

Abstract
Genetic factors are believed to account for 25% of the interindividual differences in Years of Life (YL) among humans. However, the genetic loci that have thus far been found to be associated with YL explain a very small proportion of the expected genetic variation in this trait, perhaps reflecting the complexity of the trait and the limitations of traditional association studies when applied to traits affected by a large number of small-effect genes. Using data from the Framingham Heart Study and statistical methods borrowed largely from the field of animal genetics (whole-genome prediction, WGP), we developed a WGP model for the study of YL and evaluated the extent to which thousands of genetic variants across the genome examined simultaneously can be used to predict interindividual differences in YL. We find that a sizable proportion of differences in YL—which were unexplained by age at entry, sex, smoking and BMI—can be accounted for and predicted using WGP methods. The contribution of genomic information to prediction accuracy was even higher than that of smoking and body mass index (BMI) combined; two predictors that are considered among the most important life-shortening factors. We evaluated the impacts of familial relationships and population structure (as described by the first two marker-derived principal components) and concluded that in our dataset population structure explained partially, but not fully the gains in prediction accuracy obtained with WGP. Further inspection of prediction accuracies by age at death indicated that most of the gains in predictive ability achieved with WGP were due to the increased accuracy of prediction of early mortality, perhaps reflecting the ability of WGP to capture differences in genetic risk to deadly diseases such as cancer, which are most often responsible for early mortality in our sample.
Introduction
Agricultural and biomedical research has shown through controlled experiments and familial studies that many complex traits are highly heritable, suggesting that in principle, such traits could be predicted early in life from knowledge of individuals' genotypes. Human longevity is not an exception: empirical evidence from twin and familial studies indicate that approximately 25% of inter-individual differences in human lifespan can be attributed to genetic factors [1]–[3].
Research with model organisms offers several examples of genetic polymorphisms having a sizable effect on lifespan [4]. However, although genome-wide association studies (GWAS) and linkage scans in humans have uncovered several regions significantly associated with longevity and aging traits [5]–[9], only a few of these associations have been consistently confirmed, and our ability to predict inter-individual differences in expected Years of Life (YL) remains limited [7].
Several diseases (e.g., cancer, cardiovascular disease) and biological events (e.g., stroke, heart failure) can lead to death, and the genetic architecture (i.e., the set of genes having an effect on the trait and the ways they interact) of each of these mortality-related traits is expected to be disorder-specific. Therefore, the genetic architecture of YL is likely to include a large number, perhaps thousands, of possibly interacting genes.
Recent articles [10], [11] have suggested that the limited advances in our ability to predict complex human traits and diseases using genomic information may partially reflect the limitations of traditional GWAS to detect significant associations with complex genetic architectures. These authors have suggested that Whole Genome Prediction (WGP) may be better suited than traditional GWAS to the prediction of complex traits.
Whole genome prediction exploits multi-locus linkage-disequilibrium (LD) between quantitative trait loci (QTL) and genome-wide markers (e.g., SNPs) to predict inter-individual differences in a quantitative trait that are attributable to genetic factors. Unlike traditional association studies, in which the association between markers and phenotypes is tested one marker at a time, WGP uses all available markers to regress phenotype onto genomic information. This methodology was first proposed in the field of animal breeding by Meuwissen Hayes and Goddard in 2001 [12]. Since then, several simulation [12], [13] and empirical studies have demonstrated its predictive power with plant [14], [15] and animal [16]–[19] data.
More recently, research with human height showed that much of the so-called missing heritability of complex traits could be recovered using genome-wide panels of common variants [11] and, more importantly, that regression using WGP methods can improve the prediction of yet-to-be observed human phenotypes [20]. A next logical question is whether these findings apply to traits of greater medical or practical importance. Here, we: (a) extend WGP methods, which were originally developed for continuous un-censored outcomes, to accommodate censoring, a feature commonly encountered in applications with human data, (b) developed a WGP model for YL and (c) quantified the ability of this model to account for and to predict inter-individual differences in human YL that are not accounted for by major factors such as sex, Body Mass Index (BMI, kg/m2) and smoking.
Materials and Methods
Model
Many outcomes in human-health studies are either binary (e.g., presence/absence of diseases) or are subject to censoring (i.e., bounds of the outcome are known, but the exact value of outcome remains unknown). And it is well established that ignoring censoring yields biased estimates [21]. The linear models commonly used for WGP can be easily extended to accommodate binary or censored outcomes. Here, we present an extension that accommodates censoring. Similar ideas can be used to model binary outcomes as well [22].
In our WGP models, we describe YL (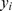 , i = 1,…,n) as the sum of individual-specific means (
, i = 1,…,n) as the sum of individual-specific means (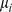 ) which, as we explain below, will be a function of genetic and non-genetic factors, and of a model residual (
) which, as we explain below, will be a function of genetic and non-genetic factors, and of a model residual (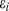 ) which is assumed to be a normal random variable with mean zero and variance
) which is assumed to be a normal random variable with mean zero and variance 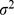 ; therefore
; therefore 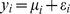 . For individuals with known YL, we observe
. For individuals with known YL, we observe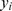 ; for individuals with censoring at age equal to
; for individuals with censoring at age equal to 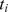 , the observed event is
, the observed event is 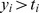 . In our WGP model, expected YL (
. In our WGP model, expected YL (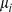 ) was described using a linear regression,
) was described using a linear regression,
| (1) |
which had three components: 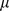 , an effect common to all subjects;
, an effect common to all subjects; 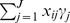 , a regression component accounting for the effects of non genetic covariates (sex, smoking and BMI covariates in our application); and
, a regression component accounting for the effects of non genetic covariates (sex, smoking and BMI covariates in our application); and 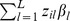 , a regression on SNP genotypes
, a regression on SNP genotypes 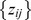 where
where 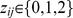 counts the number of copies of the least frequent allele at the jth SNP. By combining (1) with the normal assumptions described above, we derived the likelihood function for censored and un-censored individuals (see Methods S1 for further details).
counts the number of copies of the least frequent allele at the jth SNP. By combining (1) with the normal assumptions described above, we derived the likelihood function for censored and un-censored individuals (see Methods S1 for further details).
