL. Inhibition of target of rapamycin signaling and stress activate autophagy in Chlamydomonas reinhardtii. Plant Physiol. 2010;152:1874–88. doi: 10.1104/pp.109.152520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Koike M, Shibata M, Waguri S, Yoshimura K, Tanida I, Kominami E, et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease) Am J Pathol. 2005;167:1713–28. doi: 10.1016/S0002-9440(10)61253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ost A, Svensson K, Ruishalme I, Brännmark C, Franck N, Krook H, et al. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol Med. 2010;16:235–46. doi: 10.2119/molmed.2010.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–92. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gniadek TJ, Warren G. WatershedCounting3D: a new method for segmenting and counting punctate structures from confocal image data. Traffic. 2007;8:339–46. doi: 10.1111/j.1600-0854.2007.00538.x. [DOI] [PubMed] [Google Scholar]
- 212.Decuypere J-P, Welkenhuyzen K, Luyten T, Ponsaerts R, Dewaele M, Molgó J, et al. IP 3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy. 2011;7:1472–89. doi: 10.4161/auto.7.12.17909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Amer AO, Swanson MS. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell Microbiol. 2005;7:765–78. doi: 10.1111/j.1462-5822.2005.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–66. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 215.Ogawa M, Sasakawa C. Intracellular survival of Shigella. Cell Microbiol. 2006;8:177–84. doi: 10.1111/j.1462-5822.2005.00652.x. [DOI] [PubMed] [Google Scholar]
- 216.Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, et al. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics. 2011;27:1179–80. doi: 10.1093/bioinformatics/btr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Wu JQ, Pollard TD. Counting cytokinesis proteins globally and locally in fission yeast. Science. 2005;310:310–4. doi: 10.1126/science.1113230. [DOI] [PubMed] [Google Scholar]
- 218.Geng J, Baba M, Nair U, Klionsky DJ. Quantitative analysis of autophagy-related protein stoichiometry by fluorescence microscopy. J Cell Biol. 2008;182:129–40. doi: 10.1083/jcb.200711112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Brady NR, Hamacher-Brady A, Yuan H, Gottlieb RA. The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. FEBS J. 2007;274:3184–97. doi: 10.1111/j.1742-4658.2007.05849.x. [DOI] [PubMed] [Google Scholar]
- 220.Qadir MA, Kwok B, Dragowska WH, To KH, Le D, Bally MB, et al. Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res Treat. 2008;112:389–403. doi: 10.1007/s10549-007-9873-4. [DOI] [PubMed] [Google Scholar]
- 221.Dolloff NG, Ma X, Dicker DT, Humphreys RC, Li LZ, El-Deiry WS. Spectral imaging-based methods for quantifying autophagy and apoptosis. Cancer Biol Ther. 2011;12:349–56. doi: 10.4161/cbt.12.4.17175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Phadwal K, Alegre-Abarrategui J, Watson AS, Pike L, Anbalagan S, Hammond EM, et al. A novel method for autophagy detection in primary cells: Impaired levels of macroautophagy in immunosenescent T cells. Autophagy. 2012;8:677–89. doi: 10.4161/auto.18935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 224.Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 2007;3:323–8. doi: 10.4161/auto.4012. [DOI] [PubMed] [Google Scholar]
- 225.Szeto J, Kaniuk NA, Canadien V, Nisman R, Mizushima N, Yoshimori T, et al. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy. 2006;2:189–99. doi: 10.4161/auto.2731. [DOI] [PubMed] [Google Scholar]
- 226.Kaniuk NA, Kiraly M, Bates H, Vranic M, Volchuk A, Brumell JH. Ubiquitinated-protein aggregates form in pancreatic beta-cells during diabetes-induced oxidative stress and are regulated by autophagy. Diabetes. 2007;56:930–9. doi: 10.2337/db06-1160. [DOI] [PubMed] [Google Scholar]
- 227.Fujita K, Maeda D, Xiao Q, Srinivasula SM. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci U S A. 2011;108:1427–32. doi: 10.1073/pnas.1014156108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Pierre P. Dendritic cells, DRiPs, and DALIS in the control of antigen processing. Immunol Rev. 2005;207:184–90. doi: 10.1111/j.0105-2896.2005.00300.x. [DOI] [PubMed] [Google Scholar]
- 229.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 230.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 231.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 232.Calvo-Garrido J, Escalante R. Autophagy dysfunction and ubiquitin-positive protein aggregates in Dictyostelium cells lacking Vmp1. Autophagy. 2010;6:100–9. doi: 10.4161/auto.6.1.10697. [DOI] [PubMed] [Google Scholar]
- 233.Bjørkøy G, Lamark T, Johansen T. p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy. 2006;2:138–9. doi: 10.4161/auto.2.2.2405. [DOI] [PubMed] [Google Scholar]
- 234.Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279–89. doi: 10.1016/j.molcel.2011.07.039. [DOI] [PubMed] [Google Scholar]
- 235.Köchl R, Hu XW, Chan EYW, Tooze SA. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 2006;7:129–45. doi: 10.1111/j.1600-0854.2005.00368.x. [DOI] [PubMed] [Google Scholar]
- 236.Eng KE, Panas MD, Hedestam GB, McInerney GM. A novel quantitative flow cytometry-based assay for autophagy. Autophagy. 2010;6:634–41. doi: 10.4161/auto.6.5.12112. [DOI] [PubMed] [Google Scholar]
- 237.Ciechomska IA, Tolkovsky AM. Non-autophagic GFP-LC3 puncta induced by saponin and other detergents. Autophagy. 2007;3:586–90. doi: 10.4161/auto.4843. [DOI] [PubMed] [Google Scholar]
- 238.Tsvetkov AS, Miller J, Arrasate M, Wong JS, Pleiss MA, Finkbeiner S. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A. 2010;107:16982–7. doi: 10.1073/pnas.1004498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Joo JH, Dorsey FC, Joshi A, Hennessy-Walters KM, Rose KL, McCastlain K, et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol Cell. 2011;43:572–85. doi: 10.1016/j.molcel.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008;19:4651–9. doi: 10.1091/mbc.E08-03-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Decuypere JP, Bultynck G, Parys JB. A dual role for Ca2+ in autophagy regulation. Cell Calcium. 2011;50:242–50. doi: 10.1016/j.ceca.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 242.Nyfeler B, Bergman P, Triantafellow E, Wilson CJ, Zhu Y, Radetich B, et al. Relieving autophagy and 4EBP1 from rapamycin resistance. Mol Cell Biol. 2011;31:2867–76. doi: 10.1128/MCB.05430-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.García-Martínez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM, et al. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR) Biochem J. 2009;421:29–42. doi: 10.1042/BJ20090489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Pereira GJ, Hirata H, Fimia GM, do Carmo LG, Bincoletto C, Han SW, et al. Nicotinic acid adenine dinucleotide phosphate (NAADP) regulates autophagy in cultured astrocytes. J Biol Chem. 2011;286:27875–81. doi: 10.1074/jbc.C110.216580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68:8022–30. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 246.Liu TJ, Koul D, LaFortune T, Tiao N, Shen RJ, Maira SM, et al. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol Cancer Ther. 2009;8:2204–10. doi: 10.1158/1535-7163.MCT-09-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem. 2010;285:9100–13. doi: 10.1074/jbc.M109.060061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Puissant A, Auberger P. AMPK- and p62/SQSTM1-dependent autophagy mediate Resveratrol-induced cell death in chronic myelogenous leukemia. Autophagy. 2010;6:655–7. doi: 10.4161/auto.6.5.12126. [DOI] [PubMed] [Google Scholar]
- 249.Vingtdeux V, Chandakkar P, Zhao H, d’Abramo C, Davies P, Marambaud P. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-β peptide degradation. FASEB J. 2011;25:219–31. doi: 10.1096/fj.10-167361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Gordon PB, Holen I, Fosse M, Røtnes JS, Seglen PO. Dependence of hepatocytic autophagy on intracellularly sequestered calcium. J Biol Chem. 1993;268:26107–12. [PubMed] [Google Scholar]
- 251.Ganley IG, Wong PM, Gammoh N, Jiang X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell. 2011;42:731–43. doi: 10.1016/j.molcel.2011.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–32. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–52. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 254.Casarejos MJ, Solano RM, Gómez A, Perucho J, de Yébenes JG, Mena MA. The accumulation of neurotoxic proteins, induced by proteasome inhibition, is reverted by trehalose, an enhancer of autophagy, in human neuroblastoma cells. Neurochem Int. 2011;58:512–20. doi: 10.1016/j.neuint.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 255.Carpenter JE, Jackson W, Benetti L, Grose C. Autophagosome formation during varicella-zoster virus infection following endoplasmic reticulum stress and the unfolded protein response. J Virol. 2011;85:9414–24. doi: 10.1128/JVI.00281-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889–92. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285:10850–61. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]
- 259.Tormo D, Checin´ska A, Alonso-Curbelo D, Pérez-Guijarro E, Cañón E, Riveiro-Falkenbach E, et al. Targeted activation of innate immunity for therapeutic induction of autophagy and apoptosis in melanoma cells. Cancer Cell. 2009;16:103–14. doi: 10.1016/j.ccr.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010;32:227–39. doi: 10.1016/j.immuni.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Tamura N, Oku M, Sakai Y. Atg8 regulates vacuolar membrane dynamics in a lipidation-independent manner in Pichia pastoris. J Cell Sci. 2010;123:4107–16. doi: 10.1242/jcs.070045. [DOI] [PubMed] [Google Scholar]
- 262.Strømhaug PE, Reggiori F, Guan J, Wang C-W, Klionsky DJ. Atg21 is a phosphoinositide binding protein required for efficient lipidation and localization of Atg8 during uptake of aminopeptidase I by selective autophagy. Mol Biol Cell. 2004;15:3553–66. doi: 10.1091/mbc.E04-02-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Baens M, Noels H, Broeckx V, Hagens S, Fevery S, Billiau AD, et al. The dark side of EGFP: defective polyubiquitination. PLoS One. 2006;1:e54. doi: 10.1371/journal.pone.0000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Calì T, Galli C, Olivari S, Molinari M. Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem Biophys Res Commun. 2008;371:405–10. doi: 10.1016/j.bbrc.2008.04.098. [DOI] [PubMed] [Google Scholar]
- 265.Al-Younes HM, Al-Zeer MA, Khalil H, Gussmann J, Karlas A, Machuy N, et al. Autophagy-independent function of MAP-LC3 during intracellular propagation of Chlamydia trachomatis. Autophagy. 2011;7:814–28. doi: 10.4161/auto.7.8.15597. [DOI] [PubMed] [Google Scholar]
- 266.Shroff H, Galbraith CG, Galbraith JA, White H, Gillette J, Olenych S, et al. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc Natl Acad Sci U S A. 2007;104:20308–13. doi: 10.1073/pnas.0710517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–34. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Cherra SJ, III, Kulich SM, Uechi G, Balasubramani M, Mountzouris J, Day BW, et al. Regulation of the autophagy protein LC3 by phosphorylation. J Cell Biol. 2010;190:533–9. doi: 10.1083/jcb.201002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Sarkar S, Korolchuk VI, Renna M, Imarisio S, Fleming A, Williams A, et al. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell. 2011;43:19–32. doi: 10.1016/j.molcel.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Rosado CJ, Mijaljica D, Hatzinisiriou I, Prescott M, Devenish RJ. Rosella: a fluorescent pH-biosensor for reporting vacuolar turnover of cytosol and organelles in yeast. Autophagy. 2008;4:205–13. doi: 10.4161/auto.5331. [DOI] [PubMed] [Google Scholar]
- 271.Mijaljica D, Rosado CJ, Devenish RJ, Prescott M. Biosensors for monitoring autophagy In: Serra PA, ed. Biosensors-Emerging Materials and Applications. Croatia: InTech, 2011:383-400. [Google Scholar]
- 272a.Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 2010;90:1103–63. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]
- 272b.Zhou C, Zhong W, Zhou J, Sheng F, Fang Z, et al. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy. 2012 doi: 10.4161/auto.20284. In press. [DOI] [PubMed] [Google Scholar]
- 273.Zhou J, Lin J, Zhou C, Deng X, Xia B. Cytotoxicity of red fluorescent protein DsRed is associated with the suppression of Bcl-xL translation. FEBS Lett. 2011;585:821–7. doi: 10.1016/j.febslet.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 274.Loos B, Genade S, Ellis B, Lochner A, Engelbrecht AM. At the core of survival: autophagy delays the onset of both apoptotic and necrotic cell death in a model of ischemic cell injury. Exp Cell Res. 2011;317:1437–53. doi: 10.1016/j.yexcr.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 275.Shvets E, Fass E, Elazar Z. Utilizing flow cytometry to monitor autophagy in living mammalian cells. Autophagy. 2008;4:621–8. doi: 10.4161/auto.5939. [DOI] [PubMed] [Google Scholar]
- 276.Hundeshagen P, Hamacher-Brady A, Eils R, Brady NR. Concurrent detection of autolysosome formation and lysosomal degradation by flow cytometry in a high-content screen for inducers of autophagy. BMC Biol. 2011;9:38. doi: 10.1186/1741-7007-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.de la Calle C, Joubert PE, Law HK, Hasan M, Albert ML. Simultaneous assessment of autophagy and apoptosis using multispectral imaging cytometry. Autophagy. 2011;7:1045–51. doi: 10.4161/auto.7.9.16252. [DOI] [PubMed] [Google Scholar]
- 278.Gannagé M, Dormann D, Albrecht R, Dengjel J, Torossi T, Rämer PC, et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe. 2009;6:367–80. doi: 10.1016/j.chom.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Kaminskyy V, Abdi A, Zhivotovsky B. A quantitative assay for the monitoring of autophagosome accumulation in different phases of the cell cycle. Autophagy. 2011;7:83–90. doi: 10.4161/auto.7.1.13893. [DOI] [PubMed] [Google Scholar]
- 280.Kirkin V, Lamark T, Sou YS, Bjørkøy G, Nunn JL, Bruun JA, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–16. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 281.Larsen KB, Lamark T, Øvervatn A, Harneshaug I, Johansen T, Bjørkøy G. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy. 2010;6:784–93. doi: 10.4161/auto.6.6.12510. [DOI] [PubMed] [Google Scholar]
- 282.Huang JJ, Li HR, Huang Y, Jiang WQ, Xu RH, Huang HQ, et al. Beclin 1 expression: a predictor of prognosis in patients with extranodal natural killer T-cell lymphoma, nasal type. Autophagy. 2010;6:777–83. doi: 10.4161/auto.6.6.12784. [DOI] [PubMed] [Google Scholar]
- 283.Sivridis E, Koukourakis MI, Zois CE, Ledaki I, Ferguson DJ, Harris AL, et al. LC3A-positive light microscopy detected patterns of autophagy and prognosis in operable breast carcinomas. Am J Pathol. 2010;176:2477–89. doi: 10.2353/ajpath.2010.090049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Sivridis E, Giatromanolaki A, Liberis V, Koukourakis MI. Autophagy in endometrial carcinomas and prognostic relevance of ‘stone-like’ structures (SLS): what is destined for the atypical endometrial hyperplasia? Autophagy. 2011;7:74–82. doi: 10.4161/auto.7.1.13947. [DOI] [PubMed] [Google Scholar]
- 285.Giatromanolaki A, Koukourakis MI, Koutsopoulos A, Chloropoulou P, Liberis V, Sivridis E. High Beclin 1 expression defines a poor prognosis in endometrial adenocarcinomas. Gynecol Oncol. 2011;123:147–51. doi: 10.1016/j.ygyno.2011.06.023. [DOI] [PubMed] [Google Scholar]
- 286.Chen Y, Lu Y, Lu C, Zhang L. Beclin-1 expression is a predictor of clinical outcome in patients with esophageal squamous cell carcinoma and correlated to hypoxia-inducible factor (HIF)-1α expression. Pathol Oncol Res. 2009;15:487–93. doi: 10.1007/s12253-008-9143-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Wan X-B, Fan X-J, Chen M-Y, Xiang J, Huang P-Y, Guo L, et al. Elevated Beclin 1 expression is correlated with HIF-1α in predicting poor prognosis of nasopharyngeal carcinoma. Autophagy. 2010;6:395–404. doi: 10.4161/auto.6.3.11303. [DOI] [PubMed] [Google Scholar]
- 288.Shi YH, Ding ZB, Zhou J, Qiu SJ, Fan J. Prognostic significance of Beclin 1-dependent apoptotic activity in hepatocellular carcinoma. Autophagy. 2009;5:380–2. doi: 10.4161/auto.5.3.7658. [DOI] [PubMed] [Google Scholar]
- 289.Ding ZB, Shi YH, Zhou J, Qiu SJ, Xu Y, Dai Z, et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008;68:9167–75. doi: 10.1158/0008-5472.CAN-08-1573. [DOI] [PubMed] [Google Scholar]
- 290.Pirtoli L, Cevenini G, Tini P, Vannini M, Oliveri G, Marsili S, et al. The prognostic role of Beclin 1 protein expression in high-grade gliomas. Autophagy. 2009;5:930–6. doi: 10.4161/auto.5.7.9227. [DOI] [PubMed] [Google Scholar]
- 291.Karpathiou G, Sivridis E, Koukourakis MI, Mikroulis D, Bouros D, Froudarakis ME, et al. Light-chain 3A autophagic activity and prognostic significance in non-small cell lung carcinomas. Chest. 2011;140:127–34. doi: 10.1378/chest.10-1831. [DOI] [PubMed] [Google Scholar]
- 292.Fujii S, Mitsunaga S, Yamazaki M, Hasebe T, Ishii G, Kojima M, et al. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci. 2008;99:1813–9. doi: 10.1111/j.1349-7006.2008.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Li BX, Li CY, Peng RQ, Wu XJ, Wang HY, Wan DS, et al. The expression of beclin 1 is associated with favorable prognosis in stage IIIB colon cancers. Autophagy. 2009;5:303–6. doi: 10.4161/auto.5.3.7491. [DOI] [PubMed] [Google Scholar]
