

Abstract
The use of hybridization-based methods for DMD mutation analysis is increasingly common. We report a case of Becker Muscular Dystrophy in which discrepant results between a PCR-based (SCAIP) and a comparative genomic hybridization assay incompletely characterized the mutation (an inversion of exons 23 and 24). These results demonstrate the limits of sensitivity and specificity of both tests, and highlight the need for more detailed analysis when intronic deletions are detected by comparative genome hybridization methods.
Keywords: Becker muscular dystrophy, inversion mutation, SCAIP, MLPA, CGH
Becker muscular dystrophy (BMD) and the more severe Duchenne muscular dystrophy (DMD) are both caused by mutations in the DMD gene that can be detected in genomic DNA samples in 93-96% of cases 1,2. Approximately 65% of DMD mutations are deletions of one or more exons, and another 5% are exonic duplications that require methods for detecting exon dosage. Most non-deletion/non-duplication mutations are point mutations, including nonsense mutations, subexonic frameshifting insertions and deletions, and rarely missense mutations 3,4.
For many years the standard diagnostic test utilized multiplex PCR of a limited number of exons within deletion “hotspots”, where 98% of deletions occur 5. Relatively inexpensive and reliable for detection of deletions in hemizygous males, the multiplex PCR test has been largely replaced by methods that interrogate all exons and also readily allow dosage measurements in female carriers. These include multiplex ligation-dependent probe amplification (MLPA) that detects exon copy number 6, and comparative genomic hybridization (CGH) arrays that may also detect copy number changes in non-exonic regions 7-9. Meanwhile, detection of point mutations has become feasible through the routine use of direct sequencing methods from genomic template DNA 3 or chip-based resequencing. Sequencing of cDNA derived from muscle mRNA remains useful for detecting all mutation classes and for validating the implications of certain mutations detected at the genomic level (such as putative splice site mutations). In addition, cDNA sequencing is necessary for detecting the small percentage of mutations, such as deep intronic point mutations, that are not readily detectable with current genomic screening methods but result in inclusion of intronic sequence as pseudoexons within the mRNA transcript 10.
Here we report the characterization of an unusual DMD mutation that led to discrepant results between PCR-based and CGH array-based diagnostic methods. Both methods were performed accurately, and their combined results gave a clue to the presence of the actual mutation, an inversion of exons 23 and 24 within the gene.
Case Report
The patient was initially found to have an incidental elevation of serum transaminases that led to a gastroenterological evaluation around age 18 months. By age 5, he was found to have an elevated serum creatine kinase (1035 iu/L by report; exact upper limit for the lab used unknown). At age 12 (his most recent examination), he has myalgias with exertion and decreased stamina compared to peers, but only minimal limb-girdle weakness (modified Medical Research Council grade 5-/5 in hip flexors and deltoids, with strength otherwise 5/5).
At age 5 he underwent a clinical diagnostic DMD mutation analysis that included multiplex PCR and Southern blot analysis using eight cDNA probes covering the DMD coding region. The clinical report noted that (1) Southern blot with probe 30-2 (covering exons 20 through 30) revealed a band shift of the HindIII fragment that normally contains exons 22-25 to a position that corresponds with a slightly lower molecular weight, and (2) exons 22 through 25 were not included in the multiplex PCR reaction.
Based upon the clinical features and this result, the patient (DR43488) was eligible for enrollment in the United Dystrophinopathy Project database (http://dystrophy.genetics.utah.edu). Under an Institutional Review Board approved protocol and following informed consent, his initial testing in our lab was performed under the UDP protocol using the SCAIP method, as described elsewhere 3. Amplification of all exons (as the first step in SCAIP analysis 3) from a genomic DNA template revealed the absence of an amplification product for exon 24 alone. As per standard laboratory protocol, amplification was repeated using alternate internal primer sets flanking exon 24 in order to exclude the possibility of polymorphisms at the primer hybridization site. No product was seen by agarose gel electrophoresis using the standard (LxRx) or three alternate exon 24 primer configurations (Figure 1A).
Figure 1.