The Bayesian model is completed by assigning a prior density to the collection of model unknowns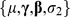 . Here, we structure the prior density using a modified version of the Bayesian LASSO (BL) [23]. This model has been effectively used for WGP in plants [14], [15], [24], animals [14], [18], [19], [25] and humans [20]. We extend this model to accommodate censoring as well as effects other than those of markers. In our model, we assigned independent vague prior densities to the intercept (
. Here, we structure the prior density using a modified version of the Bayesian LASSO (BL) [23]. This model has been effectively used for WGP in plants [14], [15], [24], animals [14], [18], [19], [25] and humans [20]. We extend this model to accommodate censoring as well as effects other than those of markers. In our model, we assigned independent vague prior densities to the intercept (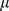 ) and to the effects of sex, smoking and BMI (
) and to the effects of sex, smoking and BMI (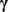 ). This treatment yields estimates of the effects of these non-genetic factors that are similar to those obtained with likelihood-based methods. For the remaining unknowns we adopt the prior-specification of the BL of Park and Casella [23] (see Methods S1 for further details). The joint prior-density (see expression 2 in the Methods S1) is indexed by a set of four hyper-parameters, including the prior degree of freedom and scale assigned to the residual variance (denoted as df and S, respectively), and the rate and shape parameters (dentoed as
). This treatment yields estimates of the effects of these non-genetic factors that are similar to those obtained with likelihood-based methods. For the remaining unknowns we adopt the prior-specification of the BL of Park and Casella [23] (see Methods S1 for further details). The joint prior-density (see expression 2 in the Methods S1) is indexed by a set of four hyper-parameters, including the prior degree of freedom and scale assigned to the residual variance (denoted as df and S, respectively), and the rate and shape parameters (dentoed as  and s, respectively) assigned to the regularization parameter of the BL. A discussion of how these can be chosen is given in Perez et al. [26]. Here, following those guidelines, we set
and s, respectively) assigned to the regularization parameter of the BL. A discussion of how these can be chosen is given in Perez et al. [26]. Here, following those guidelines, we set 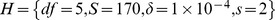 . Given the characteristics of our data (sample size, number of markers and allele frequencies and observed variability on YL), these values provide priors with small influences on predictions.
. Given the characteristics of our data (sample size, number of markers and allele frequencies and observed variability on YL), these values provide priors with small influences on predictions.
Implementation
Models were fitted using a modified version of the BLR package [27] of R [28] which handles censoring (right, left and interval) according to the model described above. In addition to BLR, R-packages bayesm [29], splines [28] and SuppDists [30] were used to implement the sampler.
Data
(N = 5,117) were from the original (N = 1,493) and offspring (N = 3,624) cohorts of the Framingham Heart Study. Data and material distributions from this study are made in accordance with the individual consent history of each participant (see http://www.framinghamheartstudy.org/research/consentfms.html for further details about consent forms). And the current study has been approved by the Internal Review Board of University of Alabama at Birmingham (IRB Protocol Number: X090720002). The criteria for inclusion in the study included being 18 years or older at time of recruitment, having survival information as of 2007, and having complete information for covariates (sex, smoking and BMI).
Average age at entry was 37 with a standard deviation (SD) of 9.0 years. Of the participants, two thirds (N = 3,390) were censored (i.e., at the time at which survival records were defined, these individuals were still alive), 55% were female, and 36% never smoked. Mean BMI at first exam was 25.0 with a SD of 4.1 kg/m2. Subjects were genotyped using the Affymetrix GeneChip Human Mapping 500K Array Set. For details on the genotyping method, please refer to Framingham SHARe at the NCBI dbGaP website (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000007.v3.p2). Other editing and genotyping quality control and imputation procedures were as described in Makowsky et al. [20].
Primary Data Analysis
Using the specification of equation (3) we generated a sequence of nested models by changing the predictors included in the right-hand side of the linear predictor (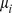 ). Our baseline model (denoted as MA-0) includes an intercept, sex, and age at entry; the latter modeled nonparametrically using a 4-df natural spline [31] with interior and boundary knots chosen using the default specifications of the natural spline (ns) function of the spline package of R [28]; with 4-df, interior knots were placed at the 25th, 50th and 75th sample percentiles of the predictor variables. We extended this model by adding smoking and BMI (also modeled nonparameterically using a 4-df natural spline [31]) this model is denoted as M
B-0. Subsequently, models MA-0 and MB-0 were then extended by adding subsets of evenly spaced SNPs, from 2.5K (K = thousand) to 80K; these models were denoted as M(.)-2.5K, M(.)-5K, M(.)-10K, M(.)-20K, M(.)-40K and M(.)-80K, where (.) was either A or B.
). Our baseline model (denoted as MA-0) includes an intercept, sex, and age at entry; the latter modeled nonparametrically using a 4-df natural spline [31] with interior and boundary knots chosen using the default specifications of the natural spline (ns) function of the spline package of R [28]; with 4-df, interior knots were placed at the 25th, 50th and 75th sample percentiles of the predictor variables. We extended this model by adding smoking and BMI (also modeled nonparameterically using a 4-df natural spline [31]) this model is denoted as M
B-0. Subsequently, models MA-0 and MB-0 were then extended by adding subsets of evenly spaced SNPs, from 2.5K (K = thousand) to 80K; these models were denoted as M(.)-2.5K, M(.)-5K, M(.)-10K, M(.)-20K, M(.)-40K and M(.)-80K, where (.) was either A or B.