- 294.Koukourakis MI, Giatromanolaki A, Sivridis E, Pitiakoudis M, Gatter KC, Harris AL. Beclin 1 over- and underexpression in colorectal cancer: distinct patterns relate to prognosis and tumour hypoxia. Br J Cancer. 2010;103:1209–14. doi: 10.1038/sj.bjc.6605904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Giatromanolaki A, Koukourakis MI, Harris AL, Polychronidis A, Gatter KC, Sivridis E. Prognostic relevance of light chain 3 (LC3A) autophagy patterns in colorectal adenocarcinomas. J Clin Pathol. 2010;63:867–72. doi: 10.1136/jcp.2010.079525. [DOI] [PubMed] [Google Scholar]
- 296.Sivridis E, Koukourakis MI, Mendrinos SE, Karpouzis A, Fiska A, Kouskoukis C, et al. Beclin-1 and LC3A expression in cutaneous malignant melanomas: a biphasic survival pattern for beclin-1. Melanoma Res. 2011;21:188–95. doi: 10.1097/CMR.0b013e328346612c. [DOI] [PubMed] [Google Scholar]
- 297.Giatromanolaki AN, Charitoudis GS, Bechrakis NE, Kozobolis VP, Koukourakis MI, Foerster MH, et al. Autophagy patterns and prognosis in uveal melanomas. Mod Pathol. 2011;24:1036–45. doi: 10.1038/modpathol.2011.63. [DOI] [PubMed] [Google Scholar]
- 298.McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM, Statistics Subcommittee of the NCI-EORTC Working Group on Cancer Diagnostics Reporting recommendations for tumor marker prognostic studies (REMARK) J Natl Cancer Inst. 2005;97:1180–4. doi: 10.1093/jnci/dji237. [DOI] [PubMed] [Google Scholar]
- 299.Kuwahara Y, Oikawa T, Ochiai Y, Roudkenar MH, Fukumoto M, Shimura T, et al. Enhancement of autophagy is a potential modality for tumors refractory to radiotherapy. Cell Death Dis. 2011;2:e177. doi: 10.1038/cddis.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Hou YJ, Dong LW, Tan YX, Yang GZ, Pan YF, Li Z, et al. Inhibition of active autophagy induces apoptosis and increases chemosensitivity in cholangiocarcinoma. Lab Invest. 2011;91:1146–57. doi: 10.1038/labinvest.2011.97. [DOI] [PubMed] [Google Scholar]
- 301.O’Donovan TR, O’Sullivan GC, McKenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy. 2011;7:509–24. doi: 10.4161/auto.7.5.15066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Yoshimura K, Shibata M, Koike M, Gotoh K, Fukaya M, Watanabe M, et al. Effects of RNA interference of Atg4B on the limited proteolysis of LC3 in PC12 cells and expression of Atg4B in various rat tissues. Autophagy. 2006;2:200–8. doi: 10.4161/auto.2744. [DOI] [PubMed] [Google Scholar]
- 303.Tamura H, Shibata M, Koike M, Sasaki M, Uchiyama Y. Atg9A protein, an autophagy-related membrane protein, is localized in the neurons of mouse brains. J Histochem Cytochem. 2010;58:443–53. doi: 10.1369/jhc.2010.955690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Cui J, Bai XY, Shi S, Cui S, Hong Q, Cai G, et al. Age-related changes in the function of autophagy in rat kidneys. Age (Dordr) 2011;34:329–39. doi: 10.1007/s11357-011-9237-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Mellén MA, de la Rosa EJ, Boya P. The autophagic machinery is necessary for removal of cell corpses from the developing retinal neuroepithelium. Cell Death Differ. 2008;15:1279–90. doi: 10.1038/cdd.2008.40. [DOI] [PubMed] [Google Scholar]
- 306.Mellén MA, de la Rosa EJ, Boya P. Autophagy is not universally required for phosphatidyl-serine exposure and apoptotic cell engulfment during neural development. Autophagy. 2009;5:964–72. doi: 10.4161/auto.5.7.9292. [DOI] [PubMed] [Google Scholar]
- 307.Shibata M, Yoshimura K, Furuya N, Koike M, Ueno T, Komatsu M, et al. The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun. 2009;382:419–23. doi: 10.1016/j.bbrc.2009.03.039. [DOI] [PubMed] [Google Scholar]
- 308.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–63. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 309.Germain M, Nguyen AP, Le Grand JN, Arbour N, Vanderluit JL, Park DS, et al. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J. 2011;30:395–407. doi: 10.1038/emboj.2010.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr., Iwata J, Kominami E, et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104:14489–94. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Wang QJ, Ding Y, Kohtz DS, Mizushima N, Cristea IM, Rout MP, et al. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26:8057–68. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Nezis IP, Simonsen A, Sagona AP, Finley K, Gaumer S, Contamine D, et al. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol. 2008;180:1065–71. doi: 10.1083/jcb.200711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Bartlett BJ, Isakson P, Lewerenz J, Sanchez H, Kotzebue RW, Cumming RC, et al. p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy. 2011;7:572–83. doi: 10.4161/auto.7.6.14943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–58. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10:507–15. doi: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 316.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–4. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44:134–46. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–23. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 319.Jain A, Lamark T, Sjøttem E, Larsen KB, Awuh JA, Øvervatn A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–91. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Bardag-Gorce F, Francis T, Nan L, Li J, He Lue Y, French BA, et al. Modifications in P62 occur due to proteasome inhibition in alcoholic liver disease. Life Sci. 2005;77:2594–602. doi: 10.1016/j.lfs.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 321.Myeku N, Figueiredo-Pereira ME. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J Biol Chem. 2011;286:22426–40. doi: 10.1074/jbc.M110.149252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Norman JM, Cohen GM, Bampton ET. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy. 2010;6:1042–56. doi: 10.4161/auto.6.8.13337. [DOI] [PubMed] [Google Scholar]
- 323.Lelouard H, Schmidt EK, Camosseto V, Clavarino G, Ceppi M, Hsu HT, et al. Regulation of translation is required for dendritic cell function and survival during activation. J Cell Biol. 2007;179:1427–39. doi: 10.1083/jcb.200707166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–7. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- 325.Nakaso K, Yoshimoto Y, Nakano T, Takeshima T, Fukuhara Y, Yasui K, et al. Transcriptional activation of p62/A170/ZIP during the formation of the aggregates: possible mechanisms and the role in Lewy body formation in Parkinson’s disease. Brain Res. 2004;1012:42–51. doi: 10.1016/j.brainres.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 326.Zheng Q, Su H, Ranek MJ, Wang X. Autophagy and p62 in cardiac proteinopathy. Circ Res. 2011;109:296–308. doi: 10.1161/CIRCRESAHA.111.244707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Colosetti P, Puissant A, Robert G, Luciano F, Jacquel A, Gounon P, et al. Autophagy is an important event for megakaryocytic differentiation of the chronic myelogenous leukemia K562 cell line. Autophagy. 2009;5:1092–8. doi: 10.4161/auto.5.8.9889. [DOI] [PubMed] [Google Scholar]
- 328.Toepfer N, Childress C, Parikh A, Rukstalis D, Yang W. Atorvastatin induces autophagy in prostate cancer PC3 cells through activation of LC3 transcription. Cancer Biol Ther. 2011;12:691–9. doi: 10.4161/cbt.12.8.15978. [DOI] [PubMed] [Google Scholar]
- 329.BenYounès A, Tajeddine N, Tailler M, Malik SA, Shen S, Métivier D, et al. A fluorescence-microscopic and cytofluorometric system for monitoring the turnover of the autophagic substrate p62/SQSTM1. Autophagy. 2011;7:883–91. doi: 10.4161/auto.7.8.15538. [DOI] [PubMed] [Google Scholar]
- 330.Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–97. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- 331.Lim J, Kim HW, Youdim MB, Rhyu IJ, Choe KM, Oh YJ. Binding preference of p62 towards LC3-ll during dopaminergic neurotoxin-induced impairment of autophagic flux. Autophagy. 2011;7:51–60. doi: 10.4161/auto.7.1.13909. [DOI] [PubMed] [Google Scholar]
- 332.Waguri S, Komatsu M. Biochemical and morphological detection of inclusion bodies in autophagy-deficient mice. Methods Enzymol. 2009;453:181–96. doi: 10.1016/S0076-6879(08)04009-3. [DOI] [PubMed] [Google Scholar]
- 333.Hocking LJ, Lucas GJ, Daroszewska A, Mangion J, Olavesen M, Cundy T, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet. 2002;11:2735–9. doi: 10.1093/hmg/11.22.2735. [DOI] [PubMed] [Google Scholar]
- 334.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–30. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Maloverjan A, Piirsoo M, Michelson P, Kogerman P, Osterlund T. Identification of a novel serine/threonine kinase ULK3 as a positive regulator of Hedgehog pathway. Exp Cell Res. 2010;316:627–37. doi: 10.1016/j.yexcr.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 336.Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Chan EY, Tooze SA. Evolution of Atg1 function and regulation. Autophagy. 2009;5:758–65. doi: 10.4161/auto.8709. [DOI] [PubMed] [Google Scholar]
- 338.Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. 2007;282:25464–74. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- 339.Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Carling D, Mayer FV, Sanders MJ, Gamblin SJ. AMP-activated protein kinase: nature’s energy sensor. Nat Chem Biol. 2011;7:512–8. doi: 10.1038/nchembio.610. [DOI] [PubMed] [Google Scholar]
- 341.Samari HR, Møller MT, Holden L, Asmyhr T, Seglen PO. Stimulation of hepatocytic AMP-activated protein kinase by okadaic acid and other autophagy-suppressive toxins. Biochem J. 2005;386:237–44. doi: 10.1042/BJ20040609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Chiacchiera F, Matrone A, Ferrari E, Ingravallo G, Lo Sasso G, Murzilli S, et al. p38alpha blockade inhibits colorectal cancer growth in vivo by inducing a switch from HIF1alpha- to FoxO-dependent transcription. Cell Death Differ. 2009;16:1203–14. doi: 10.1038/cdd.2009.36. [DOI] [PubMed] [Google Scholar]
- 344.Kovács AL, Seglen PO. Inhibition of hepatocytic protein degradation by methylaminopurines and inhibitors of protein synthesis. Biochim Biophys Acta. 1981;676:213–20. doi: 10.1016/0304-4165(81)90189-6. [DOI] [PubMed] [Google Scholar]
- 345.Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, et al. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem. 2009;284:31484–92. doi: 10.1074/jbc.M109.033936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–71. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 347.Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud PD, Yu RT, Alvarez JG, et al. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell. 2011;145:607–21. doi: 10.1016/j.cell.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Pfisterer SG, Mauthe M, Codogno P, Proikas-Cezanne T. Ca2+/calmodulin-dependent kinase (CaMK) signaling via CaMKI and AMP-activated protein kinase contributes to the regulation of WIPI-1 at the onset of autophagy. Mol Pharmacol. 2011;80:1066–75. doi: 10.1124/mol.111.071761. [DOI] [PubMed] [Google Scholar]
- 349.Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1α and SIRT1 pathways. FEBS Lett. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Samari HR, Seglen PO. Inhibition of hepatocytic autophagy by adenosine, aminoimidazole-4-carboxamide riboside, and N6-mercaptopurine riboside. Evidence for involvement of amp-activated protein kinase. J Biol Chem. 1998;273:23758–63. doi: 10.1074/jbc.273.37.23758. [DOI] [PubMed] [Google Scholar]
- 351.Sanchez AM, Csibi A, Raibon A, Cornille K, Gay S, Bernardi H, et al. AMPK promotes skeletal muscle autophagy through activation of Forkhead FoxO3a and interaction with Ulk1. J Cell Biochem. 2012;113:695–710. doi: 10.1002/jcb.23399. [DOI] [PubMed] [Google Scholar]
- 352.Inoki K, Zhu T, Guan K-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/S0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 353.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Egan D, Kim J, Shaw RJ, Guan K-L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7:643–4. doi: 10.4161/auto.7.6.15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Emerling BM, Viollet B, Tormos KV, Chandel NS. Compound C inhibits hypoxic activation of HIF-1 independent of AMPK. FEBS Lett. 2007;581:5727–31. doi: 10.1016/j.febslet.2007.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Vucicevic L, Misirkic M, Janjetovic K, Vilimanovich U, Sudar E, Isenovic E, et al. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy. 2011;7:40–50. doi: 10.4161/auto.7.1.13883. [DOI] [PubMed] [Google Scholar]
- 360.Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, et al. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281:34870–9. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 361.Grotemeier A, Alers S, Pfisterer SG, Paasch F, Daubrawa M, Dieterle A, et al. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal. 2010;22:914–25. doi: 10.1016/j.cellsig.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 362.Williams T, Forsberg LJ, Viollet B, Brenman JE. Basal autophagy induction without AMP-activated protein kinase under low glucose conditions. Autophagy. 2009;5:1155–65. doi: 10.4161/auto.5.8.10090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A. 2011;108:4788–93. doi: 10.1073/pnas.1100844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–83. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol. 2010;191:155–68. doi: 10.1083/jcb.201002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Tang HW, Wang YB, Wang SL, Wu MH, Lin SY, Chen GC. Atg1-mediated myosin II activation regulates autophagosome formation during starvation-induced autophagy. EMBO J. 2011;30:636–51. doi: 10.1038/emboj.2010.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Jung CH, Jun CB, Ro S-H, Kim Y-M, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Chang Y-Y, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol Biol Cell. 2009;20:2004–14. doi: 10.1091/mbc.E08-12-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Chan EYW, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. 2009;29:157–71. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Jung CH, Seo M, Otto NM, Kim DH. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy. 2011;7:1212–21. doi: 10.4161/auto.7.10.16660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Löffler AS, Alers S, Dieterle AM, Keppeler H, Franz-Wachtel M, Kundu M, et al. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy. 2011;7:696–706. doi: 10.4161/auto.7.7.15451. [DOI] [PubMed] [Google Scholar]
- 373.Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis. 2007;26:86–93. doi: 10.1016/j.nbd.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 374.Lavieu G, Scarlatti F, Sala G, Carpentier S, Levade T, Ghidoni R, et al. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J Biol Chem. 2006;281:8518–27. doi: 10.1074/jbc.M506182200. [DOI] [PubMed] [Google Scholar]
- 375.Brunn GJ, Hudson CC, Sekulicá A, Williams JM, Hosoi H, Houghton PJ, et al. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science. 1997;277:99–101. doi: 10.1126/science.277.5322.99. [DOI] [PubMed] [Google Scholar]
- 376.Yip CK, Murata K, Walz T, Sabatini DM, Kang SA. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol Cell. 2010;38:768–74. doi: 10.1016/j.molcel.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Cheong H, Nair U, Geng J, Klionsky DJ. The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae. Mol Biol Cell. 2008;19:668–81. doi: 10.1091/mbc.E07-08-0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell. 2005;16:2544–53. doi: 10.1091/mbc.E04-08-0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150:1507–13. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Scott SV, Nice DC, III, Nau JJ, Weisman LS, Kamada Y, Keizer-Gunnink I, et al. Apg13p and Vac8p are part of a complex of phosphoproteins that are required for cytoplasm to vacuole targeting. J Biol Chem. 2000;275:25840–9. doi: 10.1074/jbc.M002813200. [DOI] [PubMed] [Google Scholar]
- 381.Mao K, Wang K, Zhao M, Xu T, Klionsky DJ. Two MAPK-signaling pathways are required for mitophagy in Saccharomyces cerevisiae. J Cell Biol. 2011;193:755–67. doi: 10.1083/jcb.201102092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Yeh YY, Wrasman K, Herman PK. Autophosphorylation within the Atg1 activation loop is required for both kinase activity and the induction of autophagy in Saccharomyces cerevisiae. Genetics. 2010;185:871–82. doi: 10.1534/genetics.110.116566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Singh K, Matsuyama S, Drazba JA, Almasan A. Autophagy-dependent senescence in response to DNA damage and chronic apoptotic stress. Autophagy. 2012;8:236–51. doi: 10.4161/auto.8.2.18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Djavaheri-Mergny M, Amelotti M, Mathieu J, Besançon F, Bauvy C, Souquère S, et al. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem. 2006;281:30373–82. doi: 10.1074/jbc.M602097200. [DOI] [PubMed] [Google Scholar]
- 385.Liu Z, Lenardo MJ. Reactive oxygen species regulate autophagy through redox-sensitive proteases. Dev Cell. 2007;12:484–5. doi: 10.1016/j.devcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 386.Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A, et al. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem. 2004;279:18384–91. doi: 10.1074/jbc.M313561200. [DOI] [PubMed] [Google Scholar]