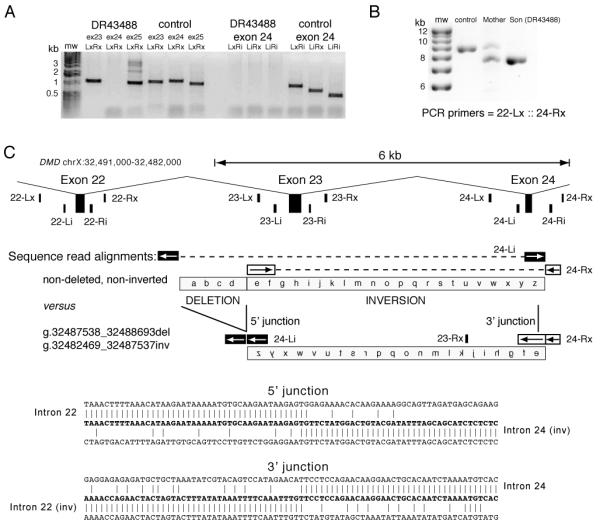
PCR and sequencing confirmation of the DMD deletion/inversion mutation. (A) PCR amplicons from SCAIP deletion analysis of exons 23, 24 and 25 in the proband (DR43488) and a normal control. (B) PCR amplicons using primers 22-Lx and exon 24-Rx for a normal control, the mother of DR43488, and patient DR43488. (C) Locations of PCR/sequencing primers in a 9 kb interval of the DMD gene (coordinates chrX:32,491,000-32,482,000), including exons 22, 23 and 24. Diagrammatic alignment of sequence reads generated from primers 24-Li and 24-Rx on normal versus inverted exon 23 to exon 24 segments. The breakpoint sequence from the 22-Lx::24-Rx amplicon from patient DR43488 defines the 5′ junction (sequence from 24-Li primer) and 3′ junction (sequence from 24-Rx primer).
In order to reconfirm the results, and to ascertain the mother’s carrier status, genomic DNA samples from the boy and his mother were tested at a clinical laboratory using a DMD Oligonucleotide Array Comparative Genomic Hybridization array 9. This revealed a normal gene dosage signal for all exons, including exon 24 in both the mother and the patient. However, it revealed a deletion in intron 22 in a heterozygous fashion in the mother, and a hemizygous fashion in the patient. This 1.1 kb deletion included nucleotides 32,487,572 through 32,488,656 of the genomic sequence (chrX reference sequence from GRCh37 assembly); this intron 22 segment is found approximately 1.6 kb from the 3′ end of exon 22, and 0.7 kb from the 5′ end of exon 23.
We presumed that both results were performed in a technically correct fashion and postulated that a complex mutation comprised of an inversion of exons 23 to 24 and an intron 22 deletion could account for both results. Repeat blood samples were obtained, and another round of diagnostic testing was therefore undertaken in our laboratory. MLPA analysis 11 revealed the presence of both exons 23 and 24 in the patient and his mother, with normal dosage for all DMD exons. PCR amplification with primers 23-Rx and 24-Rx resulted in a 1.6 kb amplification product that would be expected only in the setting of an inversion (data not shown). We then undertook long range PCR (Roche) using the manufacturer’s recommended conditions with primers flanking exon 22 (22-Lx) and exon 24 (24-Rx), and successfully amplified a band of approximately 8 kb from DNA from both the boy and his mother, and a band of approximately 9 kb from a wild-type control and the boy’s mother (see Figure 1B).
Sequencing of this ~ 8 kb PCR product from the patient revealed an inversion of exon 23 through exon 24 associated with the intron 22 deletion breakpoint (Figure 1C and D). The 5′ inversion breakpoint is at the site of the 1.16 kb deletion first detected by CGH array within intron 22. The 3′ inversion breakpoint is located within intron 24 between the splice donor site of exon 24 and the location of the internal PCR primer site (24-Ri). The sequence generated from the 24-Li primer aligns with intron 23, exon 24 and intron 24 (196 nt. to nucleotide 32,482,469) before aligning with intron 22 at the 5′ junction of the deletion (nucleotide 32,488,694). The sequence generated from the 24-Rx primer begins in intron 24, aligning through to nucleotide 32,482,468 and then continues aligning in intron 22 beginning at nucleotide 32,487,537. These alignments are consistent with a complex deletion/inversion: intron 22 deletion (g.32487538_32488693del, 1.16 kb) plus an exon 23 to 24 inversion (g.32482469_32487537inv, 5.07 kb).
Discussion
These results show limitations inherent in CGH, MLPA, and SCAIP, methods which are in widespread clinical use. The parsimonious explanation for the discrepant results from CGH and SCAIP testing was an inversion, and an amplification using predicted primer pairs resulted in the confirmation of that explanation and characterization of the breakpoints. The reported absence of exon 24 by SCAIP is explainable by the location of the 3′ break point between exon 24 and flanking PCR primer site (24-Ri).