Models were first fitted to the entire dataset to obtain parameter estimates (estimated posterior means of effects and of variance parameters) and to evaluate the goodness of fit and the Deviance Information Criterion [32]. Subsequently, the prediction accuracy of each of the models was assessed using a 10-fold cross-validation (CV). Prediction accuracy was evaluated using two different metrics: a CV R-squared ( ) and the area under Longitudinal Receiving Operating Characteristic Curves (AUC(τ) [33]). The
) and the area under Longitudinal Receiving Operating Characteristic Curves (AUC(τ) [33]). The 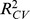 measures the proportion of inter-individual differences in years of life that can be accounted by CV-predictions, this was calculated as
measures the proportion of inter-individual differences in years of life that can be accounted by CV-predictions, this was calculated as 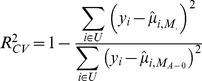 where:
where: 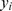 denotes observed YL of the ith individual,
denotes observed YL of the ith individual, 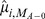 is a 10-fold CV-prediction of YL derived from model
is a 10-fold CV-prediction of YL derived from model 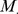 and
and 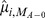 is the estimated average YL, derived from a model that only included an intercept and the effect of age at entry, which is taken here as our baseline model. This statistic can be evaluated only with subjects that have already died; therefore, in the 10-fold CV, the summation in the formula for
is the estimated average YL, derived from a model that only included an intercept and the effect of age at entry, which is taken here as our baseline model. This statistic can be evaluated only with subjects that have already died; therefore, in the 10-fold CV, the summation in the formula for 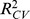 uses only data from subjects with an observed age at death. Un-censored subjects do not constitute a random sample of individuals and this may induce bias in our estimate of R-squared. Because of this, we consider a second measure of prediction performance based on longitudinal AUCs [33]. To this end we defined a sequence of thresholds (τ = 60, 65, 70, 75, 80, 85, 90, 95 YL) and for each of these thresholds we generated survival indicator variables
uses only data from subjects with an observed age at death. Un-censored subjects do not constitute a random sample of individuals and this may induce bias in our estimate of R-squared. Because of this, we consider a second measure of prediction performance based on longitudinal AUCs [33]. To this end we defined a sequence of thresholds (τ = 60, 65, 70, 75, 80, 85, 90, 95 YL) and for each of these thresholds we generated survival indicator variables 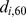 ,
,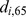 ,…,
,…,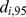 where:
where: 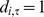 if individual i had YL<τ;
if individual i had YL<τ; 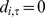 if individual i was still alive at time τ, and un-determined if individual i had an age at censoring smaller than τ. The number of individuals for which
if individual i was still alive at time τ, and un-determined if individual i had an age at censoring smaller than τ. The number of individuals for which 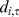 was determined (i.e., those that had known YL or age at censoring greater than τ) were 4495, 3836, 3262, 2773, 2366, 2102, 1889 and 1762 for
was determined (i.e., those that had known YL or age at censoring greater than τ) were 4495, 3836, 3262, 2773, 2366, 2102, 1889 and 1762 for 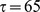 ,…,
,…, 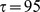 , respectively. Using these survival indicator variables and CV predictions of YL (
, respectively. Using these survival indicator variables and CV predictions of YL (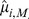 ) derived from the models above described we computed the AUC(τ) for every threshold using the R-package pROC [34].
) derived from the models above described we computed the AUC(τ) for every threshold using the R-package pROC [34].
Evaluation of the effects of population structure and familial relationships on prediction accuracy
The distribution of genotypes, their allele frequency, levels of LD, etc., can be affected by factors such as population structure, admixture or familial relationships. Therefore, a certain proportion of the prediction accuracy of WGP could be attributed to those factors. To further explore this, a series of additional analysis were carried out. First, in order to account for population structure, we extended the model including age, sex, smoking and BMI as predictors (MB-0) by adding the effects of the first two principal components (PCs) derived from the same set of 80K SNPs used in M(.)-80K. Second, to quantify the relative importance of familial relationships on prediction accuracy we carried out two additional analyses: (a) we extended MB-0 by adding an effect representing a regression on the pedigree. This was done using the standards of the additive infinitesimal model of quantitative genetics [35], and this model is denoted as MB-PED. And (b) we fitted models MA-0K, MB-0K and MB-80K in a 10fold CV where entire families, as opposed to individuals, were assigned to folds; therefore, in this CV predictions are derived from nominally-unrelated individuals.
Results
Full data analysis
Using MB-0, we estimated an average (± posterior SD) difference in YL between females and males of 3.1 (±0.42) years and between smokers and nonsmokers of −4.1 (±0.44) years. Using estimates from MB-0, we computed the expected YL of a nonsmoking 35-year-old by sex and BMI; the results are displayed in (Figure S1). Expected YL was greatest within the range 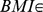 [20], [25]; extreme BMI values, lower than 20 or higher than 25, were associated with a decrease in YL. Using MB-0 we estimate an expected decrease in YL of 0.43 year per extra unit of BMI in the range
[20], [25]; extreme BMI values, lower than 20 or higher than 25, were associated with a decrease in YL. Using MB-0 we estimate an expected decrease in YL of 0.43 year per extra unit of BMI in the range 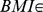 [25], [40]. Overall, these patterns are in agreement with what has been reported previously for the effect of sex [36]–[38], smoking [36], [39] and BMI [21], [36] on YL.
[25], [40]. Overall, these patterns are in agreement with what has been reported previously for the effect of sex [36]–[38], smoking [36], [39] and BMI [21], [36] on YL.