- 387.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–60. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Zeng X, Kinsella TJ. Mammalian target of rapamycin and S6 kinase 1 positively regulate 6-thioguanine-induced autophagy. Cancer Res. 2008;68:2384–90. doi: 10.1158/0008-5472.CAN-07-6163. [DOI] [PubMed] [Google Scholar]
- 389.Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–6. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Mochizuki H, Toda H, Ando M, Kurusu M, Tomoda T, Furukubo-Tokunaga K. Unc-51/ATG1 controls axonal and dendritic development via kinesin-mediated vesicle transport in the Drosophila brain. PLoS One. 2011;6:e19632. doi: 10.1371/journal.pone.0019632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Wairkar YP, Toda H, Mochizuki H, Furukubo-Tokunaga K, Tomoda T, Diantonio A. Unc-51 controls active zone density and protein composition by downregulating ERK signaling. J Neurosci. 2009;29:517–28. doi: 10.1523/JNEUROSCI.3848-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Loh SH, Francescut L, Lingor P, Bähr M, Nicotera P. Identification of new kinase clusters required for neurite outgrowth and retraction by a loss-of-function RNA interference screen. Cell Death Differ. 2008;15:283–98. doi: 10.1038/sj.cdd.4402258. [DOI] [PubMed] [Google Scholar]
- 393.Zhou X, Babu JR, da Silva S, Shu Q, Graef IA, Oliver T, et al. Unc-51-like kinase 1/2-mediated endocytic processes regulate filopodia extension and branching of sensory axons. Proc Natl Acad Sci U S A. 2007;104:5842–7. doi: 10.1073/pnas.0701402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Tomoda T, Kim JH, Zhan C, Hatten ME. Role of Unc51.1 and its binding partners in CNS axon outgrowth. Genes Dev. 2004;18:541–58. doi: 10.1101/gad.1151204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Okazaki N, Yan J, Yuasa S, Ueno T, Kominami E, Masuho Y, et al. Interaction of the Unc-51-like kinase and microtubule-associated protein light chain 3 related proteins in the brain: possible role of vesicular transport in axonal elongation. Brain Res Mol Brain Res. 2000;85:1–12. doi: 10.1016/S0169-328X(00)00218-7. [DOI] [PubMed] [Google Scholar]
- 396.Young ARJ, Chan EYW, Hu XW, Köchl R, Crawshaw SG, High S, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006;119:3888–900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 397.Reggiori F, Shintani T, Nair U, Klionsky DJ. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy. 2005;1:101–9. doi: 10.4161/auto.1.2.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, Reggiori F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol. 2010;190:1005–22. doi: 10.1083/jcb.200912089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Reggiori F, Tucker KA, Stromhaug PE, Klionsky DJ. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell. 2004;6:79–90. doi: 10.1016/S1534-5807(03)00402-7. [DOI] [PubMed] [Google Scholar]
- 400.Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116:1679–88. doi: 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- 401.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–68. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Thompson AR, Doelling JH, Suttangkakul A, Vierstra RD. Autophagic nutrient recycling in Arabidopsis directed by the ATG8 and ATG12 conjugation pathways. Plant Physiol. 2005;138:2097–110. doi: 10.1104/pp.105.060673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–32. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 404.Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152:519–30. doi: 10.1083/jcb.152.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–96. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 406.Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11:468–76. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2008;105:19211–6. doi: 10.1073/pnas.0810452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–72. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Fan W, Nassiri A, Zhong Q. Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L) Proc Natl Acad Sci U S A. 2011;108:7769–74. doi: 10.1073/pnas.1016472108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol. 2010;190:511–21. doi: 10.1083/jcb.200911141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12:747–57. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Guan J, Stromhaug PE, George MD, Habibzadegah-Tari P, Bevan A, Dunn WA, Jr., et al. Cvt18/Gsa12 is required for cytoplasm-to-vacuole transport, pexophagy, and autophagy in Saccharomyces cerevisiae and Pichia pastoris. Mol Biol Cell. 2001;12:3821–38. doi: 10.1091/mbc.12.12.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Barth H, Meiling-Wesse K, Epple UD, Thumm M. Autophagy and the cytoplasm to vacuole targeting pathway both require Aut10p. FEBS Lett. 2001;508:23–8. doi: 10.1016/S0014-5793(01)03016-2. [DOI] [PubMed] [Google Scholar]
- 414.Proikas-Cezanne T, Waddell S, Gaugel A, Frickey T, Lupas A, Nordheim A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene. 2004;23:9314–25. doi: 10.1038/sj.onc.1208331. [DOI] [PubMed] [Google Scholar]
- 415.Monastyrska I, Klionsky DJ. Autophagy in organelle homeostasis: peroxisome turnover. Mol Aspects Med. 2006;27:483–94. doi: 10.1016/j.mam.2006.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Nair U, Klionsky DJ. Molecular mechanisms and regulation of specific and nonspecific autophagy pathways in yeast. J Biol Chem. 2005;280:41785–8. doi: 10.1074/jbc.R500016200. [DOI] [PubMed] [Google Scholar]
- 417.Tallóczy Z, Virgin HW, IV, Levine B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy. 2006;2:24–9. doi: 10.4161/auto.2176. [DOI] [PubMed] [Google Scholar]
- 418.Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbé S, Clague MJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6:506–22. doi: 10.4161/auto.6.4.11863. [DOI] [PubMed] [Google Scholar]
- 419.Proikas-Cezanne T, Ruckerbauer S, Stierhof YD, Berg C, Nordheim A. Human WIPI-1 puncta-formation: a novel assay to assess mammalian autophagy. FEBS Lett. 2007;581:3396–404. doi: 10.1016/j.febslet.2007.06.040. [DOI] [PubMed] [Google Scholar]
- 420.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–76. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Mauthe M, Jacob A, Freiberger S, Hentschel K, Stierhof YD, Codogno P, et al. Resveratrol-mediated autophagy requires WIPI-1 regulated LC3 lipidation in the absence of induced phagophore formation. Autophagy. 2011;7:1448–61. doi: 10.4161/auto.7.12.17802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Lu Q, Yang P, Huang X, Hu W, Guo B, Wu F, et al. The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev Cell. 2011;21:343–57. doi: 10.1016/j.devcel.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 423.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res. 2007;17:839–49. doi: 10.1038/cr.2007.78. [DOI] [PubMed] [Google Scholar]
- 425.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 426.Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009;10:285–92. doi: 10.1038/embor.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 2008;4:949–51. doi: 10.4161/auto.6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–88. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Lossi L, Gambino G, Ferrini F, Alasia S, Merighi A. Posttranslational regulation of BCL2 levels in cerebellar granule cells: A mechanism of neuronal survival. Dev Neurobiol. 2009;69:855–70. doi: 10.1002/dneu.20744. [DOI] [PubMed] [Google Scholar]
- 430.Lossi L, Gambino G, Salio C, Merighi A. Autophagy regulates the post-translational cleavage of BCL-2 and promotes neuronal survival. ScientificWorldJournal. 2010;10:924–9. doi: 10.1100/tsw.2010.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Scarlatti F, Maffei R, Beau I, Codogno P, Ghidoni R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ. 2008;15:1318–29. doi: 10.1038/cdd.2008.51. [DOI] [PubMed] [Google Scholar]
- 432.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–80. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001;2:330–5. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Amritraj A, Peake K, Kodam A, Salio C, Merighi A, Vance JE, et al. Increased activity and altered subcellular distribution of lysosomal enzymes determine neuronal vulnerability in Niemann-Pick type C1-deficient mice. Am J Pathol. 2009;175:2540–56. doi: 10.2353/ajpath.2009.081096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Castino R, Bellio N, Follo C, Murphy D, Isidoro C. Inhibition of PI3k class III-dependent autophagy prevents apoptosis and necrosis by oxidative stress in dopaminergic neuroblastoma cells. Toxicol Sci. 2010;117:152–62. doi: 10.1093/toxsci/kfq170. [DOI] [PubMed] [Google Scholar]
- 436.Yue Z, Horton A, Bravin M, DeJager PL, Selimi F, Heintz N. A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron. 2002;35:921–33. doi: 10.1016/S0896-6273(02)00861-9. [DOI] [PubMed] [Google Scholar]
- 437.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 439.Valbuena A, Castro-Obregón S, Lazo PA. Downregulation of VRK1 by p53 in response to DNA damage is mediated by the autophagic pathway. PLoS One. 2011;6:e17320. doi: 10.1371/journal.pone.0017320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Lorin S, Pierron G, Ryan KM, Codogno P, Djavaheri-Mergny M. Evidence for the interplay between JNK and p53-DRAM signalling pathways in the regulation of autophagy. Autophagy. 2010;6:153–4. doi: 10.4161/auto.6.1.10537. [DOI] [PubMed] [Google Scholar]
- 441.Nara A, Mizushima N, Yamamoto A, Kabeya Y, Ohsumi Y, Yoshimori T. SKD1 AAA ATPase-dependent endosomal transport is involved in autolysosome formation. Cell Struct Funct. 2002;27:29–37. doi: 10.1247/csf.27.29. [DOI] [PubMed] [Google Scholar]
- 442.Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, et al. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol. 1999;147:435–46. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230–9. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- 444.Moussay E, Kaoma T, Baginska J, Muller A, Van Moer K, Nicot N, et al. The acquisition of resistance to TNFα in breast cancer cells is associated with constitutive activation of autophagy as revealed by a transcriptome analysis using a custom microarray. Autophagy. 2011;7:760–70. doi: 10.4161/auto.7.7.15454. [DOI] [PubMed] [Google Scholar]
- 445.Mitroulis I, Kourtzelis I, Kambas K, Rafail S, Chrysanthopoulou A, Speletas M, et al. Regulation of the autophagic machinery in human neutrophils. Eur J Immunol. 2010;40:1461–72. doi: 10.1002/eji.200940025. [DOI] [PubMed] [Google Scholar]
- 446.Rodríguez-Muela N, Germain F, Mariño G, Fitze PS, Boya P. Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ. 2012;19:162–9. doi: 10.1038/cdd.2011.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Vázquez P, Arroba AI, Cecconi F, de la Rosa EJ, Boya P, De Pablo F. Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy. 2012;8:187–99. doi: 10.4161/auto.8.2.18535. [DOI] [PubMed] [Google Scholar]
- 448.Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120:127–41. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Gorski SM, Chittaranjan S, Pleasance ED, Freeman JD, Anderson CL, Varhol RJ, et al. A SAGE approach to discovery of genes involved in autophagic cell death. Curr Biol. 2003;13:358–63. doi: 10.1016/S0960-9822(03)00082-4. [DOI] [PubMed] [Google Scholar]
- 450.Lee C-Y, Clough EA, Yellon P, Teslovich TM, Stephan DA, Baehrecke EH. Genome-wide analyses of steroid- and radiation-triggered programmed cell death in Drosophila. Curr Biol. 2003;13:350–7. doi: 10.1016/S0960-9822(03)00085-X. [DOI] [PubMed] [Google Scholar]
- 451.Denton D, Shravage B, Simin R, Baehrecke EH, Kumar S. Larval midgut destruction in Drosophila: not dependent on caspases but suppressed by the loss of autophagy. Autophagy. 2010;6:163–5. doi: 10.4161/auto.6.1.10601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Franzetti E, Huang ZJ, Shi YX, Xie K, Deng XJ, Li JP, et al. Autophagy precedes apoptosis during the remodeling of silkworm larval midgut. Apoptosis. 2012;17:305–24. doi: 10.1007/s10495-011-0675-0. [DOI] [PubMed] [Google Scholar]
- 453.Juhász G, Puskás LG, Komonyi O, Erdi B, Maróy P, Neufeld TP, et al. Gene expression profiling identifies FKBP39 as an inhibitor of autophagy in larval Drosophila fat body. Cell Death Differ. 2007;14:1181–90. doi: 10.1038/sj.cdd.4402123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Barth JM, Szabad J, Hafen E, Köhler K. Autophagy in Drosophila ovaries is induced by starvation and is required for oogenesis. Cell Death Differ. 2011;18:915–24. doi: 10.1038/cdd.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- 456.Phillips AR, Suttangkakul A, Vierstra RD. The ATG12-conjugating enzyme ATG10 Is essential for autophagic vesicle formation in Arabidopsis thaliana. Genetics. 2008;178:1339–53. doi: 10.1534/genetics.107.086199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Seiliez I, Gutierrez J, Salmerón C, Skiba-Cassy S, Chauvin C, Dias K, et al. An in vivo and in vitro assessment of autophagy-related gene expression in muscle of rainbow trout (Oncorhynchus mykiss) Comp Biochem Physiol B Biochem Mol Biol. 2010;157:258–66. doi: 10.1016/j.cbpb.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 458.Lapierre LR, Gelino S, Meléndez A, Hansen M. Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr Biol. 2011;21:1507–14. doi: 10.1016/j.cub.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Sandri M. Autophagy in health and disease. 3. Involvement of autophagy in muscle atrophy. Am J Physiol Cell Physiol. 2010;298:C1291–7. doi: 10.1152/ajpcell.00531.2009. [DOI] [PubMed] [Google Scholar]
- 460.Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11:1305–14. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- 461.Ropolo A, Grasso D, Pardo R, Sacchetti ML, Archange C, Lo Re A, et al. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J Biol Chem. 2007;282:37124–33. doi: 10.1074/jbc.M706956200. [DOI] [PubMed] [Google Scholar]
- 462.Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell. 2010;141:1042–55. doi: 10.1016/j.cell.2010.04.034. [DOI] [PubMed] [Google Scholar]
- 463.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–33. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–7. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 465.Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–66. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 466.Kang YA, Sanalkumar R, O’Geen H, Linnemann AK, Chang CJ, Bouhassira EE, et al. Autophagy driven by a master regulator of hematopoiesis. Mol Cell Biol. 2012;32:226–39. doi: 10.1128/MCB.06166-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Ma D, Panda S, Lin JD. Temporal orchestration of circadian autophagy rhythm by C/EBPβ. EMBO J. 2011;30:4642–51. doi: 10.1038/emboj.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet. 2011;43:242–5. doi: 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- 469.Meenhuis A, van Veelen PA, de Looper H, van Boxtel N, van den Berge IJ, Sun SM, et al. MiR-17/20/93/106 promote hematopoietic cell expansion by targeting sequestosome 1-regulated pathways in mice. Blood. 2011;118:916–25. doi: 10.1182/blood-2011-02-336487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Roccaro AM, Sacco A, Jia X, Azab AK, Maiso P, Ngo HT, et al. microRNA-dependent modulation of histone acetylation in Waldenstrom macroglobulinemia. Blood. 2010;116:1506–14. doi: 10.1182/blood-2010-01-265686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Martinet W, De Meyer GR, Andries L, Herman AG, Kockx MM. In situ detection of starvation-induced autophagy. J Histochem Cytochem. 2006;54:85–96. doi: 10.1369/jhc.5A6743.2005. [DOI] [PubMed] [Google Scholar]
- 472.Pattingre S, Petiot A, Codogno P. Analyses of Galpha-interacting protein and activator of G-protein-signaling-3 functions in macroautophagy. Methods Enzymol. 2004;390:17–31. doi: 10.1016/S0076-6879(04)90002-X. [DOI] [PubMed] [Google Scholar]
- 473.Bauvy C, Meijer AJ, Codogno P. Assaying of autophagic protein degradation. Methods Enzymol. 2009;452:47–61. doi: 10.1016/S0076-6879(08)03604-5. [DOI] [PubMed] [Google Scholar]
- 474.Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283:22847–57. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- 475.Kabuta T, Furuta A, Aoki S, Furuta K, Wada K. Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem. 2008;283:23731–8. doi: 10.1074/jbc.M801918200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, et al. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–24. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–92. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 478.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–63. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 479.Tomek K, Wagner R, Varga F, Singer CF, Karlic H, Grunt TW. Blockade of fatty acid synthase induces ubiquitination and degradation of phosphoinositide-3-kinase signaling proteins in ovarian cancer. Mol Cancer Res. 2011;9:1767–79. doi: 10.1158/1541-7786.MCR-10-0467. [DOI] [PubMed] [Google Scholar]
- 480.Zimmermann AC, Zarei M, Eiselein S, Dengjel J. Quantitative proteomics for the analysis of spatio-temporal protein dynamics during autophagy. Autophagy. 2010;6:1009–16. doi: 10.4161/auto.6.8.12786. [DOI] [PubMed] [Google Scholar]
- 481.Kristensen AR, Schandorff S, Høyer-Hansen M, Nielsen MO, Jäättelä M, Dengjel J, et al. Ordered organelle degradation during starvation-induced autophagy. Mol Cell Proteomics. 2008;7:2419–28. doi: 10.1074/mcp.M800184-MCP200. [DOI] [PubMed] [Google Scholar]
- 482.Furuya N, Kanazawa T, Fujimura S, Ueno T, Kominami E, Kadowaki M. Leupeptin-induced appearance of partial fragment of betaine homocysteine methyltransferase during autophagic maturation in rat hepatocytes. J Biochem. 2001;129:313–20. doi: 10.1093/oxfordjournals.jbchem.a002859. [DOI] [PubMed] [Google Scholar]