The 5′ deletion/inversion junction occurs in unique sequence, while the 3′ inversion junction occurs within a 235 nt. DNA transposon-like medium reiterated frequency repeat element (MER20) located in intron 24. The nucleotides encompassing the 1.16 kb deletion are derived from intron 22, with no additional insertion of nucleotides at either the 5′ or 3′ inversion junction. These breakpoint sequence features suggest that the mechanism underlying this rearrangement was nonhomologous end joining (NHEJ) rather than nonallelic homologous recombination (NAHR). DMD deletions account for ~65% of mutations, and studies involving sequencing of dozens of deletion breakpoints have found that NHEJ appears to be the major mechanism, often with the breakpoint junction exhibiting a few nucleotides of homology between 5′ and 3′ flanking sequences or the addition of a few extraneous bases 12,13. A sequenced nonrecurrent deletion breakpoint involving the 17p11.2 Smith–Magenis syndrome region involved a MER5B element, and it was suggested that double-strand break substrates within low density MER elements may not be able to be repaired by homologous recombination in a timely manner, thus leading to error-prone NHEJ 14. The DMD exon 23-24 inversion/deletion observed here may be another example of a complex rearrangement involving the NHEJ pathway.
This case – only the fifth intragenic DMD inversion in the literature 15-18 – provides a cautionary note to the interpretation of any individual gene dosage test. Both MLPA and CGH revealed normal exon dosage. In our standard diagnostic algorithm, the absence of a detectable mutation by gene dosage and SCAIP methods leads us to recommend muscle biopsy to determine mRNA structure. In this case, a biopsy had not been performed as part of the diagnostic workup; furthermore, even after recontacting the patient and his family, we were unable to obtain tissue for mRNA or protein analysis, as they declined any tissue sampling whatsoever. This refusal included skin biopsy, from which fibroblast cultures could have been established for MyoD-forced myogenesis and subsequent DMD expression; this option is less invasive than muscle biopsy yet still allows reliable characterization of DMD mRNA structure. Nevertheless, we would predict that the mRNA contains an in-frame deletion of exons 23 and 24, which would be consistent with the patient’s relatively mild clinical course. Such a mild course suggests that these exons encode protein regions that of limited significance, and that exon skipping of small mutations in this region may prove beneficial. As in all of the previously reported cases, the inversion mutation was flanked by a deletion, in this case the smallest (1.16 kb) yet reported; this pattern suggests that the deletion event is intrinsic to the formation of the inversion. We emphasize that the report of a novel intronic deletion of any size on a diagnostic CGH test should lead to more detailed analysis by additional genomic or mRNA studies. Neuromuscular clinicians should have a high index of suspicion for the presence of an inversion mutation when interpreting CGH results.
Acknowledgements
The authors wish to acknowledge the assistance of K. Hak and C. Hamil. This work was supported by R01 NS043264 (KMF and RBW).
References
- 1.Yan J, Feng J, Buzin CH, Scaringe W, Liu Q, Mendell JR, den Dunnen J, Sommer SS. Three-tiered noninvasive diagnosis in 96% of patients with Duchenne muscular dystrophy (DMD) Hum Mutat. 2004;23(2):203–204. doi: 10.1002/humu.10307. [DOI] [PubMed] [Google Scholar]
- 2.Dent KM, Dunn DM, von Niederhausern AC, Aoyagi AT, Kerr L, Bromberg MB, Hart KJ, Tuohy T, White S, den Dunnen JT, Weiss RB, Flanigan KM. Improved molecular diagnosis of dystrophinopathies in an unselected clinical cohort. Am J Med Genet A. 2005 doi: 10.1002/ajmg.a.30617. [DOI] [PubMed] [Google Scholar]
- 3.Flanigan KM, von Niederhausern A, Dunn DM, Alder J, Mendell JR, Weiss RB. Rapid direct sequence analysis of the dystrophin gene. Am J Hum Genet. 2003;72(4):931–939. doi: 10.1086/374176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prior TW, Bartolo C, Pearl DK, Papp AC, Snyder PJ, Sedra MS, Burghes AH, Mendell JR. Spectrum of small mutations in the dystrophin coding region. Am J Hum Genet. 1995;57(1):22–33. [PMC free article] [PubMed] [Google Scholar]