Table 1 shows estimates of residual variance and DIC by model. The intercept-only model (not included in Table 1) yielded an estimate of variance of YL of 135, and the estimated residual variance of MA-0 was 104.1; therefore, approximately 23%, computed as 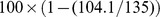 , of observed variability in YL in our dataset can be explained by differences in age at entry and sex. Model MB-0 yielded an estimate of residual variance of 98.7, indicating that BMI and smoking accounted for about 5% of inter-individual differences in YL that were not accounted for by age at entry and sex; this was computed as
, of observed variability in YL in our dataset can be explained by differences in age at entry and sex. Model MB-0 yielded an estimate of residual variance of 98.7, indicating that BMI and smoking accounted for about 5% of inter-individual differences in YL that were not accounted for by age at entry and sex; this was computed as 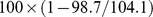 . Adding SNPs to MA-0 or MB-0 resulted in a marked increase in goodness of fit, and this is reflected in a substantial reduction in the estimated residual variance (Table 1). For instance, MB-80K yielded an estimate of the residual variance that was 65% smaller than that of the MB-0K, computed as
. Adding SNPs to MA-0 or MB-0 resulted in a marked increase in goodness of fit, and this is reflected in a substantial reduction in the estimated residual variance (Table 1). For instance, MB-80K yielded an estimate of the residual variance that was 65% smaller than that of the MB-0K, computed as 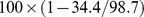 .
.
Table 1. Estimated posterior mean of residual variance and Deviance Information Criterion (DIC, ‘smaller is better’) by number of SNPs (rows) and nongenetic covariates (columns) included in the model.
| Residual | Variance* | Deviance Information Criterion (DIC) | ||
| Thousands of SNPs in the Model | Age+Sex | Age+Sex+BMI+Smoking | Age+Sex | Age+Sex+BMI+Smoking |
| 0 | 104.1 | 98.7 | 14,744 | 14,625 |
| 2.5 | 79.7 | 75.5 | 14,268 | 14,158 |
| 5.0 | 68.3 | 64.1 | 14,130 | 14,007 |
| 10.0 | 57.1 | 554.5 | 13,951 | 13,845 |
| 20.0 | 48.1 | 46.2 | 13,772 | 13,673 |
| 40.0 | 40.3 | 39.6 | 13,540 | 13,479 |
| 80.0 | 34.6 | 34.4 | 13,337 | 13,289 |
: Posterior mean of the residual variance, the estimate of this parameter can be regarded as a proxy of goodness of fit to the data used to train the model.
Due to the curse of dimensionality [40], the increase in goodness of fit achieved by adding SNPs to the model may reflect genetic variability captured by SNPs, over-fitting, or a combination of both. However, DIC, a model comparison criterion that balances goodness of fit and model complexity, decreased monotonically with the number of SNPs, suggesting that information is being added as marker density increases.
Evaluation of prediction accuracy in cross validation
Figure 1 shows estimated 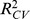 versus marker density (from 0 to 80K) by model. The
versus marker density (from 0 to 80K) by model. The 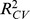 of a model including age at entry and sex,
of a model including age at entry and sex, 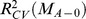 , was approximately 6%. The addition of smoking and BMI resulted in a doubling of
, was approximately 6%. The addition of smoking and BMI resulted in a doubling of 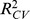 , from
, from 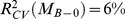 to
to 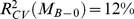 ; as expected, the addition of smoking and BMI increased prediction accuracy by a sizable amount. Prediction accuracy increased monotonically with the number of markers both in models with and without BMI and smoking covariates. These results confirm that markers are capturing information about expected YL that cannot be predicted using major factors such as age at entry, sex, smoking and BMI. Using 80K markers, we were able to increase
; as expected, the addition of smoking and BMI increased prediction accuracy by a sizable amount. Prediction accuracy increased monotonically with the number of markers both in models with and without BMI and smoking covariates. These results confirm that markers are capturing information about expected YL that cannot be predicted using major factors such as age at entry, sex, smoking and BMI. Using 80K markers, we were able to increase 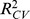 from 6% to 11% for the model without smoking and BMI (MA-(.)) and from 12% to 21% for the model including smoking and BMI (MB-(.)). (Table S1) shows
from 6% to 11% for the model without smoking and BMI (MA-(.)) and from 12% to 21% for the model including smoking and BMI (MB-(.)). (Table S1) shows 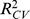 for models MA-0, MB-0 and MB-80K by fold of the CV. The variability in
for models MA-0, MB-0 and MB-80K by fold of the CV. The variability in 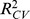 across folds reflects uncertainty about our estimates due to sampling of training and testing datasets. Although we observed an overall superiority of MB-0 over MA-0 this superiority did not occur in every fold of the CV. However, MB-80K outperformed models without SNP information (MA-0 and MB-0) consistently across folds indicating that SNPs are capturing important and consistent patterns of variability in human lifespan.
across folds reflects uncertainty about our estimates due to sampling of training and testing datasets. Although we observed an overall superiority of MB-0 over MA-0 this superiority did not occur in every fold of the CV. However, MB-80K outperformed models without SNP information (MA-0 and MB-0) consistently across folds indicating that SNPs are capturing important and consistent patterns of variability in human lifespan.
Figure 1. Cross-validation R-squared (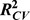 ) by number of markers and model.
) by number of markers and model.

Circles represent the 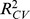 obtained in a 10-fold CV.
obtained in a 10-fold CV.
The above results indicate that markers can explain a sizable proportion of inter-individual differences in YL that are not accounted for by age at entry, sex, smoking and BMI. To obtain further insights into the source of this improvement in prediction accuracy, we present in Figure 2 the average absolute value of the CV prediction error (from the 10-fold CV) and its SE by range of YL for models MB-0K and MB-80K. As expected, for both models, the absolute value prediction error was lowest for people dying around median age (80 YL) and increased for people dying early or late in life. Predictions derived from model MB-80K were much more accurate than those of MB-0 for the prediction of YL of people dying early in life; however, the prediction accuracy of the model with markers was slightly higher than that of MB-80K for subjects dying at intermediate ages. This suggests that the overall higher predictive ability of MB-80K is due mostly to improvements in prediction of early mortality.
Figure 2. Absolute value CV prediction error versus range of YL.