- 483.Ueno T, Ishidoh K, Mineki R, Tanida I, Murayama K, Kadowaki M, et al. Autolysosomal membrane-associated betaine homocysteine methyltransferase. Limited degradation fragment of a sequestered cytosolic enzyme monitoring autophagy. J Biol Chem. 1999;274:15222–9. doi: 10.1074/jbc.274.21.15222. [DOI] [PubMed] [Google Scholar]
- 484.Øverbye A, Sætre F, Hagen LK, Johansen HT, Seglen PO. Autophagic activity measured in whole rat hepatocytes as the accumulation of a novel BHMT fragment (p10), generated in amphisomes by the asparaginyl proteinase, legumain. Autophagy. 2011;7:1011–27. doi: 10.4161/auto.7.9.16436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Seglen PO, Øverbye A, Saetre F. Sequestration assays for mammalian autophagy. Methods Enzymol. 2009;452:63–83. doi: 10.1016/S0076-6879(08)03605-7. [DOI] [PubMed] [Google Scholar]
- 486.Mercer CA, Kaliappan A, Dennis PB. Macroautophagy-dependent, intralysosomal cleavage of a betaine homocysteine methyltransferase fusion protein requires stable multimerization. Autophagy. 2008;4:185–94. doi: 10.4161/auto.5275. [DOI] [PubMed] [Google Scholar]
- 487.Nimmerjahn F, Milosevic S, Behrends U, Jaffee EM, Pardoll DM, Bornkamm GW, et al. Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur J Immunol. 2003;33:1250–9. doi: 10.1002/eji.200323730. [DOI] [PubMed] [Google Scholar]
- 488.Taylor GS, Long HM, Haigh TA, Larsen M, Brooks J, Rickinson AB. A role for intercellular antigen transfer in the recognition of EBV-transformed B cell lines by EBV nuclear antigen-specific CD4+ T cells. J Immunol. 2006;177:3746–56. doi: 10.4049/jimmunol.177.6.3746. [DOI] [PubMed] [Google Scholar]
- 489.Klionsky DJ, Emr SD. Membrane protein sorting: biosynthesis, transport and processing of yeast vacuolar alkaline phosphatase. EMBO J. 1989;8:2241–50. doi: 10.1002/j.1460-2075.1989.tb08348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Venerando R, Miotto G, Kadowaki M, Siliprandi N, Mortimore GE. Multiphasic control of proteolysis by leucine and alanine in the isolated rat hepatocyte. Am J Physiol. 1994;266:C455–61. doi: 10.1152/ajpcell.1994.266.2.C455. [DOI] [PubMed] [Google Scholar]
- 491.Häussinger D, Hallbrucker C, vom Dahl S, Lang F, Gerok W. Cell swelling inhibits proteolysis in perfused rat liver. Biochem J. 1990;272:239–42. doi: 10.1042/bj2720239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.vom Dahl S, Häussinger D. Cell hydration and proteolysis control in liver. Biochem J. 1995;312:988–9. doi: 10.1042/bj3120988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–5. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 494.Reggiori F, Monastyrska I, Shintani T, Klionsky DJ. The actin cytoskeleton is required for selective types of autophagy, but not nonspecific autophagy, in the yeast Saccharomyces cerevisiae. Mol Biol Cell. 2005;16:5843–56. doi: 10.1091/mbc.E05-07-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Manjithaya R, Jain S, Farré JC, Subramani S. A yeast MAPK cascade regulates pexophagy but not other autophagy pathways. J Cell Biol. 2010;189:303–10. doi: 10.1083/jcb.200909154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Journo D, Mor A, Abeliovich H. Aup1-mediated regulation of Rtg3 during mitophagy. J Biol Chem. 2009;284:35885–95. doi: 10.1074/jbc.M109.048140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Kanki T, Klionsky DJ. Mitophagy in yeast occurs through a selective mechanism. J Biol Chem. 2008;283:32386–93. doi: 10.1074/jbc.M802403200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Kanki T, Wang K, Baba M, Bartholomew CR, Lynch-Day MA, Du Z, et al. A genomic screen for yeast mutants defective in selective mitochondria autophagy. Mol Biol Cell. 2009;20:4730–8. doi: 10.1091/mbc.E09-03-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell. 2009;17:98–109. doi: 10.1016/j.devcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 501.Sakai Y, Koller A, Rangell LK, Keller GA, Subramani S. Peroxisome degradation by microautophagy in Pichia pastoris: identification of specific steps and morphological intermediates. J Cell Biol. 1998;141:625–36. doi: 10.1083/jcb.141.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Nazarko TY, Nicaud JM, Sibirny AA. Observation of the Yarrowia lipolytica peroxisome-vacuole dynamics by fluorescence microscopy with a single filter set. Cell Biol Int. 2005;29:65–70. doi: 10.1016/j.cellbi.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 503.Roetzer A, Gratz N, Kovarik P, Schüller C. Autophagy supports Candida glabrata survival during phagocytosis. Cell Microbiol. 2010;12:199–216. doi: 10.1111/j.1462-5822.2009.01391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Bormann C, Sahm H. Degradation of microbodies in relation to activities of alcohol oxidase and catalase in Candida boidinii. Arch Microbiol. 1978;117:67–72. doi: 10.1007/BF00689353. [DOI] [PubMed] [Google Scholar]
- 505.Stasyk OV, Nazarko TY, Sibirny AA. Methods of plate pexophagy monitoring and positive selection for ATG gene cloning in yeasts. Methods Enzymol. 2008;451:229–39. doi: 10.1016/S0076-6879(08)03216-3. [DOI] [PubMed] [Google Scholar]
- 506.Hutchins MU, Veenhuis M, Klionsky DJ. Peroxisome degradation in Saccharomyces cerevisiae is dependent on machinery of macroautophagy and the Cvt pathway. J Cell Sci. 1999;112:4079–87. doi: 10.1242/jcs.112.22.4079. [DOI] [PubMed] [Google Scholar]
- 507.Mukaiyama H, Oku M, Baba M, Samizo T, Hammond AT, Glick BS, et al. Paz2 and 13 other PAZ gene products regulate vacuolar engulfment of peroxisomes during micropexophagy. Genes Cells. 2002;7:75–90. doi: 10.1046/j.1356-9597.2001.00499.x. [DOI] [PubMed] [Google Scholar]
- 508.Tuttle DL, Dunn WA., Jr. Divergent modes of autophagy in the methylotrophic yeast Pichia pastoris. J Cell Sci. 1995;108:25–35. doi: 10.1242/jcs.108.1.25. [DOI] [PubMed] [Google Scholar]
- 509.Nazarko TY, Huang J, Nicaud JM, Klionsky DJ, Sibirny AA. Trs85 is required for macroautophagy, pexophagy and cytoplasm to vacuole targeting in Yarrowia lipolytica and Saccharomyces cerevisiae. Autophagy. 2005;1:37–45. doi: 10.4161/auto.1.1.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Veenhuis M, Douma A, Harder W, Osumi M. Degradation and turnover of peroxisomes in the yeast Hansenula polymorpha induced by selective inactivation of peroxisomal enzymes. Arch Microbiol. 1983;134:193–203. doi: 10.1007/BF00407757. [DOI] [PubMed] [Google Scholar]
- 511.Monosov EZ, Wenzel TJ, Lüers GH, Heyman JA, Subramani S. Labeling of peroxisomes with green fluorescent protein in living P. pastoris cells. J Histochem Cytochem. 1996;44:581–9. doi: 10.1177/44.6.8666743. [DOI] [PubMed] [Google Scholar]
- 512.Wiemer EA, Wenzel T, Deerinck TJ, Ellisman MH, Subramani S. Visualization of the peroxisomal compartment in living mammalian cells: dynamic behavior and association with microtubules. J Cell Biol. 1997;136:71–80. doi: 10.1083/jcb.136.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Monastyrska I, van der Heide M, Krikken AM, Kiel JAKW, van der Klei IJ, Veenhuis M. Atg8 is essential for macropexophagy in Hansenula polymorpha. Traffic. 2005;6:66–74. doi: 10.1111/j.1600-0854.2004.00252.x. [DOI] [PubMed] [Google Scholar]
- 514.Devenish RJ, Prescott M, Turcic K, Mijaljica D. Monitoring organelle turnover in yeast using fluorescent protein tags. Methods Enzymol. 2008;451:109–31. doi: 10.1016/S0076-6879(08)03209-6. [DOI] [PubMed] [Google Scholar]
- 515.Farré JC, Manjithaya R, Mathewson RD, Subramani S. PpAtg30 tags peroxisomes for turnover by selective autophagy. Dev Cell. 2008;14:365–76. doi: 10.1016/j.devcel.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Kanki T, Klionsky DJ. The molecular mechanism of mitochondria autophagy in yeast. Mol Microbiol. 2010;75:795–800. doi: 10.1111/j.1365-2958.2009.07035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Tal R, Winter G, Ecker N, Klionsky DJ, Abeliovich H. Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. J Biol Chem. 2007;282:5617–24. doi: 10.1074/jbc.M605940200. [DOI] [PubMed] [Google Scholar]
- 518.Abeliovich H. Stationary-phase mitophagy in respiring Saccharomyces cerevisiae. Antioxid Redox Signal. 2011;14:2003–11. doi: 10.1089/ars.2010.3807. [DOI] [PubMed] [Google Scholar]
- 519.Aksam EB, Koek A, Kiel JAKW, Jourdan S, Veenhuis M, van der Klei IJ. A peroxisomal lon protease and peroxisome degradation by autophagy play key roles in vitality of Hansenula polymorpha cells. Autophagy. 2007;3:96–105. doi: 10.4161/auto.3534. [DOI] [PubMed] [Google Scholar]
- 520.Roberts P, Moshitch-Moshkovitz S, Kvam E, O’Toole E, Winey M, Goldfarb DS. Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae. Mol Biol Cell. 2003;14:129–41. doi: 10.1091/mbc.E02-08-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Krick R, Muehe Y, Prick T, Bremer S, Schlotterhose P, Eskelinen E-L, et al. Piecemeal microautophagy of the nucleus requires the core macroautophagy genes. Mol Biol Cell. 2008;19:4492–505. doi: 10.1091/mbc.E08-04-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Farré JC, Krick R, Subramani S, Thumm M. Turnover of organelles by autophagy in yeast. Curr Opin Cell Biol. 2009;21:522–30. doi: 10.1016/j.ceb.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Kvam E, Goldfarb DS. Structure and function of nucleus-vacuole junctions: outer-nuclear-membrane targeting of Nvj1p and a role in tryptophan uptake. J Cell Sci. 2006;119:3622–33. doi: 10.1242/jcs.03093. [DOI] [PubMed] [Google Scholar]
- 524.Millen JI, Krick R, Prick T, Thumm M, Goldfarb DS. Measuring piecemeal microautophagy of the nucleus in Saccharomyces cerevisiae. Autophagy. 2009;5:75–81. doi: 10.4161/auto.5.1.7181. [DOI] [PubMed] [Google Scholar]
- 525.Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281:30299–304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Klionsky DJ, Cuervo AM, Dunn WA, Jr., Levine B, van der Klei I, Seglen PO. How shall I eat thee? Autophagy. 2007;3:413–6. doi: 10.4161/auto.4377. [DOI] [PubMed] [Google Scholar]
- 528.Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, et al. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II) Hum Mol Genet. 2007;16:618–29. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- 529.Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol. 2008;10:602–10. doi: 10.1038/ncb1723. [DOI] [PubMed] [Google Scholar]
- 530.Yorimitsu T, Klionsky DJ. Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Mol Biol Cell. 2005;16:1593–605. doi: 10.1091/mbc.E04-11-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Shintani T, Huang W-P, Stromhaug PE, Klionsky DJ. Mechanism of cargo selection in the cytoplasm to vacuole targeting pathway. Dev Cell. 2002;3:825–37. doi: 10.1016/S1534-5807(02)00373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Abeliovich H, Darsow T, Emr SD. Cytoplasm to vacuole trafficking of aminopeptidase I requires a t-SNARE-Sec1p complex composed of Tlg2p and Vps45p. EMBO J. 1999;18:6005–16. doi: 10.1093/emboj/18.21.6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Brown CR, Chiang H-L. A selective autophagy pathway that degrades gluconeogenic enzymes during catabolite inactivation. Commun Integr Biol. 2009;2:177–83. doi: 10.4161/cib.7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Schüle T, Rose M, Entian KD, Thumm M, Wolf DH. Ubc8p functions in catabolite degradation of fructose-1, 6-bisphosphatase in yeast. EMBO J. 2000;19:2161–7. doi: 10.1093/emboj/19.10.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Schork SM, Thumm M, Wolf DH. Catabolite inactivation of fructose-1,6-bisphosphatase of Saccharomyces cerevisiae. Degradation occurs via the ubiquitin pathway. J Biol Chem. 1995;270:26446–50. doi: 10.1074/jbc.270.44.26446. [DOI] [PubMed] [Google Scholar]
- 536.Regelmann J, Schüle T, Josupeit FS, Horak J, Rose M, Entian KD, et al. Catabolite degradation of fructose-1,6-bisphosphatase in the yeast Saccharomyces cerevisiae: a genome-wide screen identifies eight novel GID genes and indicates the existence of two degradation pathways. Mol Biol Cell. 2003;14:1652–63. doi: 10.1091/mbc.E02-08-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 537.Hung GC, Brown CR, Wolfe AB, Liu J, Chiang HL. Degradation of the gluconeogenic enzymes fructose-1,6-bisphosphatase and malate dehydrogenase is mediated by distinct proteolytic pathways and signaling events. J Biol Chem. 2004;279:49138–50. doi: 10.1074/jbc.M404544200. [DOI] [PubMed] [Google Scholar]
- 538.Chiang H-L, Schekman R, Hamamoto S. Selective uptake of cytosolic, peroxisomal, and plasma membrane proteins into the yeast lysosome for degradation. J Biol Chem. 1996;271:9934–41. doi: 10.1074/jbc.271.17.9934. [DOI] [PubMed] [Google Scholar]
- 539.Huang PH, Chiang H-L. Identification of novel vesicles in the cytosol to vacuole protein degradation pathway. J Cell Biol. 1997;136:803–10. doi: 10.1083/jcb.136.4.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Alibhoy AA, Giardina BJ, Dunton DD, Chiang H-L. Vid30 is required for the association of Vid vesicles and actin patches in the vacuole import and degradation pathway. Autophagy. 2012;8:29–46. doi: 10.4161/auto.8.1.18104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Brown CR, Wolfe AB, Cui D, Chiang H-L. The vacuolar import and degradation pathway merges with the endocytic pathway to deliver fructose-1,6-bisphosphatase to the vacuole for degradation. J Biol Chem. 2008;283:26116–27. doi: 10.1074/jbc.M709922200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Chiang MC, Chiang H-L. Vid24p, a novel protein localized to the fructose-1, 6-bisphosphatase-containing vesicles, regulates targeting of fructose-1,6-bisphosphatase from the vesicles to the vacuole for degradation. J Cell Biol. 1998;140:1347–56. doi: 10.1083/jcb.140.6.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Brown CR, Dunton D, Chiang H-L. The vacuole import and degradation pathway utilizes early steps of endocytosis and actin polymerization to deliver cargo proteins to the vacuole for degradation. J Biol Chem. 2010;285:1516–28. doi: 10.1074/jbc.M109.028241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Vida TA, Emr SD. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol. 1995;128:779–92. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Brown CR, Hung GC, Dunton D, Chiang H-L. The TOR complex 1 is distributed in endosomes and in retrograde vesicles that form from the vacuole membrane and plays an important role in the vacuole import and degradation pathway. J Biol Chem. 2010;285:23359–70. doi: 10.1074/jbc.M109.075143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Geisler S, Holmström KM, Treis A, Skujat D, Weber SS, Fiesel FC, et al. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy. 2010;6:871–8. doi: 10.4161/auto.6.7.13286. [DOI] [PubMed] [Google Scholar]
- 547.Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–31. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 548.Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–40. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18:1042–52. doi: 10.1016/j.chembiol.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 550.Yang JY, Yang WY. Spatiotemporally controlled initiation of Parkin-mediated mitophagy within single cells. Autophagy. 2011;7:1230–8. doi: 10.4161/auto.7.10.16626. [DOI] [PubMed] [Google Scholar]
- 551.Luiken JJ, van den Berg M, Heikoop JC, Meijer AJ. Autophagic degradation of peroxisomes in isolated rat hepatocytes. FEBS Lett. 1992;304:93–7. doi: 10.1016/0014-5793(92)80596-9. [DOI] [PubMed] [Google Scholar]
- 552.Yokota S. Formation of autophagosomes during degradation of excess peroxisomes induced by administration of dioctyl phthalate. Eur J Cell Biol. 1993;61:67–80. [PubMed] [Google Scholar]
- 553.Huybrechts SJ, Van Veldhoven PP, Brees C, Mannaerts GP, Los GV, Fransen M. Peroxisome dynamics in cultured mammalian cells. Traffic. 2009;10:1722–33. doi: 10.1111/j.1600-0854.2009.00970.x. [DOI] [PubMed] [Google Scholar]
- 554.Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol. 2010;189:671–9. doi: 10.1083/jcb.201001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–37. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–80. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–40. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558.Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15:887–900. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Lyamzaev KG, Nepryakhina OK, Saprunova VB, Bakeeva LE, Pletjushkina OY, Chernyak BV, et al. Novel mechanism of elimination of malfunctioning mitochondria (mitoptosis): formation of mitoptotic bodies and extrusion of mitochondrial material from the cell. Biochim Biophys Acta. 2008;1777:817–25. doi: 10.1016/j.bbabio.2008.03.027. [DOI] [PubMed] [Google Scholar]
- 560.Hara-Kuge S, Fujiki Y. The peroxin Pex14p is involved in LC3-dependent degradation of mammalian peroxisomes. Exp Cell Res. 2008;314:3531–41. doi: 10.1016/j.yexcr.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 561.Ezaki J, Kominami E, Ueno T. Peroxisome degradation in mammals. IUBMB Life. 2011;63:1001–8. doi: 10.1002/iub.537. [DOI] [PubMed] [Google Scholar]
- 562.Øverbye A, Fengsrud M, Seglen PO. Proteomic analysis of membrane-associated proteins from rat liver autophagosomes. Autophagy. 2007;3:300–22. doi: 10.4161/auto.3910. [DOI] [PubMed] [Google Scholar]