- 5.Beggs AH, Koenig M, Boyce FM, Kunkel LM. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet. 1990;86(1):45–48. doi: 10.1007/BF00205170. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz M, Duno M. Improved molecular diagnosis of dystrophin gene mutations using the multiplex ligation-dependent probe amplification method. Genet Test. 2004;8(4):361–367. doi: 10.1089/gte.2004.8.361. [DOI] [PubMed] [Google Scholar]
- 7.Hegde MR, Chin EL, Mulle JG, Okou DT, Warren ST, Zwick ME. Microarray-based mutation detection in the dystrophin gene. Hum Mutat. 2008;29(9):1091–1099. doi: 10.1002/humu.20831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saillour Y, Cossee M, Leturcq F, Vasson A, Beugnet C, Poirier K, Commere V, Sublemontier S, Viel M, Letourneur F, Barbot JC, Deburgrave N, Chelly J, Bienvenu T. Detection of exonic copy-number changes using a highly efficient oligonucleotide-based comparative genomic hybridization-array method. Hum Mutat. 2008;29(9):1083–1090. doi: 10.1002/humu.20829. [DOI] [PubMed] [Google Scholar]
- 9.del Gaudio D, Yang Y, Boggs BA, Schmitt ES, Lee JA, Sahoo T, Pham HT, Wiszniewska J, Chinault AC, Beaudet AL, Eng CM. Molecular diagnosis of Duchenne/Becker muscular dystrophy: enhanced detection of dystrophin gene rearrangements by oligonucleotide array-comparative genomic hybridization. Hum Mutat. 2008;29(9):1100–1107. doi: 10.1002/humu.20841. [DOI] [PubMed] [Google Scholar]
- 10.Gurvich OL, Tuohy TM, Howard MT, Finkel RS, Medne L, Anderson CB, Weiss RB, Wilton SD, Flanigan KM. DMD pseudoexon mutations: splicing efficiency, phenotype, and potential therapy. Ann Neurol. 2008;63(1):81–89. doi: 10.1002/ana.21290. [DOI] [PubMed] [Google Scholar]
- 11.White SJ, Aartsma-Rus A, Flanigan KM, Weiss RB, Kneppers AL, Lalic T, Janson AA, Ginjaar HB, Breuning MH, den Dunnen JT. Duplications in the DMD gene. Hum Mutat. 2006;27(9):938–945. doi: 10.1002/humu.20367. [DOI] [PubMed] [Google Scholar]
- 12.Toffolatti L, Cardazzo B, Nobile C, Danieli GA, Gualandi F, Muntoni F, Abbs S, Zanetti P, Angelini C, Ferlini A, Fanin M, Patarnello T. Investigating the mechanism of chromosomal deletion: characterization of 39 deletion breakpoints in introns 47 and 48 of the human dystrophin gene. Genomics. 2002;80(5):523–530. [PubMed] [Google Scholar]
- 13.Nobile C, Toffolatti L, Rizzi F, Simionati B, Nigro V, Cardazzo B, Patarnello T, Valle G, Danieli GA. Analysis of 22 deletion breakpoints in dystrophin intron 49. Hum Genet. 2002;110(5):418–421. doi: 10.1007/s00439-002-0721-7. [DOI] [PubMed] [Google Scholar]
- 14.Shaw CJ, Lupski JR. Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum Genet. 2005;116(1-2):1–7. doi: 10.1007/s00439-004-1204-9. [DOI] [PubMed] [Google Scholar]
- 15.Cagliani R, Sironi M, Ciafaloni E, Bardoni A, Fortunato F, Prelle A, Serafini M, Bresolin N, Comi GP. An intragenic deletion/inversion event in the DMD gene determines a novel exon creation and results in a BMD phenotype. Hum Genet. 2004;115(1):13–18. doi: 10.1007/s00439-004-1118-6. [DOI] [PubMed] [Google Scholar]
- 16.Oshima J, Magner DB, Lee JA, Breman AM, Schmitt ES, White LD, Crowe CA, Merrill M, Jayakar P, Rajadhyaksha A, Eng CM, Del Gaudio D. Regional genomic instability predisposes to complex dystrophin gene rearrangements. Hum Genet. 2009 doi: 10.1007/s00439-009-0679-9. [DOI] [PubMed] [Google Scholar]
- 17.Bovolenta M, Neri M, Fini S, Fabris M, Trabanelli C, Venturoli A, Martoni E, Bassi E, Spitali P, Brioschi S, Falzarano MS, Rimessi P, Ciccone R, Ashton E, McCauley J, Yau S, Abbs S, Muntoni F, Merlini L, Gualandi F, Ferlini A. A novel custom high density-comparative genomic hybridization array detects common rearrangements as well as deep intronic mutations in dystrophinopathies. BMC Genomics. 2008;9:572. doi: 10.1186/1471-2164-9-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madden HR, Fletcher S, Davis MR, Wilton SD. Characterization of a complex Duchenne muscular dystrophy-causing dystrophin gene inversion and restoration of the reading frame by induced exon skipping. Hum Mutat. 2009;30(1):22–28. doi: 10.1002/humu.20806. [DOI] [PubMed] [Google Scholar]