Circles represent the average absolute value prediction error for each group of YL (YL≤65, 65<YL≤70,..,YL>95); and vertical bars represent the 95% confidence interval defined by the average absolute value prediction error ±1.96×SE.
Figure 3 shows the AUC (vertical axis) for models MA-0, MB-0, and MB-80K for each of the 8 thresholds (horizontal axis). Adding BMI and smoking information to a model that included sex and age (MB-0 vs MA-0) resulted in an increase in AUC(τ) of roughly 5–7%. When 80 thousand SNPs (MB-80K) were added to a model that included age, sex, smoking and BMI as covariates we observed a substantial increase in classification performance for prediction of early stage survival status (relative to MB-0, MB-80K yielded an increase in AUC(60) of 18%), a more modest increase in AUC(τ) for survival status at ages 65–90 (MB-80K outperformed MB-0 by about 14% for AUC(65) and by 7–10% for AUC(70)–AUC(90)), and no change in AUC(95). These results are consistent with those observed with 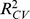 in that they indicate that genomic information can increase the prediction accuracy of lifespan, mostly due to an increase in the prediction of early mortality. Table S1 shows estimates of AUC(τ) for models MA-0, MB-0 and MB-80K by fold of the CV. Similar to what we observed for
in that they indicate that genomic information can increase the prediction accuracy of lifespan, mostly due to an increase in the prediction of early mortality. Table S1 shows estimates of AUC(τ) for models MA-0, MB-0 and MB-80K by fold of the CV. Similar to what we observed for 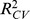 , although we found an overall superiority in the classification performance of MB-0 relative to that of MA-0 such superiority was not consistently observed in every fold. However, for early and intermediate survival status (τ≤85) model MB-80K had a classification performance that was consistently higher than that of models without genetic information (MA-0 and MB-0). For late mortality (τ>85) such superiority was not consistently observed across folds.
, although we found an overall superiority in the classification performance of MB-0 relative to that of MA-0 such superiority was not consistently observed in every fold. However, for early and intermediate survival status (τ≤85) model MB-80K had a classification performance that was consistently higher than that of models without genetic information (MA-0 and MB-0). For late mortality (τ>85) such superiority was not consistently observed across folds.
Figure 3. Area under the receiving operating characteristic curve (AUC) for survival status define at different time points (60, 65,…,95 years of life) and three models that differed on the predictor variables used to predict expected years of life.

Effects of population structure
The estimated 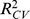 of model MB-GWPC was 15.77%, this is roughly half the way from the
of model MB-GWPC was 15.77%, this is roughly half the way from the 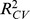 of model MB-0 (11.45%) and that of model MB-80K (21.40%). Results for AUC showed similar patterns. This indicates that a sizable proportion of inter-individual differences in YL could be attributed to genetic differences associated to population structure. On the other hand, the fact that the
of model MB-0 (11.45%) and that of model MB-80K (21.40%). Results for AUC showed similar patterns. This indicates that a sizable proportion of inter-individual differences in YL could be attributed to genetic differences associated to population structure. On the other hand, the fact that the 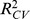 of model MB-80K was 37% higher than that of model MB-GWPC suggests that genetic factors beyond those associated with population structure account for a sizable proportion of inter-individual differences in YL.
of model MB-80K was 37% higher than that of model MB-GWPC suggests that genetic factors beyond those associated with population structure account for a sizable proportion of inter-individual differences in YL.
Effects of familial relationships
Model MB-PED, which including age at entry, sex, BMI, smoking and pedigree information showed clear signs of over-fitting (the posterior mean of the residual variance was 11.1, compared to 34.4 for model MB-80K) and, consequently, had a very poor predictive performance; even worse than our baseline model (MA-0). This is most likely to occur because of two reasons. First, the pedigree is very sparse, with 37% of nominally un-related individuals and most of the remaining individuals coming from relatively small nuclear families (74% of individuals were in families with 3 or less members). Additionally, in the great majority of nuclear families the offspring have censored YL. Therefore, in this dataset the amount of familial information available for prediction is very limited. To further illustrate this, we counted for every subject in the 10-fold CV where individuals were randomly assigned to folds the number of close-relatives (father, mother, offspring or full-sib) which were used for prediction (i.e., those which were assigned to a different fold). We found that in our CV 41.6 of the observations were predicted without having any direct relative in the training dataset (i.e. in other folds) and 70.75 were predicted without having any un-censored direct relative available for training. Only 10% of individuals had 3 or more direct relatives in the training datasets, and no-one had 3 or more direct relative with observed YL assigned to a different fold.
Our second approach to quantify the relative importance of family relationships on prediction accuracy consisted on fitting models MA-0, MB-0 and MB-80K in a10-fold CV where entire families, as opposed to individuals, were assigned to folds. Such setting guarantees that no-direct relatives are used for prediction. The 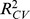 obtained in this new CV were very similar (
obtained in this new CV were very similar (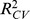 were 11.9% and 22.3% for MB-0 and MB-80K, respectively) to the ones we obtained when subjects, as opposed to entire families, were assigned to folds (here,
were 11.9% and 22.3% for MB-0 and MB-80K, respectively) to the ones we obtained when subjects, as opposed to entire families, were assigned to folds (here, 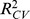 were 11.9% and 22.3% for MB-0 and MB-0, respectively). Combining all these results we conclude that in our analysis familiar relationships were not a major factor explaining the prediction accuracy obtained with WGP.
were 11.9% and 22.3% for MB-0 and MB-0, respectively). Combining all these results we conclude that in our analysis familiar relationships were not a major factor explaining the prediction accuracy obtained with WGP.
Prediction accuracy and causes of mortality
Our results suggest that genomic information can enhance prediction of lifespan, mostly by improving prediction of early mortality. This can be due to several factors, one of which may be that SNPs are capturing genetic risk to certain diseases that are most responsible for early mortality. Figure 4 presents the distribution of death by cause and range of age at death in the Framingham sample. Cancer was the leading cause of death for people dying early in life, and the relative importance of cancer as a cause of death declined with increasing YL. On the other hand, the relative importance of other causes of death was much higher for people dying at older ages.