- 563.Ju JS, Miller SE, Jackson E, Cadwell K, Piwnica-Worms D, Weihl CC. Quantitation of selective autophagic protein aggregate degradation in vitro and in vivo using luciferase reporters. Autophagy. 2009;5:511–9. doi: 10.4161/auto.5.4.7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.Fuentealba RA, Marasa J, Diamond MI, Piwnica-Worms D, Weihl CC. An aggregation sensing reporter identifies leflunomide and teriflunomide as polyglutamine aggregate inhibitors. Hum Mol Genet. 2012;21:664–80. doi: 10.1093/hmg/ddr500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Webster P. Cytoplasmic bacteria and the autophagic pathway. Autophagy. 2006;2:159–61. doi: 10.4161/auto.2826. [DOI] [PubMed] [Google Scholar]
- 566.Dubuisson JF, Swanson MS. Mouse infection by Legionella, a model to analyze autophagy. Autophagy. 2006;2:179–82. doi: 10.4161/auto.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Jordan TX, Randall G. Manipulation or capitulation: virus interactions with autophagy. Microbes Infect. 2012;14:126–39. doi: 10.1016/j.micinf.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Knodler LA, Celli J. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell Microbiol. 2011;13:1319–27. doi: 10.1111/j.1462-5822.2011.01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–35. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Deretic V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol Rev. 2011;240:92–104. doi: 10.1111/j.1600-065X.2010.00995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Kageyama S, Omori H, Saitoh T, Sone T, Guan J-L, Akira S, et al. The LC3 recruitment mechanism is separate from Atg9L1-dependent membrane formation in the autophagic response against Salmonella. Mol Biol Cell. 2011;22:2290–300. doi: 10.1091/mbc.E10-11-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183:5909–16. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 573.Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–21. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 574.Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–33. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A. 2009;106:6226–31. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Rich KA, Burkett C, Webster P. Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol. 2003;5:455–68. doi: 10.1046/j.1462-5822.2003.00292.x. [DOI] [PubMed] [Google Scholar]
- 577.Shahnazari S, Brumell JH. Mechanisms and consequences of bacterial targeting by the autophagy pathway. Curr Opin Microbiol. 2011;14:68–75. doi: 10.1016/j.mib.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 578.McLean JE, Wudzinska A, Datan E, Quaglino D, Zakeri Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J Biol Chem. 2011;286:22147–59. doi: 10.1074/jbc.M110.192500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Mao Y, Da L, Tang H, Yang J, Lei Y, Tiollais P, et al. Hepatitis B virus X protein reduces starvation-induced cell death through activation of autophagy and inhibition of mitochondrial apoptotic pathway. Biochem Biophys Res Commun. 2011;415:68–74. doi: 10.1016/j.bbrc.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 580.Orvedahl A, Alexander D, Tallóczy Z, Sun Q, Wei Y, Zhang W, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 581.Alexander DE, Ward SL, Mizushima N, Levine B, Leib DA. Analysis of the role of autophagy in replication of herpes simplex virus in cell culture. J Virol. 2007;81:12128–34. doi: 10.1128/JVI.01356-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Leib DA, Alexander DE, Cox D, Yin J, Ferguson TA. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J Virol. 2009;83:12164–71. doi: 10.1128/JVI.01676-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Zhang H, Monken CE, Zhang Y, Lenard J, Mizushima N, Lattime EC, et al. Cellular autophagy machinery is not required for vaccinia virus replication and maturation. Autophagy. 2006;2:91–5. doi: 10.4161/auto.2.2.2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584.Heaton NS, Randall G. Dengue virus and autophagy. Viruses. 2011;3:1332–41. doi: 10.3390/v3081332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Dreux M, Gastaminza P, Wieland SF, Chisari FV. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci U S A. 2009;106:14046–51. doi: 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586.Collins CA, De Mazière A, van Dijk S, Carlsson F, Klumperman J, Brown EJ. Atg5-independent sequestration of ubiquitinated mycobacteria. PLoS Pathog. 2009;5:e1000430. doi: 10.1371/journal.ppat.1000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587.Moreau K, Lacas-Gervais S, Fujita N, Sebbane F, Yoshimori T, Simonet M, et al. Autophagosomes can support Yersinia pseudotuberculosis replication in macrophages. Cell Microbiol. 2010;12:1108–23. doi: 10.1111/j.1462-5822.2010.01456.x. [DOI] [PubMed] [Google Scholar]
- 588.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–5. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Grasso D, Ropolo A, Lo Ré A, Boggio V, Molejón MI, Iovanna JL, et al. Zymophagy, a novel selective autophagy pathway mediated by VMP1-USP9x-p62, prevents pancreatic cell death. J Biol Chem. 2011;286:8308–24. doi: 10.1074/jbc.M110.197301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Al Rawi S, Louvet-Vallée S, Djeddi A, Sachse M, Culetto E, Hajjar C, et al. Allophagy: A macroautophagic process degrading spermatozoid-inherited organelles. Autophagy. 2012;8:421–3. doi: 10.4161/auto.19242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Sato M, Sato K. Maternal inheritance of mitochondrial DNA: Degradation of paternal mitochondria by allogeneic organelle autophagy, allophagy. Autophagy. 2012;8:424–5. doi: 10.4161/auto.19243. [DOI] [PubMed] [Google Scholar]
- 592.Al Rawi S, Louvet-Vallée S, Djeddi A, Sachse M, Culetto E, Hajjar C, et al. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 2011;334:1144–7. doi: 10.1126/science.1211878. [DOI] [PubMed] [Google Scholar]
- 593.Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science. 2011;334:1141–4. doi: 10.1126/science.1210333. [DOI] [PubMed] [Google Scholar]
- 594.Seglen PO, Gordon PB, Tolleshaug H, Høyvik H. Use of [3H]raffinose as a specific probe of autophagic sequestration. Exp Cell Res. 1986;162:273–7. doi: 10.1016/0014-4827(86)90446-5. [DOI] [PubMed] [Google Scholar]
- 595.Kopitz J, Kisen GO, Gordon PB, Bohley P, Seglen PO. Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J Cell Biol. 1990;111:941–53. doi: 10.1083/jcb.111.3.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Nair U, Thumm M, Klionsky DJ, Krick R. GFP-Atg8 protease protection as a tool to monitor autophagosome biogenesis. Autophagy. 2011;7:1546–50. doi: 10.4161/auto.7.12.18424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Plomp PJ, Gordon PB, Meijer AJ, Høyvik H, Seglen PO. Energy dependence of different steps in the autophagic-lysosomal pathway. J Biol Chem. 1989;264:6699–704. [PubMed] [Google Scholar]
- 598.Høyvik H, Gordon PB, Berg TO, Strømhaug PE, Seglen PO. Inhibition of autophagic-lysosomal delivery and autophagic lactolysis by asparagine. J Cell Biol. 1991;113:1305–12. doi: 10.1083/jcb.113.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599.Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy. 2006;2:39–46. doi: 10.4161/auto.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Lorenz H, Hailey DW, Lippincott-Schwartz J. Fluorescence protease protection of GFP chimeras to reveal protein topology and subcellular localization. Nat Methods. 2006;3:205–10. doi: 10.1038/nmeth857. [DOI] [PubMed] [Google Scholar]
- 601.Dagda RK, Zhu J, Kulich SM, Chu CT. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson’s disease. Autophagy. 2008;4:770–82. doi: 10.4161/auto.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.McNeil PL, Murphy RF, Lanni F, Taylor DL. A method for incorporating macromolecules into adherent cells. J Cell Biol. 1984;98:1556–64. doi: 10.1083/jcb.98.4.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603.Kim J, Huang W-P, Stromhaug PE, Klionsky DJ. Convergence of multiple autophagy and cytoplasm to vacuole targeting components to a perivacuolar membrane compartment prior to de novo vesicle formation. J Biol Chem. 2002;277:763–73. doi: 10.1074/jbc.M109134200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604.Kovács AL, László L, Kovács J. Effect of amino acids and cycloheximide on changes caused by vinblastine, leupeptin and methylamine in the autophagic/lysosomal system of mouse hepatocytes in vivo. Exp Cell Res. 1985;157:83–94. doi: 10.1016/0014-4827(85)90154-5. [DOI] [PubMed] [Google Scholar]
- 605.Swanson MS, Byrne BG, Dubuisson JF. Kinetic analysis of autophagosome formation and turnover in primary mouse macrophages. Methods Enzymol. 2009;452:383–402. doi: 10.1016/S0076-6879(08)03623-9. [DOI] [PubMed] [Google Scholar]
- 606.Beugnet A, Tee AR, Taylor PM, Proud CG. Regulation of targets of mTOR (mammalian target of rapamycin) signalling by intracellular amino acid availability. Biochem J. 2003;372:555–66. doi: 10.1042/BJ20021266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Urban J, Soulard A, Huber A, Lippman S, Mukhopadhyay D, Deloche O, et al. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol Cell. 2007;26:663–74. doi: 10.1016/j.molcel.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 608.Jomain-Baum M, Garber AJ, Farber E, Hanson RW. The effect of cycloheximide on the interaction between mitochondrial respiration and gluconeogenesis in guinea pig and rat liver. J Biol Chem. 1973;248:1536–43. [PubMed] [Google Scholar]
- 609.Garber AJ, Jomain-Baum M, Salganicoff L, Farber E, Hanson RW. The effects of cycloheximide on energy transfer in rat and guinea pig liver mitochondria. J Biol Chem. 1973;248:1530–5. [PubMed] [Google Scholar]
- 610.Mora R, Dokic I, Kees T, Hüber CM, Keitel D, Geibig R, et al. Sphingolipid rheostat alterations related to transformation can be exploited for specific induction of lysosomal cell death in murine and human glioma. Glia. 2010;58:1364–83. doi: 10.1002/glia.21013. [DOI] [PubMed] [Google Scholar]
- 611.Bright NA, Lindsay MR, Stewart A, Luzio JP. The relationship between lumenal and limiting membranes in swollen late endocytic compartments formed after wortmannin treatment or sucrose accumulation. Traffic. 2001;2:631–42. doi: 10.1034/j.1600-0854.2001.20906.x. [DOI] [PubMed] [Google Scholar]
- 612.Deter RL. Quantitative characterization of dense body, autophagic vacuole, and acid phosphatase-bearing particle populations during the early phases of glucagon-induced autophagy in rat liver. J Cell Biol. 1971;48:473–89. doi: 10.1083/jcb.48.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613.Deter RL. Analog modeling of glucagon-induced autophagy in rat liver. I. Conceptual and mathematical model of telolysosome-autophagosome-autolysosome interaction. Exp Cell Res. 1975;94:122–6. doi: 10.1016/0014-4827(75)90538-8. [DOI] [PubMed] [Google Scholar]
- 614.Deter RL. Analog modeling of glucagon-induced autophagy in rat liver. II. Evaluation of iron labeling as a means for identifying telolysosome, autophagosome and autolysosome populations. Exp Cell Res. 1975;94:127–39. doi: 10.1016/0014-4827(75)90539-X. [DOI] [PubMed] [Google Scholar]
- 615.Deter RL, Baudhuin P, de Duve C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J Cell Biol. 1967;35:C11–6. doi: 10.1083/jcb.35.2.C11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Deter RL, de Duve C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol. 1967;33:437–49. doi: 10.1083/jcb.33.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617.Strømhaug PE, Berg TO, Fengsrud M, Seglen PO. Purification and characterization of autophagosomes from rat hepatocytes. Biochem J. 1998;335:217–24. doi: 10.1042/bj3350217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618.Deter RL. Electron microscopic evaluation of subcellular fractions obtained by ultracentrifugation. In: Hayat MA, ed. Principles and Techniques of Electron Microscopy. New York: Van Nostrand Reinhold Co., 1973:199-235. [Google Scholar]
- 619.Marzella L, Ahlberg J, Glaumann H. Isolation of autophagic vacuoles from rat liver: morphological and biochemical characterization. J Cell Biol. 1982;93:144–54. doi: 10.1083/jcb.93.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Wattiaux R, Wattiaux-De Coninck S, Ronveaux-dupal M-F, Dubois F. Isolation of rat liver lysosomes by isopycnic centrifugation in a metrizamide gradient. J Cell Biol. 1978;78:349–68. doi: 10.1083/jcb.78.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621.Rodríguez-Navarro JA, Rodríguez L, Casarejos MJ, Solano RM, Gómez A, Perucho J, et al. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis. 2010;39:423–38. doi: 10.1016/j.nbd.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 622.Weibel ER, Bolender RP. Stereological techniques for electron microscopic morphometry. In: Hayat MA, ed. Principles and Techniques of Electron Microscopy. New York: Van Nostrand Reinhold Co., 1973:237-96. [Google Scholar]
- 623.Baudhuin P, Evrard P, Berthet J. Electron microscopic examination of subcellular fractions. I. The preparation of representative samples from suspensions of particles. J Cell Biol. 1967;32:181–91. doi: 10.1083/jcb.32.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Baudhuin P, Berthet J. Electron microscopic examination of subcellular fractions. II. Quantitative analysis of the mitochondrial population isolated from rat liver. J Cell Biol. 1967;35:631–48. doi: 10.1083/jcb.35.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625.Storrie B, Madden EA. Isolation of subcellular organelles. Methods Enzymol. 1990;182:203–25. doi: 10.1016/0076-6879(90)82018-W. [DOI] [PubMed] [Google Scholar]
- 626.Balch WE, Rothman JE. Characterization of protein transport between successive compartments of the Golgi apparatus: asymmetric properties of donor and acceptor activities in a cell-free system. Arch Biochem Biophys. 1985;240:413–25. doi: 10.1016/0003-9861(85)90046-3. [DOI] [PubMed] [Google Scholar]
- 627.Graham JM. Isolation of lysosomes from tissues and cells by differential and density gradient centrifugation. In: Bonifacino JS, Dasso M, Harfod JB, Lippincott-Schwartz J and Yamada KM, eds. Current Protocols in Cell Biology: John Wiley & Sons, Inc., 2000:Unit 3.6. [DOI] [PubMed] [Google Scholar]
- 628.Cuervo AM, Dice JF, Knecht E. A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem. 1997;272:5606–15. doi: 10.1074/jbc.272.9.5606. [DOI] [PubMed] [Google Scholar]
- 629.Iwai-Kanai E, Yuan H, Huang C, Sayen MR, Perry-Garza CN, Kim L, et al. A method to measure cardiac autophagic flux in vivo. Autophagy. 2008;4:322–9. doi: 10.4161/auto.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–93. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–52. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Chiarelli R, Agnello M, Roccheri MC. Sea urchin embryos as a model system for studying autophagy induced by cadmium stress. Autophagy. 2011;7:1028–34. doi: 10.4161/auto.7.9.16450. [DOI] [PubMed] [Google Scholar]
- 633.Morici G, Agnello M, Spagnolo F, Roccheri MC, Di Liegro CM, Rinaldi AM. Confocal microscopy study of the distribution, content and activity of mitochondria during Paracentrotus lividus development. J Microsc. 2007;228:165–73. doi: 10.1111/j.1365-2818.2007.01860.x. [DOI] [PubMed] [Google Scholar]
- 634.Martinet W, De Meyer GR, Andries L, Herman AG, Kockx MM. Detection of autophagy in tissue by standard immunohistochemistry: possibilities and limitations. Autophagy. 2006;2:55–7. doi: 10.4161/auto.2217. [DOI] [PubMed] [Google Scholar]
- 635.Holt SV, Wyspianska B, Randall KJ, James D, Foster JR, Wilkinson RW. The development of an immunohistochemical method to detect the autophagy-associated protein LC3-II in human tumor xenografts. Toxicol Pathol. 2011;39:516–23. doi: 10.1177/0192623310396903. [DOI] [PubMed] [Google Scholar]
- 636.Kimura S, Fujita N, Noda T, Yoshimori T. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol. 2009;452:1–12. doi: 10.1016/S0076-6879(08)03601-X. [DOI] [PubMed] [Google Scholar]
- 637.Dehay B, Bové J, Rodríguez-Muela N, Perier C, Recasens A, Boya P, et al. Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci. 2010;30:12535–44. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638.Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13:655–67. doi: 10.1016/j.cmet.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–98. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640.Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med. 2010;16:1313–20. doi: 10.1038/nm.2247. [DOI] [PubMed] [Google Scholar]
- 641.Haspel J, Shaik RS, Ifedigbo E, Nakahira K, Dolinay T, Englert JA, et al. Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy. 2011;7:629–42. doi: 10.4161/auto.7.6.15100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 642.Uchiyama Y. Autophagic cell death and its execution by lysosomal cathepsins. Arch Histol Cytol. 2001;64:233–46. doi: 10.1679/aohc.64.233. [DOI] [PubMed] [Google Scholar]
- 643.Biolo G, Fleming RY, Maggi SP, Wolfe RR. Transmembrane transport and intracellular kinetics of amino acids in human skeletal muscle. Am J Physiol. 1995;268:E75–84. doi: 10.1152/ajpendo.1995.268.1.E75. [DOI] [PubMed] [Google Scholar]
- 644.Svanberg E, Möller-Loswick AC, Matthews DE, Körner U, Andersson M, Lundholm K. The role of glucose, long-chain triglycerides and amino acids for promotion of amino acid balance across peripheral tissues in man. Clin Physiol. 1999;19:311–20. doi: 10.1046/j.1365-2281.1999.00183.x. [DOI] [PubMed] [Google Scholar]