Figure 4. Proportion of deaths by cause, and range of age at death.

Causes included cancer, coronary heart disease (CHD), cardio-vascular accident (CVA), other cardio-vascular diseases (Other CVD) and other causes.
Discussion
Familial studies suggest that roughly 25% of the inter-individual differences in YL can be attributed to genetic factors [7]. Although linkage and association studies have reported several variants associated with human lifespan and aging-related traits [6], [8], [9], [41], the individual effects of these variants is usually small and our ability to use genetic information to predict human lifespan remains very limited. Recent studies [10], [11], [20] suggest that WGP is effective at predicting complex traits. Here, we developed a WGP model for the prediction of YL and evaluated its predictive power using data from the Framingham longitudinal study.
When genetic markers were added to a model accounting for age at entry, sex, smoking, and BMI, the increase in 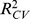 obtained by adding 80K SNPs (∼9–10% of inter-individual differences in YL) was greater than the increase obtained by adding smoking and BMI (∼6% of inter-individual differences in YL), indicating that genetic markers are making a relatively important contribution to predictive ability. Similar results were obtained when prediction accuracy was evaluated using longitudinal AUC's.
obtained by adding 80K SNPs (∼9–10% of inter-individual differences in YL) was greater than the increase obtained by adding smoking and BMI (∼6% of inter-individual differences in YL), indicating that genetic markers are making a relatively important contribution to predictive ability. Similar results were obtained when prediction accuracy was evaluated using longitudinal AUC's.
As anticipated, our results suggest that the genetic basis of YL involves a large number of variants. The observation that DIC and prediction accuracy improved with marker density suggests that a large number of markers spread across the genome are needed to account for differences at QTLs affecting YL, and this is consistent with what one would expect for a trait that conforms to an “infinitesimal” model [42], [43]. This pattern is also consistent with empirical evidence obtained for traits that conform to the infinitesimal model, such as human height [20] or production traits in dairy cattle [19].
Our results are also consistent with those of Yashin et al. [44] who, using a subset of the dataset used here (1,173 individuals of the original cohort), found that a sizable proportion of inter-individual differences in YL (20% in the training dataset) can be explained by the joint influence of 168 small-effect genetic variants which were pre-selected using p-values derived from single-marker regressions. Although the study by Yashin et al. [44] and the one presented here both suggest that a large number of variants is needed to account for interindividual differences in YL, the two studies differ in many respects: (a) our study uses a larger sample size (N = 5,117, versus N = 1,173) and incorporates both uncensored and censored observations, (b) unlike the Yashin study, where markers were pre-selected using statistics derived from single marker regression, here we used a much larger number of markers (up to 80K), spread along the whole genome, (c) although the two studies used an additive linear score to predict YL, the two scores are different. In the Yashin study the score consist of a sum of so-called “longevity alleles”, while in our study the predictive score is a weighted sum of allele dosage, with weights given by estimates of marker effects, (d) in some of our models we account for the effects of BMI and smoking, while these covariates were not accounted for in the Yashin study. Finally (d) in our study we focused on prediction accuracy of yet-to be observed outcomes, while the study by Yashin et al. reports the proportion of interindividual differences in YL that could be accounted for in the same dataset that was used to derive the predictive score. Nevertheless, despite the differences in the datasets and methods used, both studies provide consistent evidence that an important proportion of differences in YL can be predicted using genomic information and that capturing those patterns requires considering a large number of small-effects variants.
In addition to demonstrating that a sizable proportion differences in YL can be predicted using genomic information, we found that most of the gains in prediction accuracy obtained with use of genetic information came from increased accuracy of prediction of early mortality. Further examination of the distribution of causes of death by age at death reveled that cancer was the leading cause of death for people dying early in life. Therefore, a possible explanation of our results is that the ability of our WGP to capture cancer risk (indirectly through YL) was higher than for other death-related disorders. Further studies, using disorder-specific responses (e.g., presence/absence or onset of cancer) and case-control datasets will be needed to confirm this conjecture.
The Framingham dataset has a familial design and exhibits some level of population structure, much of which can be described through PCA of genome-wide SNPs. Whole-Genome Prediction exploits multi-locus LD between markers and QTL. These patterns of LD are likely to change across sub-groups in the population and because of this, models fitted using WGP cannot be regarded as ‘universal equations’. The validity across sub-groups of the patterns captured by a WGP model will depend on the extent to which genetic features (e.g., stratification) present in training samples are also present in those used for validation.
Including the first two marker-derived PCs increased prediction accuracy markedly, indicating that YL covariates with ancestry, as described by the first 2 PCs. However, the level of prediction accuracy attained by models using the first two marker-derived PCs was substantially lower than that of the model using 80K genome-wide SNPs, suggesting that the genetic factors affecting YL cannot be fully described by features such as population structure. The effects of familial relationships on the prediction accuracy of WGP are well established [13], [20]. However, in our study, the pedigree is relatively sparse and when families with more than one subject exist the offspring are highly likely to be censored; therefore, familiar relationships are not very informative to begin with, explaining why in this study familial relationships did not show a strong effect on the prediction accuracy of WGP.
Supporting Information
Describes the Bayesian model used.
(DOCX)
Estimated expected years of life versus Body Mass Index (BMI) by sex (estimates derived from a model which included sex, age at entry, smoking and BMI as predictors).
(TIFF)
Cross-validation R-squared and Area Under Longitudinal Receiver Operating Characteristic Curves by model and fold of a 10-fold cross-validation.
(DOC)
Acknowledgments
The authors would like to thank the participants and organizers of the Framingham Heart Study, Sir David Cox and Drs. Henry Robertson and Emily Dhurandhar for comments and suggestions provided on an early draft of the manuscript and Vinodh Srinivasasainagendra for assistance in downloading the dataset. Insightful suggestions made by the associate editor and two anonymous reviewers are gratefully acknowledged.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This project had funding from National Institutes of Health grants P30CA13148, P30DK056336, R01GM077490, R01DK076771, T32HL105346 and KRAFT-grant “University of Alabama at Birmingham doctorate Training Program in Obesity and Nutrition Research.” The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Hjelmborg JB, Iachine I, Skytthe A, Vaupel JW, McGue M, et al. Genetic influence on human lifespan and longevity. Human genetics. 2006;119:312–321. doi: 10.1007/s00439-006-0144-y. [DOI] [PubMed] [Google Scholar]
- 2.Herskind AM, McGue M, Holm NV, S\örensen TI, Harvald B, et al. The heritability of human longevity: a population-based study of 2872 Danish twin pairs born 1870–1900. Human Genetics. 1996;97:319–323. doi: 10.1007/BF02185763. [DOI] [PubMed] [Google Scholar]
- 3.Iachine IA, Holm NV, Harris JR, Begun AZ, Iachina MK, et al. How heritable is individual susceptibility to death? The results of an analysis of survival data on Danish, Swedish and Finnish twins. Twin research. 1998;1:196–205. doi: 10.1375/136905298320566168. [DOI] [PubMed] [Google Scholar]
- 4.Braeckman BP, Vanfleteren JR. Genetic control of longevity in C. elegans. Experimental Gerontology. 2007;42:90–98. doi: 10.1016/j.exger.2006.04.010. doi: doi:10.1016/j.exger.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 5.Puca AA, Daly MJ, Brewster SJ, Matise TC, Barrett J, et al. A genome-wide scan for linkage to human exceptional longevity identifies a locus on chromosome 4. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10505–10508. doi: 10.1073/pnas.181337598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newman AB, Walter S, Lunetta KL, Garcia ME, Slagboom PE, et al. A meta-analysis of four genome-wide association studies of survival to age 90 years or older: the Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2010;65:478–487. doi: 10.1093/gerona/glq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christensen K, Johnson TE, Vaupel JW. The quest for genetic determinants of human longevity: challenges and insights. Nat Rev Genet. 2006;7:436–448. doi: 10.1038/nrg1871. doi: 10.1038/nrg1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sebastiani P, Solovieff N, Puca A, Hartley SW, Melista E, et al. Genetic signatures of exceptional longevity in humans. Science. 2010 doi: 10.1126/science.1190532. [DOI] [PubMed] [Google Scholar]
- 9.Lunetta K, D'Agostino R, Karasik D, Benjamin E, Guo CY, et al. Genetic correlates of longevity and selected age-related phenotypes: a genome-wide association study in the Framingham Study. BMC medical genetics. 2007;8:S13. doi: 10.1186/1471-2350-8-S1-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de los Campos G, Gianola D, Allison DB. Predicting genetic predisposition in humans: the promise of whole-genome markers. Nat Rev Genet. 2010;11:880–886. doi: 10.1038/nrg2898. doi: 10.1038/nrg2898. [DOI] [PubMed] [Google Scholar]
- 11.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, et al. Common SNPs explain a large proportion of the heritability for human height. Nature genetics. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meuwissen TH, Hayes BJ, Goddard ME. Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001;157:1819–1829. doi: 10.1093/genetics/157.4.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Habier D, Fernando RL, Dekkers JCM. The impact of genetic relationship information on genome-assisted breeding values. Genetics. 2007;177:2389–2397. doi: 10.1534/genetics.107.081190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de los Campos G, Naya H, Gianola D, Crossa J, Legarra A, et al. Predicting quantitative traits with regression models for dense molecular markers and pedigree. Genetics. 2009;182:375–385. doi: 10.1534/genetics.109.101501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crossa J, de los Campos G, Perez P, Gianola D, Burgueño J, et al. Prediction of genetic values of quantitative traits in plant breeding using pedigree and molecular markers. Genetics. 2010;186:713–724. doi: 10.1534/genetics.110.118521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.VanRaden PM, Van Tassell CP, Wiggans GR, Sonstegard TS, Schnabel RD, et al. Invited review: reliability of genomic predictions for North American Holstein bulls. Journal of Dairy Science. 2009;92:16–24. doi: 10.3168/jds.2008-1514. [DOI] [PubMed] [Google Scholar]
- 17.Hayes BJ, Bowman PJ, Chamberlain AJ, Goddard ME. Invited review: Genomic selection in dairy cattle: Progress and challenges. Journal of Dairy Science. 2009;92:433–443. doi: 10.3168/jds.2008-1646. [DOI] [PubMed] [Google Scholar]
- 18.Weigel KA, de Los Campos G, Vazquez AI, Rosa GJM, Gianola D, et al. Accuracy of direct genomic values derived from imputed single nucleotide polymorphism genotypes in Jersey cattle. J Dairy Sci. 2010;93:5423–5435. doi: 10.3168/jds.2010-3149. doi: 10.3168/jds.2010-3149. [DOI] [PubMed] [Google Scholar]