- 645.Volpi E, Ferrando AA, Yeckel CW, Tipton KD, Wolfe RR. Exogenous amino acids stimulate net muscle protein synthesis in the elderly. J Clin Invest. 1998;101:2000–7. doi: 10.1172/JCI939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646.Denne SC, Liechty EA, Liu YM, Brechtel G, Baron AD. Proteolysis in skeletal muscle and whole body in response to euglycemic hyperinsulinemia in normal adults. Am J Physiol. 1991;261:E809–14. doi: 10.1152/ajpendo.1991.261.6.E809. [DOI] [PubMed] [Google Scholar]
- 647.Fukagawa NK, Minaker KL, Young VR, Matthews DE, Bier DM, Rowe JW. Leucine metabolism in aging humans: effect of insulin and substrate availability. Am J Physiol. 1989;256:E288–94. doi: 10.1152/ajpendo.1989.256.2.E288. [DOI] [PubMed] [Google Scholar]
- 648.Gelfand RA, Barrett EJ. Effect of physiologic hyperinsulinemia on skeletal muscle protein synthesis and breakdown in man. J Clin Invest. 1987;80:1–6. doi: 10.1172/JCI113033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 649.Mortimore GE, Pösö AR. Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu Rev Nutr. 1987;7:539–64. doi: 10.1146/annurev.nu.07.070187.002543. [DOI] [PubMed] [Google Scholar]
- 650.Pozefsky T, Felig P, Tobin JD, Soeldner JS, Cahill GF., Jr Amino acid balance across tissues of the forearm in postabsorptive man. Effects of insulin at two dose levels. J Clin Invest. 1969;48:2273–82. doi: 10.1172/JCI106193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651.Weber SM, Levitz SM. Chloroquine interferes with lipopolysaccharide-induced TNF-α gene expression by a nonlysosomotropic mechanism. J Immunol. 2000;165:1534–40. doi: 10.4049/jimmunol.165.3.1534. [DOI] [PubMed] [Google Scholar]
- 652.Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–6. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 653.Chotechuang N, Azzout-Marniche D, Bos C, Chaumontet C, Gaudichon C, Tomé D. Down-regulation of the ubiquitin-proteasome proteolysis system by amino acids and insulin involves the adenosine monophosphate-activated protein kinase and mammalian target of rapamycin pathways in rat hepatocytes. Amino Acids. 2011;41:457–68. doi: 10.1007/s00726-010-0765-2. [DOI] [PubMed] [Google Scholar]
- 654.Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov. 2011;10:29–46. doi: 10.1038/nrd3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655.Akagi Y, Isaka Y, Akagi A, Ikawa M, Takenaka M, Moriyama T, et al. Transcriptional activation of a hybrid promoter composed of cytomegalovirus enhancer and beta-actin/beta-globin gene in glomerular epithelial cells in vivo. Kidney Int. 1997;51:1265–9. doi: 10.1038/ki.1997.172. [DOI] [PubMed] [Google Scholar]
- 656.Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol. 2011;22:902–13. doi: 10.1681/ASN.2010070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 657.Hartleben B, Gödel M, Meyer-Schwesinger C, Liu S, Ulrich T, Köbler S, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120:1084–96. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 658.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131:1137–48. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 659.Aits S, Gustafsson L, Hallgren O, Brest P, Gustafsson M, Trulsson M, et al. HAMLET (human alpha-lactalbumin made lethal to tumor cells) triggers autophagic tumor cell death. Int J Cancer. 2009;124:1008–19. doi: 10.1002/ijc.24076. [DOI] [PubMed] [Google Scholar]
- 660.Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–69. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 661.Hou YC, Hannigan AM, Gorski SM. An executioner caspase regulates autophagy. Autophagy. 2009;5:530–3. doi: 10.4161/auto.5.4.8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 662.Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjørkøy G, Johansen T, et al. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol. 2010;190:523–31. doi: 10.1083/jcb.201002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 663.Piras A, Gianetto D, Conte D, Bosone A, Vercelli A. Activation of autophagy in a rat model of retinal ischemia following high intraocular pressure. PLoS One. 2011;6:e22514. doi: 10.1371/journal.pone.0022514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 664.Schwarze PE, Seglen PO. Reduced autophagic activity, improved protein balance and enhanced in vitro survival of hepatocytes isolated from carcinogen-treated rats. Exp Cell Res. 1985;157:15–28. doi: 10.1016/0014-4827(85)90148-X. [DOI] [PubMed] [Google Scholar]
- 665.Russo R, Berliocchi L, Adornetto A, Varano GP, Cavaliere F, Nucci C, et al. Calpain-mediated cleavage of Beclin-1 and autophagy deregulation following retinal ischemic injury in vivo. Cell Death Dis. 2011;2:e144. doi: 10.1038/cddis.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 666.Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19:87–95. doi: 10.1038/cdd.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 667.Beaulaton J, Lockshin RA. Ultrastructural study of the normal degeneration of the intersegmental muscles of Anthereae polyphemus and Manduca sexta (Insecta, Lepidoptera) with particular reference of cellular autophagy. J Morphol. 1977;154:39–57. doi: 10.1002/jmor.1051540104. [DOI] [PubMed] [Google Scholar]
- 668.Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 669.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–20. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 670.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–10. doi: 10.1038/nrm2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 671.van Doorn WG, Beers EP, Dangl JL, Franklin-Tong VE, Gallois P, Hara-Nishimura I, et al. Morphological classification of plant cell deaths. Cell Death Differ. 2011;18:1241–6. doi: 10.1038/cdd.2011.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 672.Giusti C, Tresse E, Luciani MF, Golstein P. Autophagic cell death: analysis in Dictyostelium. Biochim Biophys Acta. 2009;1793:1422–31. doi: 10.1016/j.bbamcr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 673.Luciani MF, Giusti C, Harms B, Oshima Y, Kikuchi H, Kubohara Y, et al. Atg1 allows second-signaled autophagic cell death in Dictyostelium. Autophagy. 2011;7:501–8. doi: 10.4161/auto.7.5.14957. [DOI] [PubMed] [Google Scholar]
- 674.Uchikawa T, Yamamoto A, Inouye K. Origin and function of the stalk-cell vacuole in Dictyostelium. Dev Biol. 2011;352:48–57. doi: 10.1016/j.ydbio.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 675.Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16:1093–107. doi: 10.1038/cdd.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676.Guimarães CA, Benchimol M, Amarante-Mendes GP, Linden R. Alternative programs of cell death in developing retinal tissue. J Biol Chem. 2003;278:41938–46. doi: 10.1074/jbc.M306547200. [DOI] [PubMed] [Google Scholar]
- 677.Lossi L, Gambino G, Mioletti S, Merighi A. In vivo analysis reveals different apoptotic pathways in pre- and postmigratory cerebellar granule cells of rabbit. J Neurobiol. 2004;60:437–52. doi: 10.1002/neu.20032. [DOI] [PubMed] [Google Scholar]
- 678.Lossi L, Alasia S, Salio C, Merighi A. Cell death and proliferation in acute slices and organotypic cultures of mammalian CNS. Prog Neurobiol. 2009;88:221–45. doi: 10.1016/j.pneurobio.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 679.Thorburn A. I think autophagy controls the death of my cells: what do I do to get my paper published? Autophagy. 2011;7:455–6. doi: 10.4161/auto.7.5.14797. [DOI] [PubMed] [Google Scholar]
- 680.Kaushik S, Bandyopadhyay U, Sridhar S, Kiffin R, Martinez-Vicente M, Kon M, et al. Chaperone-mediated autophagy at a glance. J Cell Sci. 2011;124:495–9. doi: 10.1242/jcs.073874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 681.Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol. 2011;23:184–9. doi: 10.1016/j.ceb.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 682.Kaushik S, Cuervo AM. Methods to monitor chaperone-mediated autophagy. Methods Enzymol. 2009;452:297–324. doi: 10.1016/S0076-6879(08)03619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 683.Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15:305–9. doi: 10.1016/0968-0004(90)90019-8. [DOI] [PubMed] [Google Scholar]
- 684.Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–3. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 685.Cuervo AM, Dice JF. Unique properties of lamp2a compared to other lamp2 isoforms. J Cell Sci. 2000;113:4441–50. doi: 10.1242/jcs.113.24.4441. [DOI] [PubMed] [Google Scholar]
- 686.Finn PF, Mesires NT, Vine M, Dice JF. Effects of small molecules on chaperone-mediated autophagy. Autophagy. 2005;1:141–5. doi: 10.4161/auto.1.3.2000. [DOI] [PubMed] [Google Scholar]
- 687.Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol. 2008;28:5747–63. doi: 10.1128/MCB.02070-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 688.Aniento F, Emans N, Griffiths G, Gruenberg J. Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J Cell Biol. 1993;123:1373–87. doi: 10.1083/jcb.123.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 689.Salvador N, Aguado C, Horst M, Knecht E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J Biol Chem. 2000;275:27447–56. doi: 10.1074/jbc.M001394200. [DOI] [PubMed] [Google Scholar]
- 690.Koga H, Martinez-Vicente M, Macian F, Verkhusha VV, Cuervo AM. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat Commun. 2011;2:386. doi: 10.1038/ncomms1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 691.Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011;20:131–9. doi: 10.1016/j.devcel.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692.Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010;20:143–8. doi: 10.1016/j.cub.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 693.Niemann A, Baltes J, Elsässer HP. Fluorescence properties and staining behavior of monodansylpentane, a structural homologue of the lysosomotropic agent monodansylcadaverine. J Histochem Cytochem. 2001;49:177–85. doi: 10.1177/002215540104900205. [DOI] [PubMed] [Google Scholar]
- 694.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439–44. [PubMed] [Google Scholar]
- 695.Florez-McClure ML, Linseman DA, Chu CT, Barker PA, Bouchard RJ, Le SS, et al. The p75 neurotrophin receptor can induce autophagy and death of cerebellar Purkinje neurons. J Neurosci. 2004;24:4498–509. doi: 10.1523/JNEUROSCI.5744-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 696.Moriyasu Y, Hattori M, Jauh G-Y, Rogers JC. Alpha tonoplast intrinsic protein is specifically associated with vacuole membrane involved in an autophagic process. Plant Cell Physiol. 2003;44:795–802. doi: 10.1093/pcp/pcg100. [DOI] [PubMed] [Google Scholar]
- 697.Biederbick A, Kern HF, Elsässer HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol. 1995;66:3–14. [PubMed] [Google Scholar]
- 698.Høyer-Hansen M, Bastholm L, Mathiasen IS, Elling F, Jäättelä M. Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ. 2005;12:1297–309. doi: 10.1038/sj.cdd.4401651. [DOI] [PubMed] [Google Scholar]
- 699.Munafó DB, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114:3619–29. doi: 10.1242/jcs.114.20.3619. [DOI] [PubMed] [Google Scholar]
- 700.Gutierrez MG, Munafó DB, Berón W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117:2687–97. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- 701.Freundt EC, Czapiga M, Lenardo MJ. Photoconversion of Lysotracker Red to a green fluorescent molecule. Cell Res. 2007;17:956–8. doi: 10.1038/cr.2007.80. [DOI] [PubMed] [Google Scholar]
- 702.Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–12. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 703.Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20:355–62. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 704.Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600–6. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 705.Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol. 2009;186:773–82. doi: 10.1083/jcb.200907014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 706.Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280:20722–9. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- 707.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–8. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 708.Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286:9587–97. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 709.Crişan TO, Plantinga TS, van de Veerdonk FL, Farcaş MF, Stoffels M, Kullberg BJ, et al. Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS One. 2011;6:e18666. doi: 10.1371/journal.pone.0018666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 710.Kleinnijenhuis J, Oosting M, Plantinga TS, van der Meer JW, Joosten LA, Crevel RV, et al. Autophagy modulates the Mycobacterium tuberculosis-induced cytokine response. Immunology. 2011;134:341–8. doi: 10.1111/j.1365-2567.2011.03494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 711.Cantino D, Mosso R, Baccino FM. Changes induced by fasting and cycloheximide in the vacuolar apparatus of rat hepatocytes. A morphometric investigation. Boll Soc Ital Biol Sper. 1979;55:1884–9. [PubMed] [Google Scholar]
- 712.Kovács J. Morphometric study of the effect of leupeptin, vinblastine, estron acetate and cycloheximide on the autophagic vacuole-lysosomal compartments in mouse seminal vesicle cells. Virchows Arch B Cell Pathol Incl Mol Pathol. 1983;42:83–93. doi: 10.1007/BF02890372. [DOI] [PubMed] [Google Scholar]
- 713.Papadopoulos T, Pfeifer U. Regression of rat liver autophagic vacuoles by locally applied cycloheximide. Lab Invest. 1986;54:100–7. [PubMed] [Google Scholar]
- 714.Rumpelt HJ, Albring M, Thoenes W. Prevention of D-galactosamine-induced hepatocellular autophagocytosis by cycloheximide. Virchows Arch B Cell Pathol. 1974;16:195–203. doi: 10.1007/BF02894074. [DOI] [PubMed] [Google Scholar]
- 715.Rumpelt HJ, Weisbach T. Effect of cycloheximide on glucagon-induced autophagy. Quantitative examinations on hepatocytes in the rat. Am J Pathol. 1978;91:49–56. [PMC free article] [PubMed] [Google Scholar]
- 716.Kovács AL, Kovács J. Autophagocytosis in mouse seminal vesicle cells in vitro. Temperature dependence and effects of vinblastine and inhibitors of protein synthesis. Virchows Arch B Cell Pathol Incl Mol Pathol. 1980;32:97–104. doi: 10.1007/BF02889018. [DOI] [PubMed] [Google Scholar]
- 717.Rodemann HP, Dittmann K, Toulany M. Radiation-induced EGFR-signaling and control of DNA-damage repair. Int J Radiat Biol. 2007;83:781–91. doi: 10.1080/09553000701769970. [DOI] [PubMed] [Google Scholar]
- 718.Chaachouay H, Ohneseit P, Toulany M, Kehlbach R, Multhoff G, Rodemann HP. Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother Oncol. 2011;99:287–92. doi: 10.1016/j.radonc.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 719.Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008;68:1485–94. doi: 10.1158/0008-5472.CAN-07-0562. [DOI] [PubMed] [Google Scholar]
- 720.Eng CH, Yu K, Lucas J, White E, Abraham RT. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal. 2010;3:ra31. doi: 10.1126/scisignal.2000911. [DOI] [PubMed] [Google Scholar]
- 721.Seglen PO, Gordon PB. Effects of lysosomotropic monoamines, diamines, amino alcohols, and other amino compounds on protein degradation and protein synthesis in isolated rat hepatocytes. Mol Pharmacol. 1980;18:468–75. [PubMed] [Google Scholar]
- 722.Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci U S A. 2011;108:11121–6. doi: 10.1073/pnas.1107969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 723.Wei P, Zhang L, Lu Y, Man N, Wen L. C60(Nd) nanoparticles enhance chemotherapeutic susceptibility of cancer cells by modulation of autophagy. Nanotechnology. 2010;21:495101. doi: 10.1088/0957-4484/21/49/495101. [DOI] [PubMed] [Google Scholar]
- 724.Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8:397–403. doi: 10.1016/S0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 725.Mehdi S. Cell-penetrating inhibitors of calpain. Trends Biochem Sci. 1991;16:150–3. doi: 10.1016/0968-0004(91)90058-4. [DOI] [PubMed] [Google Scholar]
- 726.Holen I, Gordon PB, Seglen PO. Inhibition of hepatocytic autophagy by okadaic acid and other protein phosphatase inhibitors. Eur J Biochem. 1993;215:113–22. doi: 10.1111/j.1432-1033.1993.tb18013.x. [DOI] [PubMed] [Google Scholar]
- 727.Sasaki K, Murata M, Yasumoto T, Mieskes G, Takai A. Affinity of okadaic acid to type-1 and type-2A protein phosphatases is markedly reduced by oxidation of its 27-hydroxyl group. Biochem J. 1994;298:259–62. doi: 10.1042/bj2980259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 728.Robinson DG, Albrecht S, Moriysu Y. The V-ATPase inhibitors concanamycin A and bafilomycin A lead to Golgi swelling in tobacco BY-2 cells. Protoplasma. 2004;224:255–60. doi: 10.1007/s00709-004-0070-6. [DOI] [PubMed] [Google Scholar]
- 729.Wu YC, Wu WK, Li Y, Yu L, Li ZJ, Wong CC, et al. Inhibition of macroautophagy by bafilomycin A1 lowers proliferation and induces apoptosis in colon cancer cells. Biochem Biophys Res Commun. 2009;382:451–6. doi: 10.1016/j.bbrc.2009.03.051. [DOI] [PubMed] [Google Scholar]
- 730.Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- 731.Ostenfeld MS, Høyer-Hansen M, Bastholm L, Fehrenbacher N, Olsen OD, Groth-Pedersen L, et al. Anti-cancer agent siramesine is a lysosomotropic detergent that induces cytoprotective autophagosome accumulation. Autophagy. 2008;4:487–99. doi: 10.4161/auto.5774. [DOI] [PubMed] [Google Scholar]
- 732.Matsuoka K, Higuchi T, Maeshima M, Nakamura K. A vacuolar-type H+-ATPase in a nonvacuolar organelle is required for the sorting of soluble vacuolar protein precursors in tobacco cells. Plant Cell. 1997;9:533–46. doi: 10.1105/tpc.9.4.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 733.Arstila AU, Nuuja IJ, Trump BF. Studies on cellular autophagocytosis. Vinblastine-induced autophagy in the rat liver. Exp Cell Res. 1974;87:249–52. doi: 10.1016/0014-4827(74)90477-7. [DOI] [PubMed] [Google Scholar]
- 734.Hirsimäki Y, Arstila AU, Trump BF. Autophagocytosis: in vitro induction by microtuble poisons. Exp Cell Res. 1975;92:11–4. doi: 10.1016/0014-4827(75)90630-8. [DOI] [PubMed] [Google Scholar]
- 735.Kominami E, Hashida S, Khairallah EA, Katunuma N. Sequestration of cytoplasmic enzymes in an autophagic vacuole-lysosomal system induced by injection of leupeptin. J Biol Chem. 1983;258:6093–100. [PubMed] [Google Scholar]
- 736.Réz G, Fellinger E, Réti M, Biczó I, Kovács AL. Time course of quantitative morphological changes of the autophagic-lysosomal compartment of murine seminal vesicle epithelial cells under the influence of vinblastine. J Submicrosc Cytol Pathol. 1990;22:529–34. [PubMed] [Google Scholar]
- 737.Oliva O, Réz G, Pálfia Z, Fellinger E. Dynamics of vinblastine-induced autophagocytosis in murine pancreatic acinar cells: influence of cycloheximide post-treatments. Exp Mol Pathol. 1992;56:76–86. doi: 10.1016/0014-4800(92)90025-7. [DOI] [PubMed] [Google Scholar]
- 738.von Haefen C, Sifringer M, Menk M, Spies CD. Ethanol enhances susceptibility to apoptotic cell death via down-regulation of autophagy-related proteins. Alcohol Clin Exp Res. 2011;35:1381–91. doi: 10.1111/j.1530-0277.2011.01473.x. [DOI] [PubMed] [Google Scholar]
- 739.Krüger U, Wang Y, Kumar S, Mandelkow EM. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging. 2012 doi: 10.1016/j.neurobiolaging.2011.11.009. In press. [DOI] [PubMed] [Google Scholar]
- 740.Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 741.Palomo GM, Cerrato T, Gargini R, Diaz-Nido J. Silencing of frataxin gene expression triggers p53-dependent apoptosis in human neuron-like cells. Hum Mol Genet. 2011;20:2807–22. doi: 10.1093/hmg/ddr187. [DOI] [PubMed] [Google Scholar]
- 742.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 743.Fleming A, Noda T, Yoshimori T, Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol. 2011;7:9–17. doi: 10.1038/nchembio.500. [DOI] [PubMed] [Google Scholar]
- 744.Yu K, Toral-Barza L, Shi C, Zhang WG, Lucas J, Shor B, et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009;69:6232–40. doi: 10.1158/0008-5472.CAN-09-0299. [DOI] [PubMed] [Google Scholar]
- 745.Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010;70:288–98. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 746.Roscic A, Baldo B, Crochemore C, Marcellin D, Paganetti P. Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. J Neurochem. 2011;119:398–407. doi: 10.1111/j.1471-4159.2011.07435.x. [DOI] [PubMed] [Google Scholar]
- 747.Fan QW, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T, et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal. 2010;3:ra81. doi: 10.1126/scisignal.2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 748.Sakagami H, Kawase M, Wakabayashi H, Kurihara T. Factors that affect the type of cell death induced by chemicals. Autophagy. 2007;3:493–5. doi: 10.4161/auto.4594. [DOI] [PubMed] [Google Scholar]
- 749.Doelling JH, Walker JM, Friedman EM, Thompson AR, Vierstra RD. The APG8/12-activating enzyme APG7 is required for proper nutrient recycling and senescence in Arabidopsis thaliana. J Biol Chem. 2002;277:33105–14. doi: 10.1074/jbc.M204630200. [DOI] [PubMed] [Google Scholar]
- 750.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–5. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 751.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 752.Zhu H, Wu H, Liu X, Li B, Chen Y, Ren X, et al. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy. 2009;5:816–23. doi: 10.4161/auto.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 753.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, et al. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–57. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 754.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–78. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 755.Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, Morskaya SS, et al. 5′-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med. 2008;14:1256–63. doi: 10.1038/nm.1887. [DOI] [PubMed] [Google Scholar]
- 756.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27:1110–21. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 757.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 758.Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2008;4:309–14. doi: 10.4161/auto.5474. [DOI] [PubMed] [Google Scholar]
- 759.Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. 2009;11:1355–62. doi: 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 760.Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009;186:255–68. doi: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 761.Kimball SR, Siegfried BA, Jefferson LS. Glucagon represses signaling through the mammalian target of rapamycin in rat liver by activating AMP-activated protein kinase. J Biol Chem. 2004;279:54103–9. doi: 10.1074/jbc.M410755200. [DOI] [PubMed] [Google Scholar]
- 762.Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem. 1995;270:2320–6. doi: 10.1074/jbc.270.5.2320. [DOI] [PubMed] [Google Scholar]
- 763.Klionsky DJ, Meijer AJ, Codogno P, Neufeld TP, Scott RC. Autophagy and p70S6 kinase. Autophagy. 2005;1:59–61. doi: 10.4161/auto.1.1.1536. [DOI] [PubMed] [Google Scholar]
- 764.Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273:3963–6. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 765.Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170:1101–11. doi: 10.1083/jcb.200504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 766.Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 767.Furuya N, Liang XH, Levine B. Autophagy and cancer. In: Klionsky DJ, ed. Autophagy. Georgetown, TX: Landes Bioscience, 2004:241-55. [Google Scholar]
- 768.de Medina P, Paillasse MR, Segala G, Khallouki F, Brillouet S, Dalenc F, et al. Importance of cholesterol and oxysterols metabolism in the pharmacology of tamoxifen and other AEBS ligands. Chem Phys Lipids. 2011;164:432–7. doi: 10.1016/j.chemphyslip.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 769.de Medina P, Payré B, Boubekeur N, Bertrand-Michel J, Tercé F, Silvente-Poirot S, et al. Ligands of the antiestrogen-binding site induce active cell death and autophagy in human breast cancer cells through the modulation of cholesterol metabolism. Cell Death Differ. 2009;16:1372–84. doi: 10.1038/cdd.2009.62. [DOI] [PubMed] [Google Scholar]
- 770.Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3:331–8. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 771.Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A. 2007;104:19023–8. doi: 10.1073/pnas.0709695104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 772.Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–83. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 773.Vicencio JM, Ortiz C, Criollo A, Jones AW, Kepp O, Galluzzi L, et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ. 2009;16:1006–17. doi: 10.1038/cdd.2009.34. [DOI] [PubMed] [Google Scholar]
- 774.Dayan F, Bilton RL, Laferrière J, Trottier E, Roux D, Pouyssegur J, et al. Activation of HIF-1alpha in exponentially growing cells via hypoxic stimulation is independent of the Akt/mTOR pathway. J Cell Physiol. 2009;218:167–74. doi: 10.1002/jcp.21584. [DOI] [PubMed] [Google Scholar]
- 775.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–81. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 776.Yamashita S, Yurimoto H, Murakami D, Yoshikawa M, Oku M, Sakai Y. Lag-phase autophagy in the methylotrophic yeast Pichia pastoris. Genes Cells. 2009;14:861–70. doi: 10.1111/j.1365-2443.2009.01316.x. [DOI] [PubMed] [Google Scholar]
- 777.van Zutphen T, Baerends RJ, Susanna KA, de Jong A, Kuipers OP, Veenhuis M, et al. Adaptation of Hansenula polymorpha to methanol: a transcriptome analysis. BMC Genomics. 2010;11:1. doi: 10.1186/1471-2164-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 778.Inoue Y, Suzuki T, Hattori M, Yoshimoto K, Ohsumi Y, Moriyasu Y. AtATG genes, homologs of yeast autophagy genes, are involved in constitutive autophagy in Arabidopsis root tip cells. Plant Cell Physiol. 2006;47:1641–52. doi: 10.1093/pcp/pcl031. [DOI] [PubMed] [Google Scholar]
- 779.Yano K, Suzuki T, Moriyasu Y. Constitutive autophagy in plant root cells. Autophagy. 2007;3:360–2. doi: 10.4161/auto.4158. [DOI] [PubMed] [Google Scholar]
- 780.Gordon PB, Kisen GO, Kovács AL, Seglen PO. Experimental characterization of the autophagic-lysosomal pathway in isolated rat hepatocytes. Biochem Soc Symp. 1989;55:129–43. [PubMed] [Google Scholar]
- 781.Poli A, Gordon PB, Schwarze PE, Grinde B, Seglen PO. Effects of insulin and anchorage on hepatocytic protein metabolism and amino acid transport. J Cell Sci. 1981;48:1–18. doi: 10.1242/jcs.48.1.1. [DOI] [PubMed] [Google Scholar]
- 782.Schliess F, Reissmann R, Reinehr R, vom Dahl S, Häussinger D. Involvement of integrins and Src in insulin signaling toward autophagic proteolysis in rat liver. J Biol Chem. 2004;279:21294–301. doi: 10.1074/jbc.M313901200. [DOI] [PubMed] [Google Scholar]
- 783.vom Dahl S, Dombrowski F, Schmitt M, Schliess F, Pfeifer U, Häussinger D. Cell hydration controls autophagosome formation in rat liver in a microtubule-dependent way downstream from p38MAPK activation. Biochem J. 2001;354:31–6. doi: 10.1042/0264-6021:3540031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 784.vom Dahl S, Stoll B, Gerok W, Häussinger D. Inhibition of proteolysis by cell swelling in the liver requires intact microtubular structures. Biochem J. 1995;308:529–36. doi: 10.1042/bj3080529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 785.Kovacs AL, Zhang H. Role of autophagy in Caenorhabditis elegans. FEBS Lett. 2010;584:1335–41. doi: 10.1016/j.febslet.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 786.Zhang Y, Yan L, Zhou Z, Yang P, Tian E, Zhang K, et al. SEPA-1 mediates the specific recognition and degradation of P granule components by autophagy in C. elegans. Cell. 2009;136:308–21. doi: 10.1016/j.cell.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 787.Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010;1:e10. doi: 10.1038/cddis.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 788.Gosai SJ, Kwak JH, Luke CJ, Long OS, King DE, Kovatch KJ, et al. Automated high-content live animal drug screening using C. elegans expressing the aggregation prone serpin α1-antitrypsin Z. PLoS One. 2010;5:e15460. doi: 10.1371/journal.pone.0015460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 789.Alberti A, Michelet X, Djeddi A, Legouis R. The autophagosomal protein LGG-2 acts synergistically with LGG-1 in dauer formation and longevity in C. elegans. Autophagy. 2010;6:622–33. doi: 10.4161/auto.6.5.12252. [DOI] [PubMed] [Google Scholar]
- 790.Kang C, You YJ, Avery L. Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev. 2007;21:2161–71. doi: 10.1101/gad.1573107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 791.Willig KI, Kellner RR, Medda R, Hein B, Jakobs S, Hell SW. Nanoscale resolution in GFP-based microscopy. Nat Methods. 2006;3:721–3. doi: 10.1038/nmeth922. [DOI] [PubMed] [Google Scholar]
- 792.Huang B, Bates M, Zhuang X. Super-resolution fluorescence microscopy. Annu Rev Biochem. 2009;78:993–1016. doi: 10.1146/annurev.biochem.77.061906.092014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 793.Alers S, Löffler AS, Paasch F, Dieterle AM, Keppeler H, Lauber K, et al. Atg13 and FIP200 act independently of Ulk1 and Ulk2 in autophagy induction. Autophagy. 2011;7:1424–33. doi: 10.4161/auto.7.12.18027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 794.Brown WR, Hubbard SJ, Tickle C, Wilson SA. The chicken as a model for large-scale analysis of vertebrate gene function. Nat Rev Genet. 2003;4:87–98. doi: 10.1038/nrg998. [DOI] [PubMed] [Google Scholar]
- 795.Baba TW, Giroir BP, Humphries EH. Cell lines derived from avian lymphomas exhibit two distinct phenotypes. Virology. 1985;144:139–51. doi: 10.1016/0042-6822(85)90312-5. [DOI] [PubMed] [Google Scholar]
- 796.Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity. 2009;30:588–98. doi: 10.1016/j.immuni.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 797a.Hou YC, Chittaranjan S, Barbosa SG, McCall K, Gorski SM. Effector caspase Dcp-1 and IAP protein Bruce regulate starvation-induced autophagy during Drosophila melanogaster oogenesis. J Cell Biol. 2008;182:1127–39. doi: 10.1083/jcb.200712091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 797b.Fouillet A, Levet C, Virgone A, Robin M, Dourlen P, Rieusset J, et al. ER stress inhibits neuronal death by promoting autophagy. Autophagy. 2012 doi: 10.4161/auto.19716. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 798.Josefsen L, Droce A, Sondergaard TE, Sørensen JL, Bormann J, Schäfer W, et al. Autophagy provides nutrients for nonassimilating fungal structures and is necessary for plant colonization but not for infection in the necrotrophic plant pathogen Fusarium graminearum. Autophagy. 2012;8:326–37. doi: 10.4161/auto.18705. [DOI] [PubMed] [Google Scholar]
- 799.Nadal M, Gold SE. The autophagy genes ATG8 and ATG1 affect morphogenesis and pathogenicity in Ustilago maydis. Mol Plant Pathol. 2010;11:463–78. doi: 10.1111/j.1364-3703.2010.00620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 800.Pollack JK, Harris SD, Marten MR. Autophagy in filamentous fungi. Fungal Genet Biol. 2009;46:1–8. doi: 10.1016/j.fgb.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 801.Richie DL, Fuller KK, Fortwendel J, Miley MD, McCarthy JW, Feldmesser M, et al. Unexpected link between metal ion deficiency and autophagy in Aspergillus fumigatus. Eukaryot Cell. 2007;6:2437–47. doi: 10.1128/EC.00224-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 802.Pinan-Lucarré B, Balguerie A, Clavé C. Accelerated cell death in Podospora autophagy mutants. Eukaryot Cell. 2005;4:1765–74. doi: 10.1128/EC.4.11.1765-1774.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 803.Asakura M, Ninomiya S, Sugimoto M, Oku M, Yamashita S, Okuno T, et al. Atg26-mediated pexophagy is required for host invasion by the plant pathogenic fungus Colletotrichum orbiculare. Plant Cell. 2009;21:1291–304. doi: 10.1105/tpc.108.060996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 804.Liu X-H, Lu J-P, Zhang L, Dong B, Min H, Lin F-C. Involvement of a Magnaporthe grisea serine/threonine kinase gene, MgATG1, in appressorium turgor and pathogenesis. Eukaryot Cell. 2007;6:997–1005. doi: 10.1128/EC.00011-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 805.Nguyen LN, Bormann J, Le GT, Stärkel C, Olsson S, Nosanchuk JD, et al. Autophagy-related lipase FgATG15 of Fusarium graminearum is important for lipid turnover and plant infection. Fungal Genet Biol. 2011;48:217–24. doi: 10.1016/j.fgb.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 806.Deng YZ, Ramos-Pamplona M, Naqvi NI. Methods for functional analysis of macroautophagy in filamentous fungi. Methods Enzymol. 2008;451:295–310. doi: 10.1016/S0076-6879(08)03220-5. [DOI] [PubMed] [Google Scholar]
- 807.Berger B, Abdalla FC, Cruz-Landim C. Effect of narcosis with CO2 on the ovarian development in queens of Apis mellifera (Hymenoptera, Apini) Sociobiology. 2005;45:261–70. [Google Scholar]
- 808.Silva-Zacarin ECM, Tomaino GA, Brocheto-Braga MR, Taboga SR, De Moraes RL. Programmed cell death in the larval salivary glands of Apis mellifera (Hymenoptera, Apidae) J Biosci. 2007;32:309–28. doi: 10.1007/s12038-007-0031-2. [DOI] [PubMed] [Google Scholar]
- 809.Gregorc A, Bowen ID. Programmed cell death in the honey-bee (Apis mellifera L.) larvae midgut. Cell Biol Int. 1997;21:151–8. doi: 10.1006/cbir.1997.0127. [DOI] [PubMed] [Google Scholar]
- 810.Navajas M, Migeon A, Alaux C, Martin-Magniette M, Robinson G, Evans J, et al. Differential gene expression of the honey bee Apis mellifera associated with Varroa destructor infection. BMC Genomics. 2008;9:301. doi: 10.1186/1471-2164-9-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 811.Spruessel A, Steimann G, Jung M, Lee SA, Carr T, Fentz AK, et al. Tissue ischemia time affects gene and protein expression patterns within minutes following surgical tumor excision. Biotechniques. 2004;36:1030–7. doi: 10.2144/04366RR04. [DOI] [PubMed] [Google Scholar]
- 812.Espina V, Edmiston KH, Heiby M, Pierobon M, Sciro M, Merritt B, et al. A portrait of tissue phosphoprotein stability in the clinical tissue procurement process. Mol Cell Proteomics. 2008;7:1998–2018. doi: 10.1074/mcp.M700596-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 813.Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol. 2010;221:117–24. doi: 10.1002/path.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 814.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 815.Vanhorebeek I, Gunst J, Derde S, Derese I, Boussemaere M, Güiza F, et al. Insufficient activation of autophagy allows cellular damage to accumulate in critically ill patients. J Clin Endocrinol Metab. 2011;96:E633–45. doi: 10.1210/jc.2010-2563. [DOI] [PubMed] [Google Scholar]