- 19.Vazquez AI, Rosa GJM, Weigel KA, de Los Campos G, Gianola D, et al. Predictive ability of subsets of single nucleotide polymorphisms with and without parent average in US Holsteins. Journal of dairy science. 2010;93:5942–5949. doi: 10.3168/jds.2010-3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makowsky R, Pajewski NM, Klimentidis YC, Vazquez AI, Duarte CW, et al. Beyond Missing Heritability: Prediction of Complex Traits. PLoS Genet. 2011;7:e1002051. doi: 10.1371/journal.pgen.1002051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fontaine KR, Redden DT, Wang C, Westfall AO, Allison DB. Years of life lost due to obesity. JAMA: The Journal of the American Medical Association. 2003;289:187–193. doi: 10.1001/jama.289.2.187. [DOI] [PubMed] [Google Scholar]
- 22.Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. The American Journal of Human Genetics. 2011 doi: 10.1016/j.ajhg.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park T, Casella G. The bayesian lasso. Journal of the American Statistical Association. 2008;103:681–686. [Google Scholar]
- 24.de los Campos G, Gianola D, Rosa GJM, Weigel KA, Crossa J. Semi-parametric genomic-enabled prediction of genetic values using reproducing kernel Hilbert spaces methods. Genetics Research. 2010;92:295–308. doi: 10.1017/S0016672310000285. [DOI] [PubMed] [Google Scholar]
- 25.Weigel KA, De Los Campos G, González-Recio O, Naya H, Wu XL, et al. Predictive ability of direct genomic values for lifetime net merit of Holstein sires using selected subsets of single nucleotide polymorphism markers. Journal of dairy science. 2009;92:5248–5257. doi: 10.3168/jds.2009-2092. [DOI] [PubMed] [Google Scholar]
- 26.Pérez P, de los Campos G, Crossa J, Gianola D. Genomic-Enabled Prediction Based on Molecular Markers and Pedigree Using the Bayesian Linear Regression Package in R. The Plant Genome Journal. 2010;3:106–116. doi: 10.3835/plantgenome2010.04.0005. doi: 10.3835/plantgenome2010.04.0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de los Campos G, Pérez P. BLR: Bayesian linear regression. 2010. R package version 1.2. R-project, available at: http://cran.r-project.org/web/packages/BLR/index.html. Accessed 2012 June 28th. [DOI] [PubMed]
- 28.R Development Core Team. R: A language and environment for statistical computing. 2010. R Foundation for Statistical Computing, Vienna, Austria. R Foundation for Statistical Computing. R-project, available at: http://www.R-project.org. Accessed 2012 June 28th.
- 29.Rossi P, McCulloch R. bayesm: Bayesian inference for marketing/micro-econometrics. R package version. 2010:2–2. [Google Scholar]
- 30.Wheeler B. SuppDists: Supplementary distributions. R package version. 2008:1–1. [Google Scholar]
- 31.Hastie TJ, Tibshirani RJ. Generalized additive models. Chapman & Hall/CRC 1990 [Google Scholar]
- 32.Spiegelhalter DJ, Best NG, Carlin BP, Linde A van der. Bayesian Measures of Model Complexity and Fit. Journal of the Royal Statistical Society. Series B (Statistical Methodology) 2002;64:583–639. [Google Scholar]
- 33.Heagerty PJ, Zheng Y. Survival Model Predictive Accuracy and ROC Curves. Biometrics. 2005;61:92–105. doi: 10.1111/j.0006-341X.2005.030814.x. doi: 10.1111/j.0006-341X.2005.030814.x. [DOI] [PubMed] [Google Scholar]
- 34.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. doi: 10.1186/1471-2105-12-77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Henderson CR. Best linear unbiased estimation and prediction under a selection model. Biometrics. 1975;31:423–447. [PubMed] [Google Scholar]
- 36.Peeters A, Barendregt JJ, Willekens F, Mackenbach JP, Mamun AA, et al. Obesity in adulthood and its consequences for life expectancy: a life-table analysis. Annals of internal medicine. 2003;138:24–32. doi: 10.7326/0003-4819-138-1-200301070-00008. [DOI] [PubMed] [Google Scholar]
- 37.Finkelstein EA, Brown DS, Wrage LA, Allaire BT, Hoerger TJ. Individual and aggregate years-of-life-lost associated with overweight and obesity. Obesity. 2009;18:333–339. doi: 10.1038/oby.2009.253. [DOI] [PubMed] [Google Scholar]
- 38.Arias E, Rostron BL, Tejada-Vera B. National vital statistics reports. 2010. National Vital Statistics Reports 58. [PubMed]
- 39.Mamun AA, Peeters A, Barendregt J, Willekens F, Nusselder W, et al. Smoking decreases the duration of life lived with and without cardiovascular disease: a life course analysis of the Framingham Heart Study. European heart journal. 2004;25:409–415. doi: 10.1016/j.ehj.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 40.Drineas P, Lewis J, Paschou P. Inferring Geographic Coordinates of Origin for Europeans Using Small Panels of Ancestry Informative Markers. PLoS ONE. 2010;5:e11892. doi: 10.1371/journal.pone.0011892. doi: 10.1371/journal.pone.0011892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poduslo SE, Huang R, Spiro A. A genome screen of successful aging without cognitive decline identifies LRP1B by haplotype analysis. Am J Med Genet. 2010;153B:114–119. doi: 10.1002/ajmg.b.30963. doi: 10.1002/ajmg.b.30963. [DOI] [PubMed] [Google Scholar]
- 42.Goddard M. Genomic selection: prediction of accuracy and maximisation of long term response. Genetica. 2009;136:245–257. doi: 10.1007/s10709-008-9308-0. [DOI] [PubMed] [Google Scholar]
- 43.Goddard ME, Hayes BJ. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nature Reviews Genetics. 2009;10:381–391. doi: 10.1038/nrg2575. [DOI] [PubMed] [Google Scholar]
- 44.Yashin AI, Wu D, Arbeev KG, Ukraintseva SV. Joint influence of small-effect genetic variants on human longevity. Aging (Albany NY) 2010;2:612–620. doi: 10.18632/aging.100191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Describes the Bayesian model used.
(DOCX)
Estimated expected years of life versus Body Mass Index (BMI) by sex (estimates derived from a model which included sex, age at entry, smoking and BMI as predictors).
(TIFF)
Cross-validation R-squared and Area Under Longitudinal Receiver Operating Characteristic Curves by model and fold of a 10-fold cross-validation.
(DOC)