- 816.Nyman E, Brännmark C, Palmér R, Brugård J, Nyström FH, Strålfors P, et al. A hierarchical whole-body modeling approach elucidates the link between in Vitro insulin signaling and in Vivo glucose homeostasis. J Biol Chem. 2011;286:26028–41. doi: 10.1074/jbc.M110.188987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 817.Buzgariu W, Chera S, Galliot B. Methods to investigate autophagy during starvation and regeneration in hydra. Methods Enzymol. 2008;451:409–37. doi: 10.1016/S0076-6879(08)03226-6. [DOI] [PubMed] [Google Scholar]
- 818.Chera S, Buzgariu W, Ghila L, Galliot B. Autophagy in Hydra: a response to starvation and stress in early animal evolution. Biochim Biophys Acta. 2009;1793:1432–43. doi: 10.1016/j.bbamcr.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 819.Chera S, de Rosa R, Miljkovic-Licina M, Dobretz K, Ghila L, Kaloulis K, et al. Silencing of the hydra serine protease inhibitor Kazal1 gene mimics the human SPINK1 pancreatic phenotype. J Cell Sci. 2006;119:846–57. doi: 10.1242/jcs.02807. [DOI] [PubMed] [Google Scholar]
- 820.Galliot B. Autophagy and self-preservation: a step ahead from cell plasticity? Autophagy. 2006;2:231–3. doi: 10.4161/auto.2706. [DOI] [PubMed] [Google Scholar]
- 821.Galliot B, Miljkovic-Licina M, de Rosa R, Chera S. Hydra, a niche for cell and developmental plasticity. Semin Cell Dev Biol. 2006;17:492–502. doi: 10.1016/j.semcdb.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 822.Sala-Mercado JA, Wider J, Undyala VV, Jahania S, Yoo W, Mentzer RM, Jr., et al. Profound cardioprotection with chloramphenicol succinate in the swine model of myocardial ischemia-reperfusion injury. Circulation. 2010;122:S179–84. doi: 10.1161/CIRCULATIONAHA.109.928242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 823.Sobolewska A, Motyl T, Gajewska M. Role and regulation of autophagy in the development of acinar structures formed by bovine BME-UV1 mammary epithelial cells. Eur J Cell Biol. 2011;90:854–64. doi: 10.1016/j.ejcb.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 824.Motyl T, Gajewska M, Zarzynska J, Sobolewska A, Gajkowska B. Regulation of autophagy in bovine mammary epithelial cells. Autophagy. 2007;3:484–6. doi: 10.4161/auto.4491. [DOI] [PubMed] [Google Scholar]
- 825.Sobolewska A, Gajewska M, Zarzyn´ska J, Gajkowska B, Motyl T. IGF-I, EGF, and sex steroids regulate autophagy in bovine mammary epithelial cells via the mTOR pathway. Eur J Cell Biol. 2009;88:117–30. doi: 10.1016/j.ejcb.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 826.Facey CO, Lockshin RA. The execution phase of autophagy associated PCD during insect metamorphosis. Apoptosis. 2010;15:639–52. doi: 10.1007/s10495-010-0499-3. [DOI] [PubMed] [Google Scholar]
- 827.Malagoli D, Abdalla FC, Cao Y, Feng Q, Fujisaki K, Gregorc A, et al. Autophagy and its physiological relevance in arthropods: Current knowledge and perspectives. Autophagy. 2010;6:575–88. doi: 10.4161/auto.6.5.11962. [DOI] [PubMed] [Google Scholar]
- 828.Mpakou VE, Nezis IP, Stravopodis DJ, Margaritis LH, Papassideri IS. Programmed cell death of the ovarian nurse cells during oogenesis of the silkmoth Bombyx mori. Dev Growth Differ. 2006;48:419–28. doi: 10.1111/j.1440-169X.2006.00878.x. [DOI] [PubMed] [Google Scholar]
- 829.Mpakou VE, Nezis IP, Stravopodis DJ, Margaritis LH, Papassideri IS. Different modes of programmed cell death during oogenesis of the silkmoth Bombyx mori. Autophagy. 2008;4:97–100. doi: 10.4161/auto.5205. [DOI] [PubMed] [Google Scholar]
- 830.Sumithra P, Britto CP, Krishnan M. Modes of cell death in the pupal perivisceral fat body tissue of the silkworm Bombyx mori L. Cell Tissue Res. 2010;339:349–58. doi: 10.1007/s00441-009-0898-3. [DOI] [PubMed] [Google Scholar]
- 831.Tettamanti G, Grimaldi A, Casartelli M, Ambrosetti E, Ponti B, Congiu T, et al. Programmed cell death and stem cell differentiation are responsible for midgut replacement in Heliothis virescens during prepupal instar. Cell Tissue Res. 2007;330:345–59. doi: 10.1007/s00441-007-0449-8. [DOI] [PubMed] [Google Scholar]
- 832.Goncu E, Parlak O. Some autophagic and apoptotic features of programmed cell death in the anterior silk glands of the silkworm, Bombyx mori. Autophagy. 2008;4:1069–72. doi: 10.4161/auto.6953. [DOI] [PubMed] [Google Scholar]
- 833.Zhou S, Zhou Q, Liu Y, Wang S, Wen D, He Q, et al. Two Tor genes in the silkworm Bombyx mori. Insect Mol Biol. 2010;19:727–35. doi: 10.1111/j.1365-2583.2010.01026.x. [DOI] [PubMed] [Google Scholar]
- 834.Zhang X, Hu ZY, Li WF, Li QR, Deng XJ, Yang WY, et al. Systematic cloning and analysis of autophagy-related genes from the silkworm Bombyx mori. BMC Mol Biol. 2009;10:50. doi: 10.1186/1471-2199-10-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 835.Li Q, Deng X, Huang Z, Zheng S, Tettamanti G, Cao Y, et al. Expression of autophagy-related genes in the anterior silk gland of the silkworm (Bombyx mori) during metamorphosis. Can J Zool. 2011;89:1019–26. doi: 10.1139/z11-075. [DOI] [Google Scholar]
- 836.Thomé RG, Santos HB, Arantes FP, Domingos FF, Bazzoli N, Rizzo E. Dual roles for autophagy during follicular atresia in fish ovary. Autophagy. 2009;5:117–9. doi: 10.4161/auto.5.1.7302. [DOI] [PubMed] [Google Scholar]
- 837.Santos HB, Thomé RG, Arantes FP, Sato Y, Bazzoli N, Rizzo E. Ovarian follicular atresia is mediated by heterophagy, autophagy, and apoptosis in Prochilodus argenteus and Leporinus taeniatus (Teleostei: Characiformes) Theriogenology. 2008;70:1449–60. doi: 10.1016/j.theriogenology.2008.06.091. [DOI] [PubMed] [Google Scholar]
- 838.Santos HB, Sato Y, Moro L, Bazzoli N, Rizzo E. Relationship among follicular apoptosis, integrin beta1 and collagen type IV during early ovarian regression in the teleost Prochilodus argenteus after induced spawning. Cell Tissue Res. 2008;332:159–70. doi: 10.1007/s00441-007-0540-1. [DOI] [PubMed] [Google Scholar]
- 839.Santos HB, Rizzo E, Bazzoli N, Sato Y, Moro L. Ovarian regression and apoptosis in the South American teleost Leporinus taeniatus Lutken (Characiformes, Anostomidae) from the São Francisco Basin. J Fish Biol. 2005;67:1446–59. doi: 10.1111/j.1095-8649.2005.00854.x. [DOI] [Google Scholar]
- 840.Couve E, Schmachtenberg O. Autophagic activity and aging in human odontoblasts. J Dent Res. 2011;90:523–8. doi: 10.1177/0022034510393347. [DOI] [PubMed] [Google Scholar]
- 841.González-Estévez C. Autophagy in freshwater planarians. Methods Enzymol. 2008;451:439–65. doi: 10.1016/S0076-6879(08)03227-8. [DOI] [PubMed] [Google Scholar]
- 842.González-Estévez C, Felix DA, Aboobaker AA, Saló E. Gtdap-1 promotes autophagy and is required for planarian remodeling during regeneration and starvation. Proc Natl Acad Sci U S A. 2007;104:13373–8. doi: 10.1073/pnas.0703588104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 843.Toyooka K, Moriyasu Y, Goto Y, Takeuchi M, Fukuda H, Matsuoka K. Protein aggregates are transported to vacuoles by a macroautophagic mechanism in nutrient-starved plant cells. Autophagy. 2006;2:96–106. doi: 10.4161/auto.2.2.2366. [DOI] [PubMed] [Google Scholar]
- 844.Svenning S, Lamark T, Krause K, Johansen T. Plant NBR1 is a selective autophagy substrate and a functional hybrid of the mammalian autophagic adapters NBR1 and p62/SQSTM1. Autophagy. 2011;7:993–1010. doi: 10.4161/auto.7.9.16389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 845.Zientara-Rytter K, Lukomska J, Moniuszko G, Gwozdecki R, Surowiecki P, Lewandowska M, et al. Identification and functional analysis of Joka2, a tobacco member of the family of selective autophagy cargo receptors. Autophagy. 2011;7:1145–58. doi: 10.4161/auto.7.10.16617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 846.Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol. 1992;119:301–11. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 847.Moriyasu Y, Ohsumi Y. Autophagy in tobacco suspension-cultured cells in response to sucrose starvation. Plant Physiol. 1996;111:1233–41. doi: 10.1104/pp.111.4.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 848.Calvo-Garrido J, Carilla-Latorre S, Kubohara Y, Santos-Rodrigo N, Mesquita A, Soldati T, et al. Autophagy in Dictyostelium: genes and pathways, cell death and infection. Autophagy. 2010;6:686–701. doi: 10.4161/auto.6.6.12513. [DOI] [PubMed] [Google Scholar]
- 849.Tung SM, Unal C, Ley A, Peña C, Tunggal B, Noegel AA, et al. Loss of Dictyostelium ATG9 results in a pleiotropic phenotype affecting growth, development, phagocytosis and clearance and replication of Legionella pneumophila. Cell Microbiol. 2010;12:765–80. doi: 10.1111/j.1462-5822.2010.01432.x. [DOI] [PubMed] [Google Scholar]
- 850.Bozzaro S, Eichinger L. The professional phagocyte Dictyostelium discoideum as a model host for bacterial pathogens. Curr Drug Targets. 2011;12:942–54. doi: 10.2174/138945011795677782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 851.Duszenko M, Ginger ML, Brennand A, Gualdrón-López M, Colombo MI, Coombs GH, et al. Autophagy in protists. Autophagy. 2011;7:127–58. doi: 10.4161/auto.7.2.13310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 852.Barquilla A, Crespo JL, Navarro M. Rapamycin inhibits trypanosome cell growth by preventing TOR complex 2 formation. Proc Natl Acad Sci U S A. 2008;105:14579–84. doi: 10.1073/pnas.0802668105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 853.Morais P, Lamas J, Sanmartín ML, Orallo F, Leiro J. Resveratrol induces mitochondrial alterations, autophagy and a cryptobiosis-like state in scuticociliates. Protist. 2009;160:552–64. doi: 10.1016/j.protis.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 854.Yakisich JS, Kapler GM. The effect of phosphoinositide 3-kinase inhibitors on programmed nuclear degradation in Tetrahymena and fate of surviving nuclei. Cell Death Differ. 2004;11:1146–9. doi: 10.1038/sj.cdd.4401473. [DOI] [PubMed] [Google Scholar]
- 855.Akematsu T, Pearlman RE, Endoh H. Gigantic macroautophagy in programmed nuclear death of Tetrahymena thermophila. Autophagy. 2010;6:901–11. doi: 10.4161/auto.6.7.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 856.Thorgaard GH, Bailey GS, Williams D, Buhler DR, Kaattari SL, Ristow SS, et al. Status and opportunities for genomics research with rainbow trout. Comp Biochem Physiol B Biochem Mol Biol. 2002;133:609–46. doi: 10.1016/S1096-4959(02)00167-7. [DOI] [PubMed] [Google Scholar]
- 857.Govoroun M, Le Gac F, Guiguen Y. Generation of a large scale repertoire of Expressed Sequence Tags (ESTs) from normalised rainbow trout cDNA libraries. BMC Genomics. 2006;7:196. doi: 10.1186/1471-2164-7-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 858.Rexroad CE, III, Lee Y, Keele JW, Karamycheva S, Brown G, Koop B, et al. Sequence analysis of a rainbow trout cDNA library and creation of a gene index. Cytogenet Genome Res. 2003;102:347–54. doi: 10.1159/000075773. [DOI] [PubMed] [Google Scholar]
- 859.Rise ML, von Schalburg KR, Brown GD, Mawer MA, Devlin RH, Kuipers N, et al. Development and application of a salmonid EST database and cDNA microarray: data mining and interspecific hybridization characteristics. Genome Res. 2004;14:478–90. doi: 10.1101/gr.1687304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 860.Salem M, Rexroad CE, III, Wang J, Thorgaard GH, Yao J. Characterization of the rainbow trout transcriptome using Sanger and 454-pyrosequencing approaches. BMC Genomics. 2010;11:564. doi: 10.1186/1471-2164-11-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 861.Polakof S, Panserat S, Craig PM, Martyres DJ, Plagnes-Juan E, Savari S, et al. The metabolic consequences of hepatic AMP-kinase phosphorylation in rainbow trout. PLoS One. 2011;6:e20228. doi: 10.1371/journal.pone.0020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 862.Seiliez I, Gabillard JC, Skiba-Cassy S, Garcia-Serrana D, Gutie´rrez J, Kaushik S, et al. An in vivo and in vitro assessment of tor signaling cascade in rainbow trout (oncorhynchus mykiss) Am J Physiol Regul Integr Comp Physiol. 2008;295:r329–35. doi: 10.1152/ajpregu.00146.2008. [DOI] [PubMed] [Google Scholar]
- 863.Seiliez I, Gabillard J-C, Riflade M, Sadoul B, Dias K, Ave´rous J, et al. Amino acids downregulate the expression of several autophagy-related genes in rainbow trout myoblasts. autophagy. 2012;8:364–75. doi: 10.4161/auto.18863. [DOI] [PubMed] [Google Scholar]
- 864.Umemiya R, Matsuo T, Hatta T, Sakakibara S, Boldbaatar D, Fujisaki K. Cloning and characterization of an autophagy-related gene, ATG12, from the three-host tick Haemaphysalis longicornis. Insect Biochem Mol Biol. 2007;37:975–84. doi: 10.1016/j.ibmb.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 865.Kawano S, Umemiya-Shirafuji R, Boldbaatar D, Matsuoka K, Tanaka T, Fujisaki K. Cloning and characterization of the autophagy-related gene 6 from the hard tick, Haemaphysalis longicornis. Parasitol Res. 2011;109:1341–9. doi: 10.1007/s00436-011-2429-x. [DOI] [PubMed] [Google Scholar]
- 866.Umemiya-Shirafuji R, Matsuo T, Liao M, Boldbaatar D, Battur B, Suzuki HI, et al. Increased expression of ATG genes during nonfeeding periods in the tick Haemaphysalis longicornis. Autophagy. 2010;6:473–81. doi: 10.4161/auto.6.4.11668. [DOI] [PubMed] [Google Scholar]
- 867.Piggott N, Cook MA, Tyers M, Measday V. Genome-wide fitness profiles reveal a requirement for autophagy during yeast fermentation. Genes Genomes Genetics. 2011;1:353–67. doi: 10.1534/g3.111.000836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 868.Cebollero E, Gonzalez R. Induction of autophagy by second-fermentation yeasts during elaboration of sparkling wines. Appl Environ Microbiol. 2006;72:4121–7. doi: 10.1128/AEM.02920-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 869.Marks VD, Ho Sui SJ, Erasmus D, van der Merwe GK, Brumm J, Wasserman WW, et al. Dynamics of the yeast transcriptome during wine fermentation reveals a novel fermentation stress response. FEMS Yeast Res. 2008;8:35–52. doi: 10.1111/j.1567-1364.2007.00338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 870.Mendes-Ferreira A, Sampaio-Marques B, Barbosa C, Rodrigues F, Costa V, Mendes-Faia A, et al. Accumulation of non-superoxide anion reactive oxygen species mediates nitrogen-limited alcoholic fermentation by Saccharomyces cerevisiae. Appl Environ Microbiol. 2010;76:7918–24. doi: 10.1128/AEM.01535-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 871.Rossignol T, Dulau L, Julien A, Blondin B. Genome-wide monitoring of wine yeast gene expression during alcoholic fermentation. Yeast. 2003;20:1369–85. doi: 10.1002/yea.1046. [DOI] [PubMed] [Google Scholar]
- 872.Teixeira MC, Raposo LR, Mira NP, Lourenço AB, Sá-Correia I. Genome-wide identification of Saccharomyces cerevisiae genes required for maximal tolerance to ethanol. Appl Environ Microbiol. 2009;75:5761–72. doi: 10.1128/AEM.00845-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 873.Yoshikawa K, Tanaka T, Furusawa C, Nagahisa K, Hirasawa T, Shimizu H. Comprehensive phenotypic analysis for identification of genes affecting growth under ethanol stress in Saccharomyces cerevisiae. FEMS Yeast Res. 2009;9:32–44. doi: 10.1111/j.1567-1364.2008.00456.x. [DOI] [PubMed] [Google Scholar]
- 874.Hazan R, Levine A, Abeliovich H. Benzoic acid, a weak organic acid food preservative, exerts specific effects on intracellular membrane trafficking pathways in Saccharomyces cerevisiae. Appl Environ Microbiol. 2004;70:4449–57. doi: 10.1128/AEM.70.8.4449-4457.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 875.Winter G, Hazan R, Bakalinsky AT, Abeliovich H. Caffeine induces macroautophagy and confers a cytocidal effect on food spoilage yeast in combination with benzoic acid. Autophagy. 2008;4:28–36. doi: 10.4161/auto.5127. [DOI] [PubMed] [Google Scholar]
- 876.Singletary K, Milner J. Diet, autophagy, and cancer: a review. Cancer Epidemiol Biomarkers Prev. 2008;17:1596–610. doi: 10.1158/1055-9965.EPI-07-2917. [DOI] [PubMed] [Google Scholar]
- 877.Su CL, Chen FN, Won SJ. Involvement of apoptosis and autophagy in reducing mouse hepatoma ML-1 cell growth in inbred BALB/c mice by bacterial fermented soybean products. Food Chem Toxicol. 2011;49:17–24. doi: 10.1016/j.fct.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 878.Abeliovich H, Gonzalez R. Autophagy in food biotechnology. Autophagy. 2009;5:925–9. doi: 10.4161/auto.5.7.9213. [DOI] [PubMed] [Google Scholar]
- 879.He C, Bartholomew CR, Zhou W, Klionsky DJ. Assaying autophagic activity in transgenic GFP-Lc3 and GFP-Gabarap zebrafish embryos. Autophagy. 2009;5:520–6. doi: 10.4161/auto.5.4.7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 880.Komoike Y, Shimojima K, Liang JS, Fujii H, Maegaki Y, Osawa M, et al. A functional analysis of GABARAP on 17p13.1 by knockdown zebrafish. J Hum Genet. 2010;55:155–62. doi: 10.1038/jhg.2010.1. [DOI] [PubMed] [Google Scholar]
- 881.Dowling JJ, Low SE, Busta AS, Feldman EL. Zebrafish MTMR14 is required for excitation-contraction coupling, developmental motor function and the regulation of autophagy. Hum Mol Genet. 2010;19:2668–81. doi: 10.1093/hmg/ddq153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 882.Makky K, Tekiela J, Mayer AN. Target of rapamycin (TOR) signaling controls epithelial morphogenesis in the vertebrate intestine. Dev Biol. 2007;303:501–13. doi: 10.1016/j.ydbio.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 883.Klionsky DJ, Baehrecke EH, Brumell JH, Chu CT, Codogno P, Cuervo AM, et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition) Autophagy. 2011;7:1273–94. doi: 10.4161/auto.7.11.17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 884.Klionsky DJ, Codogno P, Cuervo AM, Deretic V, Elazar Z, Fueyo-Margareto J, et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy. 2010;6:438–48. doi: 10.4161/auto.6.4.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]